

Abstract
Adenosine receptor (ARs) and P2Y receptors (P2YRs) that respond to extracellular nucleosides/tides are associated with new directions for therapeutics. The X-ray structures of the A 2A AR complexes with agonists and antagonists are examined in relationship to the G protein-coupled receptor (GPCR) superfamily and applied to drug discovery. Much of the data on AR ligand structure from early SAR studies, now is explainable from the A 2A AR X-ray crystallography. The ligand-receptor interactions in related GPCR complexes can be identified by means of modeling approaches, e.g. molecular docking. Thus, molecular recognition in binding and activation processes has been studied effectively using homology modeling and applied to ligand design. Virtual screening has yielded new nonnucleoside AR antagonists, and existing ligands have been improved with knowledge of the receptor interactions. New agonists are being explored for CNS and peripheral therapeutics based on in vivo activity, such as chronic neuropathic pain. Ligands for receptors more distantly related to the X-ray template, i.e. P2YRs, have been introduced and are mainly used as pharmacological tools for elucidating the physiological role of extracellular nucleotides. Other ligand tools for drug discovery include fluorescent probes, radioactive probes, multivalent probes, and functionalized nanoparticles.
Keywords: G protein-coupled receptor , purines , molecular modeling , adenosine receptor , P2Y receptor
I. Introduction: background on purine/pyrimidine receptors – new directions for therapeutics
Intense efforts by medicinal chemists in academia, government and industry have focused on the development of both agonist and antagonist ligands for the adenosine receptors (ARs) and for the P2Y receptors (P2YRs) that respond to extracellular nucleotides ( Figure 1 ). All of these receptors belong to the Family A of G protein-coupled receptors (GPCRs). 1 , 2 The ARs are now at center stage in the exploration of general methodology for GPCR structure-based design. These purine/pyrimidine receptor families constitute 12 subtypes total, including the larger (P2Y) family that has two subgroups based on similarity of receptor structure and second messenger coupling. The P2YRs are activated by a range of native agonists, i.e. ATP, ADP, UTP, UDP, and UDP-sugars, while the four subtypes of ARs are more narrow in native agonist specificity, responding almost exclusively to extracellular adenosine. Of the hundreds of GPCRs under consideration for pharmaceutical development, these purine and pyrimidine receptors are among the most intriguing because of their widespread distribution and the dual use by nature of key important molecular mediators both intracellularly and for signaling purposes outside the cell. This molecular centrality tends to place the purine and pyrimidine receptors in important regulatory functions throughout the body. It has been noted that purinergic receptors are “the most abundant receptors in living organisms and appeared early in evolution”. 3 ARs and P2YRs can be considered an important conserved vestige of the RNA world, which is also supported by the requirement, for activation of either class, of a 2′-hydroxyl group present in the native ribonucleoside and ribonucleotide agonists.
Figure 1 .

A. The time-dependent progressive activation (white arrows) of receptors for nucleosides and nucleotides on the cell surface. The 4 subtypes of ARs and 8 subtypes of P2YRs are arranged in a dendrogram according to their sequence homology. Although the ligands have commonality, the ARs and 8 subtypes of P2YRs are on distant branches of the larger dendrogram of Family A GPCRs. B. General characteristics of adenosine as a GPCR ligand and its receptors as therapeutic targets in both chemical (A) and biological (B) contexts.
Our basic research lab at NIH has introduced many of the important ligand tools for these GPCRs used by pharmacologists to determine the role of purine/pyrimidine signaling. Ligands of the ARs include clinical candidate drugs and typically have a well explored structure activity relationship (SAR). 4 , 5 However, P2YRs in some cases lack selective ligands altogether. 6 Also, many of the known P2YR ligands suffer from limited stability and bioavailability because of phosphate groups. In the P2YR field, only P2Y 12 R antagonists as antithrombotic agents are well validated clinically. 7 Nevertheless, there are many new concepts, at various stages of development, of how to apply both ARs and P2YRs toward healing or diagnostic purposes. In general, to overcome the problem of widespread distribution of these two classes of GPCRs, novel drug delivery, 8 prodrug approaches, 9 allosteric modulation 10 , 11 and biased agonism 12 are needed to avoid ligand side effects. The pharmacokinetic and other reasons for failure of some clinical trials, even with receptor subtype-selective AR ligands, have been explained. 13
A dendrogram arranged according to the degree of amino acid sequence homology of the twelve GPCRs that respond to extracellular nucleosides and nucleotides is shown in Figure 1A . Although the native ligands are chemically related, the ARs (similar to biogenic amine receptors) and P2YRs (similar to lipid receptors) are located on separate branches of the larger dendrogram of Family A GPCRs. 14 The four ARs (red traces) are coupled either to G i protein to inhibit the formation of cyclic AMP (A 1 and A 3 ARs) or to G s protein to stimulate the formation of cyclic AMP (A 2A and A 2B ARs). 1 There are three P2YRs (green traces; P2Y 12 , P2Y 13 and P2Y 14 ) that are coupled preferentially to G i and five P2YRs (yellow traces; P2Y 1 , P2Y 2 , P2Y 4 , P2Y 6 and P2Y 11 ) that are coupled preferentially to G q for activation of phospholipase C β (PLCβ). 2 G protein-independent signaling has also been studied within this family. 12 , 13b The action of extracellular signaling nucleotides and nucleosides can be viewed in the context of a temporal sequence. Nucleotides released from intracellular sources by injury, stress, or from vesicles, transporters or hemichannels provide either an immediate or a delayed response to a local challenge to tissues or cells. 15 For example, a vesicular nucleotide transporter (VNUT) can regulate uptake of nucleotides into granules and their release. 11 , 16 Adenosine can also originate in intracellular sources. The released nucleotides can act on the same or on neighboring cells, either to open fast cation channels (P2XRs, adenosine 5′-triphosphates) or for signaling on a slower time scale at metabotropic GPCRs (P2YRs, adenosine or uridine 5′-triphosphates or 5′-diphosphates). On a more prolonged time scale, the adenine nucleotides are converted by the sequential action of ectonucleotidases (for example, CD39 that hydrolyzes ATP/ADP and CD73 that hydrolyzes AMP) to the nucleoside adenosine for activation of the ARs. 11 , 16 A further metabolic step of conversion of adenosine to inosine by adenosine deaminase can create a secondary, weak agonist of the A 3 AR. 11 , 17 These transformations and sequential receptor effects serve overall biological functions dictated by circumstances, such as cytoprotection, vascular adaptations and immune effects, which may be oppositely affected at different points in the time sequence. 18 The rapid effects of nucleotides on P2 receptors tend to boost the immune response, while the slower AR effects generally attenuate the immune response.
General features of adenosine as a GPCR ligand are shown in Figure 1B , in both the chemical and the biological context. The concept of separate portions of a molecule encoding clearly delineated message and address regions originated in the study of peptide GPCRs. 27
We extend this structural division of labor within a given molecule from peptide sequences to a small molecule, i.e. adenosine. The planar adenine moiety is akin to the address region, because many adenine-like heterocycles are known to be AR antagonists 28 and also because the receptor subtype selectivity of adenosine congeners can be engineered largely by their substitution at the N 6 and C2 positions of adenine. In contrast, the three-dimensional ribose moiety with its built-in flexibility at sp 3 -hybridized atoms facilitates the conformational change of the receptor needed for activation. We know this role of the ribose from both the perspective of the ligand modification 29 and from X-ray crystallographic complexes of the A 2A AR and its agonists. 20 , 21 In the biological realm, adenosine acting at ARs or nucleotide ligands acting at P2YRs can both be likened to adjusting the volume control on a radio, rather than flipping a power switch(with the multiplicity of signaling pathways that are differentially modulated, notwithstanding). There is a basal activation of many of these receptors that helps establish the set point to maintain homeostasis in a given tissue. 1 , 2 The stimulation or antagonism of the receptor can serve to counteract a signaling imbalance in a disease state to figuratively re-adjust the set point. The levels of expression and activity of these receptors can be greatly altered in diseased tissue, providing a justification for pharmacologic intervention. This is the basis of much of the intended therapeutic use of ligands of the ARs and of the P2YRs.
There are many currents therapeutic concepts at various stages of development for using selective adenosine agonists and antagonists. 1 , 5 , 30 , 31 For example, our lab has invented two agonists of the A 3 AR that have progressed to advanced clinical trials for autoimmune inflammatory disease and cancer. 32 A total of ten such trials at various phases (I – III) by Can-Fite Biopharma are currently listed in clinicaltrials. gov. These include trials for rheumatoid arthritis, psoriasis, dry eye syndrome, and hepatocellular carcinoma. The compounds have already been administered (orally) to >700 individuals, and there are indications of efficacy with no serious adverse effects.
This perspective will cover three modes of application of structural knowledge to the design of new GPCR ligands: 1. modification of known ligands or a search for new chemotypes to modulate a given receptor using newly available X-ray structures, such as A 2A AR; 2. for other receptors in the same family, but not identical to a determined X-ray template, such as A 1 and A 3 ARs; 3. for receptors, such as P2YRs, more distantly related to X-ray templates. For the ARs, the luxury of having an A 2A AR X-ray structure available empowers many studies of structure-based ligand design at this subtype. Both agonist-bound and antagonist-bound forms of the A 2A AR are available, including one complex with inverse agonist 4-[2-[7-amino-2-(2-furyl)-1,2,4-triazolo[1,5-a][1,3,5]triazin-5-yl-amino]ethylphenol 1 (ZM241,385, Figure 2A ) having exceptionally high resolution showing details of water molecules and a sodium ion bound to an allosteric site on transmembrane helix (TM2). 19 – 23 For studying molecular recognition at the remaining ARs that have not been crystallized, homology modeling with the closely related template, i.e. A 2A AR has proven successful. 24 – 26 The P2YRs still lack a close template for homology modeling, but studies are underway in that direction, i.e. to determine the structure of a P2YR.
Figure 2 .


A. Structures of selected AR ligands discussed in the text, including those that have been co-crystallized with the A 2A AR ( 1 , 2 , 5 , and 6 ). Dashed lines indicated the major H-bonding and π-bonding contacts between the receptor and ligand present in X-ray structures. Other ligands shown include pharmacological probes ( 4 and 9–12 ) and: advanced clinical candidates 3a and 3b for Parkinson’s disease; diagnostic agents for myocardial perfusion imaging 7 (approved and in trials for sickle cell anemia and ischemic conditions) and 8 (clinical candidate). B. The helical bundle of Family A GPCRs defines a cavity for the recognition of diverse ligands. The phospholipid bilayer is not shown. Figure courtesy of Stefano Costanzi (American Univ., Washington, DC). C. Historical progression of knowledge of the AR binding site(s). 19 , 20 , 37 , 39 , 41 , 45 Arrows in upper panels indicate the following interactions: Yellow, position of terminal amino group intended for covalent linking 37 ; orange, H-bonding interaction predicted between the exocyclic amine of adenine and a conserved Asn6.55 39 ; white, proximity of 3′ and 2′-hydroxyl groups with conserved His7.43 predicted using a neoceptor approach 41 . Lower panels: left, predicted docking of agonist 5 using the antagonist-bound X-ray structure 19 , 45 ; right, actual position of agonist 6 in X-ray structure. 20 , 24
II. Determination of structures of A 2A AR complexes and relationship to the GPCR superfamily
The helical bundle of Family A GPCRs defines a cavity for the recognition of diverse ligands ( Figure 2B ). The precise chemical characteristics, size, shape, and accessibility to the extracellular milieu are tailored to each receptor family. 17 Nevertheless, the overall similarity of the architecture of these binding sites is evident in this graphical overlay. Within the rhodopsin-like Family A GPCRs, the transmembrane (TM) domain is more highly conserved than the ELs. Ligand binding occurs in the third of the TM region proximal to the exofacial side, with varying degrees of involvement of the ELs. 33 Despite variation in the orientation and depth of placement of orthosteric ligands of GPCRs, some residue positions are conserved in their role in coordinating the bound ligand (i.e. 6.55, using standard numbering convention 34 ).
At present, the structure of the only member of the twelve purine/pyrimidine receptors to be solved is the A 2A AR. 19 – 23 This receptor is a drug target for Parkinson’s disease treatment with selective antagonists, such as nonxanthines 3a and 3b that are in advanced clinical trials. 35 , 121 The structure of the receptor was determined in multiple forms with different ligands. Both antagonist-bound and agonist-bound states of the receptor were solved initially by the Stevens group using the stabilizing fusion protein T4 lysozyme. 19 , 20 A 2A AR complexes were crystallized by Heptares Therapeutics using receptors that are thermostabilized by systematic scanning mutagenesis (StaRs) include several other agonists and antagonists, including one with the antagonist 8-[4-[[[[(2-aminoethyl)amino]carbonyl]methyl)oxy]phenyl]-1,3-dipropylxanthine 2 (xanthine amine congener, XAC). 22 We introduced this high affinity ligand in 1985 as a functionalized congener for attachment to carriers and reporter groups without losing receptor recognition by coupling to the terminal amine. 36 Points of contact between the receptor and these ligands are shown in Figures 2B, C .
