

Abstract
AT2Rs [AngII (angiotensin II) type 2 receptors] contribute to the cardioprotective effects of angiotensin II receptor blockers, possibly via kinins acting on the B1R (B1 receptor) and B2R (B2 receptor). Recent studies have shown that a lack of B2R up-regulates B1R and AT2R; however, the pathophysiological relevance of such an event remains unclear. We hypothesized that up-regulation of AT2R and B1R compensates for the loss of B2R. Blockade of AT2R and/or B1R worsens cardiac remodelling and dysfunction following MI (myocardial infarction) in B2R−/− (B2-receptor-knockout mice). B2R−/− mice and WT (wild-type) controls were subjected to sham MI or MI and treated for 4 weeks with (i) vehicle, (ii) a B1R-ant (B1R antagonist; 300 μg/kg of body weight per day), (iii) an AT2R-ant [AT2 receptor antagonist (PD123319); 20 mg/kg of body weight per day], or (iv) B1R-ant + AT2R-ant. B2R−/− mice had a greater MCSA (myocyte cross-sectional area) and ICF (interstitial collagen fraction) at baseline and after MI compared with WT controls. Cardiac function and increase in macrophage infiltration, TGFβ1 (transforming growth factor β1) expression and ERK1/2 (extracellular-signal-regulated kinase 1/2) phosphorylation post-MI were similar in both strains. Blockade of AT2R or B1R worsened cardiac remodelling, hypertrophy and dysfunction associated with increased inflammation and ERK1/2 phosphorylation and decreased NO excretion in B2R−/− mice, which were exacerbated by dual blockade of B1R and AT2R. No such effects were seen in WT mice. Our results suggest that, in the absence of B2R, both B1R and AT2R play important compensatory roles in preventing deterioration of cardiac function and remodelling post-MI possibly via suppression of inflammation, TGFβ1 and ERK1/2 signalling.
Keywords: angiotensin II type 2 receptor (AT2 receptor), B1 kinin receptor, B2 kinin receptor, heart failure, inflammation, myocardial infarction, renin–angiotensin system
INTRODUCTION
CHF (chronic heart failure) due to myocardial ischaemia is a major cause of death in the U.S.A. and worldwide. Activation of the RAS (renin–angiotensin system) and release of AngII (angiotensin II) acting on the AT1Rs (AngII type 1 receptors) both play a crucial role in the pathophysiology of CHF. Blockade of AT1R with ARBs (angiotensin receptor blockers) has been shown to improve LV (left ventricular) function, regress LV remodelling and prolong survival in patients with CHF [1,2]. We and others [3–5] have demonstrated that the cardioprotective effects of ARBs are mediated in part via activation of AT2Rs (AngII type 2 receptors) due to increased circulating AngII during AT1R blockade. AngII in turn acts on AT2R and affords cardioprotection via a kinin-dependent pathway [6,7].
Two types of kinin receptors have been identified: B1R (B1 receptor) and B2R. Most known cardiovascular actions of kinins are generally attributed to the constitutively expressed B2R, whereas the role of B1R is not well defined. Although B1R is weakly expressed under physiological conditions, it is markedly increased in response to pathological stimuli such as myocardial ischaemia [8,9]. We have reported previously that both B1R and B2R contribute to the cardioprotective effects of ARBs [3,8,10]. Up-regulation of the B1R is also demonstrated when the B2R is absent [11], which may compensate for the loss of B2R. Indeed, we have shown that blockade or deletion of B2R alone does not have a significant impact on cardiac morphology and function either under basal conditions or in animals with MI (myocardial infarction); however, blocking both B2R and B1R exacerbates cardiac remodelling and dysfunction post-MI [8]. Kakoki et al. [12] have also shown that renal ischaemia/reperfusion injury becomes more severe in mice lacking both B1R and B2R compared with a lack of B2R alone. Taken together, these studies suggest that up-regulation of B1R during deletion or blockade of B2R may represent a compensatory mechanism.
A lack of the B2R also reportedly up-regulates the AT2R. Shariat-Mader et al. [13] showed that, in B2R−/− mice, AT2R expression increased 8-fold in association with increased B1R. However, the functional or pathophysiological relevance of B1R and AT2R up-regulation in B2R−/− mice is not well understood. The present study is designed to test the hypothesis that, in the absence of B2R, AT2R and B1R are up-regulated, which compensates for the loss of B2R. Using B2R−/− mice subjected to MI, we studied whether blockade of AT2R and/or B1R in the absence of B2R increases inflammation and aggravates cardiac hypertrophy, fibrosis, dilatation and dysfunction. The roles of TGFβ1 (transforming growth factor β1) and ERK (extracellular-signal-regulated kinase)1/2 signalling were also investigated.
MATERIALS AND METHODS
Animals
Male B2R−/− mice (B6; 129S7-bdkrb2tm1Jfh/J; stock number 002641) and WT (wild-type) controls (B6129SF2/J; stock number 101045) were purchased from Jackson Laboratories. Both strains were on a mixed genetic background of C57BL/6J and 129S7/SvEv. Animals were housed in an air-conditioned room with a 12 h light/12 h dark cycle, received standard chow and drank tap water. This study was approved by the IACUC (Institutional Animal Care and Use Committee) of the Henry Ford Health System. All studies were conducted in accordance with the NIH (National Institutes of Health) Guidelines for the Care and Use of Laboratory Animals.
