

Abstract
Mouse strain background can influence vulnerability to excitotoxic neuronal cell death and potentially modulate phenotypes in transgenic mouse models of human disease. Evidence supports a contribution of excitotoxicity to the selective death of medium spiny neurons in Huntington’s disease (HD). Here, we assess whether strain differences in excitotoxic vulnerability influence striatal cell death in a knock-in mouse model of HD. Previous studies that evaluated resistance to excitotoxic lesions in several mouse models of HD had variable outcomes. In the present study, we directly compare one model on two different background strains to test the contribution of strain to excitotoxicity-mediated neurodegeneration. Mice of the FVB/N strain, which are highly vulnerable to excitotoxicity, become extremely resistant to quinolinic acid-induced striatal neurodegeneration with age, when carrying a huntingtin (Htt) allele expressing a HD transgene (CAG140). The resistance is much greater than the age-dependent resistance that has been previously reported in YAC128 mice. By 12 months of age, both heterozygous and homozygous FVB.CAG140 mice displayed virtually complete resistance to quinolinic acid-induced striatal neurodegeneration. A similar resistance develops in CAG140 mice on a C57BL/6N background although the effect size is smaller because C57BL/6N mice are already resistant due to genetic background. In a direct comparison with the YAC128 mice, FVB.CAG140 mice have greater resistance. FVB.CAG140 mice are also resistant to neurodegeneration following kainic acid-induced status epilepticus suggesting the existence of a common cellular mechanism that provides protection against multiple types of excitotoxic insult. These findings establish FVB.CAG140 mice as a useful model to investigate the cellular and molecular mechanisms that confer neuroprotection against excitotoxicity.
Keywords: Excitotoxin, Huntington’s disease, inbred mouse strains, resistance, cell death, neurodegeneration, genetic background
INTRODUCTION
Excitotoxicity is implicated in multiple neurodegenerative diseases, although its exact role in disease progression is not fully understood. One hypothesis to explain striatal neuron death in Huntington’s disease (HD) is that excitotoxic cell death is triggered by glutamatergic input from the cerebral cortex [1–3]. In this regard, HD can be modeled in animals with an intrastriatal injection of quinolinic acid (QUIN), an excitotoxic neurotoxin that triggers the selective degeneration of spiny stellate neurons [4]. N-methyl-D-aspartate (NMDA) receptor activation of medium spiny neurons can increase apoptosis in cells that express mutant huntingtin [5, 6], which is consistent with a mechanism of excitotoxicity contributing to the pathogenesis of HD.
Mouse strain background is an important consideration when studying excitotoxicity. Certain strains of inbred mice are resistant to QUIN-induced striatal neurodegeneration as well as other forms of excitotoxic neurodegeneration [7, 8]. For example, C57BL/6N (C57BL/6) mice exhibit substantial resistance to QUIN-induced striatal neurodegeneration while FVB/N mice are highly vulnerable. FVB/N mice are more vulnerable than other strains to multiple forms of excitotoxicity, including QUIN and kainic acid (KA) [9–11].
The choice of strain background for genetic models of disease is typically driven by the advantages different strains provide for genetic manipulation, or in breeding thereafter. Significantly, genetic differences between inbred strains can affect the phenotype of the model [12, 13]. There exist many genetic mouse models of HD on different strain backgrounds [14–21], some of which involve mixed backgrounds that have not been fully characterized. Neurodegeneration due to the HD mutation is variable between these models. Although this may be due in part to the length of the repeat [22], or the type of transgenic model (i.e. fragment, knock-in, or full length) [14, 15, 23–25], genetic background likely influences neurodegeneration in HD mice [26, 27].
Several groups have studied QUIN-induced striatal neurodegeneration in different mouse models of HD with variable outcomes that are often age-dependent. N171-82Q mice on a mixed C3H/HEJ × C57BL/6J strain background also showed resistance to QUIN-induced striatal neurodegeneration compared to controls at 15 weeks of age, but other ages were not assessed [28]. The shortstop model, on an FVB/N background, has very little QUIN-induced striatal neurodegeneration compared to wild type littermates at 6 months of age [29]. In contrast, other HD mouse models either have no reported difference to QUIN-induced striatal neurodegeneration (such as the tgHD100 mice on a mixed SJL × C57BL/6 background [30]), or they display increased QUIN-induced striatal neurodegeneration compared to wild type controls (YAC72 mice on an FVB/N strain background at 6 and 10 months of age [5]). Because different studies tested mice at different ages, variability in excitotoxic vulnerability in the different HD models may be due to differences in the age of the mice at the time of assessment.
In studies that have tested multiple ages, the resistance to QUIN is variable, and usually increases with age. Brundin and colleagues reported age-dependent resistance to striatal QUIN injection compared to wild type littermates in both the R6/1 and R6/2 mouse models, which are on a mixed CBA × C57BL/6 strain background. The R6/1 mice are more vulnerable than WT mice at 3 and 8 weeks of age and more resistant than WT at 13, 18, 22, and 36 weeks of age. The R6/2 mice are more vulnerable than WT at 3 weeks of age, and more resistant than WT at 6 and 12 weeks of age [31, 32]. Similarly, the QUIN-induced striatal neurodegeneration in the YAC128 mice varies with age (YAC128 are more vulnerable than WT at 1.3, 3 and 6 months of age, and more resistant than WT at 10, 12, and 18 months of age) [33]. These models are on various strain backgrounds; including strains that are vulnerable to excitotoxicity, are resistant to excitotoxicity or have not yet been characterized for excitotoxic vulnerability. An interaction between strain background and cell death may exist in HD mouse models. However, it is difficult to draw firm conclusions from the literature due to variations in the dose, timing or type of excitotoxic lesion, the background strain, the type of HD mutation or transgenic model, and the methods of measuring the amount of QUIN-induced striatal neurodegeneration. While there have been studies of phenotype resulting from the same HD mutation on several background strains (e.g. YAC128 model) [26], no study has directly compared the susceptibility to excitotoxicity of the same HD mouse model on different background strains.
In the present study, we sought to address this issue. We show that the extent of resistance to excitotoxicity is dependent on genotype, age and strain background. CAG140 knock-in mice, which were originally characterized on a mixed genetic background [18, 23], were bred onto both FVB/NHsd and C57BL/6NHsd congenic strain backgrounds. Resistance to QUIN-induced excitotoxicity was assessed across multiple ages and over a time course of neurodegeneration and two different excitotoxic lesions (QUIN and KA) were performed in aged CAG140 mice. QUIN-induced striatal neurodegeneration in the FVB.CAG140 knock-in model was also compared to a full-length transgenic model (YAC128). These studies were designed to gain insight into the underlying mechanism behind striatal cell death and survival in HD. Our results reveal that the presence of the CAG140 transgene causes cellular changes that result in a dramatic resistance to an otherwise lethal excitotoxic insult in both FVB/NHsd and C57BL/6NHsd strain backgrounds.
MATERIALS AND METHODS
All procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Experimental protocols were approved by the Institutional Animal Care and Use Committee at the University of California, Irvine. Transgenic mice in this study were from the B6.CAG140 line, FVB.CAG140 line, or the YAC128 line. Mice from defined strains were obtained from commercial suppliers as noted below.
Mice and genotyping
FVB/NHsd and C57BL/6NHsd mice were obtained from Harlan (Indianapolis, IN, USA). CAG140 mice with the mutant huntingtin allele [18], generously donated by M.F. Chesselet (UCLA), were originally generated using a 129S1 embryonic stem cell line and maintained on a congenic C57BL/6J background. Heterozygous CAG140 animals were back-crossed with FVB/NHsd and C57BL/6NHsd mice for 10 generations to generate experimental animals and littermate (wild-type) controls used here. The CAG140 knock-in mouse model was selected for these studies because expression of the mutant HD protein is under the control of the endogenous huntingtin locus on mouse chromosome 5. The mice express one copy of mutant huntingtin protein (mHtt) and one copy of mouse wild-type huntingtin (Htt) in the case of the heterozygotes (HET) or two copies of mHtt with no wild-type Htt in the case of the homozygotes (HOM).
To transfer the mutant huntingtin allele to the FVB/NHsd strain background, B6:129-CAG140 mice were out crossed with FVB/NHsd mice. Offspring were backcrossed with FVB/NHsd mice for nine generations to produce FVB.129S1(B6)-CAG140 (N10) mice (i.e. congenic on a FVB/NHsd strain background), hereafter referred to as FVB.CAG140. These animals were intercrossed to generate homozygous and heterozygous FVB.CAG140 mice and littermate controls that were wild type at the huntingtin locus (termed WT FVB.CAG140). We also compared the extent of QUIN-induced striatal neurodegeneration in C57BL/6NHsd mice, housed at the University of California, Irvine. YAC128 mice (line 53; official name FVB-Tg (YAC128) 53Hay), were generated by microinjection of a yeast artificial chromosome (YAC) containing a human HD allele into the pronucleus of a FVB/N fertilized oocyte (Slow et al. 2003).
For controls in immunohistochemistry analyses, HD transgenic mice (line R6/2) (B6CBA-Tg (HDexon1) 62 Gpb/1J) on a CBA × C57BL/6J strain of origin were purchased from Jackson Laboratories, Bar Harbor, ME [17]. Mice were genotyped as described elsewhere [34]. The PCR products were separated in a 1% agarose gel containing 0.25 μg/ml ethidium bromide. Since the number of CAG repeats in this mouse model has been shown to vary [20, 35], repeat numbers were determined (Laragen, Culver City, CA, USA) in 10 mice from the first generation of mice after 10 generations of backcrossing. The number of CAG repeats in FVB.CAG140 N11 mice was 127 ± 12, a range consistent with variability of expansion within individual offspring.
Quinolinic acid treatment
Mice were tested for vulnerability to excitotoxicity with a unilateral striatal injection of quinolinic acid (QUIN) (Sigma-Aldrich, St. Louis, MO), as previously described [10]. Briefly, mice were anesthetized with 33 mL/kg i.p. Avertin and secured in a stereotaxic device. QUIN (15 nmols in 0.5 μL saline, pH 6.8) was injected into the right striatum (0.7 mm rostral to bregma, 2.1 mm lateral to the midline and 2.5 mm ventral to the dura mater) over a period of three minutes. Mice were perfused with 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS), pH 7.4 at seven, 10, 13 or 16 days post-injection. Brains were post-fixed overnight with 4% PFA, cryoprotected in 30% sucrose in PBS, and cut into 35 um coronal sections on a freezing microtome.