Characterization of molecular recognition at the ARs began long before their X-ray structural determination. Figure 2C outlines points along the historical progression of knowledge of the AR binding site(s), from the first molecular modeling, done by our lab in collaboration with Ad IJzerman (Leiden Univ.) in 1992. 37 Conserved residues and nucleoside ligand structures make it possible to apply knowledge gleaned about one AR subtype to the recognition at another subtype. Prior to the report of the crystallographic structure of the A 2A AR, 19 the only method to represent the 3D structure of the ARs was through molecular modeling. 38 This approach relied on information from convergent sources: homology modeling of the protein and ligand docking, receptor mutagenesis (1995), 39 and both qualitative and quantitative SAR of ligands.
The progression of AR structural definition was greatly advanced by application of the neoceptor approach, which is a rationally designed receptor mutant that is orthogonally activated by a strategically modified agonist. 40 , 41 This approach is conceptually akin to RASSLs/DREADDs that have reengineered GPCR binding sites, except that the neoceptors begin with a rational design process for modification of both GPCR and tailored agonist ligands. 42 The first step in constructing neoceptors is to propose a binding hypothesis consistent with molecular modeling that predicts group proximity. The selective enhancement of affinity upon complementary favorable changes in the structures of a ligand and a receptor protein establishes a reliable anchor point for the hypothetical binding of the ligand. This was applied successfully first to the A 3 AR and later to the A 2A AR (2005) by introducing an electrostatic pair to confirm proximity of the ribose 2′ and 3′-hydroxyl groups to a conserved His278 7.43 in TM7. This H-bonding proximity later appeared in the agonist-bound A 2A AR X-ray structures. 20 , 43
We have reevaluated methods of ligand docking in light of the X-ray data, initially antagonist binding to the inactive state of the A 2A AR and subsequently agonist binding. 45 The latter predicted structure approximated well the structure of an agonist-bound stable complex (2011). 20 A comparison of different docking approaches showed that even one extra constraint on the ligand position derived from physical measurements, such as a single H-bond implicated using site-directed mutagenesis, greatly improves the accuracy of in silico docking. 45 Our predicted model of docking of agonists in the A 2A AR (2009), using the inactive state of the receptor as a template, almost exactly overlays the X-ray structure that was determined two years later. 20 The early modeling has evolved into a precise understanding of the separate processes of binding and activation; conformational changes of the ribose moiety are key to the activation process. 20 This is borne out in the general need in agonists (especially acting at the A 3 AR) for flexibility of the 5′ moiety of adenosine (CH 2 OH) or nucleosides resembling adenosine-5′- N -ethyluronamide 5 (NECA) with a CONH-small alkyl group, when present. 29
Two distinct regions within the helical bundle of the ARs could be discerned in the early modeling, corresponding to the hydrophobic address and hydrophilic message regions around adenosine. The differences in the particular architecture of agonist- and antagonist-bound conformations have been analyzed in exquisite detail for the A 2A AR and by analogy to other ARs. 20 The conformation of the A2AAR complex with 65, although lacking a G protein, displays features of an active-like conformation, e.g. spreading of the ionic lock. TMs 5, 6 and 7 engage in most of the conformation movement that leads to receptor activation, while the only pronounced movement within TMs 1–4 is a piston-like upward movement of TM3 to accommodate an agonist. The agonist-bound structure of A 2A AR features a contracted ribose binding region, in which hydrophilic residues of the pocket are drawn toward H bonding groups of the ribose. The transformation of antagonist-bound to an agonist-bound structure of the A 2A AR (a movie simulation in the Supporting Information of Xu et al. 20 is recommended for viewing) shows how residues in the hydrophilic regions of TM3 and TM7, primarily, approach the ribose moiety of the highly appended agonist 6-(2,2-diphenylethylamino)-9-((2R,3R,4S,5S)-5-(ethylcarbamoyl)-3,4-dihydroxytetrahydrofuran-2-yl)- N -(2-(3-(1-(pyridin-2-yl)piperidin-4-yl)ureido)ethyl)-9H-purine-2-carboxamide 6 (UK432097). This high affinity agonist from Pfizer was in clinical trials for chronic obstructive pulmonary disease with the expectation of a localized action in the lungs precisely because of its large size and extensive H bonding, which also contributed to A 2A AR crystal stabilization. 13 The receptor-bound 6 is very similar to the structures of other bound AR agonists, obtained using StaRs. 21 , 42 , 47 There is a pattern of conserved H bonding of both the ribose and adenine moieties to key constraining amino acid residues, many of which are conserved in other AR subtypes. For example, the side chain amide of Asn254 6.55 (using the nomenclature of Ballesteros and Weinstein 34 ) acts as a stabilizing H bond donor to the adenine N7 and acceptor from N 6 H, consistent with the previously established SAR that did not tolerate 7-deaza or disubstituted N 6 analogues ( Figure 2A ). Agonist binding expels water from the ribose binding region and thus provides an entropic stabilization of the agonist-bound state consistent with earlier thermodynamic measurements. 46 This is consistent with the early observation that adenosine and its various monosubstituted analogues displayed exceptionally high affinity (nM range) in receptor binding, unlike some other receptors, such as muscarinic acetylcholine receptors, which feature more weak native agonist binding, i.e. μM. Agonist-specific changes include hydrophilic residues that approach the ribose and tighten around it like a belt, leading to characteristic movements of TM7 and TM6, similar to activation of opsin and the β 2 -adrenergic receptor. 48 , 49 His278 7.43 moves inward to contact the 2′ and 3′ hydroxyl groups of ribose, and hydroxylic residues Thr88 3.36 and Ser2.77 7.42 are attracted to the 3′- and 5′-hydroxyl groups, respectively. Trp248 6.48 undergoes a minor movement. EL3 is folded outward in the 6 -A 2A AR complex as a consequence of a bulky N 6 substituent. Thus, some of the movements are common with other GPCRs, and others movements are particular to the AR or even to a certain ligand, e.g. the outward movement of His264 in EL3. Most of the critical amino acid interactions in the hA 2A AR crystal structures were predicted in mutagenesis/modeling studies. 20
III. Use of GPCR structures for drug discovery
Several different approaches were used earlier to discover new AR antagonist ligands and to achieve receptor selectivity: a. Optimization of the affinity and selectivity of a known antagonist by empirical methods. An example is the elaboration of analogues of the prototypical naturally occurring and widely ingested xanthines, caffeine and theophylline, as AR antagonists beginning in the early 1980s. 36 b. Later, a search was conducted for new leads from the screening of structurally diverse chemical libraries for AR binding activity. The first examples were from small (by today’s standards) libraries assembled from available flat heterocycles and many known bioactives. For example, the widely used antianginal and antihypertensive drug nifedipine bound to the A 3 AR at μM concentrations. From that effort came the identification of 1,4-dihydropyridines and flavonoids as AR antagonists that could be structurally optimized to eliminate the activity at their conventional target sites, e.g. Ca 2+ channels, and optimize their AR activity. 50 Consequently, the highly selective A 3 AR antagonist 1,4-dihydro-2-methyl-6-phenyl-4-(phenylethynyl)-3,5-pyridinedicarboxylic acid, 3-ethyl 5-(phenylmethyl) ester 4 (MRS1191), containing an elongated, rigid C4 substituent, was reported in 1996. 50 c. Combination of efficacy-lowering structural changes of nucleosides to convert a receptor subtype-selective agonist into an antagonist of similar selectivity. 29 This required a conceptual separation of the processes of binding and receptor activation, which we now know has a clear basis in the receptor structure. An example of the success of this approach is sterically constrained nucleoside analogues, such as (2 R ,3 R ,4 S ,5 S )-2-[ N 6 -3-Iodobenzyl)adenos-9′-yl]-7-aza-1-oxa-6-oxospiro[4.4]-nonan-4,5-diol 17 (R 1 = I, MRS1292), or truncated nucleosides, such as (2 R ,3 R ,4 S )-2-(2-chloro-6-(3-iodobenzylamino)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol ( 19 , R 1 = I, R 2 = Cl, LJ1251) that act as selective A 3 AR antagonists. 52 , 53 d. In silico screening of diverse libraries was carried out first based solely on ligand-defined pharmacophores and later on docking of the compounds to receptor structures or models. 51 , 52
The ARs can be considered an early example of the effective use of X-ray structures for both the selection of new chemotypes from chemical libraries and in the stepwise design of new ligands from known ligands. For example, biophysical mapping and fragment screening explored using StaRs, which can be stabilized in either agonist-preferring or antagonist-preferring conformations, are elegant and systematic approaches to this effort. 21 , 47 , 54
The X-ray structure of the A 2A AR has made possible a new paradigm in drug discovery for the ARs. Figure 3A is a flowchart of steps in the structure-based design approach that we have followed for the ARs and other GPCRs. 40 Structure-based virtual screening can be performed using the inactive A 2A AR structure, which has proven useful for the discovery of new antagonists. Fragment-based approaches, in which only a portion of a known agonist is systematically replaced, have been reported for the discovery of novel agonists using an agonist-bound A 2A AR structure. 20 This structure is representative of the activated state, especially in the vicinity of the bound ligand, because of the distancing of residues in the “ionic lock” for the inactive state, resemblance to other activated GPCR structures and the characteristic sodium-induced shift of agonist affinity observed pharmacologically, although it is not possible to directly determine the degree of activation of this structure because of the presence of the crystal-stabilizing T4 lysozyme in the 3 rd intracellular loop (IL3). Homology modeling is useful in extending both the antagonist-bound and agonist-bound A 2A AR structures to other AR subtypes or even other GPCR types. The reliability of these molecular models of GPCRs, in general, can be validated by comparing their predictions with the results of SAR and mutagenesis studies.
Figure 3 .

A. A flowchart depicting current modeling and docking approaches for closely related GPCR structures (Reprinted from ref. 40 , Copyright (2013), with permission from Elsevier). The key to footnotes:
- Selection of template is based on sequence identities, conserved key residues, binding site similarities, consideration of disulfide bridges, and shared structural features.
- DSModeler, Prime, ICM, and MOE are commercial software packages widely used for homology modeling with the structure-based alignment and the template structure as inputs and root main square deviation (RMSD) of Cα, side chain, and heavy atoms as criteria for homology modeling success.
- Ligand-supported homology modeling, energy minimization or/and molecular dynamics serve to relax unfavorable contacts.
- Automated docking in rigid binding site, induced-fit docking, or docking by Monte Carlo simulations may be used. Among the available docking software packages are: AutoDock, DOCK, Glide, GOLD, ICM, and OEDocking. SAR information can help to identify the binding pocket in the receptor.
- Modification of the lead compounds is based on the property of the binding site to attain binding and specificity for the target protein. Approaches include: fragment addition, fragment replacement, connection of fragments with new scaffolds, and other techniques.
- Database preparation with several physical and chemical filters, e.g. number of rotatable bonds in the compounds, polar surface area, and application of variations of Lipinski’s rule-of-five based on lipophilicity, hydrophobicity, and molecular weight.
- Structure-based virtual screening (SBVS) with high-throughput docking of compound database to the 3D structure of the target. Glide, Gold, ICM, AutoDock are common docking tools for SBVS. The post-docking analysis for the selection of the hits to be tested includes shape complementarity, cluster analysis, consensus scoring, geometric analysis, and visualization of binding modes.”
B. Representative structurally diverse antagonists and fragments that bind to ARs as reported: 22 26 , 23 55 , 24 58 , 25 55 , 26 21a , 27 and 28 59 , 29 2? , 30 123 , 31 – 33 60 , and 34 26 .
A. Library screening for nonnucleoside AR antagonists: ligand-based and target-based
Various recent publications have performed structure-based drug discovery of novel antagonists at the A 2A AR. 51 , 55 – 59 Some of the novel ligands discovered by this process are shown in Figure 3B . In collaboration with Jens Carlsson (now at Stockholm Univ.) and Brian Shoichet (then at Univ. of California, San Francisco), we screened the ZINC library ( http://zinc.docking.org/ , 1.6 million compounds at the time) and discovered novel chemotypes for the human A 2A AR with a hit rate of 35%. 51
Kolb et al. 26 found that the in silico screening of chemically diverse libraries at the A 1 AR had considerable overlap in reactivity with associated AR subtypes. Over two million lead-like compounds were docked in the A 1 AR homology models (based initially on the A 2A AR inactive structure), and a screen of high-ranking molecules provided a hit rate of 21% of validated AR ligands, mostly of novel chemotype, but there was considerable cross reactivity at the A 2A and A 3 ARs. The most potent hit 22 had a K i of 400 nM at the A 1 AR, but bound similarly at the A 2A AR. Thus, X-ray-based in silico screening at GPCRs is likely to identify ligands for closely related families.
Structurally diverse screens have discovered new antagonists, but have been less successful at identifying novel agonists, except for a few reports such as fragment screening using target (StaRs) immobilized NMR screening and other biophysical methods. 60 A more narrowly defined 3D pharmacophore is needed for agonists than for antagonists, because of the more restrictive spatial and H bonding requirements of the flexible ribose moiety. In light of the multiple rearrangements of amino acid side chains in that region as shown in the X-ray structures, there is likely a family of conformations of the ligand in the ribose region that must be satisfied.