Induction of MI and experimental protocols
Mice 8–10 weeks of age were anaesthetized with sodium pentobarbital (50 mg/kg of body weight, intraperitoneal). An analgesic, buprenorphine (2.5 mg/kg of body weight, subcutaneous), was given 1 h before the operation and 6 h afterward. MI was induced by ligating the coronary artery [3,14]. Briefly, a left thoracotomy was performed via the fourth intercostal space, the heart exposed and the pericardium opened. The left anterior descending coronary artery was ligated with an 8-0 silk suture. Acute myocardial ischaemia was deemed successful when the anterior wall of the LV became cyanotic and the ECG showed obvious ST segment elevation. The lungs were then inflated by increasing positive end-expiratory pressure and the thoracotomy site closed in layers. Mice were kept on a heating pad until they recovered. After the MI or sham MI, each strain was divided into (i) sham MI (n=13 WT, n=12 B2R−/−); (ii) MI+vehicle (saline) (n=9 WT, n=9 B2R−/−); (iii) MI+AT2 receptor antagonist (AT2R-ant, PD123319; 20 mg/kg of body weight per day) (n=10 WT, n=9 B2R−/−); (iv) MI+B1 receptor antagonist (B1R-ant, R892, 300 μg/kg of body weight per day) (n=7 WT, n=7 B2R−/−); and (v) MI+B1R-ant+AT2R-ant (n=8 WT, n=9 B2R−/−). Doses of the antagonists were based on previously published work of ourselves and others [10,15–18]. All drugs were infused via an osmotic mini-pump intraperitoneally and started on the same day as coronary ligation and continued for 4 weeks.
Blood pressure and cardiac function and remodelling
SBP (systolic blood pressure) and HR (heart rate) were measured in conscious mice using a non-invasive computerized tail-cuff system (BP-2000; Visitech) [19]. LV dimension and function were evaluated in awake mice using a Doppler echocardiograph equipped with a 15-MHz linear transducer (Acuson c256) as described previously [19,20].
Histopathological study
At 4 weeks after MI, the heart was stopped at diastole by intraventricular injection of 15% KCl (50 μl). The heart, lungs and liver were weighed, corrected by BW (body weight) and expressed as mg/10 g of BW to assess LV hypertrophy and lung congestion. The LV was sectioned transversely into three slices for assessment of infarct size, MCSA (myocyte cross-sectional area) and ICF (interstitial collagen fraction) as previously described [19]. Images were taken at ×400 magnification and analysed with a MicroSuite computerized image analysis system (Olympus).
Immunohistochemical staining for macrophages
Frozen sections of the left ventricle (6 μm) were stained with a rat anti-(mouse CD68) monoclonal antibody (1:200 dilution; AbD Serotec), a marker for macrophages, as described previously [19,21]. CD68-positive cells (reddish/brown staining) were recognized as macrophages and are expressed as number of cells per mm2 of myocardium.
ERK1/2 phosphorylation and TGFβ1 protein expression
LV tissue was homogenized in lysis buffer with protease inhibitors. pERK1/2 (phosphor-ERK1/2) and TGFβ1 protein expression was analysed by Western blot [19,22]. pERK1/2 was normalized by total ERK1/2 and TGFβ1 proteins by GAPDH (glyceraldehyde-3-phosphate dehydrogenase). Results were expressed as the ratio of the density of specific bands to the corresponding internal controls.
Urinary NO and creatinine
Urine was collected from the bladder under anaesthesia 4 weeks after MI to measure NOx (combined nitrate + nitrite), which are stable end products of NO [23]. NOx was measured with a colorimetric assay kit (Cayman Chemical). Urinary creatinine was measured using a commercial kit (Sigma). Urinary NOx excretion was corrected by creatinine and expressed as the ratio of NOx to creatinine.
Real-time PCR of B1R, B1R2 and AT2R mRNA expression
Additional sham-operated mice (six WT and six B2R−/−) and mice with MI (seven WT and five B2R−/−) were used to determine B1R, B1R2 and AT2R mRNA expression. Total RNA was isolated from mouse hearts using an RNeasy fibrous tissue kit (Qiagen). TaqMan gene expression assays from Applied Biosystems were used to assess expression of B1R (Mm04207315), B2R (Mm00437788), AT2R (Mm01341373) and GAPDH (Mm99999915). Real-time PCR conditions were: one cycle at 95°C for 10 min followed by 40 cycles at 95°C for 15 s and at 60°C for 1 min. Gene expression was quantified and analysed using the comparative Ct (cycle threshold value) method as described in the Applied Biosystems user bulletin. All values were corrected to GAPDH as an internal control and the value in sham-operated WT mice was set to a value of 1.0. Changes in mRNA expression after MI were expressed as 2−ΔΔCt.
Data analysis
All results are expressed as means ± S.E.M. A two-sample Student’s t test was used to compare differences between strains or between treatments within strains. When multiple comparisons were performed, Hochberg’s step-up procedure was used to adjust P values. The family-wise type I error rate was set at 0.05.
RESULTS
SBP, HR, BW and tissue weight and infarct size
SBP, HR and BW were similar among groups with or without MI. In the sham-operated mice, LV and total heart weight were heavier in B2R−/− than in WT and MI increased heart weight the same in both strains. Lung weight was similar among groups with or without MI in either strain. However, liver weight tended to be higher in all B2R−/− groups, although the increase reached statistical significance only in mice treated with B1R-ant with or without AT2R-ant. Infarct size was similar between strains with or without treatment (Table 1).
Table 1. Effect of blockade of AT2R and/or B1R on SBP, HR, tissue weight and infarct size in WT and B2R−/− mice post-MI.
*P<0.05, **P<0.01, compared with sham MI within strain; †P<0.05 and ††P<0.01 compared with WT; #P<0.05 compared with vehicle within strain. LVW, LV weight corrected by body weight; THW, total heart weight corrected by body weight; lung and liver weight were corrected by body weight; IS, infarct size.
