

Abstract
Neuronal fractalkine acts via its receptor, CX3CR1, on microglia to regulate neuroinflammation. Conflicting results have been reported in studies employing CX3CR1 deficient (Cx3cr1-/-) mice. Here, compared to wild-type, endotoxin-treated neuron-glial Cx3cr1-/- cultures produced less TNF-α, nitric oxide and superoxide; however, fractalkine treatment inhibited the release of pro-inflammatory factors compared to wild-type and BV-2 cell cultures. Furthermore, endotoxin-treated BV-2 cells expressing siRNA against CX3CR1 increased nitric oxide and TNF-α production. We hypothesize that CX3CL1-CX3CR1 signaling is neuroprotective and propose that the reduced production of pro-inflammatory signals in Cx3cr1-/- microglia may result from compensatory mechanisms and not be the direct result of CX3CR1 deficiency.
Keywords: Parkinson’s disease, Alzheimer’s disease, stroke, microglia, fractalkine, chemokines, CX3CR1, neuroinflammation
INTRODUCTION
Microglia, when activated, can produce trophic and anti-inflammatory factors, all of which serve to protect against potential insults in the central nervous system (CNS). Overactivated microglia, however, release pro-inflammatory, neurotoxic factors such as superoxide, nitric oxide (NO) and cytokines that contribute to the pathology of neurodegenerative conditions such as Parkinson’s disease (PD). The conditions governing whether microglial activation is neuroprotective or neurotoxic are largely unknown.
Fractalkine (FKN, CX3CL1) is a unique chemokine that can exist either as a static membrane-bound glycoprotein mediating cell adhesion or, after proteolytic cleavage, a soluble chemoattractant. Fractalkine, primarily expressed by neurons, is the sole ligand for its specific receptor, CX3CR1, which is present only on microglia (Harrison et al., 1998; Nishiyori et al., 1998; Ransohoff & Cardona, 2010). Fractalkine has been proposed to be an inhibitory signal, tonically regulating the activation of microglia to maintain them in a quiescent state (Ransohoff and Perry, 2009).
Several studies have demonstrated that fractalkine acts to protect neurons in vitro in lipopolysaccharide (LPS)-activated microglia by limiting their release of inflammatory factors (Mizuno et al., 2003). Most notably, treatment of cultured microglia with fractalkine has been demonstrated to dose-dependently suppress LPS-induced activation by reducing their production of NO, IL-6, TNF-α (Lyons et al., 2009; Mizuno et al., 2003). While there is a consensus among studies for a neuroprotective role of fractalkine signaling in vitro, in vivo studies of CX3CR1-deficient mice have proven controversial. Although a neuroprotective role for CX3CL1-CX3CR1 signaling has been supported in studies of CX3CR1-deficient mouse models of amyotrophic lateral sclerosis (ALS), PD (Cardona et al., 2006), and multiple sclerosis (MS) (Huang et al., 2006), a neurotoxic role for fractalkine signaling has been demonstrated in studies using transgenic mice lacking CX3CR1 as animal models for Alzheimer’s Disease (AD) (Liu et al.), stroke (Denes et al., 2008), and spinal cord injury (Donnelly et al.). The reasons for these contradictory reports are unclear.
To control for differences in preparation and treatment that limit cross-study comparisons, we compared microglial responsiveness to LPS in mixed neuron-glia cultures from Cx3cr1-/- mice, wild-type mice, and BV-2 (immortalized microglia) cells. Our study confirmed a neuroprotective role for CX3CL1-CX3CR1 signaling in primary neuron-glia cultures from wild-type mice and BV-2 cells expressing siRNA against CX3CR1, but not in mixed cultures from Cx3cr1-/- mice, which exhibited a reduced production of TNF-α, NO and superoxide. These in vitro results are in agreement with in vivo studies demonstrating a neurotoxic role for CX3CL1-CX3CR1 signaling. Because the application of exogenous fractalkine to both wild-type neuron-glia cultures and BV-2 cells led to a reduction in the LPS-induced production of TNF-α and NO while BV-2 cells expressing siRNA-CX3CR1 increased production of TNF-α and NO, we conclude that CX3CL1-CX3CR1 signaling is neuroprotective. These findings have led us to further propose that the reduced production of TNF-α, NO and superoxide from microglia of Cx3cr1-/- may be due to compensatory mechanisms that result in a reduction in the release of these pro-inflammatory signals.