For comparison of QUIN-induced striatal neurodegeneration, all three genotypes (WT, HET and HOM) of mice on either the FVB/NHsd or C57BL/6NHsd strain backgrounds were given injections of QUIN at 1, 2, 4 or 12 months of age, and tissue was harvested 7 days later. Final group size varied due to attrition from various causes (see Table 1). Starting group sizes in the 12 month old group were smaller, as male mice sometimes fought with each other and had to be euthanized due to injuries prior to being injected with QUIN. Some animals were excluded from the statistical analysis due to tissue quality or lesions not meeting inclusion criteria. Group numbers used in statistical analysis are reported in the figure legend.
Table 1.
Group numbers and attrition
| Age (mo.) | Strain | Genotype | No. mice injected | No. mice euthanized | Deaths due to: |
|---|---|---|---|---|---|
| Experiment: QUIN, 7 day survival
| |||||
| 1 | FVB | WT | 12 | 11 | |
| 1 | FVB | HET | 13 | 12 | Anesthesia |
| 1 | FVB | HOM | 13 | 10 | Surgery, anesthesia |
| 2 | FVB | WT | 10 | 10 | |
| 2 | FVB | HET | 14 | 12 | Recovery |
| 2 | FVB | HOM | 10 | 10 | |
| 4 | FVB | WT | 10 | 10 | |
| 4 | FVB | HET | 10 | 10 | |
| 4 | FVB | HOM | 8 | 8 | |
| 12 | FVB | WT | 8 | 6 | Anesthesia, recovery |
| 12 | FVB | HET | 8 | 6 | Anesthesia, recovery |
| 12 | FVB | HOM | 7 | 6 | Recovery |
| 1 | C57BL/6 | WT | 11 | 10 | Anesthesia |
| 1 | C57BL/6 | HET | 11 | 9 | Anesthesia, recovery |
| 1 | C57BL/6 | HOM | 11 | 11 | |
| 2 | C57BL/6 | WT | 12 | 9 | Anesthesia, recovery |
| 2 | C57BL/6 | HET | 13 | 10 | Anesthesia, recovery |
| 2 | C57BL/6 | HOM | 12 | 9 | Anesthesia, recovery |
| 4 | C57BL/6 | WT | 11 | 9 | Anesthesia |
| 4 | C57BL/6 | HET | 12 | 10 | Anesthesia, recovery |
| 4 | C57BL/6 | HOM | 9 | 7 | Anesthesia |
| 12 | C57BL/6 | WT | 11 | 8 | Anesthesia, recovery |
| 12 | C57BL/6 | HET | 8 | 7 | Recovery |
| 12 | C57BL/6 | HOM | 9 | 4 | Anesthesia, recovery |
|
| |||||
| Experiment: QUIN time course, 10, 13 or 16 day survival
| |||||
| 6 or 12 | FVB | WT | 13 | 12 | Recovery |
| 12 | FVB | HET | 13 | 12 | Recovery |
|
| |||||
| Experiment: QUIN, 7 day survival, comparing HD mouse models
| |||||
| 12 | FVB | WT | 15 | 10 | Anesthesia, recovery |
| 12 | FVB | YAC128 | 14 | 14 | |
| 12 | FVB | HET | 3 | 3 | |
| 12 | FVB | HOM | 1 | 1 | |
|
| |||||
| Experiment: Kainic acid treatment, 3 day survival
| |||||
| 12 | FVB | WT | 2 | 2 | |
| 12 | FVB | HET | 10 | 7 | Within 3 hrs post KA |
| 12 | FVB | HOM | 5 | 1 | Within 3 hrs post KA |
To define the time course of QUIN-induced striatal neurodegeneration, mice were injected as above and killed 10, 13, and 16 days post QUIN (for group sizes see table). For consistency with previous studies, a time-point of 7 days was used for most experiments although we also assessed neurodegeneration at time points following 7 days. WT mice used in the time course experiment were WT FVB/NHsd.CAG140 N10Fx (i.e. non-CAG140-Htt allele containing) littermates and FVB/NHsd (i.e. non-transgenic) mice ordered from Harlan (the only time these mice were used in the study). For clarity, we distinguish these mice as WT FVB.CAG140 and FVB/NHsd hereafter. Both sources of ‘WT’ mice had considerable QUIN-induced striatal neurodegeneration that was indistinguishable and thus quantitative data from the two groups were pooled.
We also compared the extent of QUIN-induced striatal neurodegeneration in FVB.CAG140 mice with YAC128 animals, another HD model produced and maintained on a FVB/N strain background. For this comparison, 12 month old YAC128 mice and WT, HET and HOM FVB.CAG140 animals received QUIN injections as described above and mice were killed 7 days post injection. QUIN-induced striatal neurodegeneration in the four FVB.CAG140 mice injected in this experiment was not significantly different from the five animals in the first experiment, so data from the two groups were pooled.
Kainic acid treatment for inducing status epilepticus
To determine whether resistance extends to another type of excitotoxic neurodegeneration, we assessed hippocampal neurodegeneration after an episode of status epilepticus. Twelve month old FVB.CAG140 mice (n = 10 HET and 5 HOM) and WT littermates (n = 2) received subcutaneous injections of kainic acid (KA), which causes seizures that lead to hippocampal cell death. Mice were given 30 mg/kg KA, with supplemental doses of 15 mg/kg every 45 minutes until the mice exhibited level five seizures [9]. Seizure duration and intensity was scored according to a six-point seizure scale [8] adapted for mice. Seizures were rated at 5 minute intervals over a continuous four hour period. Two HET and three HOM FVB.CAG140 mice died during the status epilepticus; one HET and one HOM FVB.CAG140 mouse died during the post-injection survival period. A total of two WT, seven HET and one HOM FVB.CAG140 mice survived following extended status epilepticus, and were euthanized 3 days post KA by anesthetic overdose. Five age and sex matched WT FVB.CAG140 mice that did not receive injections of KA were used as non-injected controls (NIC) for cresyl violet cell quantification. Mice were perfused and brains were prepared as described above.
Assessing neurodegeneration and survival
To detect degenerating neurons, every fourth coronal section was mounted on a slide and stained with FluoroJadeB, an anionic fluorescein derivative (FJB, Histochem) [36]. Slides were immersed in 100%, 70% ethanol and dH20 followed by 0.06% potassium permanganate for 10 minutes at room temperature (RT). After rinsing, slides were immersed in 0.001% FJB/0.1% acetic acid for 20 minutes at RT, cleared in xylenes and mounted with DPX.
Degenerating (FJB-positive) neurons were counted using Image J (v1.40 g, NIH). The injections produced a roughly spherical area containing degenerating neurons. To estimate the number of degenerating neurons in a section, the area of the striatum was delineated, a threshold for determining cell size was set, and the total number of FJB-positive cells within each section was determined. The numbers represent the number of cells counted only in the one-in-four series of sections analyzed for each animal. To compare manual cell counts to counts collected automatically by Image J, four sections were chosen for both manual and automated cell counts. The manual (136, 140, 637, and 2216) and automated (115, 121, 670 and 1999) counts for the same four sections were significantly correlated to each other (P = 0.0016, R2 = 0.997).
Neuronal degeneration in the hippocampus after KA-induced status epilepticus was assessed using NIH Image J to count FJB-positive neurons and measure the length of the cell layers in the dentate gyrus (DG), CA1, CA3 and the hilus in four sections spaced at 40 μm intervals beginning approximately 1.5 mm caudal to bregma. Values were averaged and divided by the length of the subfield to obtain counts of FJB-positive neurons per unit length of the cell body layers in each region for each mouse.
To assess neuronal survival in the hippocampus after KA-induced seizures, coronal sections (40 μm) were collected and every fourth section was processed for cresyl violet staining. Tissue was first rinsed in PBS, followed by 0.05% cresyl violet (15 min at RT) After rinsing, slides were immersed in 70%, 95%, and 100% ethanol then allowed to dry before being cleared in xylenes and cover slipped with DPX. Cell counts were made in areas CA1, CA3 and the dentate gyrus. Images were obtained using a 60×, 1.4 NA objective on an Olympus A × 80 microscope with the aid of Magnafire-SP software in every alternately stained section (~320 μm separation distance). Four counting frames (100 × 100 μm) per section were randomly selected and the number of cresyl violet stained cells was evaluated using Image J. Criteria used for counting neurons were (1) presence of a nucleus; (2) a characteristically healthy neuronal morphology and (3) a cell body at least 10 μm in diameter. Cell counts were averaged per animal and final counts were shown as percentages compared to control mice.
Striatal and lesion volume measurements
Striatal and lesion volume were analyzed planimetrically in every eighth section in FJB stained tissue using NIH Image J. The striatum was defined using the corpus callosum and lateral ventricle as the rostral, dorsal and medial boundaries and the anterior commissure as the caudal endpoint. The lesion was defined as the area containing FJB-positive neurons. Total striatal volume on each side or lesion volume on the injected side was then calculated using the Cavalieri equation.
Immunohistochemistry
Immunostaining for mHtt aggregates was performed using mouse monoclonal anti-human Htt antibody (EM48, 1:200, MAB5374, Chemicon). The EM48 monoclonal antibody was generated against a GST fusion protein composed of the first 256 amino acids of human huntingtin lacking the polyglutamine tract. EM48 recognizes an epitope in human huntingtin exon 1 that is included in the Htt CAG140 knock-in allele. Endogenous peroxidases were blocked in frozen sections (35 μm) by immersion in 4% hydrogen peroxide/10% methanol for 10 minutes at RT. Sections were then blocked by incubation in PBS containing 5% normal goat serum (NGS) and 0.1% Triton-X 100 at RT for 2 hr. Sections were reacted with antibody in blocking buffer by static incubation in a humidified chamber overnight at RT. Western blots using lysate from HEK293 cells with EM48 produces a single band at a molecular weight of >250 kDa (Millipore product information), and stains mHtt aggregates in transgenic animals that express 82 to 150 CAG repeats in the human Htt protein. As a positive control, sections from transgenic R6/2 mice were processed in parallel to tissue from this study. A key control for antibody specificity is that immunostaining for EM48 is absent in FVB.CAG140 WT mice. No immunostaining was seen on control sections in which the primary antibody was omitted.