| WT |
B2R−/− |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MI |
MI |
|||||||||
| Parameter | Sham MI (n=13) |
Vehicle (n=9) |
AT2R-ant (n=10) |
B1R-ant (n=7) |
AT2Rant+ B1R-ant (n=8) |
Sham MI (n=12) |
Vehicle (n=9) |
AT2R-ant (n=9) |
B1R-ant (n=7) |
AT2Rant+ B1R-ant (n=9) |
| BW (g) | 31±2 | 31±2 | 31±1 | 31±1 | 31±1 | 33±11 | 32±1 | 31±1 | 35±1 | 33±2 |
| SBP (mmHg) | 111±4 | 106±5 | 110±4 | 108±5 | 106±4 | 123±5 | 120±7 | 111±3 | 119±4 | 117±5 |
| HR (beats/min) | 673±12 | 642±25 | 670±14 | 646±20 | 673±23 | 654±18 | 618±19 | 601±29 | 661±15 | 626±31 |
| LVW (mg/10 g) | 31±1 | 41±1** | 38±2 | 39±2 | 38±2 | 35±2† | 42±2** | 41±3 | 44±3 | 43±3 |
| THW (mg/10 g) | 42±2 | 53±2** | 51±3 | 52±2 | 51±3 | 47±2† | 58±4* | 56±4 | 60±4 | 58±4 |
| Lungs (mg/10 g) | 60±4 | 61±4 | 70±6 | 60±7 | 61±5 | 60±2 | 67±7 | 71±7 | 68±4 | 70±4 |
| Liver (mg/10 g) | 392±18 | 381±14 | 409±11 | 364±22 | 384±15 | 443±18 | 420±15 | 454±25 | 469±10††# | 463±14††# |
| IS (%) | – | 34±3 | 32±3 | 34±3 | 32±2 | – | 37±3 | 38±3 | 36±4 | 36±3 |
Cardiac function and remodelling
Knockout of B2R had no effect on basal LV chamber size and function. MI increased LVDd (LV diastolic dimension) and decreased EF (ejection fraction) similarly in both strains. Blocking AT2R or B1R further increased LVDd and decreased EF in B2R−/− mice, and these changes were exaggerated when both AT2R and B1R were blocked. None of these effects were seen in the WT mice (Figure 1).
Figure 1. Effect of AT2R and/or B1R blockade on LV EF and LVDd in WT and kinin B2R−/− mice post-MI.

(A) Representative two-dimensional M-mode ECGs of conscious WT and B2R−/− mice with sham MI or MI treated with vehicle, the AT2R antagonist PD123319 (AT2R-ant), the kinin B1 receptor antagonist R892 (B1R-ant) or AT2R-ant plus B1R-ant for 4 weeks. (B) Quantitative analysis of LVDd (top) and EF (bottom). ***P < 0.001 compared with sham within strain; †P < 0.05 and ††P < 0.01 compared with vehicle within strain; #P < 0.05 compared with AT2R-ant or B1R-ant alone within strain.
Myocyte hypertrophy and cardiac interstitial fibrosis
Under basal conditions (sham MI), B2R−/− mice exhibited increased MCSA and ICF compared with WT. MI increased MCSA and ICF in both strains and this increase was greater in B2R−/− mice. AT2R-ant, B1R-ant or the combination of these two had no significant effect on MCSA and ICF in WT. However, in B2R−/− mice both AT2R-ant and B1R-ant alone increased MCSA and ICF further and these responses were exacerbated by dual blockade of AT2R and B1R (Figure 2).
Figure 2. Effect of AT2R and/or B1R blockade on MCSA and ICF in WT and B2R−/− mice post-MI.

(A) Representative images showing MCSA (delineated by the interstitial space) and interstitial collagen deposition (green staining) in WT and B2R−/− given vehicle, AT2R-ant, B1R-ant or AT2R-ant + B1R-ant. (B) Quantitative analysis of MCSA (top) and ICF (bottom). ***P < 0.001 compared with sham within strain; †P < 0.05 and †††P < 0.001 compared with vehicle within strain; #P < 0.05 compared with AT2R-ant or B1R-ant alone within strain.
Macrophage infiltration, TGFβ1 protein expression and ERK1/2 phosphorylation in the heart
A few macrophages were seen infiltrating the myocardium in both WT and B2R−/− mice with sham MI. Macrophage infiltration was increased after MI and no strain difference was noticed. Blocking either AT2R or B1R increased the number of infiltrated macrophages only in B2R−/− mice. Dual blockade of AT2R and B1R markedly elevated macrophage infiltration, which was also only seen in B2R−/− (Figure 3). Cardiac TGFβ1 protein expression did not differ between sham-operated WT and B2R−/− animals and rose similarly in response to MI in both strains. AT2R-ant or B1R-ant significantly enhanced TGF-β1 protein expression in B2R−/− mice but not in WT mice, whereas dual blockade of AT2R and B1R markedly enhanced TGF-β1 expression in both strains, but the response was greater in B2R−/− mice (Figure 4). ERK1/2 phosphorylation was similar between sham-operated groups and increased similarly after MI in both strains. AT2R-ant or B1R-ant significantly increased ERK1/2 phosphorylation further only in B2R−/− mice, while blocking both AT2R and B1R enhanced this effect in B2R−/− but not in WT mice (Figure 5).
Figure 3. Effect of AT2R and/or B1R blockade on macrophage infiltration in the LV myocardium in WT and B2R−/− post-MI.

(A) Representative images showing infiltrating macrophages (reddish-brown stained) in WT and B2R−/− given vehicle, AT2R-ant, B1R-ant or AT2R-ant + B1R-ant. (B) Quantification of the number of macrophages infiltrating the LV myocardium. *P < 0.05 compared with sham within strain; †P < 0.05 and ††P < 0.01 compared with vehicle within strain; #P < 0.05 compared with AT2R-ant or B1R-ant alone within strain.
Figure 4. Effect of AT2R and/or B1R blockade on TGFβ1 protein expression in WT and B2R−/− mice post-MI.
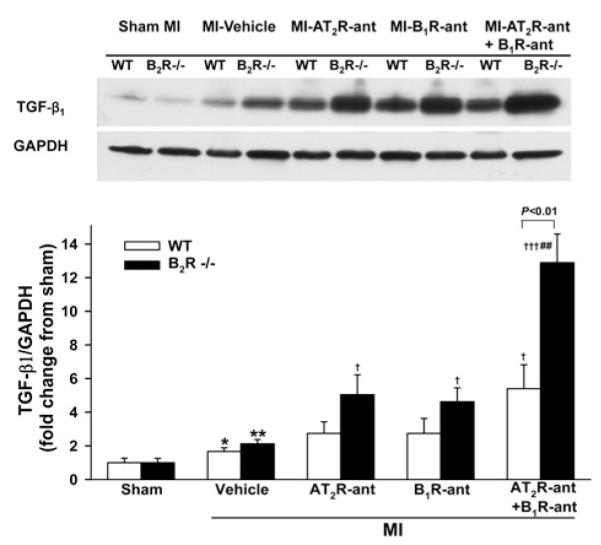
(A) Representative Western blots of TGFβ1 and GAPDH. (B) Semi-quantitative analysis of TGFβ1 protein corrected by GAPDH and expressed as fold change relative to sham controls within strain. *P < 0.05 and **P < 0.01 compared with sham within strain; †P < 0.05 and †††P < 0.001 compared with vehicle within strain; ##P < 0.01 compared with AT2R-ant or B1R-ant alone within strain.