B. Design of new ligands by modification of known agonists and antagonists
1. Docking and modification of known ligands at the A 2A AR
Most in silico screens using GPCR structures or homology models are for antagonists (including AR screens 51 , 55 ), but a significant number of GPCR drugs are agonists (e.g. opioids, 5HT 1B for migraines, etc.). Therefore, we need methods for rational design of agonists and their analogues that might have diverse pharmacological properties, including effector-biased agonism. 12 , 61
Three regions of adenosine (C2, 5′, and N 6 ) have been modified structurally based on insights from each binding subpocket in the A 2A AR X-ray structures. Each of these three regions, highly modified in past SAR studies, has its own considerations given the new structural insights, and different approaches have been applied: the C2 region has the most freedom of substitution, because it is open to the extracellular region; the 5′ region is a small pocket that is limited in size; and the N 6 region is intermediate in its ability to be extended. 4 The N 6 region is partly accessible to the extracellular space; there are also strict stereochemical considerations in proximity to the exocyclic amine. 62
A recommended first step in establishing a binding model for predictive use for new derivatives is to see if the model explains the SAR of known ligands. Model docking of a test set of nucleoside agonists to an active form of the A 2A AR explains structurally much of the previous, empirically-derived SAR. 24 In order to take into account the ELs, we used the X-ray structure of the 5 -A 2A AR complex determined using StaRs that includes more of these portions of the receptor. 21 , 43 Three residues of the second extracellular loop (EL2) seem to be in proximity of important terminal functionality on several adenosine agonists that are extended at the C2. The terminal carboxylate group of 2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′- N -ethylcarboxamidoadenosine (CGS21680, 11a ) is predicted to interact with K153 EL2 ; the terminal alkylamino group of the corresponding ethylenediamine derivative 11b (APEC) is predicted to interact with E169, a residue in the C-terminal half of EL2 that folds into the main adenosine binding region. The same carboxylate group is shared with the exocyclic adenine in many non N 6 -substituted analogues, and it was shown by earlier mutagenesis to be important in ligand recognition. 63 It was evident previously that addition of terminal amines led to enhanced potency of both AR agonists and antagonists, but the structural basis of such functionalized congeners was unknown. 36 The terminal isothiocyanate group of affinity label 11c , made by elongation of an amine-functionalized A 2A AR agonist 11b with a bifunctional cross-linking moiety, p -phenylene diisothiocyanate (DITC), is predicted to react covalently with K150 EL2 to form a thiourea anchor on the receptor. This terminal isothiocyanate group is required for potent irreversible inhibition of the A 2A AR by 11c and its consequent prolonged vasodilation of the coronary artery that is not diminished upon washout. 64
Based on the docking modes, we prepared a variety of amino acid conjugates of 11a by amide formation at the terminal carboxylate. 24 The interactions of the carboxylate group of a high affinity D-His conjugate 12 with both K150 EL2 and K153 EL2 and of the imidazole ring with E169 EL2 were predicted. 24 Now various predicted interactions can be tested using site-directed mutagenesis.
Fragment-based searching of new AR agonists was performed by Katritch et al. 65 The focus was on the sterically restricted 5′ region, which consists of an N -ethylcarboxamide in 5 . Substitutions of the amine component of the amides were screened in silico at an active state of the A 2A AR 20 using the ICM molecular modeling package, 66 with the restriction of MW <150, and the resulting hits included both known and novel fragments at the 5′ amide. A receiver operating characteristic (ROC) curve, reflecting the degree of predictive accuracy of the model for selecting genuine ligands from a training set, validated the use of an agonist-bound A 2A AR structure to select true agonists. An alternative template in silico that was adapted from the inactive A 2A AR was less successful for retrieving agonists. Novel 5′-carboxamide moieties were also identified, for example, a pyrazole amide 9a was predicted to be favored in interaction with this region of the receptor. When the ligand was synthesized, it proved to be a moderately potent A 2A AR agonist with a K i value of 230 nM. Gain of affinity at the A 1 AR was noted for some of the derivatives such as an oxetane 9b , which was explained by putative H bonding to Asn 5.42 . The change in a single residue at 5.46 between A 2A AR and A 1 AR from Cys to Trp enlarged that region sterically by pushing TM5 away from TM3 and TM4. This allows a larger 5′-carboxamido group of partial agonist 9b to be accommodated in the A 1 AR, leading to a K i value of 12 nM.
2. Ligands for receptors closely related to the X-ray template: A 1 and A 3 ARs
The effects of truncation of hydrophilic functionality in the ‘message’ region of adenosine derivatives were analyzed pharmacologically. A correlation of changes at the N 6 position in two parallel series of nucleosides has revealed that the A 3 AR affinity is better preserved than A 1 AR affinity in 4′-truncated nucleoside analogues. 62 Diverse N 6 -alkyl andarylalkyl groups that favor A 1 AR affinity in the ribose series were applied to the 5′-truncated series of (N)-methanocarba analogues. At the A 3 AR affinity was largely maintained, while at the A 1 AR there was a loss of affinity of generally two orders of magnitude. The structural basis for this difference between the subtypes was probed using homology modeling. Recognition of agonists at the A 3 AR is less dependent on H-bonding groups about the 5′ position of ribose than is ligand recognition at the A 1 AR. This correlates with the H-bond donating ability of His250 6.52 in the A 2A AR that is conserved in the A 1 AR (His251 6.52 ) but is absent in the A 3 AR (replaced by Ser247 6.52 that is predicted not to be in direct contact with the ligand).
Activation of the A 1 AR in the truncated (N)-methanocarba series is highly variable, ranging from full efficacy to low efficacy partial agonism depending on subtle differences in N 6 substitution. 62 Thus, there is compensation at a remote region of the receptor for the loss of an important element of recognition and receptor activation in the 5′ region, as shown by docking of many nucleosides in an A 1 AR homology model, derived from the closely related X-ray crystallographic structure of A 2A AR. We are now studying this N 6 interaction in more detail. Agonist (1 R ,2 R ,3 S ,4 R ,5 S )-4-(2-chloro-6-((dicyclopropylmethyl)amino)-9 H -purin-9-yl)bicyclo[3.1.0]hexane-2,3-diol 10 (MRS5474) is a truncated N 6 -dicyclopropylmethyl (N)-methanocarba analogue that fully and selectively activates the A 1 AR because of the addition of the N 6 -dicyclopropylmethyl group. 62 Even minor deviations from the structure of 10 lose the pharmacologically favorable combination of A 1 AR affinity, selectivity and efficacy. According to our homology modeling, there are two separate subpockets in the A 1 AR for branched N 6 substituents, but only one of these subpockets exists in the A 3 AR. This is our structural hypothesis for the A 1 /A 3 selectivity of 8 . In light of this pharmacologically optimized structure having more drug-like physical properties, 10 occupies a sweet spot as an A 1 AR agonist among nucleoside structures.
Nucleoside derivatives as optimized A 3 AR ligands
Although the two prototypical agonists of the A 3 AR of nanomolar affinity have shown numerous selective actions in model systems and apparently in humans, as evidenced by the lack of cardiovascular side effects at low to moderate doses of N 6 -(3-iodobenzyl)-5′- N -methylcarboxamidoadenosine 13 (IB-MECA), 32 questions of their pharmacological selectivity remain. There are examples of these two agonists activating other AR subtypes; including A 1 , A 2A and even the low affinity A 2B AR depending on concentration. 67 High concentrations (~30 μM) of 2-chloro- N 6 -(3-iodobenzyl)-5′- N -methylcarboxamidoadenosine 14 (Cl-IB-MECA) 32 have been noted to have AR-independent actions that affect survival, for example blockage of cell cycle progression at G1/S and G2/M transitions. 68 Consequently, we embarked on an effort to enhance the A 3 AR selectivity by further structural modification of this series of nucleosides.
To address the question of cross reactivity of these agonists at unrelated sites, we collected data for biological activities of these agents, as listed in the PubChem publicly available database. 69 The results provided an initial indication that the relatively few off target and unexpected interactions (i.e. not related to ARs) occur only at high μM concentrations ( Figure 4A ). Therefore, the action of 13 and 14 would appear to be relatively selective for the ARs with respect to diverse cellular pathways.
Figure 4 .

A. The structures of two prototypical A 3 AR agonists ( 13 , IB-MECA, CF101) and cancer ( 14 , Cl-IB-MECA, CF102), which are in clinical trials, and a survey of biological activities listed in the PubChem database. These compounds are in clinical trials for a variety of autoimmune inflammatory diseases ( 13 ) and cancer ( 14 ). Representative examples of off-target effects at high concentration are given. B. Chronological progression of the key steps in the design of nucleosides as ligands of the A 3 AR. The letters labeling specific chemical functionality are described in the text.
Progress in the systematic molecular refinement of A 3 AR-selective nucleoside ligands can be outlined chronologically. Representative structures are shown in Figure 3B , and this diagram is not exhaustive of the total SAR of the A 3 AR, which involves numerous research labs. 38 , 70 The logic behind changes in each region highlighted is dissected separately.
The need for small amide moieties for AR binding in general was established early in the process. The superiority of a 5′- N -methyluronamide in 15 for achieving selectivity for the A 3 AR was reported in 1994. 71
In a comparison of arylalkyl groups, an unbranched N 6 -benzyl group was best suited for selectivity for the A 3 AR, although considerable affinity was seen with other groups such as N 6 -(2-phenylethyl). 3-Chloro or 3-iodo substitution ( 15 , R 1 ) was also associated with high A 3 AR affinity.
Substitution with sulfur in place of 4′-oxygen in 18 as a means of preserving or enhancing A 3 AR activity was discovered in collaboration with the lab of Prof. Lak Shin Jeong at EWHA University. 72 Initially, it seemed that sulfur substitution was more geared for maintaining A 2A AR affinity, 73 but in an extensive SAR exploration at the A 3 AR it became clear that the 4′-thio modification was especially applicable to this subtype.
-
Pharmacological theory teaches that structural modifications of an agonist ligand resulting in partial agonism, if sufficiently substantial, can be combined to achieve antagonism. There is a continuum of degrees of efficacy that implies antagonism at its lowest point, i.e. the ligand binds competitively with the native agonist but cannot induce the necessary conformational change for receptor activation. We and others have combined individual changes that produce partial agonists in a known series of agonists, notably at A 3 AR or P2Y 1 R, to remove the residual activation of the receptor. 29 , 52 , 61 , 74 , 75 For example, the conversion of selective nucleoside agonists of the A 3 AR into selective antagonists may be effected by modification of the ribose moiety or by addition of suitable groups at the C2, N 6 or C8 positions of adenine.
One of the effects of truncation of adenosine derivatives at the 4′ position, e.g. 19 , is that A 3 AR agonists are converted into antagonists. 52 Numerous nucleosides exist at the boundary of A 3 AR agonism and antagonism. 29 Truncation of the 5′ group, i.e. removal of either CH 2 OH for native 7-riboside structures or a 5′- N -uronamide in 4 -like analogues, tended to preserve the affinity but not efficacy at the A 3 AR. Thus, it was possible to generate a wide range of selectively binding A 3 AR antagonists. An advantage of this modification in the design of antagonists in the nucleoside series in comparison to chemically diverse antagonists was the closer agreement in A 3 AR affinity at different species.
A comparison of the A 2A AR X-ray structure with residues in the A 1 /A 3 binding sites around the ribose moiety, which is important for the activation process, helps explain the effects of truncation. 20 , 24 , 25 , 62 The A 3 AR is less dependent on the 5′ region for recognition (likely related to the absence of His250 6.52 ), but still dependent on remaining hydrophilic residues for activation. The effects of truncation on recognition at the various AR subtypes can be understood in terms of interactions with specific amino acid residues in the binding site and subsites ( Figure 5 ). Hydrophilic residues are especially important in this process. The effects on binding and activation can be analyzed separately in conjunction with changes in the ligand structure.