Medium spiny neurons were identified by using an antibody against dopamine and cAMP regulated phosphoprotein, 32 kD (DARPP-32, 1:2000, AB1656, Millipore), performed as for EM48 immunostaining (without Triton). This polyclonal antiserum was generated against a synthetic peptide (CQVEMIR-RRRPTPAM) coupled to KLH and recognizes phosphorylated threonine 34 of the DARPP-32 protein. When used for immunohistochemistry, the DARPP-32 antibody selectively stains medium sized stellate neurons in the striatum. This selective pattern of staining is one confirmation of antibody specificity. No immunostaining was seen on control sections in which either the primary or secondary antibody was omitted.
For non-fluorescent immunohistochemistry, biotin conjugated secondary goat anti-mouse (for EM48) and goat anti-rabbit (for DARPP-32) were used. Antibodies were visualized by the Avidin-Biotin Complex (ABC) system with diaminobenzimide (DAB) (Vector Labs, Burlingame, CA) and sections stained for DARPP-32 were counterstained with 0.5% methyl green. For fluorescent immunohistochemistry (Htt and FJB), goat anti-mouse secondary antibody conjugated to AlexaFluor 594 in PBS/NGS and 0.1% Triton-X 100 was used. In the final PBS rinse after incubation with secondary antibody sections were immediately transferred to 0.06% potassium permanganate in PBS for 4 minutes, rinsed in PBS for two minutes, transferred to 0.001% FJB/0.1% acetic acid staining solution for 10 minutes, and rinsed in PBS before clearing in xylenes and coverslipping with DPX.
Stereological analysis of numbers of DARP-32 positive neurons
Uniform random sampling of the coronally cut brain tissue was performed according to standard stereological principles in sections spaced 280 μm apart. An Olympus BX51 microscope with a motorized stage and Stereo Investigator software (Microbright field) were used. Sampling parameters were determined to get a coefficient of error (CE) ratio of less than 0.05. The optical fractionator probe was used to count nucleoli of DARPP-32 positive neurons in the striatum using a 60× objective in each contour. Quantification in Stereo Investigator using mean measured thickness yielded the estimated total number of DARPP-32 positive neurons within each striatum.
Immunoblotting
YAC128 and WT littermates, and FVB.CAG140 WT, HET and HOM mice (6 or 12 months old) were euthanized with an overdose of Avertin and brains were removed. Olfactory bulbs and the cerebella were removed, and the remaining brain was divided into right and left hemispheres and snap-frozen in dry ice cooled isopentane. Proteins were extracted from the right hemisphere by homogenization and sonication in SDP lysis buffer (50 mM Tris pH8.0, 150 mM NaCl, 1% Igepal, 40 mM B-glycerophosphate, 10 mM NaF, 1X Roche complete protease inhibitor, 1 mM sodium orthovanadate and 800 mM PMSF) followed by incubation on ice for 10 min. Debris was removed by centrifugation (15 min, 20,000 g, 4°C) and the supernatant retained. Protein concentrations were determined by DC assay (Bio-Rad). Samples were prepared for immunoblotting by denaturing the lysates in lithium dodecyl sulfate (LDS) sample buffer (Life Technology, Carlsbad, CA) with 100 mM DTT and heating to 70°C for 10 min. Samples were resolved using 10% low-BIS acrylamide gels [37]. Proteins were transferred to 0.45 μm nitrocellulose. Membranes were blocked with 5% milk in PBS, and then blotted for Htt (MAB2166, Millipore or BKP1 [38]) and calnexin (Sigma C4731) loading control. Proteins were detected with IR dye 800 CW goat anti-mouse (Rockland 610-131-007) and AlexaFluor 680 goat anti-rabbit (Molecular Probes A21076)-labeled secondary antibodies, and the LiCor Odyssey Infrared Imaging system. Mutant and WT Htt intensities were normalized to calnexin loading control and then to the average level of WT Htt from WT mice on the same membrane. Three mice per age and genotype were evaluated.
Filter retardation assay
To determine the relative amount of insoluble Htt, we used a filter-retardation assay to quantify the amount of large (>0.45 μm), SDS-insoluble aggregates [39] that do not pass through a 0.22 um pore size. The left hemisphere of the brains used in immunoblotting was analyzed. Lysate (50 μg) was diluted to 200 μl with 0.01% SDS and filtered through cellulose acetate membrane (Schleicher & Schuell, 0.22-μm pore size) with a Bio-Rad dot blot filtration unit. The assay was performed as described [40]. SDS-insoluble mHtt was visualized by immunoreactions with the EM48 antibody (Millipore, 1 : 1000). Peroxidase-conjugated AffiniPure goat anti-mouse or goat anti-rabbit secondary antibody (Jackson ImmunoResearch Laboratories) was used at 1:50,000 for 1 h at room temperature. Blots were detected using PICO detection reagent (Pierce). The amount of aggregated mHtt was normalized to the background levels detected in WT mice.
Statistical analysis
All data (with the exception of the microarray data, described above) were analyzed with Prism Graph-Pad (version 5.01). Where data did not display normal distribution and lacked equal variances, statistical testing was conducted accordingly. Comparison between two groups was performed with a Student’s t-test or Mann-Whitney U depending on whether the data were normally distributed. Comparisons with three or more groups were run with a parametric one way ANOVA with Bonferroni post test or Kruskal-Wallis analysis with Dunns post test depending on whether the data were normally distributed. Additionally, two-way ANOVA with Bonferroni post test was used when analyzing two factors.
RESULTS
FVB/NHsdCAG140 (FVB.CAG140) mice develop resistance to QUIN-induced striatal neurodegeneration
To begin to address whether strain can influence susceptibility to excitotoxicity in CAG140 mice, an intrastriatal injection of QUIN (15 nmol) was administered to FVB.CAG140 mice. Consistent with previous studies [10], QUIN caused extensive degeneration of striatal neurons in the FVB.CAG140 WT mice. Seven days following QUIN injections, large numbers of FJB-positive (degenerating) neurons were evident throughout the striatum on the ipsilateral side of injection (Fig. 1). The number of degenerating neurons counted following QUIN injection in the FVB/NHsd WT mice did not vary with age (Fig. 1A, D, G, J).
Fig. 1.

FVB.CAG140 mice develop resistance to QUIN-induced striatal degeneration. Striatal sections stained with FJB from 1-month-old (A) WT, (B) HET, and (C) HOM CAG140 mice on an FVB/NHsd background demonstrate vulnerability to QUIN. Representative sections from lesion epicenter (arrow points to syringe tract) of animals displaying cell death closest to the group mean. There is a different pattern at 4 months; (D) WT, (E) HET, and (F) HOM FVB.CAG140 mice demonstrate reduced cell death in the HOM mice (insets show higher magnification of FJB-positive cells, which are representative for all times). At 12 months the QUIN lesioned area is large in (G) WT, but dramatically reduced in the (H) HET and (I) HOM FVB.CAG140 mice; verified by quantification of QUIN-induced striatal neurodegeneration (J). A reduction in the number of FJB-positive cells is observed in the HOM mice at 4 and 12 months of age compared to WT and HET. At 12 months both HET (*P < 0.001) and HOM FVB.CAG140 animals exhibit less than 4% of cell death observed in WT. Both FVB HOM and HET mice have reduced cell death at 4 and/or 12 months compared to 1 and 2 months (+P < 0.0001). Data are mean ± SEM; N = 8 HOM, 12 HET, and 9 WT at 1 month, 10 HOM, 11 HET, and 9 WT at 2 months, 8 HOM, 7 HET, and 11 WT at 4 months, and 5 HOM, 6 HET, and 6 WT at 12 months; scale bar = 500 μm (50 μm in inset).
One and two month old HET and HOM FVB.CAG140 mice exhibited extensive QUIN-induced striatal neurodegeneration (Fig. 1B, C, J). At four months of age HET FVB.CAG140 mice (Fig. 1E) remain vulnerable to QUIN, displaying cell death similar to WT FVB/NHsd mice (Fig. 1B, J). Remarkably, four-month old HOM FVB.CAG140 mice showed ~30% reduction in QUIN-induced striatal neurodegeneration compared to that observed in WT and HET FVB.CAG140 mice; (Fig. 1F, J, P = 0.001, ANOVA). Group sizes used in statistics are as follows: 8 HOM, 12 HET, and 9 WT at 1 month, 10 HOM, 11 HET, and 9 WT at 2 months, and 8 HOM, 7 HET, and 11 WT at 4 months.
Resistance becomes essentially complete as mice age
We next tested resistance in older mice. At 12 months of age, WT FVB.CAG140 mice (Fig. 1G) still exhibited a high degree of vulnerability to injection of QUIN, comparable to 1, 2 and 4 month old mice (Fig. 1J). In contrast, 12 month-old HOM FVB.CAG140 mice (Fig. 1I) were essentially completely resistant to QUIN-induced striatal neurodegeneration, exhibiting only 3.6% of the cell death seen in WT littermate controls (Fig. 1G, J). Degenerating (FJB-positive) neurons were only observed in the area immediately surrounding the injection track as expected from the physical damage produced by the syringe. Even one copy of the CAG140 allele was sufficient to confer resistance to cell death at this age, with FVB.CAG140 HET mice (Fig. 1H) exhibiting 3.7% of the cell death seen in WT littermate controls (Fig. 1G, J). Thus, knock-in mice on an otherwise highly vulnerable strain background have significantly less cell death than their WT littermates (P = 0.012, ANOVA). There was significant overall effect of both age and genotype (P < 0.0001 and P = 0.0004 respectively, two-way ANOVA) in the FVB line. Group sizes used in statistics are as follows: 5 HOM, 6 HET, and 6 WT at 12 months.