Figure 5. Effect of AT2R and/or B1R blockade on ERK1/2 phosphorylation in WT and B2R−/− mice post-MI.
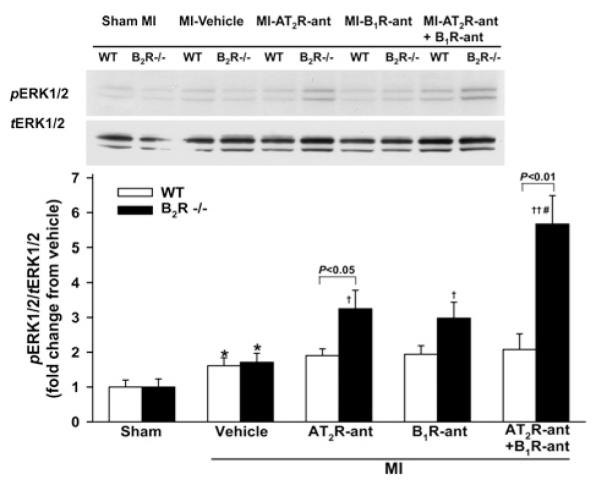
(A) Representative Western blots of pERK1/2 and total ERK1/2 (tERK1/2). (B) Semi-quantitative analysis of pERK1/2 corrected by tERK1/2 and expressed as fold change relative to sham within strain. *P<0.05 compared with sham within strain; †P < 0.05 and ††P < 0.01 compared with vehicle within strain; #P < 0.05 compared with AT2R-ant or B1R-ant alone within strain.
Urinary NOx
Urinary NOx concentration was not different in sham-operated or vehicle-treated groups between strains. AT2R-ant and B1R-ant significantly decreased urinary NOx only in B2R−/− mice. Dual inhibition of AT2R and B1R only lowered urinary NOx further in B2R−/− mice (Figure 6).
Figure 6. Effect of AT2R and/or B1R blockade on urinary NO excretion in WT and B2R−/− mice post-MI.
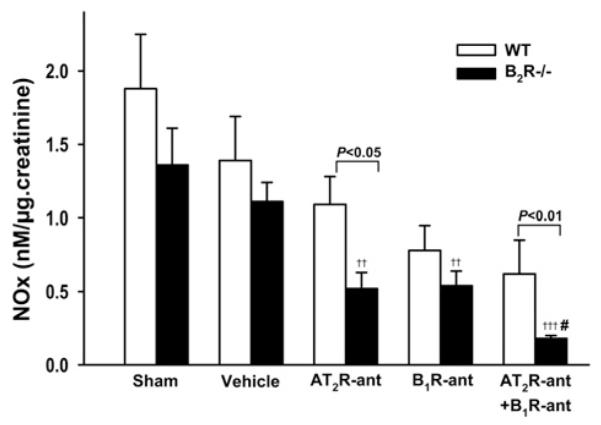
††P < 0.01 and †††P < 0.001 compared with vehicle within strain; #P < 0.05 compared with AT2R-ant or B1R-ant alone within strain.
Cardiac B1R, B2R and AT2R mRNA expression
Basal B1R and AT2R mRNA expression were significantly higher in B2R−/− mice compared with WT. B2R mRNA was undetectable in B2R−/− mice. MI increased B1R, B2R and AT2R mRNA expression in WT mice, whereas in B2R−/− mice expression of B1R and AT2R tended to be higher in response to MI, although these changes did not reach statistical significance (Figure 7).
Figure 7. Effect of B2R deletion on AT2R and B1R mRNA expression evaluated by real-time RT (reverse transcription)–PCR and normalized to GAPDH.
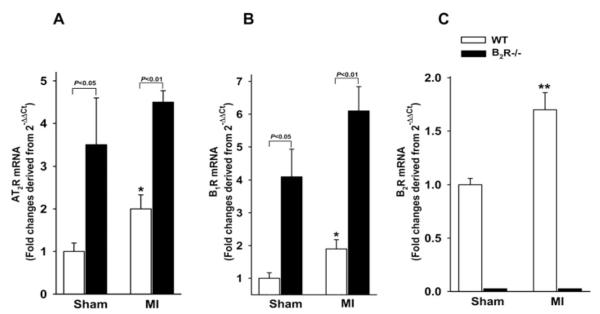
Results are presented as the fold change relative to sham WT controls. *P < 0.05 and **P < 0.001 compared with sham within strain.
DISCUSSION
In the present study, we demonstrated that deletion of B2R resulted in up-regulation of B1R and AT2R without altering SBP or cardiac function, although myocyte size and collagen deposition were slightly but significantly increased under basal conditions. Likewise B2R deletion had no significant impact on MI-induced cardiac hypertrophy, fibrosis or dysfunction compared with WT mice. These findings may suggest that up-regulation of B1R and AT2R in B2R−/− mice may have compensated for the loss of B2R, maintaining cardiac and haemodynamic homoeostasis and preventing deterioration of cardiac dysfunction and remodelling post-MI. This hypothesis is supported by our observation that blocking either AT2R or B1R exacerbated cardiac hypertrophy and dysfunction in B2R−/− mice, which was worsened by concomitant blockade of AT2R and B1R. Moreover, these detrimental cardiac effects were associated with increased macrophage infiltration, cardiac TGFβ1 protein expression and ERK1/2 phosphorylation as well as decreased urinary NOx excretion. Together these findings suggest that up-regulation of AT2R and B1R plays an important cardioprotective role in compensating for the loss of B2R, particularly in mice with myocardial ischaemia. Suppressing this compensation promotes exacerbation of MI-induced cardiac hypertrophy, fibrosis and dysfunction, possibly via an enhanced inflammatory response and ERK1/2 activation.