MATERIALS AND METHODS
Mice and primary glial cultures
To genetically delete CX3CR1 in hAPP mice, hAPP-J20 mice (C57BL/6) were crossed with CX3CR1 GFP knock-in mice in which the Cx3cr1 gene was replaced with cDNA encoding GFP (Jung et al., 2000); (Cook et al., 2001).
Mixed neuron-glia cultures were prepared from ventral mesencephalic tissues of embryonic day 13-14 Cx3cr1GFP/GFP mice or wild-type (C57BL/6J) mice, as previously described with modifications (Zhang et al., 2005; Zhou et al., 2005). Dissected mesencephalic tissue was dissociated by mechanical trituration and seeded onto poly-D-lysine-coated 24-well culture plates (4 ×105/well). Cultures were maintained in 1 ml/well DMEM/F-12 supplemented with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, and 0.1 mM nonessential amino acids and incubated at 37°C in a humidified atmosphere of 5% CO2/95% O2. The medium was changed 3 d after cultures were prepared. Immunocytochemical analysis of mixed neuron-glia cultures prepared using our culture methods revealed that the cultures contain approximately 11% microglia, 48% astrocytes, 40% neurons as previously reported (Zhang et al., 2005; Zhang et al., 2007; Wu et al., 2008). Seven-day-old cultures were used for treatment. At various time points after LPS treatment supernatant from neuron-glia cultures was collected for determining the level of TNF-α (3 h) and nitrite (24h, as an index for nitric oxide). These two time points represent the peak levels for these two immune factors.
BV-2 cell cultures and siRNA transfection
BV-2 cells are microglial-like cells from an immortalized murine-derived cell line that respond to toxic insults such as LPS similarly to primary microglial cultures (Horvath et al., 2008). Cells were thawed and passaged until logarithmic growth was achieved. Cell cultures were maintained in 25- to 75-cm2 tissue culture flasks (Corning Costar, Corning, NY) at a concentration of 5-9×105 cells/ml, in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma) supplemented with 10% heat-inactivated FBS, 2 mM glutamine (Sigma-Aldrich, S. Louis, MO) and 1 μl Pen Strep (Sigma).
Cells were passaged every 3-4 days to a density of 1×105 cells/ml. To begin the experiment, the media were removed and fresh 10% FBS-supplemented DMEM was added to the cells. Untreated cells in 10% FBS supplemented DMEM were used as control.
Cells were seeded into 24-well plates at a density of 5×104 cells/well and treated with LPS at concentrations of 10 or 15ng/ml. The plates were incubated in a tissue culture incubator at 37°C in an atmosphere of 5% CO2/95% O2 for the listed period of time. The viability was 95-98% as determined by trypan blue exclusion. Cells from each single well were withdrawn for western blotting. Aliquots of supernatant obtained from each well were used for ELISA.
The small interfering RNA (siRNA) sequence targeting CX3CR1 was developed using the siRNA Reagent System purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). BV-2 cells were transfected with siRNA-CX3CR1 or scrambled RNA using Lipofectamine 2000 (Invitrogen Inc., Carlsbad, CA). BV-2 cells were plated into a six-well plate at a density of 3×105 cells per well in antibiotic-free medium and allowed to adhere overnight. Then, 0.3 μl of Lipofectamine 2000 (Invitrogen) were added to 300 μL of Opti-MEM (OM; Invitrogen) and allowed to incubate for 15 min at room temperature. At the same time, each siRNA or scrambled RNA was added to 300 μL of OM. After 5 min, the siRNA and Lipofectamine 2000 mixtures were combined. Complexes were allowed to form for 20 min before the 600 μL mixture was added to each well. After 12 h, media was replaced with antibiotics-free culture medium and incubated overnight. The transfection procedure was repeated the next day. After incubation with culture medium for 24h, the cells were trypsinized and collected for CX3CR1 expression detection by western blotting.