Differences in QUIN-induced striatal neurodegeneration translate to differential preservation of medium spiny neurons
FJB facilitates identification of neurons that are degenerating at a given time point, but does not reveal the overall extent of neuronal loss. To address this, sections from four month old mice were stained with DARPP-32, which is a marker for spiny stellate neurons. Immunostaining for DARPP-32 was largely absent in the area surrounding the QUIN injection in the WT FVB.CAG140 mice (Fig. 2). In the area of diminished immunostaining, DARPP-32 positive neurons were absent (Fig. 2B, inset) resulting in a nearly 50% reduction in total striatal DARPP-32 positive neurons on the QUIN injected side compared to the control side (Fig. 2A, E, paired t-test, WT, control vs. QUIN injected striatum, P = 0.001). This is a substantial loss considering we are counting neurons from the rostral to the caudal boundaries of the striatum and not just within the lesion area. HET-FVB.CAG140 mice exhibited some protection from DARPP-32-loss (Fig. 2C, E) with cell death still apparent but less than in WT littermate controls (41.5% loss in WT littermate controls vs. 33.8% loss in HET mice). In keeping with the results of FJB staining, there was only a very small area of diminished DARPP-32 staining in FVB.CAG140 HOM mice (Fig. 2D) and no decrease in the total number of DARPP-32 positive neurons on the injected side (Fig. 2E, paired t-test, control vs. QUIN injected striatum, P = 0.338). Due to the DARPP-32 positive neurons outside the lesioned area, there was no significant difference in the number of neurons between the WT, HET and HOM mice in the QUIN injected striatum. To compare our method of quantifying DARPP-32 positive neurons to a measure of striatal volume, we estimated striatal volume in these same lesioned striata (Fig. 2F). Despite significant differences in the number of DARPP-32 positive neurons, there is no significant difference in striatal volume between groups 7 days post QUIN lesion. Five mice used in each group for this analysis.
Fig. 2.
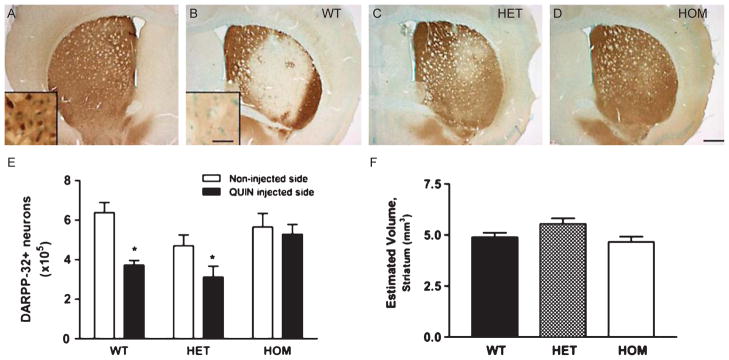
Counts of surviving medium spiny neurons confirm greater loss of neurons in WT FVB/N littermates. Striatal sections stained with DARPP-32 in 4 month old FVB.CAG140 (A, B) WT, vs. (C) HET and (D) HOM mice. Representative sections are from mice that had an amount of cell death that was closest to the group mean. (A) The section from the contralateral untreated side of the brain is from a WT animal, but is representative for all genotypes. There is no loss of DARPP-32 reactivity in knock-in mice. (E) Stereological quantification confirms that both the WT and HET groups have fewer neurons in the QUIN injected striatum than the non-injected striatum (*, P < 0.05 compared to non-injected side). The percent loss on the QUIN injected side (compared to non-injected side) was greater in the WT than the HET mice (41.5% loss in WT vs. 33.8% loss in HET FVB.CAG140 mice). In the QUIN injected striatum, DARPP-32 neuron counts were lower in the two groups that had the most cell death (WT and HET mice) compared to HOM mice. This difference was significant between the HET and the HOM mice at this time (p < 0.05). There were no differences between counts on the control side between genotypes. (F) Similarly, there are no significant differences in the striatal volume between groups. Data are mean ± SEM; N = 5 for all groups; scale bar = 500 μm (30 μm in inset).
Tissue sections were also stained for GFAP to examine astrogliosis. The area of reactive astrocytes is similar in the C57BL/6NHsd.CAG140 (B6.CAG140) WT and HOM (Fig. 3A, C), but was greater in the FVB.CAG140 WT (Fig. 3B) compared to FVB.CAG140 HOM mice (Fig. 3D). Despite the HOM mice having a small QUIN lesion, the area of reactive astrocytes (Fig. 3) was much larger than the area of FJB-positive neurons (compare to Fig. 1). For comparison, rodents given an injection of saline instead of QUIN have minimal GFAP reactivity [41]. Thus, astrocytes undergo hypertrophy even in areas in which there is minimal neuronal loss. Astrocytes have been shown to up-regulate GFAP in response to intense neuronal activity [42] and thus the large area of GFAP positive staining may be due to a direct action of QUIN on astrocytes.
Fig. 3.

Reactive astrocytes are similar in KI and WT mice. Reactive astrocytes stained with GFAP are present in both the C57BL/6 WT (A) and FVB/N WT (B) mice 7 days post QUIN injection. The area of reactive astrocytes appears slightly smaller in the HOM mice, C57BL/6 (C) and FVB/N (D) respectively, however the area of GFAP staining extends far past the border of FJB lesions for these mice at 12 months. Scale bar = 500 μm.
Time course of QUIN-induced striatal neurodegeneration
The above analysis of QUIN-induced striatal neurodegeneration was at a single time point (7 days). As FJB only labels degenerating and not dead cells, we wished to verify that cell death in FVB.CAG140 HOM mice was reduced compared with FVB.CAG140 WT mice, rather than being delayed. No reduction in surviving cells is seen in FVB.CAG140HOM mice 7 days post QUIN injection (refer to Fig. 2) indicating that cell death does not occur prior to this time point. To exclude the possibility that cell death was delayed in FVB.CAG140 HOM mice, FJB staining was assessed 10, 13 or 16 days post QUIN injection (Fig. 4). In general, the number of degenerating cells (FJB-positive cells) was considerably lower at 10–16 days than at 7 days post QUIN (compare to Fig. 1), suggesting that many degenerating neurons were either no longer FJB-positive or had been removed by phagocytosis. However, at all time points there were fewer FJB-positive cells in the FVB.CAG140 HOM mice vs. WT controls (Fig. 4, P = 0.01, 2 way ANOVA) indicating the CAG140 mice are indeed resistant to QUIN-induced striatal neurodegeneration. FJB staining localizes to the cell body, but was not present in every cell (Fig. 4H). Group sizes used in statistics are as follows: 5 WT and 4 HOM at 10 days, 4 WT and 4 HOM at 13 days, and 3 WT and 4 HOM at 16 days.
Fig. 4.

Resistance to degeneration persists 16 days post QUIN injection in aged mice. Striatal sections stained with FJB from 12 month old (A, C, E) WT and (B, D, F) HOM FVB.CAG140 mice demonstrate that there is no increase in cell death at (A, B) 10 days, (C, D) 13 days or (E, F) 16 days following time of injection. Representative sections are from mice that had an amount of cell death that was closest to the group mean. (G) Quantification shows clearly that FVB.CAG140 HOM mice consistently have less cell death than WT FVB.CAG140 mice (overall effect of genotype, P = 0.01). (H) FJB labeling is in the soma surrounding the nuclei labeled with Hoechst. Data are mean ± SEM; N = 5 WT and 4 HOM at 10 days, 4 WT and 4 HOM at 13 days, and 3 WT and 4 HOM at 16 days; scale bar = 500 μm (50 μm in H).
C57BL/6 mice carrying the CAG140 transgene also become more resistant than WT
Several previous studies have shown that mice carrying HD transgenes are more resistant to excitotoxicity [31, 33, 43]; however, a direct comparison of the effects of a particular HD transgene in different strains has not yet been performed. Accordingly, we compared the extent of striatal neurodegeneration in FVB.CAG140 (Fig. 1) vs. B6.CAG140 mice (Fig. 5) after equivalent intrastriatal injections of QUIN. Consistent with previous studies [10], B6.CAG140 WT mice had approximately half the cell death observed in FVB.CAG140 WT mice at one month (Fig. 5D vs Fig. 1J, P = 0.039, t-test, C57BL/6 WT vs. FVB/N WT). Nevertheless, B6.CAG140 HET and HOM mice became even more resistant between one to four months of age. At 1 and 2 months of age, the extent of striatal neurodegeneration was similar in WT, HET and HOM B6.CAG140 mice (P = 0.266 and P = 0.361, respectively, ANOVA). However, at four months cell death in the B6.CAG140 HET and HOM was reduced compared to WT (P = 0.003, ANOVA), similar to that observed in the FVB/N line. The HOM B6.CAG140 mice have the greatest reduction in cell death, but both the HET and HOM are significantly reduced compared to WT.B6.CAG140 HOM mice show even greater resistance compared to WT C57BL/6 mice at 12 months of age (Fig. 5B, C, P = 0.007, ANOVA).
Fig. 5.

C57BL/6 strain resistance to QUIN-induced striatal degeneration is enhanced by expression of CAG140 allele of Htt. Wild type C57BL/6 mice are much more resistant to QUIN-induced striatal neurodegeneration than FVB/N WT mice (Compare with incidence of FJB-positive cells in Fig. 1). Striatal cross sections at the epicenter of the QUIN lesion are much smaller in WT (A), HET (B), and HOM (C) B6.CAG140 mice at 12 months of age. A reduction in QUIN lesioned area can be seen in the HET and HOM mice, but the magnitude of reduction is not as great as in the FVB.CAG140 strain. The patterns of resistance are seen in the quantification of cell death (D). Four month old HOM and HET B6.CAG140 mice have a reduction in cell death compared to WT B6.CAG140 mice at this age, which persists to 12 months (*P < 0.005). The effect of age is significant in both the C57BL/6 lines as well, as the HOM starts showing resistance at 4 months compared to 1 and 2 months, and this happens at 12 months for the HET (+P < 0.001). Data are mean ± SEM; N = 8 HOM, 7 HET, and 7 WT at 1 month, 6 HOM, 8 HET, and 7 WT at 2 months, 7 HOM, 8 HET, and 9 WT at 4 months, and 3 HOM, 6 HET, and 6 WT at 12 months; scale bar = 500 μm.
