

Abstract
Salutary responses to adeno-associated viral (AAV) gene therapy have been reported in the mouse model of Sandhoff disease (SD), a neurodegenerative lysosomal storage disease caused by deficiency of β-N-acetylhexosaminidase (Hex). While untreated mice reach the humane endpoint by 4.1 months of age, mice treated by a single intracranial injection of vectors expressing human hexosaminidase may live a normal life span of 2 years. When treated with the same therapeutic vectors used in mice, two cats with SD lived to 7.0 and 8.2 months of age, compared with an untreated life span of 4.5 ± 0.5 months (n = 11). Because a pronounced humoral immune response to both the AAV1 vectors and human hexosaminidase was documented, feline cDNAs for the hexosaminidase α- and β-subunits were cloned into AAVrh8 vectors. Cats treated with vectors expressing feline hexosaminidase produced enzymatic activity >75-fold normal at the brain injection site with little evidence of an immune infiltrate. Affected cats treated with feline-specific vectors by bilateral injection of the thalamus lived to 10.4 ± 3.7 months of age (n = 3), or 2.3 times as long as untreated cats. These studies support the therapeutic potential of AAV vectors for SD and underscore the importance of species-specific cDNAs for translational research.
Introduction
The GM2 gangliosidoses are a class of lysosomal storage disorders that include Tay-Sachs disease and Sandhoff disease (SD), both causing rapid and fatal neurodegeneration in children by ~5 years of age.1 Although the pathogenic mechanism is incompletely understood, the underlying cause is catalytic deficiency of β-N-acetylhexosaminidase (Hex, EC 3.1.2.52), a lysosomal enzyme that initiates the stepwise catabolism of GM2 ganglioside by removing its terminal N-acetylgalactosamine (GalNAc) residue. Functional Hex activity requires the coordinated action of three distinct proteins, a nonhydrolytic GM2 activator protein and the two hydrolytic subunits, α and β. Subunits combine in the endoplasmic reticulum to form the two major Hex isozymes, each with different substrate specificities: HexB (ββ) or HexA (αβ), the isozyme responsible for degradation of GM2 ganglioside in humans. Therefore, GM2 gangliosidosis may be caused by a defect in either the Hex α (Tay-Sachs disease) or β (SD) subunit, or by a deficiency of the GM2 activator protein. First described in 1881,2 the GM2 gangliosidoses remain incurable today.
Hope for effective treatment of GM2 gangliosidosis arises from several recently developed experimental therapies, including adeno-associated virus (AAV) gene therapy. Because optimal production of HexA is suggested to require coexpression of both Hex subunits,3,4,5 SD mice were treated by bilateral injections of the striatum and deep cerebellar nuclei (DCN) with monocistronic vectors expressing human Hex α- and β-subunits, both with carboxyl terminal fusions to the HIV Tat protein transduction domain.6 Treated mice routinely survived to 2 years, the maximum age permitted by institutional animal welfare mandates.7,8 Hence, we contend that AAV gene therapy holds promise for successful translation to clinical trials for human GM2 gangliosidosis.
The feline model of SD, first discovered and reported in the 1970's,9 has been continuously maintained as a research colony to the present day. With stereotypical disease progression that has been characterized thoroughly in the intervening years,10,11,12,13,14,15,16,17 the feline model presents an excellent opportunity to test AAV gene therapy and address important issues to be resolved before initiating human clinical trials. For example, the cat brain is >50 times larger than the mouse brain and is more complex and anatomically similar to humans, thus presenting a better approximation of the challenges posed to widespread vector delivery in a large brain with global disease. Moreover, the human storage profiles of GM2 ganglioside, asialo-GM2 ganglioside, and myelin-enriched lipids are more effectively modeled by SD cats than SD mice, suggesting that ganglioside metabolism in cats is more similar to that in humans than in mice.18 For these reasons, the feline SD model was chosen to test AAV gene therapy as an intermediate step between mouse experiments and human clinical trials.
Results
Initial tests of AAV vector function were performed in SD cat 7-532 by injection of the right thalamus with the same AAV1 vectors expressing human Hex α- or β-subunits used to effectively treat SD mice in previous experiments7 (Table 1). Six weeks post-injection, the brain was analyzed for Hex expression and therapeutic effect. Injection of vector through a single needle tract produced substantial Hex activity spanning the entire anterior-posterior axis of the ipsilateral cerebrum, over a distance of ~2.5 cm. Fluorogenic enzyme assays with an α-subunit selective substrate (4-methylumbelliferyl 6-sulfo-2-acetamido-2-deoxy-β-D-glucopyranoside (MUGS)) measured HexA activity ranging from 236.7% of normal at the injection site to 91.4% of normal 1 cm anterior to the injection site. MUGS activity in contralateral blocks ranged from 5.0 to 10.4% normal. Untreated SD cats express 0.8 ± 1.4% (mean ± SD, n = 7) of normal MUGS activity in the cerebral cortex. Furthermore, high-performance thin-layer chromatography (HPTLC) demonstrated a reduction of GM2 ganglioside storage at the injection site in cat 7-532. In agreement with previous studies,18 GM2 comprised ~72% of all ganglioside in the untreated SD cat brain but only 23% of total ganglioside at the injection site of the treated SD cat brain (Figure 1a). Correction was not complete, however, since the normal cat brain has undetectable levels of GM2 ganglioside.