Measurement of superoxide, NO, and cytokines
The production of superoxide was determined by measuring the superoxide dismutase (SOD)-inhibitable reduction of tetrazolium salt, WST-1, as described previously, with modifications (Zhang et al., 2005). Neuron-glia cultures grown in 96-well plates were treated with LPS or LPS+FKN in 150 μl of phenol red-free treatment medium to which 50 μl of WST-1 (1 mM) with and without 800 U/ml SOD was immediately added. The absorbance at 450 nm was read with a Spectra-Max Plus microplate spectrophotometer (Molecular Devices, Sunnyvale, CA) and the data at 12-14 minutes post-treatment were analyzed. The production of NO was estimated using Griess reaction (Green et al., 1982).
The levels of TNF-α in the culture medium were measured with commercial ELISA kits from R&D Systems (Minneapolis, MN). For each experiment, three to four wells per treatment condition were used, and results from three to four independent experiments were obtained.
Gel electrophoresis and Western blotting analysis
The protein extracts from cultured cells were homogenized in radioimmunoprecipitation assay (RIPA) lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and 1:100 protease inhibitor cocktail), sonicated, and heated to 100°C for 10 min. Protein concentrations were determined using the biocinchoninic acid (BCA) assay (Pierce, Rockford, IL). Protein samples were resolved on 4–12% SDS-PAGE gels, and immunoblot analyses were performed using antibodies against CX3CR1 (1:500; Abcam, Cambridge, MA). An antibody against GAPDH (1:3000; Cell Signaling Technology, Danvers, MA) was included as an internal standard to monitor loading errors. Western blots were analyzed using the densitometry plugin in Image J.
Statistical analysis
All values are expressed as the mean ± SEM. Differences among means were analyzed using either t-tests or one-way ANOVA followed by Tukey post hoc tests. In all analyses, the null hypothesis was rejected at the 0.05 level.
RESULTS
Decrease of LPS-induced NO production and TNF-α release in microglia in CX3CR1 KO mice
The role of fractalkine signaling through CX3CR1 was first investigated by comparing LPS-induced production of pro-inflammatory factors, NO and TNF-α, in primary neuron-glia cultures prepared from the ventral mesencephalic tissues of wild-type or Cx3cr1-/- mice after LPS stimulation. When stimulated by 10 ng/ml LPS, NO production was 572±129% of baseline levels in cultures prepared from wild-type mice and was 241±72% of baseline levels in cells from Cx3cr1-/-mice (Fig. 1A). Similarly, TNF-α release was significantly higher in LPS-treated cultures from wild-type mice (3264±470 pg/ml) compared to LPS-treated cultures from Cx3cr1-/- mice (1452±118 pg/ml; Fig. 1B, p<0.05), indicating that both NO and TNF-α release are suppressed in response to immune stimulation in CX3CR1-deficient microglia compared to wild-type microglia. Thus, neuron-glia cultures with deficient CX3CL1-CX3CR1 signaling exhibit a reduction in the release of pro-inflammatory factors NO and TNF-α in response to LPS compared to wild-type, in support of a neurotoxic rather than neuroprotective role for CX3CL1-CX3CR1 signaling. Recent results from our laboratory indicate that microglia are the major source of LPS-induced release of NO and TNF-α from neuron-glia cultures (Chen et al., 2012). Therefore, these results suggest that CX3CL1-CX3CR1 signaling acts to promote microglial activation in this CX3CR1-deficient model, which is inconsistent with the conventional view of an inhibitory role for CX3CR1.