WT C57BL/6 mice are much more resistant to excitotoxic lesions than the WT FVB/N mice (Fig. 5A, D compared to Fig. 1). Because WT C57BL/6 mice are already resistant based on their genetic background, the magnitude of CAG140-induced protection is not as great. This decreased susceptibility to QUIN in the WT C57BL/6 mice with age is similar to what has been reported previously in the WT FVB/N littermates [33]. In analyzing the amount of QUIN-induced striatal neurodegeneration in all three C57BL/6 genotypes across all four ages, there is an overall effect of age, but not of genotype (P = 0.0005 and P = 0.098 respectively, two-way ANOVA). Group sizes used in statistics are as follows: 8 HOM, 7 HET, and 7 WT at 1 month, 6 HOM, 8 HET, and 7 WT at 2 months, 7 HOM, 8 HET, and 9 WT at 4 months, and 3 HOM, 6 HET, and 6 WT at 12 months. Because of this, we focused further analyses on the resistance in the FVB.CAG140 mice.
FVB.CAG140 mice also develop resistance to seizure-induced cell death in the hippocampus
To explore the scope of resistance to excitotoxins, we assessed a different form of neurodegeneration in 12 month old FVB.CAG140 mice. HET FVB.CAG140 mice exhibit full resistance to QUIN-induced striatal neurodegeneration at 12 months of age, so these mice were used to analyze hippocampal neuron death that occurs after status epilepticus seizures. Hippocampal neurodegeneration following KA-induced seizures has been well characterized in different strains of mice, and FVB/N mice are highly vulnerable [10]. The net dose of KA required to elicit persistent class 5 seizures was similar in WT and HET FVB.CAG140 mice (45 ± 21 mg/kg vs. 45 ± 12 mg/kg respectively) as was latency to the first seizure (55 ± 7 min vs. 89 ± 33 min respectively), and seizure duration (63 ± 18 min vs. 78 ± 50 min respectively).
Despite these similarities, a dramatic difference was observed in hippocampal neurodegeneration between WT and HET FVB.CAG140 12 month-old animals. Consistent with previous reports [9], WT mice exhibited extensive neuronal degeneration throughout the hippocampus evidenced by loss of neurons in cresyl violet-stained sections (Fig. 6A, B), large numbers of FJB-positive cell bodies (Fig. 6C, D), and an overall increase in FJB staining in the neuropil layers indicating degenerating axons and dendrites. In contrast, there was minimal neuronal loss (Fig. 6E, F) or FJB staining (Fig. 6G, H) in 12-month-old HET FVB.CAG140 mice.
Fig. 6.

Resistance to KA-induced hippocampal degeneration in aged FVB.CAG140 mice. Adjacent sections were stained with either cresyl violet or FJB. (A, B) Sections from WT FVB.CAG140 (WT) mice stained with cresyl violet show a marked cell loss in the dentate gyrus (DG), hilus, CA3 and CA1 of the hippocampus. (B) Individual cells can be seen in CA3, where great loss is apparent. (E, F) In contrast, the HET FVB.CAG140 (CAG140) mice have healthy neurons and no apparent loss in the same regions. (C, D) FJB-labeled degenerating neurons in WT mice are present throughout (C) the hippocampus and (D) at higher magnification in CA3. (G, H) In contrast, FJB-stained sections from HET FVB.CAG140 mice reveal few degenerating neurons. (I) Quantification of cell survival indicates greater cell survival in HET FVB.CAG140 mice compared to WT (*, P < 0.01), and fewer FJB+ neurons in CA3 (J, *, P < 0.05). N = 2 WT, 5 HET and 5 controls that did not receive KA; scale bar = 500 μm (A, C, E, G) and 100 μm (B, D, F, H).
CA3 is an area in which neuronal death is thought to occur primarily via excitotoxic mechanisms rather than ischemia [44], so we quantified cell death in this area. Counts of the number of FJB-positive neurons in CA3 confirmed the qualitative impression of reduced cell loss in CAG140 mice (P = 0.03, t-test, Fig. 6J). Nissl-stained sections reveal that by day 3 there is already extensive cell loss, so some degenerating neurons have already been eliminated indicating that counts of FJB-positive cells likely underestimate the extent of cell death. As a control, cells were also counted in WT FVB.CAG140 mice not injected with KA (NIC). WT FVB.CAG140 mice that were injected with KA had a massive loss of neurons (to a level of about 65%) in CA3 compared to both HET FVB.CAG140 mice and WT NIC mice (Fig. 6I, P = 0.005, one-way ANOVA). In contrast, cell counts in KA-treated HET FVB.CAG140 were near control levels (not significantly different with Tukey’s post-hoc test), demonstrating striking resistance of HET FVB.CAG140 mice to KA-induced neurodegeneration. Group sizes used in statistics are as follows: 2 WT, 5 HET and 5 controls that did not receive KA.
Status epilepticus in WT FVB/N mice results in neuronal death in CA3, as well as in other regions including the striatum, cerebral cortex, and thalamus. Qualitative assessments of FJB-positive cells reveal this scattered neurodegeneration in WT FVB.CAG140 mice but degeneration was minimal in all brain regions in HET FVB.CAG140 mice (Table 2).
Table 2.
Qualitative assessment of neurodegeneration following KA-induced seizures
| CA3 | CA1 | DG | Striatum | Cortex | Subiculum | Thalamus | |
|---|---|---|---|---|---|---|---|
| WT FVB | +++ | +++ | +++ | + | ++ | + | ++ |
| CAG 140.FVB | + | − | − | − | − | + | − |
Amount of FJB positive neurons were qualitatively assessed using a three point scale in several regions of the brain affected by seizure induced cell loss. −, minimal to no degeneration; +, moderate degeneration; ++, moderate degeneration in >50% of mice; +++, extensive degeneration in >50% of mice.
Resistance is greater in CAG140 mice than YAC128 mice
The susceptibility of YAC128 mice to striatal QUIN has been previously assessed, so we next compared the number of FJB-positive neurons following treatment of knock-in model mice or a full-length transgenic model with a consistent dose of QUIN. QUIN-induced striatal neurodegeneration was assessed in 12 month-old mice: WT FVB/N (Fig. 7A), hemizygous YAC128 mice (Fig. 7B), and FVB.CAG140 (Fig. 7C). Since results are very similar in HET and HOM FVB.CAG140 mice, results were pooled in these two groups. Quantitative analyses of numbers of FJB-positive neurons (Fig. 7D) revealed that YAC128 mice display less QUIN-induced striatal neurodegeneration than age matched FVB/N WT controls. This is consistent with previous reports that assessed QUIN sensitivity using lesion volume measurements [33].
Fig. 7.

FVB.CAG140 mice have significantly less QUIN-induced striatal neurodegeneration than YAC128 mice on an FVB/N strain background. Both types of HD transgenic mice show resistance to cell death compared to WT FVB/N mice. Representative striatal sections stained with FJB from 12 month old (A) WT FVB.CAG140, (B) YAC128 and (C) FVB.CAG140 (pooled HET and HOM). (D) Cell death was quantified by counting FJB-positive cells in every fourth section. The amount of FJB-positive cells is significantly lower in the FVB.CAG140 mice than both the YAC128 and WT groups (+, *P < 0.01). (E) Quantification of lesion volume. Lesion volume is progressively smaller in each group, however by post-hoc analysis lesion size in FVB.CAG140 mice were only significantly smaller than WT FVB.CAG140 mice (*P < 0.05). Data are mean ± SEM; N = 8 WT, 13 YAC128, and 9 CAG140; scalebar = 500 μm.
FVB.CAG140 mice exhibited far less cell death than YAC128 mice. The amount of cell death in the three groups differed significantly (P = 0.0004, Kruskal-Wallis), and Dunn’s post-test confirmed that FVB.CAG140 mice had significantly less cell death than both WT and YAC128 mice. For comparison to previous studies of QUIN-induced striatal neurodegeneration we also assessed lesion volume and saw similar differences between the three groups (P = 0.011, Kruskal-Wallis). Post hoc comparisons revealed that FVB.CAG140 mice had significantly smaller lesions than WT FVB.CAG140 animals, but lesion volumes between FVB.CAG140 and YAC128 mice were not significantly different. Group sizes used in statistics are as follows: 8 WT, 13 YAC128, and 9 CAG140.
Resistance develops concurrently with the appearance of mutant Htt aggregates
Mice carrying a mutant HD transgene on a genetic background that is already relatively resistant to excitotoxic neurodegeneration (i.e. C57BL/6) develop even greater resistance as the mice age [32]. It has been hypothesized that aggregates of mHtt play a role in triggering the development of resistance [45]. Accordingly, we investigated whether the development of resistance in HET and HOM FVB.CAG140 mice is related to the appearance of mHtt aggregates in striatal neurons. Sections were stained using the EM48 antibody that recognizes aggregated mHtt [46]. As reported [18], mHtt aggregates developed with age in HET and HOM FVB.CAG140 mice. In one month old FVB.CAG140 mice that are vulnerable to an injection of QUIN, no detectable striatal mHtt aggregates are visible in HET FVB.CAG140 mice (Fig. 8A), and very few in HOM mice (Fig. 8B, C). At 4 months of age when resistance is seen in the HOM mice, nuclear aggregates (indicated by arrowheads) of mHtt are present (Fig. 8E). A larger number of EM48 positive neurons are seen in the HOM FVB.CAG140 mice (Fig. 8E, F), which already exhibit induced resistance in comparison to WT (not shown) and HET mice (Fig. 8D). In 12 month-old HOM FVB.CAG140, extensive mHtt labeling is present in both the HET and HOM groups (Fig. 8G, H, I). Nuclear inclusions are indicated by an arrow. There is no detectable staining in 12 month old WT mice (Fig. 8L).
Fig. 8.