The effects of B2R deletion on regulation of SBP and cardiac structure and function remain in question. We and others reported that lack of B2R (B2R−/− mice on a C57BL/6J background) did not alter either SBP or cardiac phenotype under basal conditions, nor did it affect cardiac remodelling and function post-MI or in response to DOCA (deoxycorticosterone acetate)-salt-induced hypertension, although the therapeutic effects of ACEi (angiotensin-converting enzyme inhibitor) and ARB on cardiac remodelling and dysfunction were diminished [3,24,25]. In contrast, others have reported that lack of B2R (B2R−/− mice on a 129/J background) increased SBP and caused cardiac hypertrophy and dilated cardiomyopathy by 40 weeks of age [11,26,27]. Thus the discrepancy observed in B2R−/− mice might be attributed to differences in genetic background, age and/or the use of WT controls. In the present study, we compared B2R−/− mice on a mixed background of C57BL/6J and 129S7/SvEv with B6129SF2/J mice (WT) and found that the B2R−/− tended to have a higher SBP associated with a greater heart weight, MCSA and ICF under basal conditions, suggesting that B2R may be involved in regulation of BP and cardiac homoeostasis, which may be largely dependent on their genetic background.
Recent studies suggested that deletion of B2R up-regulates B1R and/or AT2R [11,13,28,29], which is confirmed by our current observation that both B1R and AT2R mRNA expression were increased in B2R−/− mice. However, the functional significance of B1R and/or AT2R up-regulation is not fully understood. In our study, a lack of B2R did not have a significant impact on cardiac remodelling and dysfunction post-MI, which may be partially due to up-regulation of B1R and AT2R that may assume some effects of B2R. However, blocking B1R and/or AT2R in B2R−/− mice worsened LV chamber dilatation and cardiac hypertrophy, fibrosis and dysfunction, supporting our hypothesis that up-regulation of B1 and AT2 may compensate for loss of B2R.
Whether the role of B1R expression in the cardiovascular and renal system is beneficial or harmful remains inconclusive. We and others have shown that ACEi and ARB improved cardiac function and remodelling post-MI associated with up-regulation of B1R expression and these effects were diminished in B1R−/− mice or in rats treated with a B1R antagonist, although deletion or inhibition of B1R did not have a significant impact on cardiac remodelling and dysfunction post-MI [30,31]. These findings may suggest that the cardioprotective actions of ACEi or ARB require B1R. Previously, Schulze-Topphoff et al. [32] reported that in a mouse model of EAE (experimental autoimmune encephalomyelitis), activation of B1R with the agonist, Sar-[d-Phe]des-Arg9-BK, reduced T-lymphocyte infiltration into the brain and improved the symptoms of EAE, whereas inhibition or deletion of B1R enhanced the immune response and worsened severity of the disease [32]. Merino et al. [33] reported that B1R deficiency aggravated atherosclerosis and promoted aneurysm formation in ApoE−/− (apolipoprotein E-knockout) mice [33]. All together, these reports support the concept that B1R plays a cardioprotective, anti-inflammatory and anti-atherogenic role. On the other hand, contradictory results have also been reported, showing that blocking or deleting B1R protects the heart against injury and myocyte death [34,35]. At present, we have no good explanation for these discrepancies.
It has been suggested that activation of ERK1/2 promotes cardiac inflammation, hypertrophy and fibrosis [36]. Kinins acting on B1R and/or B2R reduced inflammation by inhibiting MAPK (mitogen-activated protein kinase) activation, including ERK1/2 [37,38], whereas blocking B1R and/or B2R increases ERK1/2 phosphorylation and promotes cardiac inflammation and remodelling. Activation of AT2R also reportedly attenuated TGFβ1-induced ERK activation [39]. In the present study, we found that dual blockade of B1R and AT2R enhanced ERK1/2 phosphorylation and inflammatory responses as indicated by increased macrophage infiltration and TGFβ1 protein expression, suggesting that enhanced ERK1/2 phosphorylation and inflammation may contribute to the deterioration of cardiac remodelling and dysfunction observed in B2R−/− mice treated with both B1R and AT2R antagonists.
Studies have suggested that activation of eNOS (endothelial NO synthase) and enhanced NO production may be important mediators involved in signalling mechanisms responsible for B1R-, B2R- and/or AT2R-induced cardioprotection [40,41]. We previously reported that overexpression and activation of AT2R in cultured ECs (endothelial cells) stimulated release of NO and this stimulation was diminished by B2R blockade [42], suggesting that the kinin/NO pathway plays an important role in mediating the cardioprotective effects of AT2R. It has been suggested that NO inhibits TGFβ1, thus suppressing ERK1/2 phosphorylation, reducing proliferation, inflammation, oxidative stress and improving cardiac remodelling and dysfunction [43–45]. In the present study, we found that the levels of urinary NO in B2R−/− mice were significantly lower than in WT post-MI when treated with AT2R-ant and/or B1R-ant. It is possible that reduced NO production due to blockade of AT2R and B1R in B2R−/− enhances TGFβ1 expression and ERK1/2 signalling, which promotes inflammation and cardiac hypertrophy, fibrosis and dysfunction post-MI.
Limitations of the study
The present study has demonstrated that up-regulation of AT2R and B1R in B2R−/− mice may compensate for loss of B2R and play an important role in the cardioprotective effect of ARB. Our end points focused on functional and pathophysiological and histological remodelling of the heart; thus the detailed molecular and cellular mechanisms and signalling pathways that lead to NO synthase phosphorylation and TGFβ1 and ERK activation were not emphasized, which will be our future focus and tested in both in vivo and in vitro studies.
In summary, we have shown that deletion of B2R unregulated cardiac AT2R and B1R expression, which may compensate for loss of the beneficial effects of B2R on remodelling and dysfunction in mice with MI, since blocking both AT2R and B1R worsened cardiac remodelling, hypertrophy and dysfunction in association with enhanced inflammatory responses and ERK1/2 phosphorylation and decreased urinary NO excretion. Thus AT2R, B1R and B2R may all play an important role in protecting the heart from MI-induced damage by increasing NO production and suppressing inflammatory cell infiltration, TGFβ1 protein expression and ERK1/2 phosphorylation.