Figure 1. The pro-inflammatory response is suppressed in CX3CR1-deficient microglia.
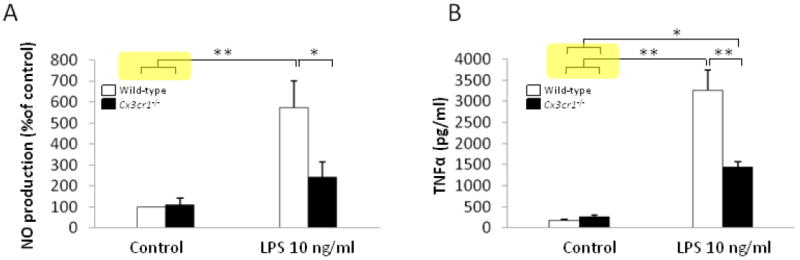
A. NO release from primary mixed neuron-glia cultures prepared from wild-type mice (C57BL/6) and Cx3cr1-/- mice was measured using Griess assays before (control) and after LPS treatment (10 ng/ml) (n=5 for cultures from both wild-type and Cx3cr1-/-; mean ± SEM; *p < 0.05, **p<0.01; one-way ANOVA followed by Tukey post hoc test). B. TNF-α levels were measured in culture supernatants by ELISA in neuron-glia cultures from wild-type mice (C57BL/6) and Cx3cr1-/- mice following LPS treatment (10 ng/ml) (n=6 for cultures from wild-type and n=4 for Cx3cr1-/- mice; mean± SEM; *p < 0.05, **p < 0.01; one-way ANOVA followed by Tukey post hoc test).
Fractalkine inhibits LPS-induced NO production and TNF-α release in primary neuron-glia cultures and murine microglial cell line, BV-2, cells
The initial results suggested that CX3CR1 signaling heightens the pro-inflammatory response of microglia in response to adverse immune stimulation and that CX3CR1-deficient microglia are less likely to be neurotoxic because of their reduced release of pro-inflammatory mediators, consistent with in vivo reports on CX3CR1-deficient microglia (Liu et al.). However, the opposite results were observed when the CX3CL1-CX3CR1 pathway was activated by the CX3CR1 agonist, fractalkine, after primary neuron-glia cultures from wild-type mice were treated with LPS. Both NO production and TNF-α release were increased after LPS stimulation compared to untreated cells (Fig. 2A; 365±21% and 2006±104 pg/ml vs. 6.77±4.78 pg/ml, respectively). However, fractalkine dose-dependently reduced the LPS-induced production of NO and TNF-α (Fig. 2A), a result consistent with what was reported previously by others (Mizuno et al., 2003). Similar results were observed when BV-2 cells, an immortalized murine-derived, microglial-like cell line, were treated with LPS alone or with both LPS and fractalkine. Fractalkine treatment (10 nM) reduced NO release by 25±0.02% (Fig. 2B) and significantly reduced TNF-α release compared to LPS-treated BV-2 cells without fractalkine treatment (Fig. 2B, p<0.05).
Figure 2. Activating CX3CR1 with fractalkine reduces the LPS-induced release of pro-inflammatory factors while reducing the expression of CX3CR1 with siRNA increases the release of pro-inflammatory factors.