Mutant HTT staining in FVB.CAG140 and YAC128 mice. Representative images of mutant HTT labeled with EM48 in the non-injected striatum. Little staining is present in1 month-old FVB.CAG140 (A) HET, but (B, C) HOM knock-in mice have light nuclear staining (arrowhead). Four month-old, (D) HET and (E, F) HOM FVB.CAG140 mice have increased nuclear staining (arrowheads) with aggregation in the HOM compared to HET mice. By 12 months of age, nuclear staining and nuclear inclusions (arrows) are apparent in both (G) HET and (H, I) HOM FVB.CAG140 mice. (J, K) Staining performed in parallel of sections from 12 month old YAC128 mice have considerably less mutant HTT staining than 4 month-old HET and HOM FVB.CAG140 mice (D–F). Nuclear staining (arrowheads) and aggregation (arrows) is denoted, although this is less intense than FVB.CAG140 knock-in mice. (L) Sections from WT FVB/N has no EM48 staining. Scale bar = 100 μm (50 μm in C, F, I, K).
Possible explanations for the difference in the extent of resistance between FVB.CAG140 and YAC128 are that either expression or aggregation of mutant protein is reduced in YAC128 mice. To test this, sections from YAC128 mice were immunostained with the EM48 antibody. We also quantified soluble mHtt, and by filter retardation assay large (>0.22 μm) SDS-insoluble mHtt, using infrared-based western analysis. Sections from 12-month-old YAC128 mice stained positively for EM48 (Fig. 8J, K), but the level of staining and number of aggregates appeared lower than in sections from HET and HOM FVB.CAG140 mice processed side by side. EM48 does not recognize mouse Htt, so this could account for the much lighter staining in the YAC128 mice. Despite the apparent differences in immunostaining, quantitative western revealed that soluble mHtt levels were similar between both 6 and 12 month CAG140 and YAC128 mice (Fig. 9A, B). As expected, FVB.CAG140 HET and HOM mice have reduced expression of WT Htt compared to YAC128 mice (Fig. 9B). The quantity of larger, insoluble mHtt species was also similar between YAC128 and CAG140 HET mice (Fig. 9C). At 12 months, the levels of insoluble mHtt are higher than at 6 months, except in the HOM mice. These results indicate that differences in the extent of resistance between FVB.CAG140 and YAC128 cannot be explained by either differences in levels of monomeric mHtt or insoluble mHtt, although it is possible that reduced level of wild-type Htt contributes to the resistance to QUIN-induced striatal neurodegeneration observed in HET and HOM FVB.CAG140 mice compared with FVB.YAC128 animals. For this section, group sizes are 3 for all groups in the western blot, and 2 for all groups in the filter retardation assay.
Fig. 9.

Mutant Htt expression is similar in FVB.CAG140 and YAC128 mice. Western blot of soluble mHtt protein from a half-brain homogenate (A) Two different anti-huntingtin antibodies demonstrate that there are similar levels of mHtt protein in YAC128 and FVB.CAG140 mice. (B) Levels are normalized to calnexin and then to endogenous Htt levels. The expression is also very similar between 6 and 12 months of age. As expected, FVB.CAG140 HET and HOM mice have reduced expression of WT Htt compared to YAC128 mice. Filter retardation-based assay to quantify the presence of insoluble mHtt protein (C) The amount of insoluble protein detected by the EM48 antibody is also similar between the two strains of mice, however the levels of insoluble protein are much higher at 12 months than 6 months. Data are mean ± SEM; N = 3 for all groups in the western blot, and 2 for all groups in the filter retardation assay.
DISCUSSION
FVB.CAG140 HOM mice develop virtually complete resistance to striatal QUIN injections with age, while WT littermates remain highly vulnerable. Resistance to QUIN is also observed in HET FVB.CAG140 mice although the resistance is acquired at a later age compared to HOM animals. This transition from vulnerability to resistance demonstrates that striatal neurons are capable of surviving an otherwise fatal excitotoxic lesion. Our results, together with previously published data, provide convincing evidence that mouse strain is an important modulator of phenotype, as is the design and integration of the genetic mutation. The differences between these HD mouse models provide an opportunity to identify genetic factors and ultimately the molecular mechanism underlying the differential cell death and survival observed. Here, we highlight the magnitude of resistance in the FVB.CAG140 compared to other HD mouse models that have been shown to demonstrate resistance to QUIN-induced striatal neurodegeneration [28, 29, 31–33]. A compelling interpretation is that the presence of the CAG140 allele, in combination with reduced expression of wild type Htt, causes cellular and molecular changes that confer resistance to excitotoxic neurodegeneration.
A possible alternative explanation for our findings is that FVB.CAG140 mice may carry some genes from the original C57BL/6 strain, which is resistant to excitotoxic cell death. The mice used here are the result of backcrossing 129S1B6-CAG140 mice with FVB/N mice (or C57BL/6 as a control). Despite backcrossing to a congenic FVB background, our colony of FVB.CAG140 mice likely carries a small residual portion of chromosome 5 from the strain background used to generate the original targeted CAG140 allele of Htt as well as the parental strain. This region could contain alleles of loci from C57BL/6J or 129S1 i.e. from the W9.5 ES cell line used to generate the targeted mutation of huntingtin [18]. Because C57BL/6 mice are resistant to QUIN-induced excitotoxic damage, it is possible that induced resistance in the FVB.CAG140 strain is due to gene(s) present on this small portion of chromosome 5 rather than to the HD transgene. After 10 generations of backcrossing, the segment of chromosome 5 flanking huntingtin that would have originated from the original CAG140 mice is expected to be about 20 cM [47]. Although some C57BL/6 genes may be present, two explanations make it unlikely that these genes account for the induced resistance.
First, C567BL/6 mice are resistant to excitotoxicity throughout their life, whereas FVB.CAG140 mice only develop resistance as they mature. This is in contrast with the YAC128 mice, which are vulnerable (compared to WT FVB/N mice) early in their life, and then develop resistance with age (compared to when they are young) [33]. This fact alone strongly suggests that resistance is not due to residual C57BL/6 genes. Second, using the same techniques as for FVB.CAG140 mice, we show here that YAC128 mice, which are on a pure FVB/N background, also develop resistance to QUIN-induced striatal neurodegeneration as they age. This is consistent with previous results [33] which showed that following injection of QUIN at roughly half the dose used here, lesion volume was less in YAC128 mice in comparison to WT controls beginning around 6 months of age.
Differences between FVB.CAG140 mice and YAC128 mice
The CAG140 knock-in model was used in this study because expression of the mHtt protein is under the control of the endogenous mouse huntingtin locus. Instead of expressing WT protein in addition to mHtt protein, HET FVB.CAG140 mice express approximately 50% less wild type Htt, while HOM FVB.CAG140 mice express no WT Htt. In contrast, YAC128 mice express full-length human mHtt under the control of the HUNTINGTON promoter and regulatory elements on the yeast artificial chromosome (YAC). In YAC128 mice, both copies of WT Htt are still present.
One possibility underlying the difference between FVB.CAG140 and YAC128 mice could be in the mutant protein itself (i.e. polymorphisms between the human and mouse protein) or in differential expression or processing of the protein in the two models. There are several identified species and proteolytic cleavage products of mHtt, and accordingly, different ways to detect these species. Data from western blot and filter retardation assays indicate that the YAC128 mice have more WT Htt than the CAG140 mice, as expected. Since full-length WT Htt has been shown to be neuroprotective [48, 49], it is counterintuitive that the CAG140 mice are more resistant than the YAC mice. This further suggests that the resistance is not simply due to levels of WT Htt, but to some property of mHtt itself.
Overall concentrations of soluble and insoluble mutant Htt was relatively comparable in the two strains. While the two assays used measure the levels of monomeric Htt (WT and mutant) and larger, insoluble mHtt, these assays, even together, cannot account for total Htt, as there are likely oligomeric species present not detectable by either of these methods [50, 51]. The relative amount of mHtt, as detected by immunohistochemistry using EM48, appeared dramatically different between FVB.CAG140 and YAC128 mice. EM48-positive nuclear aggregates were much more prominent in the FVB.CAG140 mice than the YAC128 mice, despite identical tissue treatment and staining conditions. A possible reason for this includes the continued presence of WT Htt in the YAC128 mice. This could influence reactivity to EM48, which preferentially recognizes expanded repeat and aggregated Htt.
Relationship between mHtt aggregates and resistance
If nuclear aggregation differs between these two mouse models, then it is important to consider how the presence of mHtt affects a cell’s response to an excitotoxin. The remarkable resistance to excitotoxicity described in the current study develops around the time that nuclear micro aggregates appear in the HOM FVB.CAG140 mice. In HET FVB.CAG140, both the appearance of nuclear aggregates and the development of resistance are delayed. At four months, there is a clear difference between the HET and HOM FVB.CAG140 in the number of EM48-positive aggregates. HOM mice, which have two copies of mHtt, show a resistant phenotype at this age, but the HET mice, which have one copy of mHtt and one copy of WT Htt, do not, demonstrating mHtt dose dependence of induced resistance. This would be consistent with previous hypotheses that mHtt aggregates may activate cellular mechanisms that can protect neurons from apoptosis (reviewed in [52]) and findings showing that resistance to NMDA toxicity correlates with the appearance of nuclear inclusions [32].
HD mouse models differ in the development of pathology. The time of nuclear localization, the time of aggregation, and the extent of mHtt expression varies with both genetic model and strain background [26]. Brooks et al. [53] summarized the onset of mHtt aggregation and the formation of nuclear inclusions in several mouse models that have varying degrees of susceptibility (see introduction for comparisons). However, the use of different HD mouse models to correlated aggregation with susceptibility to excitotoxicity may be confounded by different models’ unique disease progression profiles and appearance of aggregates, which may not be comparable across models.
More work is needed to fully understand how the different soluble, oligomer and aggregate forms of mHtt impact the cellular environment. Mutant Htt can be cleaved, producing an N-terminal fragment that is toxic, and the appearance of this fragment corresponds with behavioral and neurological dysfunction in mouse models of HD [1, 46, 54]. It has been proposed that aggregated mHtt is less toxic than oligomeric forms of this mutant protein and there is evidence that aggregates can protect cells from death [45]. However, the role of the protein in cell survival is complex, as there are potentially several oligomeric forms that differentially contribute to neurotoxicity [55].