Table 1. AAV-treated cats and corresponding serum antibody titers to the vectors.
Figure 1.

Therapeutic benefit of human Hex expressed from an AAV1 vector in Sandhoff disease (SD) cats. (a) Cat 7-532 was injected in the right thalamus with a 1:2 ratio of AAV1-CAG-hHEXA/B-Tat-WPRE (A:B = 1 × 1011 g.e.:2 × 1011 g.e., Table 1). Tissue was collected 6 weeks post-injection, and GM2 storage was analyzed by high-performance thin-layer chromatography. Shown are samples from the injected thalamus (Inj), the contralateral thalamus (Contra), and corresponding sections from untreated SD and normal (N) cats. GM2 levels were quantitated densitometrically and expressed as a percentage of total ganglioside on the plate (%GM2). Gangliosides were identified by comparison to known standards and labeled accordingly. The absence of a prominent GM2 band in the normal control lane caused a slight migration shift for GM1 and other bands. (b) Two SD cats treated for long-term follow-up (7-588 and 7-589) had delayed disease progression and increased life span (7.0 and 8.2 months, respectively). Before intracranial injection, cat 7-589 was treated by intravenous injection of AAV8 vectors with a liver-targeted promoter. Disease progression was scored according to a clinical rating scale developed for untreated SD cats, with average age of symptom acquisition compiled from nine separate animals (SD Avg). (c) Total brain lipids were analyzed as in a from the injection site (Inj) and the adjacent anterior (Ant) and posterior (Post) blocks from AAV-treated cats (7-588 and 7-589) at humane endpoint. GM2 (dark bar) and GA2 (light bar) levels were compared with corresponding blocks of an untreated SD cat at its humane endpoint (4.3 months of age). Data for AAV-treated cats is expressed as “fold untreated”, with the untreated level indicated by a dashed line. AAV, adeno-associated virus; g.e., genome equivalents.
After demonstrating functionality of AAV vectors expressing human Hex in the SD cat brain, long-term therapeutic experiments were conducted in two SD cats (7-588 and 7-589) using bilateral thalamic injection of a 1:1 ratio of vectors AAV1-hHEXA/B-Tat (Table 1, Supplementary Table S1). To minimize an anticipated immune response to the human Hex subunit-Tat fusion proteins, both cats were immunosuppressed with 10 mg/kg cyclosporine administered orally twice per day for the duration of the experiment. To further minimize immunity to the therapeutic proteins and to treat non-central nervous system organs, a peripheral tolerization protocol reported previously for Niemann-Pick disease mice19 was employed in one of the SD cats (7-589). Cat 7-589 was treated first by intravenous injection of 5.0 × 1012 genome equivalents (g.e.)/vector/kg of AAV8 vectors expressing the human Hex subunit-Tat fusion proteins from a liver-restricted enhancer/promoter (DC172).20 Two weeks following intravenous treatment, cat 7-589 was treated identically to cat 7-588. Due to the limited availability of SD cats, control treatments of cyclosporine or peripheral tolerization alone were not performed.
AAV-treated cats 7-588 and 7-589 lived to 7.0 and 8.2 months, respectively, compared with a humane endpoint of 4.5 ± 0.5 months for untreated SD cats (n = 11). Although treated animals progressed through disease stages typical of untreated SD cats, they acquired symptoms at older ages (Figure 1b). At end point, both AAV-treated cats expressed brain MUGS activity between 25 and 30% normal at the injection site, which diminished to 5–13% normal 1 cm anterior to the injection site. In addition, HPTLC documented mild to moderate decreases in GM2 ganglioside storage (16.3–32.9%) at the injection site with increased GM2 storage (19.5–36.3%) in adjacent brain blocks, compared with an untreated SD cat. Similarly, GA2 ganglioside levels decreased by up to 55.1% at the injection site or increased slightly in adjacent blocks (Figure 1c).
Though routine histopathology with hematoxylin and eosin staining revealed no clear evidence of a cellular infiltrate in the brain of cats 7-588 and 7-589 (data not shown), both animals demonstrated pronounced serum antibody titers against the AAV1 vectors (Table 1) and purified human HexA enzyme (Figure 2). While undetectable in pre-treatment samples, serum antibody titers against the AAV1 vectors were 1:65,536 and 1:24,576 at the humane endpoint for cats 7-588 and 7-589, respectively. Also, serum titers against semi-purified human HexA were 1:8,192 and 1:27,307 for cats 7-588 and 7-589, respectively, demonstrating a pronounced immune response to the human transgene product. To further investigate the immune response after AAV gene therapy in the cat brain, four additional SD cats and four normal cats were injected with vectors expressing the human Hex subunits with or without C-terminal Tat fusions (Table 1). For all SD cats and two normal cats, brain and other tissues were collected at 1.4–2.1 months post-injection and analyzed for enzymatic activity and evidence of an immune response. The two remaining normal cats (9-1252, 9-1253) were observed for behavioral signs of vector toxicity for 14.3 and 12.2 months, respectively, before tissues were collected. No treated cats in this study, including those followed for over 1 year, demonstrated any clinical signs of vector toxicity.
Figure 2.