In order to improve upon the selectivity and potency of the A 3 AR agonists, we examined the conformational factors of ribose in receptor recognition. Ring constrained substitutes for ribose were incorporated showing that the A 3 AR greatly favors the (N) conformation as in 16 . The (N)-methanocarba, pioneered by Victor Marquez for antiviral nucleosides, 76 was incorporated into adenosine and its A 3 AR-selective derivatives and found to be conducive to enhanced selectivity. The opposite isomer, which maintains a South (S) conformation of the ribose-like ring was >100-fold less potent at the A 3 AR. Thus, (N) lock of the ring more closely approximates an active conformation of ribose when bound to the receptor. Other forms of conformationally locked substitutions for the tetrahydrofuryl ring were incorporated in nucleosides for binding to the A 3 AR, 77 and the (N)-methanocarba is the most successful in enhancing pharmacological properties. A 3 AR antagonist 17 has a conformationally-locked spirolactam that maintains selectivity at the A 3 AR but precludes receptor activation. 29
The extended, rigid C2-arylalkynyl group in 20 was found to be compatible with both the (N)-methanocarba ring system and the preferred substituted N 6 -benzyl group. This C2 modification was previously introduced in native ribose-containing nucleosides by Cristalli and coworkers, 78 but its enhancing effect seems not to be additive in combination with large N 6 -benzyl and other groups unless the (N)-methanocarba ring system is present. This combination of relatively rigid substitutions of adenosine created a dilemma in the receptor modeling and docking of this series. A homology model was constructed based on the close template of the agonist complex of the A 2A AR. 20 In order to maintain the conserved stabilizing H-bonds within the hydrophilic ribose binding regions, it was necessary to shift TM2 away from the ligand to avoid a steric clash with the C2 substituent. 25 This shift was justified by using as template for the TM2 alone structures of several other GPCRs in active forms, i.e. the β 2 adrenergic receptor and opsin. 48 , 49 In those structures, the position of TM2 is displaced outward from its position in the A 2A AR. Thus, a hybrid homology model that takes advantage of two templates for different regions of the receptor was suitable for docking of the new series of phenylethynyl derivatives 20 . 25
The affinity of this series was sufficiently robust to accommodate a range of N 6 substitutions. Small groups, such as N 6 -methyl, are associated with high affinity at the human A 3 AR, with substantial reductions noted at the rat and mouse A 3 AR, at least in the 9-riboside series with species dependent affinities differing by as much as 100-fold. 25 In the case of rigid multiply-substituted full agonist 5 , the ratio of affinities at human to rat was ~10-fold. N 6 -Benzyl substitution in this series provided a closer correspondence of affinities at the human and mouse A 3 ARs. One such agonist was (1 S ,2 R ,3 S ,4 R ,5 S )-4-(6-((3-chlorobenzyl)amino)-2-((3,4-difluorophenyl)ethynyl)-9 H -purin-9-yl)-2,3-dihydroxy- N -methylbicyclo[3.1.0]hexane-1-carboxamide 20 (R 3 = 3-chlorobenzyl, R 4 = 3,4-difluoro, MRS5698), which is bioavailable and has in vivo activity.
The 5′- N -methyluronamide could be deleted without losing hA 3 AR selectivity in the C2-arylalkynyl series as in 21 , similar to previous truncation in other series of adenosine derivatives, but it limited the range of substitutions possible to maintain high affinity at the A 3 AR. The 5′- N -methyluronamide serves as an important stabilizing anchor of the hydrophilic ribose-binding region of the ARs. However, its loss can be tolerated if suitable compensatory features of the truncated nucleoside ligands maintain A 3 AR affinity. For example, A 3 AR selectivity of 21 is associated with small N 6 groups (i), such as methyl, ethyl, and methyloxy, but not larger groups, such as N 6 -benzyl or 2-phenylethyl in 21 . As found earlier, 51 a consequence of the loss of the flexible 5′- N -methyluronamide was reduction in the efficacy in activation of the A 3 AR. In some cases, the efficacy was sufficiently suppressed that the 4′-truncated derivatives were essentially full antagonists with respect to inhibition of adenylate cyclase (e.g. 21 , R 3 = (CH 2 ) 2 Ph, R 4 = H, K i 20 nM). The low efficacy could be modulated with changes in the C2-arylalkynyl group, such that some of the derivatives (e.g. 21 , R 3 = CH 3 O, R 4 = H, 30% efficacy, K i 13 nM) were selective partial agonists of the human A 3 AR. GPCR partial agonists might have superior pharmacological properties to full agonists for in vivo application. 44 , 79 Thus, there is a pronounced interrelationship of substitution at various sites on the adenosine structure with respect to their effects on AR affinity, selectivity and efficacy. Underlying this analysis is the observation that interactions of specific regions of potent AR ligands could be effectively modeled through ligand docking to the appropriate receptor.
Figure 5 .
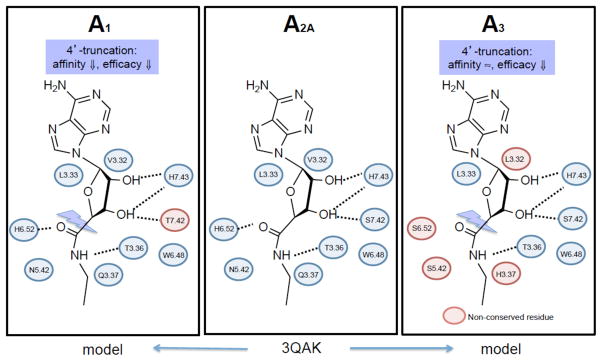
A comparison of the amino acids residues lining the subpockets of the ARs that are important for recognition of the ribose moiety. 3QAK is an X-ray structure of the 6 -A 2A AR complex, 20 which in this figure is adapted to 5 . The interactions of 3QAK were generalized by homology to A 1 AR and A 3 AR models (blue residues are conserved), and the general pharmacological effects of truncation are indicated. Many of these residues were detected as important in ligand recognition using site directed mutagenesis prior to the elucidation of the X-ray crystallographic structure.
3. Ligands for receptors more distantly related to current X-ray structural templates: P2YRs
P2YRs have a hopeful future for drug discovery, although the medicinal chemistry is more challenging than for the ARs. The correspondence of the native mononucleotide agonists to the eight subtypes of P2YRs is shown in Figure 1A . Naturally occurring dinucleotides, such as diadenosine tetraphosphate (Ap 4 A), also bind to various P2YRs. Synthesis and purification of large numbers of nucleotide derivatives is more labor intensive, and pharmacological evaluation is subject to uncertainty due to possible instability and poor availability of these ligands. Library screening of chemical libraries to identify new antagonist leads has so far only succeeded for a few of the P2YR subtypes (such as 43 , Figure 6 ). 80 In recent years, the modeling of P2YRs is also more difficult than for some other GPCRs, because of the lack of a close template. Concerning a template selection for P2YRs, this family is most closely related to CXCR4 among the X-ray structures currently existing. 81 Also worthy of consideration for their structural similarity to the P2YRs are the opioid receptors and a protease activated receptor (PAR1). 82 , 83 CXCR4 has already served as a suitable template for several of the P2YRs, 84 based 25.6% sequence identity in TMs and a Pro kink in same position in TM2 (2.58). As a result, the binding cavity in the CXCR4-based P2Y 12 R model is relatively extended and solvent-exposed. The disulfide between the N-terminus and EL3 found in the CXCR4 structure is possible due to the presence of analogous Cys residues but the does not seem to be necessary for P2Y 12 R activation. 120
Figure 6 .

Representative P2YR ligands from the NIH laboratory. 6 , 80 , 88 , 89 , 115 , 118 EC 50 or IC 50 values (nM) at the appropriate receptor are indicated. Partial agonist 37 displays 43% maximal efficacy. 117
We have studied the structure and function of the entire family of P2YRs through sequence analysis, molecular modeling, site-directed mutagenesis of one subtype, and extensive SAR probing of agonist and antagonist ligands. Novel selective agonists and antagonists of the less explored P2YRs include nucleotides and uncharged or nonhydrolyzable compounds. Only P2Y 1 and P2Y 12 Rs have extensively explored SAR of competitive antagonists, while selective nucleotide agonists have been reported for P2Y 1 , P2Y 2 , P2Y 4 , P2Y 6 , and P2Y 14 Rs. Nucleotide agonists have been effectively converted into selective antagonists by functional group manipulation at P2Y 1 (5′-diphosphates to 3′,5′-bisphosphates) and P2Y 12 Rs (5′-diphosphates to 5′-triphosphates).
The conformationally locked methanocarba analogues of the nucleotides have delineated clear conformational preferences for the subtypes linked to PLC, the P2Y 1 R-like subfamily. (1′R,2′S,4′S,5′S)-4-(2-iodo-6-methylamino-purin-9-yl)-1-[(phosphato)-methyl]-2-(phosphato)-bicyclo[3.1.0]hexane (MRS2500, 36 ), a selective P2Y 1 R antagonist of the (N)-methanocarba class, 85 was the first subnanomolar antagonist of this receptor and serves as a widely used probe for in vitro and in vivo studies, 86 , 87 for example, to test the hypothesis that such antagonists are effective antithrombotics. Although various adenosine bisphosphate derivatives were shown to antagonize the P2Y 1 R, the conformational lock of 36 greatly enhances its affinity, selectivity and antithrombotic properties. Stability in vivo is also enhanced, because of reduced hydrolysis of the 5′-phosphate by 5-nucleotidase due to the (N) conformation. A similarly locked 5′-diphosphate derivative, [[(1 R ,2 R ,3 S ,4 R ,5 S )-4-[6-amino-2-(methylthio)-9 H -purin-9-yl]-2,3-dihydroxybicyclo[3.1.0]hex-1-yl]methyl] diphosphoric acid mono ester 35 (MRS2365) is a potent full and selective agonist of the P2Y 1 R.
The P2Y 2 R and P2Y 4 R, which both respond to 5′-triphosphates, are difficult to distinguish pharmacologically, but recently selective agonists were introduced with the aid of molecular modeling. 88 , 89 The 2′-amino group on UTP analogues such as selective P2Y 2 R agonist 38 was predicted, using a rhodopsin-based model, to interact with two residues of the receptor by both H and amine-π bonding. Phosphonate 37 appears to be an allosteric partial agonist that is selective for the P2Y 2 R, which normally requires a 5′-diphosphate for activation. 117 The elongated N 4 -arylalkoxy group of selective P2Y 4 R agonist N 4 -phenylpropoxycytidine-5′-triphosphate 39 (MRS4062) was predicted to occupy an extracellular loop region that is more spacious in this subtype than in the P2Y 2 R, using CXCR4-based homology models. 89
The requirement by the P2Y 6 R, which is normally activated by UDP, of a South (S) conformation of ribose, unusual for the P2Y family, was demonstrated through receptor modeling and docking, followed by the confirmatory synthesis of (S)-methanocarba-UDP 40 . 118 The preference for the (S) conformation is so pronounced that the corresponding (N)-methanocarba-UDP is completely inactive. Certain dinucleoside triphosphates such as P 1 -(uridine 5′-)-P 4 -( N 4 -methoxycytidine 5′-)triphosphate 41 (MRS2957) are P2Y 6 R-selective agonists.
In the P2Y 12 R-like receptor subfamily, linked to inhibition of cAMP accumulation, we have not identified the preferred conformation of the ribose moiety. The SAR of UDP and UDP-glucose derivatives as agonists at the P2Y 14 R has been explored, to identify methylene phosphonate derivative 42 as a subnanomolar agonist. 115 An initial conclusion that UDP antagonizes the P2Y 14 R was later modified to characterize UDP generally as a potent agonist. The agonist glucose moiety has been functionalized for coupling to carriers by amine coupling to the carboxylate of the glucuronic acid moiety, which was predicted using molecular modeling to protrude to the extracellular region.
IV. Novel ligand tools for drug discovery – fluorescent probes, radioactive probes, multivalent probes, and functionalized nanoparticles
We have also concentrated on the introduction of specialized ligand probes for GPCR characterization that access multiple regions of a single receptor or that can possibly bridge multiple receptors. The SAR studies discussed so far are geared toward recognition by monomeric receptors, but it is recognized that stable dimers of GPCRs can have altered pharmacology. 90 The two protomeric receptor proteins in a pair communicate allosterically, typically through negative cooperativity, 119 to affect ligand binding and activation. The heterodimerization of the A 2A AR and the dopamine D 2 R occurs in the striatum. The A 2A AR/D 2 R is a well-validated heterodimer that is thought to be important in regulation of striatal pathways in the brain governing movement. 92 Ligands that have a preference for this or another (A 1 AR/A 2A AR) heterodimeric pair are sought as drugs for the CNS disorders, such as Parkinson’s disease. 91
GPCR dimers have been studied by molecular modeling 93 and pharmacologically. 91 We have designed multivalent AR ligands that are intended to be able to adopt a geometry suitable for bridging protomers of dimeric (and possibly higher order aggregates) of GPCRs. The theoretical docking of a multivalent dendrimeric conjugate of an A 2A AR agonist to the homodimer receptor complex was demonstrated. 93 A fluorescent adduct of agonist 11b with Alexa Fluor 532 served as a probe to follow the allosteric modulation within A 2A AR/D 2 R heterodimers using real-time FRET. 92 Recently, we introduced fluorescent probes for characterizing the A 3 AR and the A 2A AR, antagonist 44 and agonist 46 (demonstrated using flow cytometry) and antagonist 45 (demonstrated using fluorescence polarization), respectively ( Figure 7A ). 94 , 95 , 122 The fluorophores in each case were predicted by molecular modeling to form specific associations with the extracellular regions of the receptors that would enhance the affinity. Thus, the GPCR affinity/selectivity of a given conjugate is only partially dependent on the pharmacophore and is greatly influenced by the presence of distal groups on the ligand. The agonist fluorescent probes were shown to internalize with the receptor by agonist-induced processes.
Figure 7 .