A. NO release is increased after LPS (15 ng/ml) compared to control levels and fractalkine (FKN) treatment in microglia in primary mixed neuron-glia cultures prepared from wild-type mice. Fractalkine treatment in LPS-treated cultures dose-dependently reduces NO release at 0.1 nM, 1 nM, and 10 nM (normalized to LPS-treated cultures; n=3; mean ± SEM; **p < 0.01; one-way ANOVA followed by Tukey post hoc test). TNF-α levels are undetectable in control and FKN treated cultures, but are detectable after LPS treatment. These levels are reduced in cultures treated with both LPS and fractalkine at 0.1 nM, 1 nM, and 10 nM compared to cultures treated with LPS alone (n=3; mean ± SEM; **p < 0.01; one-way ANOVA followed by Tukey post hoc test). B. NO production is increased after LPS (15 ng/ml) compared to control levels in BV-2 cells and fractalkine treatment dose-dependently reduces NO release. (n=3; mean ± SEM; **p < 0.01; one-way ANOVA followed by Tukey post hoc test). TNF-α release is dose-dependently decreased in BV-2 cells treated with LPS and fractalkine (n=3; **p < 0.01; one-way ANOVA followed by Tukey post hoc test). C. Western blot densitometry analysis using CX3CR1 antibody revealed a 49.39±2.27% decrease in CX3XR1 expression in BV-2 cells expressing siRNA for CX3CR1 compared to BV-2 cells expressing scrambled siRNA; density is expressed as the ratio of CX3CR1/GAPDH (bar graph). D. BV-2 cells were treated with LPS (15ng/ml) and FKN (1 nM). NO production was significantly higher in BV-2 cells transfected with CX3CR1 siRNA compared to cells transfected with scrambled siRNA (n=3; mean ± SEM; *p<0.05). TNFα release was significantly higher in BV-2 cells transfected with CX3CR1 siRNA compared to cells transfected with scrambled siRNA (n=3; mean ± SEM; *p<0.05).
The effect of CX3CR1 knockdown on LPS-induced inflammatory response in BV-2 cells
To further address these contradictory results, we investigated how the loss of CX3CR1 affects the release of pro-inflammatory factors using siRNA against CX3CR1 in BV-2 cells because siRNA is toxic for primary cultured microglia. In BV-2 cells expressing siRNA-CX3CR1, CX3CR1 expression was reduced to 49.3±2.2%, as demonstrated by western blot analysis (Fig. 2C), whereas a scrambled version of CX3CR1 siRNA did not affect CX3CR1 expression in BV-2 cells or CX3CR1 siRNA (Fig. 2C). In contrast to our results in Cx3cr1-/- microglia, NO production was significantly higher in LPS-treated BV-2 cells in which CX3CR1 was knocked down compared to cells transfected with scrambled siRNA (Fig. 2D, p<0.05; 52.2±6.1 μM and 39.8±3.2, respectively). Similarly, TNF-α release (4h) was significantly higher in BV-2 cells transfected with siRNA-CX3CR1 compared to cells transfected with scrambled siRNA (Fig. 2D, p<0.05; 6999±766 and 4790±720 pg/ml, respectively). Thus, the siRNA-mediated reduction of CX3CR1, but not the genetic deletion of CX3CR1, enhances the LPS-induced production of pro-inflammatory factors.
Inhibition of CX3CR1 on LPS-induced superoxide production
The results obtained thus far indicate that neuron-glia cultures from Cx3cr1-/- mice respond with a less robust pro-inflammatory response than both cultures from wild-type mice and BV-2 cells. Because one of the major effectors of the pro-inflammatory response that results in neurotoxicity is an enhanced production of reactive oxygen species (ROS), we next investigated whether Cx3cr1-/- mice also exhibit a suppressed production of superoxide induced by LPS. As shown in Fig. 3A, superoxide production increased approximately four-fold after LPS treatment (15ng/ml) in neuron-glia cultures from wild-type mice, and this increase was dose-dependently suppressed by fractalkine (Fig. 3A). However, when superoxide production was measured in CX3CR1-deficient microglia in primary neuron-glial cultures from Cx3cr1-/- mice, we found that LPS-induced superoxide production was suppressed (Fig. 3B, p<0.05; 3.03±0.22 vs. 3.94±0.02 in WT), consistent with the decrease observed in NO and TNF-α release in CX3CR1-deficient microglia. Thus, LPS has the opposite effect on the production of immune factors by microglia from Cx3cr1-/- mice compared to wild-type microglia and BV-2 cells expressing siRNA-CX3CR1.