Possible mechanisms underlying age-dependent resistance
The mechanisms that underlie the development of resistance are currently unclear. The evidence presented here together with other reports suggests that vulnerability is specified by several factors. Identification of these factors could inform future therapeutic approaches and efforts are in progress to examine possible mechanisms, including altered expression of genes that promote survival versus excitotoxic cell death. A microarray analysis comparing expression patterns in FVB.CAG140 mice and WT controls at different ages did not reveal genes that show unique expression patterns in the aged resistant FVB.CAG140 HOM mice compared to all three of their vulnerable counterparts (manuscript in preparation). The analysis did reveal changes in gene networks, however, that occur with aging as well as in the HD mice compared to the WT littermates. For example, at 12 months, the pathways that differ between the HD and wild type mice are: N-terminal amino acid modification, regulation of homeostatic process, and apoptotic mitochondrial changes that could account for the increased survival in the FVB.CAG140 mice.
Additional mechanisms for the development of resistance could also involve alterations in synaptic signaling in CAG140 mice [23, 56, 57]. Neurons in HD mouse striatum do have altered synaptic function [58, 59] (and reviewed in [60]), which could contribute to altered susceptibility to excitotoxicity. Changes in mitochondrial function with age might also contribute to develop of resistance. Further studies are needed to examine the effect of strain background on synapses, and how that might influence excitotoxic susceptibility in the FVB.CAG140 mice.
Mutant Htt influences multiple molecular pathways [61]. For example, mHtt regulates levels and activity of a protein called striatal-enriched protein tyrosine phosphatases (STEP). The STEP pathway is down regulated in the presence of mHtt, and when this pathway is down-regulated, it could contribute to induction of compensatory mechanisms leading to the resistance of excitotoxicity [62]. Disruption of STEP also attenuates cell death due to excitotoxicity in the hippocampus [63]. In addition, mHtt affects cellular processes such as transcription, transport, metabolism, and protein degradation, any of which might lead to age-dependent resistance described here.
Susceptibility to excitotoxicity is unlikely to be cell-autonomous in HD and may involve interactions between multiple cell types. A very recent study uses chimeric mice with cells of both resistant and vulnerable strain background to show that resistant neurons become more vulnerable in the presence of cells (both neurons and glial cells) from a more vulnerable strain background, and vice versa, but the intrinsic phenotype couldn’t be completely switched [64]. Here, GFAP staining, which monitors astrocyte activity, showed a larger area of GFAP reactivity remaining, even though there was a reduction in the QUIN lesion in the FVB.CAG140 HOM mice. This could indicate that although there is no QUIN-induced striatal neurodegeneration in these mice, there is neuronal distress to which the astrocytes are responding. It is also possible that they contribute to the resistance seen after excitotoxic lesion.
Induced resistance is not unique to the striatum
In FVB.CAG140 mice, resistance extends to other types of excitotoxic neurodegeneration. Our results reveal that aged FVB.CAG140 mice are also resistant to excitotoxic cell death due to KA-induced seizures, and not just QUIN. Because these mice have resistance to multiple types of excitotoxicity, the mechanism underlying resistance is likely downstream of the particular post synaptic receptor activated by the excitotoxin. The age-dependent development of striking resistance in FVB.CAG140 mice provides a unique setting in which to investigate the cellular and molecular factors influencing vulnerability to excitotoxic cell death.
Acknowledgments
Supported by the Roman Reed SCI Program of the State of California, private donations to the RIRC, a grant from the CHDI (#39890) to OS. MKS was the recipient of (#TL1 TR000148), a grant from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) and the NIH Roadmap for Medical Research (#UL1 RR031985). We thank Dr. M.F. Chesselet (University of California, Los Angeles) for generously donating the founding CAG140 mice. Thanks to Kelly Matsudaira Yee for assistance with Htt immunostaining, Dr. Emily Mitchell Sontag for assistance with the filter retardation assay, and Crystal Doty and Kuljeet Vaid for protocols and assistance with Htt immunoblotting.
Footnotes
CONFLICT OF INTEREST
None declared.
References
- 1.DiFiglia M. Excitotoxic injury of the neostriatum: A model for Huntington’s disease. Trends Neurosci. 1990;13(7):286–9. doi: 10.1016/0166-2236(90)90111-m. [DOI] [PubMed] [Google Scholar]
- 2.Gil JM, Rego AC. Mechanisms of neurodegeneration in Huntington’s disease. Eur J Neurosci. 2008;27(11):2803–20. doi: 10.1111/j.1460-9568.2008.06310.x. [DOI] [PubMed] [Google Scholar]
- 3.Dong XX, Wang Y, Qin ZH. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin. 2009;30(4):379–87. doi: 10.1038/aps.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature. 1986;321(6066):168–71. doi: 10.1038/321168a0. [DOI] [PubMed] [Google Scholar]
- 5.Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, et al. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington’s disease. Neuron. 2002;33(6):849–60. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- 6.Zeron MM, Fernandes HB, Krebs C, Shehadeh J, Wellington CL, Leavitt BR, et al. Potentiation of NMDA receptor-mediated excitotoxicity linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington’s disease. Mol Cell Neurosci. 2004;25(3):469–79. doi: 10.1016/j.mcn.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 7.McLin JP, Thompson LM, Lusis AJ, Davis RC, Steward O. Genes on distal chromosome 18 determine vulnerability to excitotoxic neurodegeneration following status epilepticus, but not striatal neurodegeneration induced by quinolinic acid. Neurobiol Dis. 2008;29(3):391–9. doi: 10.1016/j.nbd.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 8.Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: Implications for gene targeting approaches. Proc Natl Acad Sci USA. 1997;94(8):4103–8. doi: 10.1073/pnas.94.8.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McLin JP, Steward O. Comparison of seizure phenotype and neurodegeneration induced by systemic kainic acid in inbred, outbred, and hybrid mouse strains. Eur J Neurosci. 2006;24(8):2191–202. doi: 10.1111/j.1460-9568.2006.05111.x. [DOI] [PubMed] [Google Scholar]
- 10.McLin JP, Thompson LM, Steward O. Differential susceptibility to striatal neurodegeneration induced by quinolinic acid and kainate in inbred, outbred and hybrid mouse strains. Eur J Neurosci. 2006;24(11):3134–40. doi: 10.1111/j.1460-9568.2006.05198.x. [DOI] [PubMed] [Google Scholar]
- 11.Schauwecker PE, Williams RW, Santos JB. Genetic control of sensitivity to hippocampal cell death induced by kainic acid: A quantitative trait loci analysis. J Comp Neurol. 2004;477(1):96–107. doi: 10.1002/cne.20245. [DOI] [PubMed] [Google Scholar]
- 12.Stevens JC, Banks GT, Festing MF, Fisher EM. Quiet mutations in inbred strains of mice. Trends Mol Med. 2007;13(12):512–9. doi: 10.1016/j.molmed.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Taft RA, Davisson M, Wiles MV. Know thy mouse. Trends Genet. 2006;22(12):649–53. doi: 10.1016/j.tig.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 14.Gray M, Shirasaki DI, Cepeda C, Andre VM, Wilburn B, Lu XH, et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. 2008;28(24):6182–95. doi: 10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heng MY, Tallaksen-Greene SJ, Detloff PJ, Albin RL. Longitudinal evaluation of the Hdh(CAG)150 knock-in murine model of Huntington’s disease. J Neurosci. 2007;27(34):8989–98. doi: 10.1523/JNEUROSCI.1830-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hodgson JG, Agopyan N, Gutekunst C-A, Leavitt BR, LePiane F, Singaraja R, et al. A YAC mouse model for Huntington’s Disease with full-length mutant Huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–92. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- 17.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87(3):493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 18.Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. J Comp Neurol. 2003;465(1):11–26. doi: 10.1002/cne.10776. [DOI] [PubMed] [Google Scholar]
- 19.Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet. 1999;8(3):397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- 20.Wheeler VC, Auerbach W, White JK, Srinidhi J, Auerbach A, Ryan A, et al. Length-dependent gametic CAG repeat instability in the Huntington’s disease knock-in mouse. Hum Mol Genet. 1999;8(1):115–22. doi: 10.1093/hmg/8.1.115. [DOI] [PubMed] [Google Scholar]
- 21.Woodman B, Butler R, Landles C, Lupton MK, Tse J, Hockly E, et al. The Hdh(Q150/Q150) knock-in mouse model of HD and the R6/2 exon 1 model develop comparable and widespread molecular phenotypes. Brain Res Bull. 2007;72(2–3):83–97. doi: 10.1016/j.brainresbull.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 22.Wheeler VC, White JK, Gutekunst CA, Vrbanac V, Weaver M, Li XJ, et al. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum Mol Genet. 2000;9(4):503–13. doi: 10.1093/hmg/9.4.503. [DOI] [PubMed] [Google Scholar]