Serum antibody titers to human HexA in AAV-treated cats. Sandhoff disease (SD) or normal cats treated by intracranial injection of AAV vectors expressing human Hex were assayed at experimental or humane endpoint for serum antibodies to human HexA. AAV-treated SD cats had significantly lower titers compared with AAV-treated normal cats (*P = 0.011 with the inclusion of 7-589, or P = 0.009 with omission of 7-589, which was pre-treated by intravenous injection before intracranial injection). Average titers of at least two assays are shown. AAV, adeno-associated virus.
As with cats 7-588 and 7-589, subsequent AAV1-treated cats had pronounced serum antibody titers to the vector and to human HexA (Table 1 and Figure 2, respectively). In AAV-treated normal cats, serum titers to vector ranged from 1:16,384 to 1:262,144 while in AAV-treated SD cats, anti-vector serum titers ranged from 1:65,536 to 1:196,608. The difference in serum titers to the vector between AAV-treated normal and SD cats did not achieve statistical significance (P = 0.051). In contrast, serum antibodies against the human HexA enzyme were statistically lower in AAV-treated SD versus normal cats (P = 0.011; range, 1:1,024–1:5,120 versus 1:8,192–1:87,381). When cat 7-589 was omitted from the SD group (because it was pre-treated by intravenous injection of vector as described above), significance of the lower serum titers to HexA in SD cats increased slightly (P = 0.009).
After high serum antibody titers to human HexA were documented in AAV-treated cats, feline HEXA and HEXB cDNAs were cloned for use in future therapeutic experiments and have been assigned GenBank accession numbers JF899596 and JF899597, respectively. Because high serum titers were also documented to the AAV1 vectors (presumably the capsid protein), feline Hex vectors were produced using a different serotype (AAVrh8). As demonstrated in Figure 3a and Supplementary Table S2, feline Hex α- and β-subunit cDNAs were functional in immortalized fibroblasts from a SD cat11 transduced with AAVrh8-fHEXA/B vectors at three different doses. For all doses tested, activity with the HexA-selective MUGS substrate was highest after coexpression of both α- and β-subunits and ranged from 97.7 to 3,817.8 nmol/mg/hour (0.8–30.3-fold normal, P = 0.043 when compared with the β-subunit alone). MUGS activity for untreated SD cells or immortalized fibroblasts from a normal cat was 4.9 ± 4.2 or 125.9 ± 28.8 nmol/mg/hour, respectively. Because MUGS substrate can be cleaved at low levels by HexB (in cats) and HexS, ion exchange chromatography was used to calculate the relative production of each isozyme. The ratio of HexA:HexB was 0.14 after expression of the β-subunit alone and 1.57 when α- and β-subunits were coexpressed (Figure 3b, P = 0.002). No HexS was detected after either treatment.
Figure 3.

HexA activity after expression of feline Hex α- and β-subunits. Immortalized skin fibroblasts from a Sandhoff disease (SD) cat were transduced with AAVrh8 vectors expressing the feline Hex subunits: β-subunit alone (β), or α- and β-subunits combined (α + β). The vector dose per subunit remained constant, and three doses were tested: 0.3, 1.3 or 5.0 × 106 g.e./cell. (a) Specific activity (S.A.) was calculated for the MUGS (HexA preferred) substrate at each dose and plotted on a logarithmic scale. After normalization to vector dose, specific activities were averaged (mean-normalized) and found to be 4.2-fold higher after α + β coexpression than after β-subunit expression alone (*P = 0.043). (b) DEAE cellulose anion exchange chromatography was used to separate HexA (A) and HexB (B) isozymes. Fractions of 0.5 ml eluates were collected using a 0–400 mmol/l NaCl gradient, and MUG substrate (cleaved by all Hex isozymes) was used to measure enzyme activity (nmol 4-MU/0.5 ml/hour). The total activity per isozyme was calculated by summing activities from fractions 1–8 (B) or 10–20 (A) and ratios of A:B were calculated for α + β (closed circles) or β (closed squares)(n = 3 each). MUG activity was measured in untreated cells as a control (none, gray triangles). A:B ratios were statistically higher in cells treated with α + β (1.57) than with β alone (0.14) (**P = 0.002). HexS was not detected in any sample but generally elutes after fraction 20. DEAE, diethylaminoethyl; g.e., genome equivalents; MUG, 4-MU-N-acetyl-β-D-glucosaminide; MUGS, 4-methylumbelliferyl 6-sulfo-2-acetamido-2-deoxy-β-D-glucopyranoside; NA, not applicable.
Having demonstrated in vitro functionality of the feline HEXA and HEXB cDNAs, a 1:1 formulation of AAVrh8-fHEXA/B vectors was injected into four heterozygote cats to test functionality and immunogenicity in the brain (Table 1). Because therapeutic benefit in SD mice was most dramatic after treatment of the thalamus and DCN,7,8 heterozygote cats were treated by the same routes in preparation for eventual experiments in SD cats. Three of the cats received bilateral injections of the thalamus and DCN and were euthanized 1.1 months post-treatment (7-705, 7-710) or 21 months post-treatment (7-708). Because it was reasoned that behavioral evidence of vector toxicity would be most apparent after a unilateral injection, one cat (7-707) was injected singly in the right thalamus, and tissues were collected 4.1 months post-surgery.