CLINICAL PERSPECTIVES.
CVD is the leading cause of death in the U.S.A. and world-wide. Activation of the RAS plays a crucial role in the pathophysiology of CVD and up until now the standard therapy has been inhibition of the RAS using ACEi and/or ARB.
We and others have demonstrated that activation of B1R, B2R and/or AT2R plays an important role in the cardioprotective effects of ACEi and ARBs. The present study helps to clarify the interaction between B2R, B1R and AT2R and the mechanisms by which these receptors offer cardioprotection.
From a clinical perspective, understanding these receptor-mediated mechanisms may help us to develop new therapeutic strategies to treat CVD, such as targeted activation of AT2R, B2R and/or B1R, or combining them with an ACEi or ARB.
Acknowledgments
FUNDING This work was supported the National Institutes of Health [grant number HL-28982 (Project II to X.-P.Y.)] and the Henry Ford Hospital Institutional Fund (grant to X.-P.Y.).
Abbreviations
- ACEi
angiotensin-converting enzyme inhibitor
- AngII
angiotensin II
- ARB
angiotensin receptor blocker
- AT1R
AngII type 1 receptor
- AT2R
AngII type 2 receptor
- AT2R-ant
AT2R antagonist
- B1R
B1 receptor
- B1R-ant
B1R antagonist
- B2R
B2 receptor
- BW
body weight
- CHF
chronic heart failure
- CVD
cardiovascular disease
- EAE
experimental autoimmune encephalomyelitis
- EF
ejection fraction
- ERK
extracellular-signal-regulated kinase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HR
heart rate
- ICF
interstitial collagen fraction
- LV
left ventricular
- LVDd
LV diastolic dimension
- MCSA
myocyte cross-sectional area
- MI
myocardial infarction
- NOx
combined nitrate + nitrite
- pERK1/2
phospho-ERK1/2
- TGFβ1
transforming growth factor β1
- RAS
renin–angiotensin system
- SBP
systolic blood pressure
- WT
wild-type
Footnotes
AUTHOR CONTRIBUTION Jiang Xu performed major parts of the experiments, including surgeries on animals, echocardiography, histological staining, data analysis and preparation of the paper; Oscar A. Carretero participated in the experimental design, data interpretation and critical discussion of the paper; Liping Zhu performed mRNA experiments and data analysis; Edward Shesely was in charge of breeding and genotyping the mice; Nour-Eddine Rhaleb participated in the experimental design, data interpretation and discussion of the paper; Xiangguo Dai performed histological studies, including cutting and staining samples; Luchen Wang assisted Jiang Xu with the animal protocols and performed the Western blots; James Yang performed the statistical analyses; Xiao-Ping Yang oversaw the design and performance of the experiments, performed echocardiography, and edited and approved the final format of the paper.
REFERENCES
- 1.Cohn JN, Tognoni G. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N. Engl. J. Med. 2001;345:1667–1675. doi: 10.1056/NEJMoa010713. [DOI] [PubMed] [Google Scholar]
- 2.Yusuf S, Teo K, Anderson C, Pogue J, Dyal L, Copland I, Schumacher H, Dagenais G, Sleight P. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: a randomised controlled trial. Lancet. 2008;372:1174–1183. doi: 10.1016/S0140-6736(08)61242-8. [DOI] [PubMed] [Google Scholar]
- 3.Yang XP, Liu YH, Mehta D, Cavasin MA, Shesely EG, Xu J, Liu F, Carretero OA. Diminished cardioprotective response to inhibition of angiotensin-converting enzyme and angiotensin II type 1 receptor in B2 kinin receptor gene knockout mice. Circ. Res. 2001;88:1072–1079. doi: 10.1161/hh1001.090759. [DOI] [PubMed] [Google Scholar]
- 4.Xu J, Carretero OA, Liu YH, Shesely EG, Yang F, Kapke A, Yang XP. Role of AT2 receptors in the cardioprotective effect of AT1 antagonists in mice. Hypertension. 2002;40:244–250. doi: 10.1161/01.hyp.0000029095.23198.ad. [DOI] [PubMed] [Google Scholar]
- 5.Jones ES, Black MJ, Widdop RE. Angiotensin AT2 receptor contributes to cardiovascular remodeling of aged rats during chronic AT1 receptor blockade. J. Mol. Cell. Cardiol. 2004;37:1023–1030. doi: 10.1016/j.yjmcc.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 6.Yayama K, Okamoto H. Angiotensin II-induced vasodilation via type 2 receptor: role of bradykinin and nitric oxide. Int. Immunopharmacol. 2008;8:312–318. doi: 10.1016/j.intimp.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 7.Tsutsumi Y, Matsubara H, Masaki H, Kurihara H, Murasawa S, Takai S, Miyazaki M, Nozawa Y, Ozono R, Nakagawa K, et al. Angiotensin II type 2 receptor overexpression activates the vascular kinin system and causes vasodilation. J. Clin. Invest. 1999;104:925–935. doi: 10.1172/JCI7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu J, Carretero OA, Sun Y, Shesely EG, Rhaleb NE, Liu YH, Liao TD, Yang JJ, Bader M, Yang XP. Role of the B1 kinin receptor in the regulation of cardiac function and remodeling after myocardial infarction. Hypertension. 2005;45:747–753. doi: 10.1161/01.HYP.0000153322.04859.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tschope C, Heringer-Walther S, Koch M, Spillmann F, Wendorf M, Leitner E, Schultheiss HP, Walther T. Upregulation of bradykinin B1-receptor expression after myocardial infarction. Br. J. Pharmacol. 2000;129:1537–1538. doi: 10.1038/sj.bjp.0703239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu YH, Yang XP, Shesely EG, Sankey SS, Carretero OA. Role of angiotensin II type 2 receptors and kinins in the cardioprotective effect of angiotensin II type 1 receptor antagonists in rats with heart failure. J. Am. Coll. Cardiol. 2004;43:1473–1480. doi: 10.1016/j.jacc.2003.11.044. [DOI] [PubMed] [Google Scholar]