- 23.Hickey MA, Kosmalska A, Enayati J, Cohen R, Zeitlin S, Levine MS, et al. Extensive early motor and non-motor behavioral deficits are followed by striatal neuronal loss in knock-in Huntington’s disease mice. Neuroscience. 2008;157(1):280–95. doi: 10.1016/j.neuroscience.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li JY, Popovic N, Brundin P. The use of the R6 transgenic mouse models of Huntington’s disease in attempts to develop novel therapeutic strategies. NeuroRx. 2005;2(3):447–64. doi: 10.1602/neurorx.2.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Raamsdonk JM, Warby SC, Hayden MR. Selective degeneration in YAC mouse models of Huntington disease. Brain Res Bull. 2007;72(2–3):124–31. doi: 10.1016/j.brainresbull.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 26.Van Raamsdonk JM, Metzler M, Slow E, Pearson J, Schwab C, Carroll J, et al. Phenotypic abnormalities in the YAC128 mouse model of Huntington disease are penetrant on multiple genetic backgrounds and modulated by strain. Neurobiol Dis. 2007;26(1):189–200. doi: 10.1016/j.nbd.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 27.Cowin RM, Bui N, Graham D, Green JR, Yuva-Paylor LA, Weiss A, et al. Genetic background modulates behavioral impairments in R6/2 mice and suggests a role for dominant genetic modifiers in Huntington’s disease pathogenesis. Mamm Genome. 2012;23(5–6):367–77. doi: 10.1007/s00335-012-9391-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jarabek BR, Yasuda RP, Wolfe BB. Regulation of proteins affecting NMDA receptor-induced excitotoxicity in a Huntington’s mouse model. Brain. 2004;127(Pt 3):505–16. doi: 10.1093/brain/awh058. [DOI] [PubMed] [Google Scholar]
- 29.Slow EJ, Graham RK, Osmand AP, Devon RS, Lu G, Deng Y, et al. Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc Natl Acad Sci U S A. 2005;102(32):11402–7. doi: 10.1073/pnas.0503634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petersen A, Chase K, Puschban Z, DiFiglia M, Brundin P, Aronin N. Maintenance of susceptibility to neurodegeneration following intrastriatal injections of quinolinic acid in a new transgenic mouse model of Huntington’s disease. Exp Neurol. 2002;175(1):297–300. doi: 10.1006/exnr.2002.7885. [DOI] [PubMed] [Google Scholar]
- 31.Hansson O, Petersen A, Leist M, Nicotera P, Castilho RF, Brundin P. Transgenic mice expressing a Huntington’s disease mutation are resistant to quinolinic acid-induced striatal excitotoxicity. Proc Natl Acad Sci U S A. 1999;96(15):8727–32. doi: 10.1073/pnas.96.15.8727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hansson O, Guatteo E, Mercuri NB, Bernardi G, Li XJ, Castilho RF, et al. Resistance to NMDA toxicity correlates with appearance of nuclear inclusions, behavioural deficits and changes in calcium homeostasis in mice transgenic for exon 1 of the huntington gene. Eur J Neurosci. 2001;14(9):1492–504. doi: 10.1046/j.0953-816x.2001.01767.x. [DOI] [PubMed] [Google Scholar]
- 33.Graham RK, Pouladi MA, Joshi P, Lu G, Deng Y, Wu NP, et al. Differential susceptibility to excitotoxic stress in YAC128 mouse models of Huntington disease between initiation and progression of disease. J Neurosci. 2009;29(7):2193–204. doi: 10.1523/JNEUROSCI.5473-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levine MS, Klapstein GJ, Koppel A, Gruen E, Cepeda C, Vargas ME, et al. Enhanced sensitivity to N-methyl-D-aspartate receptor activation in transgenic and knockin mouse models of Huntington’s disease. J Neurosci Res. 1999;58(4):515–32. [PubMed] [Google Scholar]
- 35.Lloret A, Dragileva E, Teed A, Espinola J, Fossale E, Gillis T, et al. Genetic background modifies nuclear mutant huntingtin accumulation and HD CAG repeat instability in Huntington’s disease knock-in mice. Hum Mol Genet. 2006;15(12):2015–24. doi: 10.1093/hmg/ddl125. [DOI] [PubMed] [Google Scholar]
- 36.Schmued LC, Hopkins KJ. Fluoro-Jade B: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874(2):123–30. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- 37.Carroll JB, Warby SC, Southwell AL, Doty CN, Greenlee S, Skotte N, et al. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene/allele-specific silencing of mutant huntingtin. Mol Ther. 2011;19(12):2178–85. doi: 10.1038/mt.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wellington CL, Ellerby LM, Gutekunst CA, Rogers D, Warby S, Graham RK, et al. Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington’s disease. J Neurosci. 2002;22(18):7862–72. doi: 10.1523/JNEUROSCI.22-18-07862.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wanker EE, Scherzinger E, Heiser V, Sittler A, Eickhoff H, Lehrach H. Membrane filter assay for detection of amyloid-like polyglutamine-containing protein aggregates. Methods Enzymol. 1999;309:375–86. doi: 10.1016/s0076-6879(99)09026-6. [DOI] [PubMed] [Google Scholar]
- 40.Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, et al. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90(3):549–58. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 41.Ryu JK, McLarnon JG. VEGF receptor antagonist Cyclo-VEGI reduces inflammatory reactivity and vascular leakiness and is neuroprotective against acute excitotoxic striatal insult. J Neuroinflammation. 2008;5:18. doi: 10.1186/1742-2094-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steward O, Torre ER, Tomasulo R, Lothman E. Neuronal activity up-regulates astroglial gene expression. Proc Natl Acad Sci U S A. 1991;88(15):6819–23. doi: 10.1073/pnas.88.15.6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim SH, Thomas CA, Andre VM, Cummings DM, Cepeda C, Levine MS, et al. Forebrain striatal-specific expression of mutant huntingtin protein in vivo induces cell-autonomous age-dependent alterations in sensitivity to excitotoxicity and mitochondrial function. ASN Neuro. 2011;3(3):e00060. doi: 10.1042/AN20110009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freund TF, Ylinen A, Miettinen R, Pitkanen A, Lahtinen H, Baimbridge KG, et al. Pattern of neuronal death in the rat hippocampus after status epilepticus. Relationship to calcium binding protein content and ischemic vulnerability. Brain Res Bull. 1992;28(1):27–38. doi: 10.1016/0361-9230(92)90227-o. [DOI] [PubMed] [Google Scholar]
- 45.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431(7010):805–10. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 46.Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, et al. Nuclear and neuropil aggregates in Huntington’s disease: Relationship to neuropathology. J Neurosci. 1999;19(7):2522–34. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Silver LM. Concepts and Applications. Oxford: University Press; 1995. Mouse Genetics Concepts and Applications. [Google Scholar]
- 48.Leavitt BR, van Raamsdonk JM, Shehadeh J, Fernandes H, Murphy Z, Graham RK, et al. Wild-type huntingtin protects neurons from excitotoxicity. J Neurochem. 2006;96(4):1121–9. doi: 10.1111/j.1471-4159.2005.03605.x. [DOI] [PubMed] [Google Scholar]
- 49.Van Raamsdonk JM, Pearson J, Murphy Z, Hayden MR, Leavitt BR. Wild-type huntingtin ameliorates striatal neuronal atrophy but does not prevent other abnormalities in the YAC128 mouse model of Huntington disease. BMC Neurosci. 2006;7:80. doi: 10.1186/1471-2202-7-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lajoie P, Snapp EL. Formation and toxicity of soluble polyglutamine oligomers in living cells. PLoS One. 2010;5(12):e15245. doi: 10.1371/journal.pone.0015245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nucifora LG, Burke KA, Feng X, Arbez N, Zhu S, Miller J, et al. Identification of novel potentially toxic oligomers formed in vitro from mammalian-derived expanded huntingtin exon-1 protein. J Biol Chem. 2012;287(19):16017–28. doi: 10.1074/jbc.M111.252577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zuchner T, Brundin P. Mutant huntingtin can paradoxically protect neurons from death. Cell Death Differ. 2008;15(3):435–42. doi: 10.1038/sj.cdd.4402261. [DOI] [PubMed] [Google Scholar]
- 53.Brooks SP, Jones L, Dunnett SB. Comparative analysis of pathology and behavioural phenotypes in mouse models of Huntington’s disease. Brain Res Bull. 2012;88(2–3):81–93. doi: 10.1016/j.brainresbull.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 54.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90(3):537–48. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 55.Miller J, Arrasate M, Brooks E, Libeu CP, Legleiter J, Hatters D, et al. Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat Chem Biol. 2011;7(12):925–34. doi: 10.1038/nchembio.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cummings DM, Cepeda C, Levine MS. Alterations in striatal synaptic transmission are consistent across genetic mouse models of Huntington’s disease. ASN Neuro. 2010;2(3):e00036. doi: 10.1042/AN20100007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cummings DM, Andre VM, Uzgil BO, Gee SM, Fisher YE, Cepeda C, et al. Alterations in cortical excitation and inhibition in genetic mouse models of Huntington’s disease. J Neurosci. 2009;29(33):10371–86. doi: 10.1523/JNEUROSCI.1592-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Joshi PR, Wu NP, Andre VM, Cummings DM, Cepeda C, Joyce JA, et al. Age-dependent alterations of corticostriatal activity in the YAC128 mouse model of Huntington disease. J Neurosci. 2009;29(8):2414–27. doi: 10.1523/JNEUROSCI.5687-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Milnerwood AJ, Gladding CM, Pouladi MA, Kaufman AM, Hines RM, Boyd JD, et al. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron. 2010;65(2):178–90. doi: 10.1016/j.neuron.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 60.Milnerwood AJ, Raymond LA. Early synaptic pathophysiology in neurodegeneration: Insights from Huntington’s disease. Trends Neurosci. 2010;33(11):513–23. doi: 10.1016/j.tins.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 61.Li SH, Li XJ. Huntingtin and its role in neuronal degeneration. Neuroscientist. 2004;10(5):467–75. doi: 10.1177/1073858404266777. [DOI] [PubMed] [Google Scholar]
- 62.Saavedra A, Giralt A, Rue L, Xifro X, Xu J, Ortega Z, et al. Striatal-enriched protein tyrosine phosphatase expression and activity in Huntington’s disease: A STEP in the resistance to excitotoxicity. J Neurosci. 2011;31(22):8150–62. doi: 10.1523/JNEUROSCI.3446-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Choi YS, Lin SL, Lee B, Kurup P, Cho HY, Naegele JR, et al. Status epilepticus-induced somatostatinergic hilar interneuron degeneration is regulated by striatal enriched protein tyrosine phosphatase. J Neurosci. 2007;27(11):2999–3009. doi: 10.1523/JNEUROSCI.4913-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu L, Hamre KM, Goldowitz D. Kainic Acid-Induced Neuronal Degeneration in Hippocampal Pyramidal Neurons Is Driven by Both Intrinsic and Extrinsic Factors: Analysis of FVB/N↔C57BL/6 Chimeras. J Neurosci. 2012;32(35):12093–101. doi: 10.1523/JNEUROSCI.6478-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]