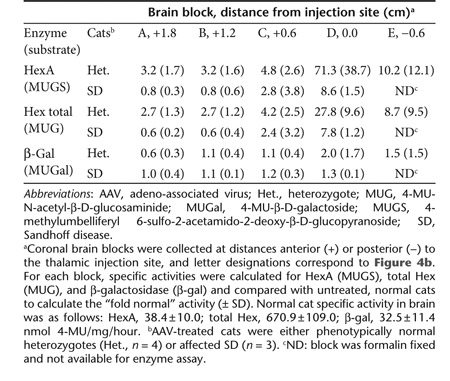
AAVrh8-fHEXA/B vectors generated high levels of enzymatic activity after injection into the SD heterozygote cat brain (Table 2). In the cerebrum, mean MUGS activity was 71.3-fold normal at the thalamic injection site, ranging from 39.8- to 127.4-fold normal in four individual cats. Over the ~2.5 cm length of the cat cerebrum, mean MUGS activity diminished with increasing distance from the injection site, yet remained greater than threefold normal at the frontal pole of the brain (1.8 cm anterior to the injection site) and greater than tenfold normal in the posterior most block (occipital lobe, 0.6 cm posterior to the injection site). MUGS activity in the cerebellum mimicked that in the cerebrum, with a mean activity of 40.4-fold normal at the DCN injection site, decreasing to 10.7-fold normal in the anterior-most cerebellar block (0.6 cm from injection, data not shown).
Table 2. Fold normal-specific activity in heterozygote and SD cat cerebrum post-AAV.
After in vivo functionality was demonstrated in heterozygote cat brains, AAVrh8 vectors expressing feline Hex were used to treat three SD cats by bilateral thalamic injections. To maximize therapeutic benefit, SD cats were injected pre-symptomatically at ~4 weeks of age with a total dose of 3.2 × 1012 g.e. (Table 1). As shown in Figure 4a, mean life span for SD cats treated with the feline Hex subunits more than doubled (10.4 ± 3.7 months) and was significantly increased compared with untreated controls (4.5 ± 0.5 months, P = 0.0064, log-rank test). As demonstrated in Supplementary Videos S1 and S2, motor function and quality of life were dramatically improved in AAV-treated versus untreated SD cats. The most debilitating symptom of SD in untreated cats (Supplementary Video S1) is whole-body tremors, which render the animal unable to eat, walk or use the litter pan independently. In all SD cats treated with AAVrh8-fHEX vectors, whole-body tremors were eliminated (Supplementary Video S2). Perhaps the best evidence of therapeutic effect is that the clinical rating scale developed for untreated SD cats (Figure 1b) was not applicable to AAV-treated SD cats. For example, AAV-treated cats did not acquire fine or whole-body tremors, and their initial and primary disease symptom was hind limb weakness, which had not been appreciated fully in untreated SD cats. In fact, two of the treated SD cats were euthanized due to hind limb weakness so pronounced that they were unable to stand, thus triggering the predetermined humane endpoint. The third SD cat was quite ambulatory at endpoint but was euthanized due to substantial weight loss (>20% maximal body weight), and at necropsy was found to have gross distension of the urinary bladder and fecal impaction. Soft-tissue compression of the spinal cord in the caudal cervical/rostral thoracic region was documented at necropsy in the two cats with pronounced hind limb weakness (7-856, 7-879), but not in the third SD cat with substantial weight loss (7-885).
Figure 4.

Therapeutic benefit in Sandhoff disease (SD) cats after intrathalamic injection of AAVrh8 vectors expressing feline Hex. SD cats (n = 3) were followed to the humane endpoint after bilateral thalamic injection of AAVrh8-fHEX vectors (1:1 ratio of α:β total dose = 3.2 × 1012 g.e.). (a) Mean survival of treated SD cats (SD + AAV) was 10.4 months, a statistically significant increase compared with nine untreated cats (SD; P = 0.0064, log-rank test). (b) Injection sites (white circles) and 0.6 cm coronal brain blocks (A–H) are shown. (c) Naphthol staining for Hex activity (red) is shown for untreated normal and SD cat brains. The fold normal Hex activity (measured by MUGS substrate) is listed below each brain block. (d) Naphthol staining for one representative AAV-treated SD cat (SD + AAV, 7-856) demonstrates Hex activity throughout the cerebrum (A–E) with minor activity in brainstem and cerebellum (F–H). MUGS activity ranged from 0.18- to 8.1-fold normal. Letter designations correspond to b. AAV, adeno-associated virus; g.e., genome equivalents; MUGS, 4-methylumbelliferyl 6-sulfo-2-acetamido-2-deoxy-β-D-glucopyranoside.
In SD cats treated bilaterally in the thalamus, Hex activity detected by postmortem histochemical staining spanned the entire cerebrum from occipital to frontal poles, though distribution was not uniform and appeared as gradients of staining intensity that diminished with distance from the injection site (Figure 4b–d). Mean MUGS activity was 8.6 ± 1.5-fold normal at the thalamic injection site and decreased to 0.8 ± 0.3-fold normal at the frontal pole (1.8 cm anterior to injection site, Table 2).