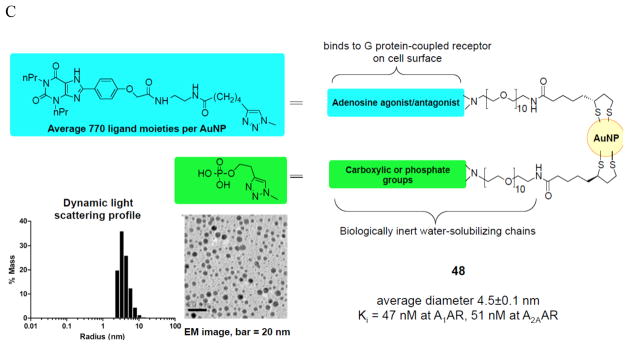
Specialized GPCR ligands linked to reporter groups or that are covalently bound to carriers in a manner that retains activity. (A) Recently reported fluorescent probes of the ARs, Alexa Fluor 488 conjugates 44 and 45 (antagonists of A 3 and A 2A ARs, respectively) and cyanine dye (Cy5) conjugate 46 (agonist of A 3 AR). 94 , 95 Tethering strategically functionalized AR ligands from quantum dots (B) and gold nanoparticles (C). 99 , 100
Macromolecular adenosine and P2Y receptor ligands have been shown to interact with and activate or antagonize their receptors. We introduced G PCR Li gand De ndrimer (GLiDe) conjugates to interact with receptor aggregates and to act as ‘smart drugs’. 96 The attachment of GPCR functionalized congeners to tree-like polyamidoamine (PAMAM) dendrimer carriers has greatly increased selectivity and affinity in novel multivalent and multifunctional probes. PAMAM dendrimers have versatile chemical functionality and properties that are compatible with biological systems. Dendritic unimolecular micelles containing tethered A 2A AR agonists and PEG (polyethylene glycol) chains were fully active in receptor activation and consequent inhibition of platelet aggregation, and the effects of pendant PEG groups on receptor recognition were relatively minor. The altered kinetics of the residual thrombotic effect is an example of qualitative differences between the monomeric and multivalent ligands. Blocking free amino groups reduced internalization. A series of conjugates of xanthine antagonist 2 tethered from PAMAM dendrimers of systematically varied stoichiometry indicated a more than additive effect of increasing ligand density on the AR affinity, which is suggestive of cooperativity of binding due to multiple binding sites on the membrane surface. 97 Nucleoside conjugates of PAMAM G4 (generation 4) dendrimers were synthesized by click chemistry ([2+3]cycloaddition of azide and alkyne) were potent A 3 AR agonists and induced potent cardioprotection. 98 Fluorescent and other reporter groups incorporated into these nucleotide conjugates provide new assay and imaging possibilities. Fluorescent imaging of the A 3 AR expressed in cultured cells was demonstrated. The receptor is not evenly distributed over the cell surface, suggesting clustering or aggregation, as is characteristic of GPCRs.
AR ligands have been covalently tethered from solid nanoparticles and shown to interact with and in some cases activate ARs. The design of adequate tethers (length, functionality, hydrophobicity, etc.) to allow small GPCR ligands on solid nanoparticles to retain receptor affinity is challenging. For example, quantum dots are solid particles of defined size and stable fluorescent properties, and they may be chemically functionalized for ligand attachment. In order to achieve aqueous solubility of highly fluorescent quantum dot conjugates of A 2A AR agonists ( 47 , Figure 7B ), it was necessary to add a highly polar, multivalent spacer consisting of a PAMAM dendron. 99 The presence of excess carboxylate groups maintained the nanoparticle conjugates in solution, which enabled the measurement of affinity at the A 2A AR. Gold nanoparticles (AuNPs) are being explored as drug carriers in the treatment and diagnosis of cancer and other diseases. Their physical properties can help target drugs, for example by active targeting or by accumulation in solid tumors by passive processes. An AuNP conjugate of XAC ( 48 , Figure 7C ) as a nonselective antagonist demonstrated binding of the functionalized solid nanoparticle to the A 2A AR, but only when a solubilizing phosphate chain was also present on the AuNP. 100 These conjugates are now suitable for further study of ARs in vitro and in vivo.
V. New directions for CNS and peripheral actions of improved AR and P2Y ligands
In the past, most of the clinical trials of AR agonists and antagonists for drug therapy have failed, partly due to nonselectivity, lack of efficacy, poor availability or the appearance of toxic side effects. 11 Although there are representative selective agents for each of the four AR subtypes, it is desirable to have AR ligands that are more drug-like in their physicochemical properties. We recently identified a nucleoside derivative that acts as an A 1 AR agonist with a distinct CNS-protective effect. Compound 8 inhibited seizures in the mouse corneal kindled seizure model with an IC 50 of 3 mg/kg at both 36 Hz and 48 Hz, which is indicative of possible application to treatment-resistant seizures. 62
A 3 AR agonists are in trials as a targeted therapy for autoimmune diseases and cancer. The development of agonists as potential therapeutic agents acting through ARs is appealing despite the risk of desensitization of the effect and subsequent receptor internalization induced by GPCR agonists. 32 A 3 AR is over expressed in cancer and inflammatory cells, whereas low or no expression is found in normal cells. The mechanism of action of a prototypical A 3 agonist 13 in models of autoimmune inflammatory disease has been ascribed apoptosis of proinflammatory cells (for example synoviocytes in rheumatoid arthritis), reduction in TNFα and its associated signaling, and preservation of normal white blood cells. The high receptor expression is reflected in the peripheral blood mononuclear cells of the patients. In a clinical study of patients with rheumatoid arthritis treated with the A 3 AR agonist 13 , a direct correlation was found between receptor expression at baseline and the patients’ response to the drug. Treatment with A 3 AR agonist 14 inhibits the development of hepatocellular carcinoma in an animal experimental model via modulation of the Wnt and NF-κB signaling pathways, resulting in apoptosis of the tumor cells, and initial patient data is encouraging. 101
Recently elucidated therapeutic directions include use of A 3 AR agonists to treat chronic neuropathic pain. 102 Three different A 3 AR agonists, including one of the more selective (N)-methanocarba derivatives ( 16 , R 1 = I, R 2 = Cl), were active in vivo. A 3 AR activation prevented the formation of the pain and also reduced the effects once it developed. The protective effects were demonstrated in two mouse models: chronic constriction injury and cancer chemotherapeutic agents having three different mechanisms (oxaliplatin, paclitaxel and bortezomib). This could provide a life-saving advantage, for instance allowing continued treatment for cancer in affected patients. The A 3 AR agonists not only reduced pain transiently when administered after it developed, but prevented its development.
Civan and coworkers have used our selective A 3 AR agonists and antagonists and receptor knockout mice to define an effect of this receptor on the formation of aqueous humor. 103 They elucidated the mechanism by which blocking an A 3 AR coupled to chloride inflow in the nonpigmented epithelial cell layer can lower intraocular pressure. Thus, topically applied A 3 AR antagonists that penetrate the cornea are promising for glaucoma treatment.
Development of P2YR agonists and antagonists for therapeutic applications is generally not as advanced as for the ARs, but nevertheless hold great potential for disease treatment. However, the use of potent P2Y 12 R antagonists as ‘blockbuster’ antithrombotic agents is well validated with three approved pharmaceutical agents. ADP activates both receptors on the platelet surface to induce components of aggregation, i.e. platelet shape change in response to P2Y 1 R and platelet adhesion, activation, and thrombus expansion and stability following P2Y 12 R activation. P2Y 1 R antagonists have shown efficacy for thrombosis in various in vivo models and possibly could impede atherosclerosis. 86 New, uncharged drug-like P2Y 1 R antagonists have been reported. 124
Among other newer ideas for P2YR-based therapeutics are: agonists of P2Y 1 R and P2Y 6 R and antagonists of P2Y 13 R and P2Y 14 R for diabetes, 104 – 108 P2Y 2 R agonists for cardiac ischemia and neurodegenerative disease, 109 , 110 P2Y 4 R agonists for cardiovascular disease and constipation. 85 , 111 In the context of diabetes, activation of the P2Y 6 R increases insulin release in mouse beta islets and beta cell lines. P2Y 6 R agonists protect mouse pancreatic MIN6 β - cells, skeletal muscle, and receptor-expressing astrocytes against TNF-induced and ischemia-induced apoptosis. Activation of the P2Y 6 R lowers intraocular pressure; thus, such agonists are of interest for treating glaucoma. 112 P2Y 6 R is also a target for neurodegeneration; the P2Y 6 R occurs in microglial cells, where it serves as a stimulus to induce phagocytosis. 113 The P2Y 14 R receptor was found to facilitate the release of histamine from mast cells and its genetic knockout increased insulin sensitivity in target tissue. Thus, selective P2Y 14 R antagonists could be a target for asthma or diabetes. 108 , 114
VI. Conclusions
Much of the data on AR ligand structure from early SAR studies, now is explainable from the A 2A AR X-ray crystallography. The ligand-receptor interactions in related GPCR complexes can be identified by means of modeling approaches, e.g. molecular docking. These insights can be used to guide the refinement of known ligands or the identification of entirely new hits. Efficacy of the native ligands as well as affinity can be systematically modulated to provide new selective antagonists, with an understanding of the receptor interactions involved in binding and activation. In this manner, new A 3 AR and P2Y 1 R antagonists have been designed. New promising directions for selective AR ligands include A 3 AR agonists for chronic neuropathic pain and improved A 2A AR antagonists for CNS-protective action. Selective ligands of P2YRs are of interest for treating immune, endocrine, neurodegenerative, and other conditions. The challenge of discovering new drug-like molecules to bind with high affinity to P2YRs has only been overcome for two of the subtypes, P2Y 1 and P2Y 12 Rs, but the expectation of increased structural knowledge promises major advances in the SAR of P2YRs.
Acknowledgments
Support from the NIDDK Intramural Research Program is acknowledged. We thank Stefano Costanzi (Dept. of Chemistry, American Univ., Washington, DC) for graphic images, and Silvia Paoletta, Evgeny Kiselev and Francesca Deflorian (NIDDK, NIH) for helpful discussions.
Abbreviations used
- AR
adenosine receptor
- cyclic AMP
adenosine 3′,5′-cyclic monophosphate
- AuNP
gold nanoparticles
- CNS
central nervous system
- EL
extracellular loop
- FRET
fluorescence resonance energy transfer
- GPCR
G protein-coupled receptor
- NECA
5′-N-ethylcarboxamidoadenosine
- PAMAM
polyamidoamine
- P2YR
P2Y receptor
- ROC
receiver operating characteristic
- SAR
structure-affinity relationship
- StaRs
thermostabilized mutated receptors
- TM
transmembrane helix
- XAC
xanthine amine congener
Biography
Kenneth A. Jacobson, Ph.D. as Chief of the Laboratory of Bioorganic Chemistry and the Molecular Recognition Section at the National Institute of Diabetes and Digestive and Kidney Diseases, NIH explores structure and pharmacology of G protein-coupled receptors. He received a B.A. from Reed College and Ph.D. in Chemistry at the University of California, San Diego. He was a Bantrell Fellow at Weizmann Institute of Science, Dept. of Organic Chemistry before joining the NIH. Recent awards include the 2003 Hillebrand Prize, appointment to the Senior Biomedical Research Service of NIH, the 2009 Sato Award and 2009 Pharmacia-ASPET Award in Experimental Therapeutics. Dr. Jacobson is an American Chemical Soc. Fellow, served as ACS Medicinal Chemistry Division Chair and is in the Division’s Medicinal Chemistry Hall of Fame.