Figure 3. Superoxide production is reduced in CX3CR1-deficient microglia.

A. LPS treatment (15 ng/ml) increases superoxide production, and this LPS-mediated increase is reduced dose-dependently by fractalkine treatment (normalized to untreated cultures; n=3; mean ± SEM; *p<0.05, **p<0.01; one-way ANOVA followed by Tukey post hoc test). B. Superoxide production is increased by LPS treatment in both wild-type mice (C57BL/6) and Cx3cr1-/- mice compared to control, but is reduced in CX3CR1-deficient microglia compared to wild-type (normalized to untreated cultures; n=3 for cultures from wild-type and Cx3cr1-/- mice; mean ± SEM;*p<0.05, **p<0.01; one-way ANOVA followed by Tukey post hoc test).
DISCUSSION
The major observations of this study are that, although the release of TNF-α and NO is markedly reduced in Cx3cr1-/- vs. WT microglia, the exogenous application of fractalkine dose-dependently reduces NO and TNF-α release in both primary cultures from WT mice and BV-2 cells. We have also shown that decreasing the expression of CX3CR1 in BV-2 cells using siRNA exacerbates the increased release of NO and TNF-α in response to LPS. These results suggest that acutely knocking down CX3CR1 promotes the release of pro-inflammatory factors while the normal release of these pro-inflammatory factors as well as superoxide is suppressed when CX3CR1 is lacking during development. These results raise the intriguing possibility that the protective results obtained in microglia from Cx3cr1-/- mice may be due to impaired responsivity that arises during development to compensate for the loss of neuron-glial communication. Indeed, in addition to the suppressed release of NO and TNF-α, CX3CR1-deficient microglia from Cx3cr1-/- also released less superoxide than WT after LPS treatment. Though the results of our experiments suggest the possibility of aberrant responsivity of microglia from Cx3cr1-/- mice, more experiments will be needed to extensively test this possibility.
Translating in vitro observations to in vivo studies can be challenging. However, our conclusion that CX3CL1-CX3CR1 plays a neuroprotective role is in agreement with in vivo studies of Cx3cr1-/- mice (Cardona et al., 2006; Huang et al., 2006). Consistent with our results, Cardona et al. demonstrated that TNFα expression was lower in Cx3cr1-/- mice than heterozygous mice after LPS injections, though, in seeming contradiction, they observed an increase in the expression of IL-1ß. On the other hand, Huang et al. found that microglial activation was increased in Cx3cr1-/- mice, as evidenced by a decrease in migration and an increase in the production of some, but not all, inflammatory factors (Huang et al., 2006).
The seemingly contradictory reports described for Cx3cr1-/- mice could potentially be explained by compensatory mechanisms nonspecific to impaired CX3CL1-CX3CR1 signaling. The interpretation of results from genetic knockout mice must take into account the possibility of confounding developmental effects, as well as the compensation of these effects by other gene products that may function in a network, or to regulate the expression of the gene of interest. These potential confounds can be exacerbated when transgenic mice are generated with multiple transgenic mutations or deletions, a commonly used approach in reports using Cx3cr1-/- mice (Fuhrmann et al.; Lee et al.). Although knockout mice have been used to investigate the role of approximately 6,500 genes (Lloyd; Lloyd, 2011), at least 15% exhibit no reported phenotypic difference from wild-type mice, suggesting that multiple mechanisms are in place to compensate for genetic deletions during development (Austin et al., 2004; Barbaric et al., 2007; Fredholm et al., 2005). Therefore, a potential consequence of the genetic knockout approach is that the disruption of one signaling pathway is likely to impact others in the same cell. This is the explanation for the unexpected result that striatopallidal neurons from mice deficient in dopamine D2 receptors (D2Rs), which are normally expressed alongside adenosine receptors (A2ARs) that oppose their tonic inhibition of GABA release, do not increase GABA release when A2ARs are pharmacologically activated (Zahniser et al., 2000). Interestingly, D2Rs and A2ARs are G-protein coupled receptors that inhibit or activate cyclic AMP, respectively, and the lack of D2Rs in D2R-/- striatopallidal neurons leads to the functional uncoupling of A2ARs from their shared effector molecule.