No clinical evidence of vector toxicity was observed in any cat treated with intracranial AAV gene therapy, even those living beyond 1 year post-treatment. However, profound vascular cuffing was observed in the brains of normal cats treated with AAV1 vectors expressing human Hex subunits (both Tat fusions and wild-types, Figure 5a). The cellular infiltrate consisted primarily of CD20+ B lymphocytes, with CD4+ T helper cells comprising <2% of the infiltrate and dispersed at low levels throughout the brain parenchyma (data not shown). A reliable antibody to feline CD8 was not available, so potential infiltrates of cytotoxic T cells were not evaluated. None of the seven cats injected with AAVrh8 vectors expressing feline Hex subunits demonstrated any behavioral or histological evidence of toxicity. Specifically, no vascular cuffing was apparent in cats treated with AAVrh8-fHEX vectors. When humoral immunity was evaluated, heterozygote and SD cats treated with AAVrh8-fHEX vectors had notable serum antibody titers to the vectors (1:4,096–1:32,768 and 1:1,024–1:3,072, respectively; Table 1). While anti-vector serum titers appear lower after treatment with AAVrh8 vectors than with AAV1 vectors, statistical significance cannot be evaluated with current cohorts and animal numbers. Though purified feline HexA was unavailable, no antibody titers were documented when sera from AAVrh8-treated heterozygotes were tested against human HexA or a feline β-subunit–specific 14-mer peptide (fHEXB 240).11
Figure 5.

Histological findings after AAV injection in the normal and Sandhoff disease (SD) cat brain. (a) Perivascular cuffing (black arrows) was apparent in the brains of normal cats expressing human Hex from AAV1 vectors, with greatest concentrations near the thalamic injection site. One representative cat (9-1254) is shown. Bar = 200 μm (inset = 100 μm). (b) Strongly eosinophilic neurons were observed in discrete areas of the brain (inset), corresponding to areas of highest Hex activity. At high magnification, granular to botryoid intracellular inclusions were apparent. Bar = 20 μm (inset = 100 μm). The region of the dorsal thalamic nuclei from cat 7-708 is shown. (c) Immunofluorescence with a monoclonal antibody to the feline β-subunit demonstrated that botryoid inclusions contained high levels of Hex (green). Dorsal thalamic nuclei of cat 7-708 were stained. Inset was taken with a standard fluorescence microscope with DAPI nuclear counterstain (bar = 100 μm). Main panel is a merged stack of 20 layers (6 μm total) from a confocal microscope (bar = 20 μm). AAV, adeno-associated virus; DAPI, 4′,6-diamidino-2-phenylindole.
The only consistent histological abnormality in cats treated with AAVrh8-fHEX vectors was the presence of strongly eosinophilic neurons with granular to botryoid intracytoplasmic inclusions (Figure 5b). Eosinophilic neurons usually appeared in discrete locations within the dorsal thalamus/hippocampus and/or cerebellar Purkinje cells, and the density of such neurons correlated with Hex-specific activity (i.e., brain blocks with the highest Hex activity generally had the highest numbers of botryoid neurons, Supplementary Table S3). Immunofluorescence confirmed that the strongly eosinophilic neurons expressed high levels of Hex, which appeared to be vesiculated within the neurons (Figure 5c). Endogenous Hex levels in normal cat brain were below the limit of detection of the immunofluorescence assay (data not shown). Table 3 summarizes the immune-related and histological findings for each treatment group in the current study.
Table 3. Summary of immune-related and histological findings.

Discussion
This study documents extraordinary levels of serum antibodies to both AAV1 and AAVrh8 vectors in normal and SD cats after direct brain injection, though pre-treatment titers were ≤1:32 in all cats tested. Maximum titers to AAV1 were at or above ~1:200,000 in both SD and normal cats, whereas the highest titer to AAVrh8 was ~1:33,000. Differences in humoral and cellular immune responses were detected between phenotypically normal and SD cats. Though both groups clearly generated serum antibodies to human HexA after AAV1 treatment, titers in normal cats (mean = 1:45,739) were statistically higher than those in SD cats (mean = 1:3,584; P = 0.009). Also, brains of normal cats treated with vectors expressing human Hex had marked cellular infiltrates apparent as vascular cuffs consisting primarily of B lymphocytes, whereas SD cats treated with the same vectors demonstrated little or no infiltrate. A differential immune response to the same AAV vectors may result from immune defects in affected animals, such as premature thymic involution caused in part by a ganglioside-mediated apoptotic cascade, as reported in cats with GM1 gangliosidosis21,22,23 and in SD mice24,25 and cats (data not shown). Though decreased thymus size is measurable only in late-stage disease, increased thymocyte apoptosis and other changes are present in early to middle disease stages, perhaps contributing to the diminished immune response of AAV-treated SD cats. It is possible that immune deficiencies in feline SD facilitate treatment response due to a reduced immune reaction to the therapeutic agent, and therefore it will be important to evaluate whether AAV treatment normalizes immune function through central or peripheral mechanisms. AAV vector and AAV-generated lysosomal enzyme have been detected in peripheral tissues after intracerebral treatment in animal models,26,27 demonstrating that AAV injected into the brain may also have peripheral effects.
Despite strong humoral immunity to both the AAV1 vectors and human HexA protein, therapeutic benefit was demonstrated in two SD cats treated by bilateral thalamic injection, with an extension of life span from 4.5 months (untreated) to 7.0 and 8.2 months. Though treated in the early postsymptomatic period, both cats demonstrated delayed disease progression (Figure 1b) and improved quality of life. It is well accepted that treatment of lysosomal storage diseases is most beneficial when performed early, preferably before the onset of disease.28 Though rare, reports of symptom reversal post-treatment have been published, as in the feline model of α-mannosidosis.29 Similarly, demonstration of substantive therapeutic benefit in postsymptomatic SD cats in the current study is encouraging for the treatment of children with GM2 gangliosidosis, most of whom are diagnosed after the onset of clinical disease.