Footnotes
This Award Address was presented at the 244th National Meeting of the American, Chemical Society in Philadelphia, Pennsylvania, August 21, 2012
References
- 1. Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller C. Nomenclature and classification of adenosine receptors – An update . Pharmacol Rev . 2011 ; 63 : 1 – 34 . doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Fumagalli M, King BF, Gachet C, Jacobson KA, Weisman GA International Union of Pharmacology LVIII . Update on the P2Y G protein-coupled nucleotide receptors: From molecular mechanisms and pathophysiology to therapy . Pharmacol Rev . 2006 ; 58 : 281 – 341 . doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Abbracchio MP, Burnstock G, Verkhratsky A, Zimmermann H. Purinergic signalling in the nervous system: an overview . Trends Neurosci . 2008 ; 32 : 19 – 29 . doi: 10.1016/j.tins.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 4. Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets . Nature Rev Drug Disc . 2006 ; 5 : 247 – 264 . doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Press NJ, Fozard JR. Progress Towards Novel Adenosine Receptor Therapeutics Gleaned From The Recent Patent Literature . Expert Opin Therap Pat . 2010 ; 20 : 987 – 1005 . doi: 10.1517/13543776.2010.495388. [DOI] [PubMed] [Google Scholar]
- 6. von Kügelgen I, Harden TK. Molecular pharmacology, physiology, and structure of the P2Y receptors . Adv Pharmacol . 2011 ; 61 : 373 – 415 . doi: 10.1016/B978-0-12-385526-8.00012-6. [DOI] [PubMed] [Google Scholar]
- 7. Oh YE, Abraham T, Saad N, Rapp JH, Vastey FL, Balmir E. A comprehensive comparative review of adenosine diphosphate receptor antagonists . Expert Opin Pharmacother . 2012 ; 13 : 175 – 191 . doi: 10.1517/14656566.2012.647683. [DOI] [PubMed] [Google Scholar]
- 8. Vaidya A, Agarwal A, Jain A, Agrawal RK, Jain SK. Bioconjugation of Polymers: A Novel Platform for Targeted Drug Delivery . Current Pharmaceutical Design . 2011 ; 17 : 1108 – 1125 . doi: 10.2174/138161211795656873. [DOI] [PubMed] [Google Scholar]
- 9. Flögel U, Burghoff S, van Lent PL, Temme S, Galbarz SL, Ding Z, El-Tayeb A, Huels S, Bönner F, Borg N, Jacoby C, Müller CE, Berg WB, Schrader J. Selective activation of adenosine A 2A receptors on immune cells by a CD73-dependent prodrug suppresses joint inflammation in experimental rheumatoid arthritis . Sci Transl Med . 2012 ; 4 : 146ra108 . doi: 10.1126/scitranslmed.3003717. [DOI] [PubMed] [Google Scholar]
- 10. Jacobson KA, Gao ZG, Göblyös A, IJzerman AP. Allosteric modulators of purine and pyrimidine receptors . Adv Pharmacol . 2011 ; 61 : 187 – 221 . doi: 10.1016/B978-0-12-385526-8.00007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gao ZG, Verzijl D, Zweemer A, Ye K, Göblyös A, IJzerman AP, Jacobson KA. Functionally biased modulation of A 3 adenosine receptor agonist efficacy and potency by imidazoquinolinamine allosteric enhancers . Biochem Pharmacol . 2011 ; 82 : 658 – 668 . doi: 10.1016/j.bcp.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular Mechanism of β-Arrestin-Biased Agonism at Seven-Transmembrane Receptors . Annu Rev Pharmacol Toxicol . 2012 ; 52 : 179 – 197 . doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. a) Mantell S, Jones R, Trevethick M. Design and application of locally delivered agonists of the adenosine A 2A receptor . Expert Rev Clin Pharmacol . 2010 ; 3 : 55 – 72 . doi: 10.1586/ecp.09.57. [DOI] [PubMed] [Google Scholar]; b) Chen JF, Eltzschig HK, Fredholm BB. Adenosine receptors as drug targets — what are the challenges? . Nature Rev Drug Disc . 2013 ; 12 : 265 – 286 . doi: 10.1038/nrd3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jacobson KA, Costanzi S. New insights for drug design from the X-ray crystallographic structures of GPCRs . Mol Pharmacol . 2012 ; 82 : 361 – 371 . doi: 10.1124/mol.112.079335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dale N, Frenguelli BG. Release of Adenosine and ATP During Ischemia and Epilepsy . Curr Neuropharmacol . 2009 ; 7 : 160 – 179 . doi: 10.2174/157015909789152146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Longhi MS, Robson SC, Bernstein SH, Serra S, Deaglio S. Biological functions of ecto-enzymes in regulating extracellular adenosine levels in neoplastic and inflammatory disease states . J Mol Med . 2013 ; 91 : 165 – 172 . doi: 10.1007/s00109-012-0991-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tilley SL, Wagoner VA, Salvatore CA, Jacobson MA, Koller BH. Adenosine and inosine increase cutaneous vasopermeability by activating A 3 receptors on mast cells . J Clin Invest . 2000 ; 105 : 361 – 367 . doi: 10.1172/JCI8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sitkovsky M. T regulatory cells: hypoxia-adenosinergic suppression and re-direction of the immune response . Trends Immunol . 2009 ; 30 : 102 – 108 . doi: 10.1016/j.it.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 19. Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, IJzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist . Science . 2008 ; 322 : 1211 – 1217 . doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. Structure of an agonist-bound human A 2A adenosine receptor . Science . 2011 ; 332 : 322 – 327 . doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. a) Andrews SP, Tehan B. Stabilised G protein-coupled receptors in structure-based drug design: a case study with adenosine A 2A receptor . Med Chem Comm . 2013 ; 4 : 52 – 67 . [Google Scholar]; b) Topiol S. X-ray structural information of GPCRs in drug design: what are the limitations and where do we go? . Expert Opin Drug Disc . 2013 doi: 10.1517/17460441.2013.783815. http://informahealthcare.com/doi/abs/10.1517/17460441.2013.783815 . [DOI] [PubMed]
- 22. Doré AS, Robertson N, Errey JC, Ng I, Hollenstein K, Tehan B, Hurrell E, Bennet K, Congreve M, Magnani F, Tate CG, Weir M, Marshall FH. Structure of the adenosine A 2A receptor in complex with ZM241385 and the xanthines XAC and caffeine . Structure . 2011 ; 19 : 1283 – 1293 . doi: 10.1016/j.str.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, Han GW, Roth CB, Heitman LH, IJzerman AP, Cherezov V, Stevens RC. Structural Basis for Allosteric Regulation of GPCRs by Sodium Ions . Science . 2012 ; 337 : 232 – 236 . doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Deflorian F, Kumar TS, Phan K, Gao ZG, Xu F, Wu H, Katritch V, Stevens RC, Jacobson KA. Evaluation of molecular modeling of agonist binding in light of the crystallographic structure of the agonist-bound A 2A adenosine receptor . J Med Chem . 2012 ; 55 : 538 – 552 . doi: 10.1021/jm201461q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tosh DK, Deflorian F, Phan K, Gao ZG, Wan TC, Gizewski E, Auchampach JA, Jacobson KA. Structure-guided design of A 3 adenosine receptor-selective nucleosides: Combination of 2-arylethynyl and bicyclo[3.1.0]hexane substitutions . J Med Chem . 2012 ; 55 : 4847 – 4860 . doi: 10.1021/jm300396n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kolb P, Phan K, Gao ZG, Marko AC, Sali A, Jacobson KA. Limits of ligand selectivity from docking to models: In silico screening for A 1 adenosine receptor antagonists . PLoS ONE . 2012 ; 7 : e49910 . doi: 10.1371/journal.pone.0049910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Valant C, Lane JR, Sexton PM, Christopoulos A. The Best of Both Worlds? Bitopic Orthosteric/Allosteric Ligands of G Protein–Coupled Receptors . Annu Rev Pharmacol Toxicol . 2012 ; 52 : 153 – 178 . doi: 10.1146/annurev-pharmtox-010611-134514. [DOI] [PubMed] [Google Scholar]
- 28. Moro S, Gao ZG, Jacobson KA, Spalluto G. Progress in pursuit of therapeutic adenosine receptor antagonists . Med Res Rev . 2006 ; 26 : 131 – 159 . doi: 10.1002/med.20048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gao ZG, Kim SK, Biadatti T, Chen W, Lee K, Barak D, Kim SG, Johnson CR, Jacobson KA. Structural determinants of A 3 adenosine receptor activation: Nucleoside ligands at the agonist/antagonist boundary . J Med Chem . 2002 ; 45 : 4471 – 4484 . doi: 10.1021/jm020211+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Riksen NP, Rongen GA. Targeting adenosine receptors in the development of cardiovascular therapeutics . Expert Rev Clin Pharmacol . 2012 ; 5 : 199 – 218 . doi: 10.1586/ecp.12.8. [DOI] [PubMed] [Google Scholar]
- 31. Gessi S, Merighi S, Fazzi D, Stefanelli A, Varani K, Borea PA. Adenosine receptor targeting in health and disease . Expert Opin Investig Drugs . 2011 ; 20 : 1591 – 1609 . doi: 10.1517/13543784.2011.627853. [DOI] [PubMed] [Google Scholar]
- 32. (a) Fishman P, Bar-Yehuda S, Liang BT, Jacobson KA. Pharmacological and therapeutic effects of A 3 adenosine receptor (A 3 AR) agonists . Drug Disc Today . 2012 ; 17 : 359 – 366 . doi: 10.1016/j.drudis.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gessi S, Merighi S, Varani K, Leung E, MacLennan S, Borea PA. The A 3 adenosine receptor: An enigmatic player in cell biology . Pharmacol Therap . 2008 ; 117 : 123 – 140 . doi: 10.1016/j.pharmthera.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 33. Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G-protein-coupled receptors . Nature . 2013 ; 494 : 185 – 194 . doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 34. Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models of structure-function relations in G protein-coupled receptors . Methods Neurosci . 1995 ; 25 : 366 – 428 . [Google Scholar]
- 35. Hodgson RA, Bedard PJ, Varty GB, Kazdoba TM, Di PT, Grzelak ME, Pond AJ, Hadjtahar A, Belanger N, Gregoire L, Dare A, Neustadt BR, Stamford AW, Hunter JC. Preladenant, a selective A 2A receptor antagonist, is active in primate models of movement disorders . Exp Neurol . 2010 ; 225 : 384 – 390 . doi: 10.1016/j.expneurol.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 36. Jacobson KA. Functionalized congener approach to the design of ligands for G protein–coupled receptors (GPCRs) . Bioconjugate Chem . 2009 ; 20 : 1816 – 1835 . doi: 10.1021/bc9000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. IJzerman AP, van Galen PJM, Jacobson KA. Molecular modeling of adenosine receptors. I. The ligand binding site on the A 1 receptor . Drug Des Discov . 1992 ; 9 : 49 – 67 . [PMC free article] [PubMed] [Google Scholar]
- 38. Cheong SL, Federico S, Venkatesan G, Mandel AL, Shao YM, Moro S, Spalluto G, Pastorin G. The A 3 Adenosine Receptor as Multifaceted Therapeutic Target: Pharmacology, Medicinal Chemistry, and In Silico Approaches . Med Res Rev . 2013 ; 33 : 235 – 335 . doi: 10.1002/med.20254. [DOI] [PubMed] [Google Scholar]
- 39. Kim J, Wess J, van Rhee AM, Shöneberg T, Jacobson KA. Site-directed mutagenesis identifies residues involved in ligand recognition in the human A 2a adenosine receptor . J Biol Chem . 1995 ; 270 : 13987 – 13997 . doi: 10.1074/jbc.270.23.13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jacobson KA, Costanzi S, Deflorian F. Probing GPCR structure: adenosine and P2Y nucleotide receptors . Methods Enzymol . 2013 ; 520 : 199 – 217 . doi: 10.1016/B978-0-12-391861-1.00009-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jacobson KA, Ohno M, Duong HT, Kim SK, Tchilibon S, Cesnek M, Holy A, Gao ZG. A neoceptor approach to unraveling microscopic interactions between the human A 2A adenosine receptor and its agonists . Chemistry and Biology . 2005 ; 12 : 237 – 247 . doi: 10.1016/j.chembiol.2004.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Conklin BR, Hsiao EC, Claeysen S, Dumuis A, Srinivasan S, Forsayeth JR, Guettier JM, Chang WC, Pei Y, McCarthy KD, Nissenson RA, Wess J, Bockaert J, Roth BL. Engineering GPCR signaling pathways with RASSLs . Nature Methods . 2008 ; 5 : 673 – 678 . doi: 10.1038/nmeth.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AG, Tate CG. Agonist-bound adenosine A 2A receptor structures reveal common features of GPCR activation . Nature . 2011 ; 474 : 521 – 525 . doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shearer J, Severson DL, Su L, Belardinelli L, Dhalla AK. Partial A 1 adenosine receptor agonist regulates cardiac substrate utilization in insulin-resistant rats in vivo . J Pharmacol Exp Ther . 2009 ; 328 : 306 – 311 . doi: 10.1124/jpet.108.143594. [DOI] [PubMed] [Google Scholar]