- 11.Duka I, Kintsurashvili E, Gavras I, Johns C, Bresnahan M, Gavras H. Vasoactive potential of the B1 bradykinin receptor in normotension and hypertension. Circ. Res. 2001;88:275–281. doi: 10.1161/01.res.88.3.275. [DOI] [PubMed] [Google Scholar]
- 12.Kakoki M, Sullivan KA, Backus C, Hayes JM, Oh SS, Hua K, Gasim AM, Tomita H, Grant R, Nossov SB, et al. Lack of both bradykinin B1 and B2 receptors enhances nephropathy, neuropathy, and bone mineral loss in Akita diabetic mice. Proc. Natl. Acad. Sci. U.S.A. 2010;107:10190–10195. doi: 10.1073/pnas.1005144107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shariat-Madar Z, Mahdi F, Warnock M, Homeister JW, Srikanth S, Krijanovski Y, Murphey LJ, Jaffa AA, Schmaier AH. Bradykinin B2 receptor knockout mice are protected from thrombosis by increased nitric oxide and prostacyclin. Blood. 2006;108:192–199. doi: 10.1182/blood-2006-01-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang F, Liu YH, Yang XP, Xu J, Kapke A, Carretero OA. Myocardial infarction and cardiac remodelling in mice. Exp. Physiol. 2002;87:547–555. doi: 10.1113/eph8702385. [DOI] [PubMed] [Google Scholar]
- 15.Liu YH, Yang XP, Sharov VG, Nass O, Sabbah HN, Peterson E, Carretero OA. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure. Role of kinins and angiotensin II type 2 receptors. J. Clin. Invest. 1997;99:1926–1935. doi: 10.1172/JCI119360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akishita M, Horiuchi M, Yamada H, Zhang L, Shirakami G, Tamura K, Ouchi Y, Dzau VJ. Inflammation influences vascular remodeling through AT2 receptor expression and signaling. Physiol. Genomics. 2000;2:13–20. doi: 10.1152/physiolgenomics.2000.2.1.13. [DOI] [PubMed] [Google Scholar]
- 17.Duka I, Duka A, Kintsurashvili E, Johns C, Gavras I, Gavras H. Mechanisms mediating the vasoactive effects of the B1 receptors of bradykinin. Hypertension. 2003;42:1021–1025. doi: 10.1161/01.HYP.0000097550.98865.35. [DOI] [PubMed] [Google Scholar]
- 18.Gobeil F, Jr, Charland S, Filteau C, Perron SI, Neugebauer W, Regoli D. Kinin B1 receptor antagonists containing α-methyl-l-phenylalanine: in vitro and in vivo antagonistic activities. Hypertension. 1999;33:823–829. doi: 10.1161/01.hyp.33.3.823. [DOI] [PubMed] [Google Scholar]
- 19.Xu J, Carretero OA, Liao TD, Peng H, Shesely EG, Xu J, Liu TS, Yang JJ, Reudelhuber TL, Yang XP. Local angiotensin II aggravates cardiac remodeling in hypertension. Am. J. Physiol. Heart. Circ. Physiol. 2010;299:H1328–H1338. doi: 10.1152/ajpheart.00538.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang XP, Liu YH, Rhaleb NE, Kurihara N, Kim HE, Carretero OA. Echocardiographic assessment of cardiac function in conscious and anesthetized mice. Am. J. Physiol. Heart. Circ. Physiol. 1999;277:H1967–H1974. doi: 10.1152/ajpheart.1999.277.5.H1967. [DOI] [PubMed] [Google Scholar]
- 21.Xu J, Carretero OA, Lin CX, Cavasin MA, Shesely EG, Yang JJ, Reudelhuber TL, Yang XP. Role of cardiac overexpression of ANG II in the regulation of cardiac function and remodeling postmyocardial infarction. Am. J. Physiol. Heart. Circ. Physiol. 2007;293:H1900–H1907. doi: 10.1152/ajpheart.00379.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhan E, Keimig T, Xu J, Peterson E, Ding J, Wang F, Yang XP. Dose-dependent cardiac effect of oestrogen replacement in mice post-myocardial infarction. Exp. Physiol. 2008;93:982–993. doi: 10.1113/expphysiol.2008.042788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N. Engl. J. Med. 1993;329:2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 24.Xu J, Carretero OA, Liu YH, Yang F, Shesely EG, Oja-Tebbe N, Yang XP. Dual inhibition of ACE and NEP provides greater cardioprotection in mice with heart failure. J. Card. Failure. 2004;10:83–89. [PubMed] [Google Scholar]
- 25.Trabold F, Pons S, Hagege AA, Bloch-Faure M, Alhenc-Gelas F, Giudicelli JF, Richer-Giudicelli C, Meneton P. Cardiovascular phenotypes of kinin B2 receptor- and tissue kallikrein-deficient mice. Hypertension. 2002;40:90–95. doi: 10.1161/01.hyp.0000021747.43346.95. [DOI] [PubMed] [Google Scholar]
- 26.Emanueli C, Maestri R, Corradi D, Marchione R, Minasi A, Tozzi MG, Salis MB, Straino S, Capogrossi MC, Olivetti G, et al. Dilated and failing cardiomyopathy in bradykinin B2 receptor knockout mice. Circulation. 1999;100:2359–2365. doi: 10.1161/01.cir.100.23.2359. [DOI] [PubMed] [Google Scholar]
- 27.Maestri R, Milia AF, Salis MB, Graiani G, Lagrasta C, Monica M, Corradi D, Emanueli C, Madeddu P. Cardiac hypertrophy and microvascular deficit in kinin B2 receptor knockout mice. Hypertension. 2003;41:1151–1155. doi: 10.1161/01.HYP.0000064180.55222.DF. [DOI] [PubMed] [Google Scholar]