Because initial experiments demonstrated high serum titers to the AAV1 vectors and purified human HexA, second-generation vectors incorporated the AAVrh8 capsid and feline HEXA/B cDNAs. AAVrh8 vectors used to transduce fibroblasts from a SD cat demonstrated that maximal Hex activity is achieved by coexpression of both subunits versus replacement of a single subunit alone, at three different doses over a 16-fold range. MUGS activity in cells expressing both subunits was 4.2-fold higher than with the β-subunit alone. Also, the ratio of HexA to HexB after coexpression of both subunits was ~11-fold higher than with expression of the β-subunit alone. Since HexA is solely responsible for degradation of GM2 ganglioside in humans, the findings are an important consideration when designing human clinical trials to be minimally invasive yet achieve the widest possible distribution of HexA throughout the brain. If optimal production of HexA is achieved at target sites chosen to provide maximal neuronal interconnectivity between brain regions (e.g., the thalamus), fewer brain injections should be required for therapeutic effect. On the other hand, supranormal HexA levels after subunit coexpression from a strong promoter (cytomegalovirus/chicken β-actin hybrid (CBA)) must not be toxic to the cells that express it.
When phenotypically normal cats were injected with AAVrh8 vectors expressing feline Hex, MUGS activity was greater than threefold normal throughout the cerebrum (Table 2) and greater than tenfold normal throughout the cerebellum (data not shown). Cats were followed for ~1, 4, and 21 months post-treatment, and none showed any clinical evidence of surgery- or vector-related toxicity. Unlike AAV1-hHEX vectors, perivascular cuffing in the brain was absent after AAVrh8-fHEX treatment. Since the choice of AAV capsid can alter the immune response to the same transgene product (reviewed in ref. 30), reduced immunity in cats expressing feline Hex may have resulted from a combination of species-specific enzyme and rh8 capsid. Though evidence of a cellular immune response was minimal, discrete regions of strongly eosinophilic neurons were found to express extraordinary levels of Hex in intracellular vesiculated bodies, and the density of these bodies correlated generally with brain blocks having the highest Hex activity. Other studies have documented brain toxicity after AAV-mediated overexpression of proteins such as green fluorescent protein or tau,31,32 manifested by neuron loss and clinical symptoms. However, no evidence of neuron loss or clinical neurological signs was found in treated cats in the current study, even those living >1 year post-injection.
This is the first report of clear therapeutic benefit from any treatment modality in a non-rodent model of GM2 gangliosidosis, with cats having a brain that is intermediate to mice and humans in complexity and size (~20-fold smaller than the brain of a human infant). In the >35-year history of the feline SD breeding colony, no experimental treatment has shown unequivocal therapeutic benefit, as reported herein for AAVrh8-fHEX vectors delivered in a surgery requiring only two injection sites and ~90 minutes to perform. In the past decade, the longest-lived untreated SD cat was 5.3 months at humane endpoint, so the average 10.4-month life span of treated cats in the current study is a significant advance (P = 0.0064) and provides real hope for future human therapy. In addition to increased longevity, quality of life was dramatically improved for animals treated with feline Hex, as demonstrated by the absence of debilitating, whole-body tremors (Supplementary Video S2). In many respects, AAV treatment converted the severe, infantile-onset phenotype in SD cats to a later-onset form of the disease described in humans, both of which exhibit muscle wasting, limb weakness, and intestinal dysfunction.33 It is hypothesized that partial restoration of enzyme activity in treated cats mimics the residual Hex activity of late-onset human patients, moderating central nervous system dysfunction and permitting the emergence of otherwise subclinical peripheral disease. These results encourage further optimization of therapy, including development of a bicistronic vector encoding Hex α and β in the same construct to ensure optimal HexA expression distal to the injection site, where co-transduction of cells with monocistronic vectors may not occur due to reduced vector concentration. In addition, improved enzyme distribution throughout the brain is needed. Though normal or above normal in most brain regions after thalamic infusion of vector, Hex activity was minimal in the temporal lobe and cerebellum (Figure 4d). Therefore, additional injection sites are expected to provide enhanced therapeutic benefit, as is being borne out in ongoing experiments.
Materials and Methods
Cloning of feline Hex α- and β-subunit cDNAs. Sequences for the feline Hex α-subunit (HEXA) and β-subunit (HEXB) open-reading frames have been deposited in the GenBank database and assigned accession numbers JF899596 and JF899597, respectively.
With cDNA prepared from cat cerebral cortex, feline HEXA cDNA was amplified using primers ORFc1 and ORFn1 (Supplementary Table S4) according to the standard protocols with Taq DNA polymerase. The fHEXA PCR product was purified with the QIAquick PCR purification kit (Qiagen, Valencia, CA), polished for 30 minutes at 72 °C with 2.5 units of Taq and 0.5 mmol/l each dNTP, then cloned into plasmid pcDNA3.3-TOPO (Invitrogen, Grand Island, NY). The fHEXA insert was ~1.8 kb.