- 45. Ivanov AA, Barak D, Jacobson KA. Evaluation of homology modeling of GPCRs in light of the A 2A adenosine receptor crystallographic structure . J Med Chem . 2009 ; 52 : 3284 – 3292 . doi: 10.1021/jm801533x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Borea PA, Dalpiaz A, Varani K, Gessi S, Gilli G. Binding thermodynamics at A 1 and A 2A addeonsine receptors . Life Sci . 1996 ; 59 : 1373 – 1388 . doi: 10.1016/0024-3205(96)00311-6. [DOI] [PubMed] [Google Scholar]
- 47. Bennett KA, Tehan G, Lebon G, Tate CG, Weir M, Marshall F, Langmead CJ. Pharmacology and Structure of Isolated Conformations of the Adenosine A 2A Receptor Define Ligand Efficacy . Mol Pharmacol . 2013 doi: 10.1124/mol.112.084509. (epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Crystal structure of opsin in its G-protein-interacting conformation . Nature . 2008 ; 455 : 497 – 502 . doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 49. Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah STA, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunihara RK, Kobilka BK. Crystal structure of the β2 adrenergic receptor-Gs protein complex . Nature . 2011 ; 477 : 549 – 555 . doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jacobson KA, Park KS, Jiang JL, Kim YC, Olah ME, Stiles GL, Ji XD. Pharmacological characterization of novel A 3 adenosine receptor-selective antagonists . Neuropharmacology . 1997 ; 36 : 1157 – 1165 . doi: 10.1016/s0028-3908(97)00104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Carlsson J, Yoo L, Gao ZG, Irwin J, Shoichet B, Jacobson KA. Structure-based discovery of adenosine A 2A receptor ligands . J Med Chem . 2010 ; 53 : 3748 – 3755 . doi: 10.1021/jm100240h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jeong LS, Pal S, Choe SA, Choi WJ, Jacobson KA, Gao ZG, Klutz AM, Hou X, Kim HO, Lee HW, Lee SK, Tosh DK, Moon HR. Structure activity relationships of truncated D- and L- 4′-thioadenosine derivatives as species-independent A 3 adenosine receptor antagonists . J Med Chem . 2008 ; 51 : 6609 – 6613 . doi: 10.1021/jm8008647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yang H, Avila MY, Peterson-Yantorno K, Coca-Prados M, Stone RA, Jacobson KA, Civan MM. The cross-species A 3 adenosine-receptor antagonist MRS 1292 inhibits adenosine-triggered human nonpigmented ciliary epithelial cell fluid release and reduces mouse intraocular pressure . Current Eye Res . 2005 ; 30 : 747 – 754 . doi: 10.1080/02713680590953147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhukov A, Andrews SP, Errey JC, Robertson N, Tehan B, Mason JS, Marshall FH, Weir M, Congreve M. Biophysical mapping of the adenosine A 2A receptor . J Med Chem . 2011 ; 54 : 4312 – 4323 . doi: 10.1021/jm2003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Katritch V, Jaakola VP, Lane JR, Lin J, IJzerman AP, Yeager M, Kufareva I, Stevens RC, Abagyan R. Structure-based discovery of novel chemotypes for adenosine A 2A receptor antagonists . J Med Chem . 2010 ; 53 : 1799 – 1809 . doi: 10.1021/jm901647p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sirci F, Goracci L, Rodriguez D, van Muijlwijk-Koezen J, Gutiérrez-de-Terán H, Mannhold R. Ligand-, structure- and pharmacophore-based molecular fingerprints: a case study on adenosine A 1 , A 2A , A 2B , and A 3 receptor antagonists . J Comput Aided Mol Des . 2012 ; 26 : 1247 – 1266 . doi: 10.1007/s10822-012-9612-8. [DOI] [PubMed] [Google Scholar]
- 57. van der Horst E, van der Pijl R, Mulder-Krieger T, Bender A, IJzerman AP. Substructure-based virtual screening for adenosine A 2A receptor ligands . ChemMedChem . 2011 ; 6 : 2302 – 2311 . doi: 10.1002/cmdc.201100369. [DOI] [PubMed] [Google Scholar]
- 58. van Westen GJ, van den Hoven OO, van der Pijl R, Mulder-Krieger T, de Vries H, Wegner JK, IJzerman AP, van Vlijmen HW, Bender A. Identifying novel adenosine receptor ligands by simultaneous proteochemometric modeling of rat and human bioactivity data . J Med Chem . 2012 ; 55 : 7010 – 7020 . doi: 10.1021/jm3003069. [DOI] [PubMed] [Google Scholar]
- 59. Langmead CJ, Andrews SP, Congreve M, Errey JC, Hurrell E, Marshall FH, Mason JS, Richardson CM, Robertson N, Zhukov A, Weir M. Identification of Novel Adenosine A 2A Receptor Antagonists by Virtual Screening . J Med Chem . 2012 ; 55 : 1904 – 1909 . doi: 10.1021/jm201455y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen D, Errey JC, Heitman LH, Marshall FH, IJzerman AP, Siegal G. Fragment Screening of GPCRs Using Biophysical Methods: Identification of Ligands of the Adenosine A 2A Receptor with Novel Biological Activity . ACS Chem Biol . 2012 ; 7 : 2064 – 2073 . doi: 10.1021/cb300436c. [DOI] [PubMed] [Google Scholar]
- 61. Gao ZG, Jacobson KA. Translocation of arrestin induced by human A 3 adenosine receptor ligands in an engineered cell line: Comparison with G protein-dependent pathways . Pharmacol Res . 2008 ; 57 : 303 – 311 . doi: 10.1016/j.phrs.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tosh DK, Paoletta S, Deflorian F, Phan K, Moss SM, Gao ZG, Jiang X, Jacobson KA. Structural sweet spot for A 1 adenosine receptor activation by truncated (N)-methanocarba nucleosides: Receptor docking and potent anticonvulsant activity . J Med Chem . 2012 ; 55 : 8075 – 8090 . doi: 10.1021/jm300965a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kim J, Jiang Q, Glashofer M, Yehle S, Wess J, Jacobson KA. Glutamate residues in the second extracellular loop of the human A 2A adenosine receptors are required for ligand recognition . Mol Pharmacol . 1996 ; 49 : 683 – 691 . [PMC free article] [PubMed] [Google Scholar]
- 64. Niiya K, Jacobson KA, Silvia SK, Olsson RA. Covalent binding of a selective agonist irreversibly activates guinea pig coronary artery A 2 adenosine receptors . Naunyn-Schmeideberg’s Arch Pharamacol . 1993 ; 347 : 521 – 526 . doi: 10.1007/BF00166745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tosh DK, Phan K, Gao ZG, Gakh A, Xu F, Deflorian F, Abagyan R, Stevens RC, Jacobson KA, Katritch V. Optimization of adenosine 5′-carboxamide derivatives as adenosine receptor agonists using structure-based ligand design and fragment-based searching . J Med Chem . 2012 ; 55 : 4297 – 4308 . doi: 10.1021/jm300095s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Abagyan RA, Orry A, Raush E, Budagyan L, Totrov M. ICM Manual, 3.0 . MolSoft LLC ; La Jolla, CA : 2011 . [Google Scholar]
- 67. Fozard JR. From Hypertension (+) to Asthma: interactions with the Adenosine A 3 Receptor from a Personal Perspective . In: Borea PA, editor. A 3 Adenosine Receptors from Cell Biology to Pharmacology and Therapeutics . Springer Science+Business Media B.V ; Dordrecht, Germany : 2010 . pp. 3 – 26 . [Google Scholar]
- 68. Mlejnek P, Dolezel P, Frydrich I. Effects of synthetic A 3 adenosine receptor agonists on cell proliferation and viability are receptor independent at micromolar concentrations . J Physiol Biochem . 2012 doi: 10.1007/s13105-012-0222-7. (epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 69. Canny SA, Cruz Y, Southern MA, Griffin PR. PubChem Promiscuity: a web resource for gathering compound promiscuity data from PubChem . Bioinformatics . 2012 ; 28 : 140 – 141 . doi: 10.1093/bioinformatics/btr622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Baraldi PG, Preti D, Borea PA, Varani K. Medicinal Chemistry of A 3 Adenosine Receptor Modulators: Pharmacological Activities and Therapeutic Implications . J Med Chem . 2012 ; 55 : 5676 – 5703 . doi: 10.1021/jm300087j. [DOI] [PubMed] [Google Scholar]
- 71. Gallo-Rodriguez C, Ji XD, Melman N, Siegman BD, Sanders LH, Orlina J, Fischer B, Pu QL, Olah ME, van Galen PJM, Stiles GL, Jacobson KA. Structure-activity relationships of N 6 -benzyladenosine-5′-uronamides as A 3 -selective adenosine agonists . J Med Chem . 1994 ; 37 : 636 – 646 . doi: 10.1021/jm00031a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jeong LS, Lee HW, Jacobson KA, Kim HO, Shin DH, Lee JA, Gao ZG, Lu C, Duong HT, Gunaga P, Lee SK, Jin DZ, Chun MW, Moon HR. Structure-activity relationships of 2-chloro- N 6 -substituted-4′-thioadenosine-5′ - uronamides as highly potent and selective agonists at the human A 3 adenosine receptor . J Med Chem . 2006 ; 49 : 273 – 281 . doi: 10.1021/jm050595e. [DOI] [PubMed] [Google Scholar]
- 73. Siddiqi SM, Jacobson KA, Esker JL, Melman N, Tiwari KN, Secrist JA, Schneller SW, Cristalli G, Johnson CR, IJzerman AP. Search for new purine- and ribose-modified adenosine analogues as selective agonists and antagonists at adenosine receptors . J Med Chem . 1995 ; 38 : 1174 – 1188 . doi: 10.1021/jm00007a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Costanzi S, Lambertucci C, Vittori S, Cristalli G. 2- and 8-Alkynyladenosines: conformational studies and docking to human adenosine A 3 receptor can explain their different biological behavior . J Mol Graph Model . 2003 ; 21 : 253 – 262 . doi: 10.1016/s1093-3263(02)00161-4. [DOI] [PubMed] [Google Scholar]
- 75. van Tilburg EW, van Frijtag Drabbe Künzel J, der Groote M, Vollinga RC, Lorenzen A, IJzerman AP. N 6 ,5 ′-disubstituted adenosine derivatives as partial agonists for the human adenosine A 3 receptor . J Med Chem . 1999 ; 42 : 1393 – 1400 . doi: 10.1021/jm981090+. [DOI] [PubMed] [Google Scholar]
- 76. Marquez VE. In: Modified Nucleosides: in Biochemistry, Biotechnology and Medicine . Herdewijn P, editor. Chapter 12 . Wiley-VCH Verlag GmbH & Co. KGaA ; Weinheim, Germany : 2008 . pp. 307 – 341 . [Google Scholar]
- 77. Ravn J, Qvortup K, Rosenbohm C, Koch T. design, synthesis, and biological evaluation of LNA nucleosides as adenosine A 3 receptor ligands . Med Chem . 2007 ; 15 : 5440 – 5447 . doi: 10.1016/j.bmc.2007.05.056. [DOI] [PubMed] [Google Scholar]
- 78. Volpini R, Buccioni M, Dal Ben D, Lambertucci C, Lammi C, Marucci G, Ramadori AT, Klotz KN, Cristalli G. Synthesis and biological evaluation of 2-alkynyl- N 6 -methyl-5′-N-methylcarboxamidoadenosine derivatives as potent and highly selective agonists for the human adenosine A 3 receptor . J Med Chem . 2009 ; 52 : 7897 – 7900 . doi: 10.1021/jm900754g. [DOI] [PubMed] [Google Scholar]
- 79. Hruba L, Ginsburg BC, McMahon LR. Apparent inverse relationship between cannabinoid agonist efficacy and tolerance/cross-tolerance produced by Δ-tetrahydrocannabinol treatment in rhesus monkeys . J Pharm Exp Therap . 2012 ; 342 : 843 – 849 . doi: 10.1124/jpet.112.196444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Costanzi S, Kumar TS, Balasubramanian R, Harden TK, Jacobson KA. Virtual screening leads to the discovery of novel non-nucleotide P2Y 1 receptor antagonists . Bioorg Med Chem . 2012 ; 20 : 5254 – 5261 . doi: 10.1016/j.bmc.2012.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists . Science . 2010 ; 330 : 1066 – 1071 . doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC. Structure of the human kappa-opioid receptor in complex with JDTic . Nature . 2012 ; 485 : 327 – 332 . doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zhang C, Srinivasan Y, Arlow DH, Fung JJ, Palmer D, Zheng Y, Green HF, Pandey A, Dror RO, Shaw DE, Weis WI, Coughlin SR, Kobilka BK. High-resolution crystal structure of human protease-activated receptor 1 . Nature . 2012 ; 492 : 387 – 392 . doi: 10.1038/nature11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Deflorian F, Jacobson KA. Comparison of three GPCR structural templates for modeling of the P2Y 12 nucleotide receptor . J Comput Aided Mol Des . 2011 ; 25 : 329 – 338 . doi: 10.1007/s10822-011-9423-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jacobson KA, Boeynaems JM. P2Y nucleotide receptors: Promise of therapeutic applications . Drug Disc Today . 2010 ; 15 : 570 – 578 . doi: 10.1016/j.drudis.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hechler B, Freund M, Ravanat C, Magnenat S, Cazenave JP, Gachet C. Reduced atherosclerotic lesions in P2Y 1 /apolipoprotein E double-knockout mice: the contribution of non-hematopoietic-derived P2Y 1 receptors . Circulation . 2008 ; 118 : 754 – 763 . doi: 10.1161/CIRCULATIONAHA.108.788927. [DOI] [PubMed] [Google Scholar]