- 28.Duka A, Kintsurashvili E, Duka I, Ona D, Hopkins TA, Bader M, Gavras I, Gavras H. Angiotensin-converting enzyme inhibition after experimental myocardial infarct: role of the kinin B1 and B2 receptors. Hypertension. 2008;51:1352–1357. doi: 10.1161/HYPERTENSIONAHA.107.108506. [DOI] [PubMed] [Google Scholar]
- 29.Tan Y, Keum JS, Wang B, McHenry MB, Lipsitz SR, Jaffa AA. Targeted deletion of B2-kinin receptors protects against the development of diabetic nephropathy. Am. J. Physiol. Renal. Physiol. 2007;293:F1026–F1035. doi: 10.1152/ajprenal.00203.2007. [DOI] [PubMed] [Google Scholar]
- 30.Xu J, Carretero OA, Shesely EG, Rhaleb NE, Yang JJ, Bader M, Yang XP. The kinin B1 receptor contributes to the cardioprotective effect of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers in mice. Exp. Physiol. 2009;94:322–329. doi: 10.1113/expphysiol.2008.045583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tschöpe C, Spillmann F, Altmann C, Koch M, Westermann D, Dhayat N, Bascands JL, Gera L, Hoffmann S, Schultheiss HP, et al. The bradykinin B1 receptor contributes to the cardioprotective effects of AT1 blockade after experimental myocardial infarction. Cardiovasc. Res. 2004;61:559–569. doi: 10.1016/j.cardiores.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 32.Schulze-Topphoff U, Prat A, Prozorovski T, Siffrin V, Paterka M, Herz J, Bendix I, Ifergan I, Schadock I, Mori MA, et al. Activation of kinin receptor B1 limits encephalitogenic T lymphocyte recruitment to the central nervous system. Nat. Med. 2009;15:788–793. doi: 10.1038/nm.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Merino VF, Todiras M, Mori MA, Sales VM, Fonseca RG, Saul V, Tenner K, Bader M, Pesquero JB. Predisposition to atherosclerosis and aortic aneurysms in mice deficient in kinin B1 receptor and apolipoprotein E. J. Mol. Med. 2009;87:953–963. doi: 10.1007/s00109-009-0501-0. [DOI] [PubMed] [Google Scholar]
- 34.Lagneux C, Bader M, Pesquero JB, Demenge P, Ribuot C. Detrimental implication of B1 receptors in myocardial ischemia: evidence from pharmacological blockade and gene knockout mice. Int. Immunopharmacol. 2002;2:815–822. doi: 10.1016/s1567-5769(02)00022-x. [DOI] [PubMed] [Google Scholar]
- 35.Yin H, Chao J, Bader M, Chao L. Differential role of kinin B1 and B2 receptors in ischemia-induced apoptosis and ventricular remodeling. Peptides. 2007;28:1383–1389. doi: 10.1016/j.peptides.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kehat I, Molkentin JD. Extracellular signal-regulated kinase 1/2 (ERK1/2) signaling in cardiac hypertrophy. Ann. N. Y. Acad Sci. 2010;1188:96–102. doi: 10.1111/j.1749-6632.2009.05088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chao J, Li HJ, Yao YY, Shen B, Gao L, Bledsoe G, Chao L. Kinin infusion prevents renal inflammation, apoptosis, and fibrosis via inhibition of oxidative stress and mitogen-activated protein kinase activity. Hypertension. 2007;49:490–497. doi: 10.1161/01.HYP.0000255925.01707.eb. [DOI] [PubMed] [Google Scholar]
- 38.Cellier E, Mage M, Duchêne J, Pécher C, Couture R, Bascands JL, Girolami JP. Bradykinin reduces growth factor-induced glomerular ERK1/2 phosphorylation. Am. J. Physiol. Renal. Physiol. 2003;284:F282–F292. doi: 10.1152/ajprenal.00115.2002. [DOI] [PubMed] [Google Scholar]
- 39.Habashi JP, Doyle JJ, Holm TM, Aziz H, Schoenhoff F, Bedja D, Chen Y, Modiri AN, Judge DP, Dietz HC. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science. 2011;332:361–365. doi: 10.1126/science.1192152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bove CM, Yang Z, Gilson WD, Epstein FH, French BA, Berr SS, Bishop SP, Matsubara H, Carey RM, Kramer CM. Nitric oxide mediates benefits of angiotensin II type 2 receptor overexpression during post-infarct remodeling. Hypertension. 2004;43:680–685. doi: 10.1161/01.HYP.0000115924.94236.91. [DOI] [PubMed] [Google Scholar]
- 41.Kakoki M, McGarrah RW, Kim HS, Smithies O. Bradykinin B1 and B2 receptors both have protective roles in renal ischemia/reperfusion injury. Proc. Natl. Acad. Sci. U.S.A. 2007;104:7576–7581. doi: 10.1073/pnas.0701617104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu L, Carretero OA, Liao TD, Harding P, Li H, Sumners C, Yang XP. Role of prolylcarboxypeptidase in angiotensin II type 2 receptor-mediated bradykinin release in mouse coronary artery endothelial cells. Hypertension. 2010;56:384–390. doi: 10.1161/HYPERTENSIONAHA.110.155051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moniwa N, Agata J, Hagiwara M, Ura N, Shimamoto K. The role of bradykinin B1 receptor on cardiac remodeling in stroke-prone spontaneously hypertensive rats (SHR-SP) Biol. Chem. 2006;387:203–209. doi: 10.1515/BC.2006.027. [DOI] [PubMed] [Google Scholar]
- 44.Ying WZ, Sanders PW. The interrelationship between TGF-β1 and nitric oxide is altered in salt-sensitive hypertension. Am. J. Physiol. Renal. Physiol. 2003;285:F902–F908. doi: 10.1152/ajprenal.00177.2003. [DOI] [PubMed] [Google Scholar]
- 45.Hayashida T, Decaestecker M, Schnaper HW. Cross-talk between ERK MAP kinase and Smad signaling pathways enhances TGF-β-dependent responses in human mesangial cells. FASEB J. 2003;17:1576–1578. doi: 10.1096/fj.03-0037fje. [DOI] [PubMed] [Google Scholar]