A previously published feline HEXB cDNA (GenBank no. S70340, ref. 34) is truncated at the 5′ end by only four base pairs. From S70340, gene assembly primers 5.1–5.6 (Supplementary Table S4) were designed using DNAWorks (http://helixweb.nih.gov/dnaworks)35 to generate the appropriate amino acid sequence of the 5′ end using codons chosen to reduce the GC content of the fragment. The gene assembled product was amplified with primers 5.7 (coding strand) and 5.8 (noncoding strand), which contain flanking NheI and BsmBI restriction sites, respectively. The remainder of the feline HEXB sequence was amplified from plasmid fHEXB#2,11 containing a clone truncated at the 5′ end by ~95 bp, using primers fHEXB-3 and fHEXB2-NotI, which contain BsmBI and NotI restriction sites, respectively. The gene assembled 5′ end of fHEXB was joined to the remainder of the fHEXB cDNA by restriction digestion with BsmBI.
AAV vectors. AAV vectors and their abbreviations are listed in Supplementary Table S1. AAV1 vectors containing human HEXA/B cDNAs with 3′ Tat fusions have been described previously.7 For liver-targeted expression in cat 7-589, the vectors referenced above were modified by substituting the AAV8 capsid in place of AAV1 and the DC172 promoter20 in place of the CAG promoter. Vectors containing wild-type (without Tat) cDNAs were constructed as follows. Human HEXA and HEXB cDNAs were PCR amplified from Mammalian Gene Collection (MGC) clones 14125 (IMAGE ID: 3353424; GenBank ID: BC018927) and 1725 (IMAGE ID: 2967035; GenBank ID: BC017378) obtained from the American Type Culture Collection (Manassas, VA) using primer pairs hHexA-1 + hHexA-2 and hHexB-1 + hHexB-2, respectively, which introduce SpeI and XhoI sites flanking the coding region. The feline HEXA cDNA was amplified with primers fHexA-1 and fHexA-4 that introduce NheI and NotI sites flanking the coding region. The feline HEXB cDNA was amplified in two fragments as indicated above. All PCR products were cloned into pAAV-CBA-MBG-W36 in place of the mouse β-gal (β-galactosidase) cDNA. Transgene expression in these vectors is mediated by the hybrid CBA promoter (comprised of the CMV immediate-early enhancer fused to the CBA promoter)37 and the woodchuck hepatitis virus post-transcriptional regulatory element (WPRE). AAV1 and AAVrh8 vector stocks encoding wild-type human or feline HEX subunits were produced as previously described.38
Animals and surgeries. All animal procedures were approved by the Auburn University Institutional Animal Care and Use Committee. Table 1 summarizes all AAV-treated cats. In untreated SD cats, neurological disease onset occurs at 1.7 months of age with a slight head tremor that progresses to an inability to stand (humane endpoint) by 4.5 ± 0.5 months (mean ± SD, n = 11). Disease progression was scored from 10 (normal) to 1 (lateral recumbency) according to a clinical rating scale developed for untreated SD cats (Figure 1b). Animals at an intermediate stage of disease progression were assigned a half-integer score (e.g., 5.5). Affected animals express 9.2–22.1% of normal levels of the Hex β-subunit protein, which contains three amino acid substitutions at the carboxyl terminus of the protein and a translational stop that is eight amino acids premature.11
Cats were anesthetized with ketamine (10 mg/kg) and dexmedetomidine (0.04 mg/kg) using an intravenous catheter and maintained with isofluorane (0.5–1.5%) in oxygen delivered through an endotracheal tube. Stereotaxic delivery of vector was performed using a Horsley-Clark stereotaxic apparatus (David Kopf Instruments, Tujunga, CA). Coordinates (in cm) for the thalamus relative to bregma were anterior-posterior (AP) −0.7, mediolateral (ML) ±0.4, dorsoventral (DV) −1.6, and for the DCN relative to lambda were AP 0.0, ML ±0.4, DV −1.25. The DV zero point was marked at meninges. The thalamus was injected either unilaterally or bilaterally with or without bilateral injection of the DCN. Single entry sites above each thalamus were made through the skull and dura with a 20G hypodermic needle. For DCN injections, craniotomy was performed directly above the target by inserting the needle vertically (i.e., at an angle perpendicular to the base of the stereotaxic apparatus) through the skull and then through the cerebellar tentorium, passing just caudal to the occipital pole of the cerebrum.
Vector was delivered using a Hamilton syringe (Harvard Apparatus, Holliston, MA) with a non-coring needle (22–25G). In each thalamus, 70 μl was delivered in 10–20 μl aliquots at a rate of 2 μl/minute. Between each aliquot, the injection needle was raised 0.15 cm, so that the final needle position was 1.15 cm ventral to meninges (~2.5 mm ventral to the lateral ventricle). For each DCN, a total of 20 μl was injected at a single site at a rate of 2 μl/minute.
Tissue and serum analysis. After euthanasia by pentobarbital overdose (100 mg/kg), animals were transcardially perfused with cold, heparinized saline. Each brain was divided into coronal blocks of ~0.5 cm from the frontal pole through the cerebellum. Tissues were processed as appropriate for each type of downstream analysis.