Similar to D2Rs, fractalkine receptors are Gαi-coupled receptors, and because our results from pharmacologically activating CX3CR1 conflict with our results in Cx3cr1-/-neuron-glia cultures, it is possible that the uncoupling of proteins that regulate or are regulated by CX3CL1-CX3CR1 signaling from their effector molecules occurs, leading to the inhibition of the production of pro-inflammatory molecules in CX3CR1-deficient cultures. This functional uncoupling may occur to compensate for the loss of the neuroprotective receptor, CX3CR1, thereby effectively muting the release of pro-inflammatory factors NO, TNFα, and superoxide.
In summary, we hypothesize that CX3CL1-CX3CR1 signaling is ultimately neuroprotective and propose that microglia from Cx3cr1-/-mice may not be functionally comparable to microglia from wild-type mice, possibly due to differences in the expression of important signaling molecules downstream of CX3CR1 that may arise during development. Moving forward, it will be necessary to develop transgenic mice that allow for the conditional knockout of CX3CR1, allowing investigators to more specifically study the role of fractalkine signaling and signaling pathways in future studies. Alternatively, viral-mediated knockdown of the gene can also be used to study the mechanisms underlying fractalkine signaling.
Acknowledgments
This research was supported by intramural program of NIEHS (Dr. JS Hong) as well as NIH grants to Dr. J Zhang [ES016873, ES019277; NS057567; NS062684-(6221), ES007033 (6364), ES004696 (5897), AG033398, and NS082137]
Footnotes
JS H, JZ and HN designed the experiments. HN, HM G and HZ performed the experiments, and HN, HM G and HAM analyzed the data. HAM drafted and finalized the paper with JZ and JSH.
The authors declare they have no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
LITERATURE CITED
- Austin CP, Battey JF, Bradley A, Bucan M, Capecchi M, Collins FS, Dove WF, Duyk G, Dymecki S, Eppig JT, Grieder FB, Heintz N, Hicks G, Insel TR, Joyner A, Koller BH, Lloyd KC, Magnuson T, Moore MW, Nagy A, Pollock JD, Roses AD, Sands AT, Seed B, Skarnes WC, Snoddy J, Soriano P, Stewart DJ, Stewart F, Stillman B, Varmus H, Varticovski L, Verma IM, Vogt TF, von Melchner H, Witkowski J, Woychik RP, Wurst W, Yancopoulos GD, Young SG, Zambrowicz B. The knockout mouse project. Nat Genet. 2004;36:921–924. doi: 10.1038/ng0904-921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbaric I, Miller G, Dear TN. Appearances can be deceiving: phenotypes of knockout mice. Brief Funct Genomic Proteomic. 2007;6:91–103. doi: 10.1093/bfgp/elm008. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Bilousova TV, Puntambekar SS, Melchior B, Doose JM, Ethell IM. A rose by any other name? The potential consequences of microglial heterogeneity during CNS health and disease. Neurotherapeutics. 2007;4:571–579. doi: 10.1016/j.nurt.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DN, Chen SC, Sullivan LM, Manfra DJ, Wiekowski MT, Prosser DM, Vassileva G, Lira SA. Generation and analysis of mice lacking the chemokine fractalkine. Mol Cell Biol. 2001;21:3159–3165. doi: 10.1128/MCB.21.9.3159-3165.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denes A, Ferenczi S, Halasz J, Kornyei Z, Kovacs KJ. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab. 2008;28:1707–1721. doi: 10.1038/jcbfm.2008.64. [DOI] [PubMed] [Google Scholar]