- 87. Gallego D, Gil V, Martínez-Cutillas M, Mañé N, Martín MT, Jiménez M. Purinergic neuromuscular transmission is absent in the colon of P2Y 1 knocked out mice . J Physiol . 2012 ; 590 : 1943 – 1956 . doi: 10.1113/jphysiol.2011.224345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ivanov AA, Ko H, Cosyn L, Maddileti S, Besada P, Fricks I, Costanzi S, Harden TK, Van Calenbergh S, Jacobson KA. Molecular modeling of the human P2Y 2 receptor and design of a selective agonist, 2′-amino-2′-deoxy-2-thiouridine 5′-triphosphate . J Med Chem . 2007 ; 50 : 1166 – 1176 . doi: 10.1021/jm060903o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Maruoka H, Jayasekara MPS, Barrett MO, Franklin DE, de Castro S, Kim N, Costanzi S, Harden TK, Jacobson KA. Pyrimidine nucleotides with 4-alkyloxyimino and terminal tetraphosphate δ-ester modifications as selective agonists of the P2Y 4 receptor . J Med Chem . 2011 ; 54 : 4018 – 4033 . doi: 10.1021/jm101591j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Orru M, Bakesová J, Brugarolas M, Quiroz C, Beaumont V, Goldberg SR, Lluís C, Cortés A, Franco R, Casadó V, Canela EI, Ferré S. Striatal Pre- and Postsynaptic Profile of Adenosine A 2A Receptor Antagonists . PLoS ONE . 2011 ; 6 : e16088 . doi: 10.1371/journal.pone.0016088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ferré S, Baler R, Bouvier M, Caron MG, Devi LA, Durroux T, Fuxe K, George SR, Javitch JA, Lohse MJ, Mackie K, Milligan G, Pfleger KD, Pin JP, Volkow ND, Waldhoer M, Woods AS, Franco R. Building a new conceptual framework for receptor heteromers . Nature Chem Biol . 2009 ; 5 : 131 – 134 . doi: 10.1038/nchembio0309-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Fernández-Dueñas V, Gómez-Soler M, Jacobson KA, Kumar TS, Fuxe K, Borroto-Escuela DO, Ciruela F. Molecular determinants of the adenosine A 2A R-dopamine D 2 receptor-receptor allosterism: Role of the intracellular loop 3 of the dopamine D 2 receptor . J Neurochem . 2012 ; 123 : 373 – 384 . doi: 10.1111/j.1471-4159.2012.07956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ivanov AA, Jacobson KA. Molecular modeling of a PAMAM-CGS21680 dendrimer bound to an A 2A adenosine receptor homodimer . Bioorg Med Chem Lett . 2008 ; 18 : 4312 – 4315 . doi: 10.1016/j.bmcl.2008.06.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kozma E, Kumar TS, Federico S, Phan K, Balasubramanian R, Gao ZG, Paoletta S, Moro S, Spalluto G, Jacobson KA. Novel fluorescent antagonist as a molecular probe in A 3 adenosine receptor binding assays using flow cytometry . Biochem Pharmacol . 2012 ; 83 : 1552 – 1561 . doi: 10.1016/j.bcp.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kecskés M, Kumar TS, Yoo L, Gao ZG, Jacobson KA. Novel Alexa Fluor-488 labeled antagonist of the A 2A adenosine receptor: Application to a fluorescence polarization-based receptor binding assay . Biochem Pharmacol . 2010 ; 80 : 506 – 511 . doi: 10.1016/j.bcp.2010.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Jacobson KA. GPCR ligand-dendrimer (GLiDe) conjugates: future smart drugs? . Trends Pharmacol Sci . 2010 ; 31 : 575 – 579 . doi: 10.1016/j.tips.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kecskés A, Tosh DK, Wei Q, Gao ZG, Jacobson KA. GPCR ligand dendrimer (GLiDe) conjugates: Adenosine receptor interactions of a series of multivalent xanthine antagonists . Bioconjugate Chem . 2011 ; 22 : 1115 – 1127 . doi: 10.1021/bc1005812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wan TC, Tosh DT, Du L, Gizewski E, Jacobson KA, Auchampach JA. Polyamidoamine (PAMAM) dendrimer conjugate specifically activates the A 3 adenosine receptor to improve post-ischemic/reperfusion functions in isolated mouse hearts . BMC Pharmacol . 2011 ; 11 : 11 . doi: 10.1186/1471-2210-11-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Das A, Sanjayan GJ, Kecskés M, Kumar TS, Yoo L, Gao ZG, Jacobson KA. Nucleoside conjugates of quantum dots for characterization of G protein-coupled receptors: Strategies for immobilizing A 2A adenosine receptor agonists . J Nanobiotechnol . 2010 ; 8 : 11 . doi: 10.1186/1477-3155-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Jayasekara PS, Phan K, Tosh DK, Kumar TS, Moss SM, Zhang G, Barchi JJ, Gao ZG, Jacobson KA. Modulation of G protein-coupled adenosine receptors by strategically functionalized agonists and antagonists immobilized on gold nanoparticles . Purinergic Signalling . doi: 10.1007/s11302-012-9338-z. in press . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Stemmer SM, Benjaminov O, Medalia G, Ciuraru NB, Silverman MH, Bar-Yehuda S, Fishman S, Harpaz Z, Farbstein M, Cohen S, Patoka R, Singer B, Kerns WD, Fishman P. CF102 for the Treatment of Hepatocellular Carcinoma: APhase I/II, Open-Label, Dose-Escalation Study . Oncologist . 2013 ; 18 : 25 – 26 . doi: 10.1634/theoncologist.2012-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Chen Z, Janes K, Chen C, Doyle T, Tosh DK, Jacobson KA, Salvemini D. Controlling murine and rat chronic pain through A 3 adenosine receptor activation . FASEB J . 2012 ; 26 : 1855 – 1865 . doi: 10.1096/fj.11-201541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Avila MY, Stone RA, Civan MM. Knockout of A 3 adenosine receptors reduces mouse intraocular pressure . Invest Ophthalmol Vis Sci . 2002 ; 43 : 3021 – 3026 . [PubMed] [Google Scholar]
- 104. Yelovitch S, Barr HM, Camden J, Weisman GA, Shai E, Varon D, Fischer B. Identification of a Promising Drug Candidate for the Treatment of Type 2 Diabetes Based on a P2Y 1 Receptor Agonist . J Med Chem . 2012 ; 55 : 7623 – 7635 . doi: 10.1021/jm3006355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Balasubramanian R, Maruoka H, Jayasekara PS, Gao ZG, Jacobson KA. AMP-activated protein kinase as regulator of P2Y 6 receptor-induced secretion in MIN6 mouse pancreatic β cells . Biochem Pharmacol . 2013 ; 85 : 991 – 998 . doi: 10.1016/j.bcp.2012.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Parandeh F, Abaraviciene SM, Amisten S, Erlinge D, Salehi A. Uridine diphosphate (UDP) stimulates insulin secretion by activation of P2Y 6 receptors . Biochem Biophys Res Commun . 2008 ; 370 : 499 – 503 . doi: 10.1016/j.bbrc.2008.03.119. [DOI] [PubMed] [Google Scholar]
- 107. Tan C, Voss U, Svensson S, Erlinge D, Olde B. High glucose and free fatty acids induce beta cell apoptosis via autocrine effects of ADP acting on the P2Y 13 receptor . Purinergic Signal . 2013 ; 9 : 67 – 79 . doi: 10.1007/s11302-012-9331-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Xu J, Morinaga H, Oh D, Li P, Chen A, Talukdar S, Lazarowski ER, Olefsky JM, Kim JJ. GPR105 ablation prevents inflammation and improves insulin sensitivity in mice with diet-induced obesity . J Immunol . 2012 ; 189 : 1992 – 1999 . doi: 10.4049/jimmunol.1103207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Cohen R, Shainberg A, Hochhauser E, Cheporko Y, Tobar A, Birk E, Pinhas L, Leipziger J, Don J, Porat E. UTP reduces infarct size and improves mice heart function after myocardial infarct via P2Y 2 receptor . Biochem Pharmacol . 2011 ; 82 : 1126 – 1133 . doi: 10.1016/j.bcp.2011.07.094. [DOI] [PubMed] [Google Scholar]
- 110. Chorna NE, Santiago-Pérez LI, Erb L, Seye CI, Neary JT, Sun GY, Weisman GA, González FA. P2Y 2 receptors activate neuroprotective mechanisms in astrocytic cells . J Neurochem . 2004 ; 91 : 1471 – 4159 . doi: 10.1111/j.1471-4159.2004.02699.x. [DOI] [PubMed] [Google Scholar]
- 111. Horckmans M, Léon-Gómez E, Robaye B, Balligand JL, Boeynaems JM, Dessy C, Communi D. Gene deletion of P2Y 4 receptor lowers exercise capacity and reduces myocardial hypertrophy with swimming exercise . Am J Physiol Heart Circ Physiol . 2012 ; 303 : H835 – H843 . doi: 10.1152/ajpheart.00256.2012. [DOI] [PubMed] [Google Scholar]
- 112. Ginsburg-Shmuel T, Haas M, Grbic D, Arguin G, Nadel Y, Gendron FP, Reiser G, Fischer B. UDP made a highly promising stable, potent, and selective P2Y 6 -receptor agonist upon introduction of a boranophosphate moiety . Bioorg Med Chem . 2012 ; 20 : 5483 – 5495 . doi: 10.1016/j.bmc.2012.07.042. [DOI] [PubMed] [Google Scholar]
- 113. Koizumi S, Shigemoto-Mogam Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, Joshi BV, Jacobson KA, Kohsaka S, Inoue K. UDP acting at P2Y 6 receptors is a novel mediator of microglial phagocytosis . Nature . 2007 ; 446 : 1091 – 1095 . doi: 10.1038/nature05704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Gao ZG, Wei Q, Jayasekara MPS, Jacobson KA. The role of P2Y 14 and other P2Y receptors in degranulation of human LAD2 mast cells . Purinergic Signalling . 2013 ; 9 : 31 – 40 . doi: 10.1007/s11302-012-9325-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Das A, Ko H, Burianek LE, Barrett MO, Harden TK, Jacobson KA. Human P2Y 14 receptor agonists: Truncation of the hexose moiety of uridine-5′-diphosphoglucose and its replacement with alkyl and aryl groups . J Med Chem . 2010 ; 53 : 471 – 480 . doi: 10.1021/jm901432g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Sesma JI, Kreda SM, Okada SF, van Heusden C, Moussa L, Jones LC, O’Neal WK, Togawa N, Hiasa M, Moriyama Y, Lazarowski ER. The vesicular nucleotide transporter (VNUT) regulates the nucleotide content in airway epithelial mucin granules . Am J Physiol, Cell Physiol . 2013 Mar ; doi: 10.1152/ajpcell.00371.2012. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Van Poecke S, Barrett MO, Kumar TS, Sinnaeve D, Martins JC, Jacobson KA, Harden TK, Van Calenbergh S. Synthesis and P2Y 2 receptor agonist activity of nucleotide 5′-phosphonate analogues . Bioorg Med Chem . 2012 ; 20 : 2304 – 2315 . doi: 10.1016/j.bmc.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Maruoka H, Barrett MO, Ko H, Tosh DK, Melman A, Burianek LE, Balasubramanian R, Berk B, Costanzi S, Harden TK, Jacobson KA. Pyrimidine ribonucleotides with enhanced selectivity as P2Y 6 receptor agonists: Novel 4-alkyloxyimino, (S)-methanocarba, and 5′-triphosphate γ-ester modifications . J Med Chem . 2010 ; 53 : 4488 – 4501 . doi: 10.1021/jm100287t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Han Y, Moreira IS, Urizar E, Weinstein H, Javitch JA. Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation . Nat Chem Biol . 2009 ; 5 : 688 – 695 . doi: 10.1038/nchembio.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Algaier I, Jakubowski JA, Asai F, Kügelgen I. Interaction of the active metabolite of prasugrel, R-138727, with cysteine 97 and cysteine 175 of the human P2Y 12 receptor . J Thromb Haemostasis . 2008 ; 6 : 1908 – 1914 . doi: 10.1111/j.1538-7836.2008.03136.x. [DOI] [PubMed] [Google Scholar]
- 121. Lane SD, Green CE, Steinberg JL, Ma L, Schmitz JM, Rathnayaka N, Bandak SD, Ferre S, Moeller FG. Cardiovascular and Subjective Effects of the Novel Adenosine A 2A Receptor Antagonist SYN115 in Cocaine Dependent Individuals . J Addict Res Ther . 2012 March ; S1 : 009 . doi: 10.4172/2155-6105.S1-009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Kozma E, Gizewski ET, Tosh DK, Squarcialupi L, Auchampach JA, Jacobson KA. Characterization by flow cytometry of fluorescent, selective agonist probes of the A 3 adenosine receptor . Biochem Pharmacol . 2013 ; 85 : 1171 – 1181 . doi: 10.1016/j.bcp.2013.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Carlsson J, Tosh DK, Phan K, Gao ZG, Jacobson KA. Structure-activity relationships and molecular modeling of 1,2,4-triazoles as antagonists of the A 2A adenosine receptor . ACS Med Chem Lett . 2012 ; 3 : 715 – 720 . doi: 10.1021/ml300097g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang TC, Qiao JX, Clark CG, Jua J, Price LA, Wu Q, Chang M, Zheng J, Huang CS, Everlof G, Schumacher WA, Wong PC, Seiffert DA, Stewart AB, Bostwick JS, Crain EJ, Watson CA, Rehfuss R, Wexler RR, Lam S, PY Discovery of diarylurea P2Y 1 antagonists with improved aqueous solubility. Bioorg Med Chem Lett. 2013;23:3239–3243. doi: 10.1016/j.bmcl.2013.03.125. [DOI] [PubMed] [Google Scholar]