- Donnelly DJ, Longbrake EE, Shawler TM, Kigerl KA, Lai W, Tovar CA, Ransohoff RM, Popovich PG. Deficient CX3CR1 signaling promotes recovery after mouse spinal cord injury by limiting the recruitment and activation of Ly6Clo/iNOS+ macrophages. J Neurosci. 2010;31:9910–9922. doi: 10.1523/JNEUROSCI.2114-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Chen JF, Masino SA, Vaugeois JM. Actions of adenosine at its receptors in the CNS: insights from knockouts and drugs. Annu Rev Pharmacol Toxicol. 2005;45:385–412. doi: 10.1146/annurev.pharmtox.45.120403.095731. [DOI] [PubMed] [Google Scholar]
- Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, Haass C, LaFerla FM, Kretzschmar H, Herms J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat Neurosci. 13:411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, Botti P, Bacon KB, Feng L. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath RJ, Nutile-McMenemy N, Alkaitis MS, Deleo JA. Differential migration, LPS-induced cytokine, chemokine, and NO expression in immortalized BV-2 and HAPI cell lines and primary microglial cultures. J Neurochem. 2008;107:557–569. doi: 10.1111/j.1471-4159.2008.05633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Shi FD, Jung S, Pien GC, Wang J, Salazar-Mather TP, He TT, Weaver JT, Ljunggren HG, Biron CA, Littman DR, Ransohoff RM. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. Faseb J. 2006;20:896–905. doi: 10.1096/fj.05-5465com. [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am J Pathol. 177:2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Condello C, Schain A, Harb R, Grutzendler J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-beta phagocytosis. J Neurosci. 2010;30:17091–17101. doi: 10.1523/JNEUROSCI.4403-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd KC. A knockout mouse resource for the biomedical research community. Ann N Y Acad Sci. 2011;1245:24–26. doi: 10.1111/j.1749-6632.2011.06311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons A, Lynch AM, Downer EJ, Hanley R, O’Sullivan JB, Smith A, Lynch MA. Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. J Neurochem. 2009;110:1547–1556. doi: 10.1111/j.1471-4159.2009.06253.x. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Kawanokuchi J, Numata K, Suzumura A. Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. 2003;979:65–70. doi: 10.1016/s0006-8993(03)02867-1. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Wu X, Chen PS, Dallas S, Wilson B, Block ML, Wang CC, Kinyamu H, Lu N, Gao X, Leng Y, Chuang DM, Zhang W, Lu RB, Hong JS. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol. 2008;11:1123–1134. doi: 10.1017/S1461145708009024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahniser NR, Simosky JK, Mayfield RD, Negri CA, Hanania T, Larson GA, Kelly MA, Grandy DK, Rubinstein M, Low MJ, Fredholm BB. Functional uncoupling of adenosine A(2A) receptors and reduced responseto caffeine in mice lacking dopamine D2 receptors. J Neurosci. 2000;20:5949–5957. doi: 10.1523/JNEUROSCI.20-16-05949.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Dallas S, Zhang D, Guo JP, Pang H, Wilson B, Miller DS, Chen B, McGeer PL, Hong JS, Zhang J. Microglial PHOX and Mac-1 are essential to the enhanced dopaminergic neurodegeneration elicited by A30P and A53T mutant alpha-synuclein. Glia. 2007;55:1178–1188. doi: 10.1002/glia.20532. [DOI] [PubMed] [Google Scholar]
- Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhou Y, Hong JS, Zhang J. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. Faseb J. 2005;19:533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Wang Y, Kovacs M, Jin J, Zhang J. Microglial activation induced by neurodegeneration: a proteomic analysis. Mol Cell Proteomics. 2005;4:1471–1479. doi: 10.1074/mcp.M500114-MCP200. [DOI] [PubMed] [Google Scholar]