Before immunostaining on 6 μm paraffin sections, antigen unmasking was performed by heat treatment in Tris-EDTA buffer (10 mmol/l Tris, 1 mmol/l EDTA, 0.05% Tween 20, pH 9.0), and sections were blocked for 1 hour with 5% normal donkey serum in phosphate-buffered saline containing 0.05% Tween 20. Sections were stained with a monoclonal antibody to the feline Hex β-subunit11 (1:1,000) followed by 488-conjugated donkey anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA; 1:100).
For serum antibody titers, 100 ng of AAV vectors or purified human HexA (Sigma, St Louis, MO) were coated onto ELISA plates (Nunc, Rochester, NY) and incubated overnight at 4 °C. Plates were washed and then blocked with 5% non-fat powdered milk in phosphate-buffered saline for 90 minutes at room temperature; 100 μl of twofold serial dilutions of feline serum samples were added. Goat anti-feline IgG:HRP (Jackson ImmunoResearch; 1:20,000) was used for color development with tetramethylbenzidine (Pierce, Rockford, IL).
Brain lipid analysis. As described in greater detail previously,18 total lipids were extracted with chloroform (CHCl3) and methanol (MeOH) 1:1 by volume and purified from the lyophilized brain tissue.39,40,41 Neutral and acidic lipids were separated using DEAE-Sephadex (A-25; Pharmacia Biotech, Uppsala, Sweden) column chromatography.42 The total lipid extract was applied to a DEAE-Sephadex column after suspension in solvent A (CHCl3:CH3OH:dH20, 30:60:8 by volume), which also was used to collect the neutral lipid fraction containing sphingomyelin and neutral glycosphingolipids to include asialo-GM2 (GA2). Next, acidic lipids were eluted from the column with CHCl3:CH3OH:0.8 mol/l Na acetate (30:60:8 by volume), dried by rotary evaporation and then partitioned so that acidic lipids were in the lower organic phase and gangliosides were in the upper aqueous phase.43,44,45 The resorcinol assay was used to measure the amount of sialic acid in the ganglioside fraction, which then was further purified with base treatment and desalting. Neutral lipids were dried by rotary evaporation and resuspended in CHCl3:CH3OH (2:1 by volume). To further purify GA2, an aliquot of the neutral lipid fraction was evaporated under a stream of nitrogen, treated with 1 N NaOH, and Folch partitioned.41
All lipids were analyzed qualitatively by HPTLC with an internal standard (oleoyl alcohol) added to the neutral and acidic lipids to enhance precision.41,42,43,46 Purified lipid standards were either purchased from Matreya (Pleasant Gap, PA), Sigma, or were a gift from Dr Robert Yu (Medical College of Georgia, Augusta, GA). HPTLC plates were developed, visualized, and quantitated as described.18,41 The total brain ganglioside distribution was normalized to 100% and the percentage distribution was used to calculate sialic acid concentration of individual gangliosides.40 The density value for GA2 was fit to a standard curve of known lipid to calculate concentration.
Cell culture and transfection. Fibroblasts immortalized with the Simian Virus 40 large T antigen were generated using primary skin cultures from SD (GM2/SV3 cells) or normal cats (N/SV3 cells) and have been described elsewhere.11,47 Transfection was performed in 6-well plates or 25 cm2 flasks with cells at 25–50% confluency using AAVrh8 vectors expressing feline HEXA or HEXB cDNAs. Doses in g.e./vector/cell were calculated based on cell count, and vector was incubated for 3–5 days with cells in standard growth media (Dulbecco's modified Eagle medium, 4.5 g/l glucose; 10% fetal bovine serum; 100 units/ml penicillin, 0.1 mg/ml streptomycin, and 0.25 μg/ml amphotericin B).
Enzyme assays and staining. Lysosomal enzyme activity was determined according to a previously published protocol48 with the appropriate 4-methylumbelliferyl (4-MU) substrates: β-gal, 0.5 mmol/l 4-MU-β-D-galactoside, pH 3.8; HexA, 1 mmol/l MUGS, pH 4.2; and total Hex, 1 mmol/l 4-MU-N-acetyl-β-D-glucosaminide (MUG), pH 4.3. Fluorescence of the samples was measured on a BioTek Synergy HT plate reader (BioTek, Winooski, VT) with excitation at 360 nm and emission at 450 nm. Protein concentrations were measured by the method of Lowry, and specific activity was expressed as nmol 4-MU cleaved/mg protein/hour. Qualitative detection of Hex activity in the brain was performed on 40 μm cryosections with 0.25 mmol/l naphthol AS-BI-N-acetyl-β-D-glucosaminide (Sigma) in the presence of hexazotized pararosaniline.49
DEAE cellulose anion exchange chromatography. Lysosomal enzymes were isolated from frozen brain tissue in a 10 mmol/l sodium phosphate buffer (pH 6.0) containing 0.1% Triton-X 100. Tissue homogenate was centrifuged at 15,000g for 20 minutes at 4 °C and filtered at 0.2 μm. Supernatant was applied to a DEAE cellulose column (Sigma) and fractions 1–3 were collected (0.5 ml each). A total of 26 fractions (0.5 ml each) were collected by washing the column with buffer (above) containing sodium chloride at increasing concentrations ranging from 0 to 400 mmol/l, as follows: fractions 4–8 (0 mmol/l), 9 (10 mmol/l), 10 (20 mmol/l), 11–24 (increasing concentrations of 20 mmol/l each, from 40 to 300 mmol/l), 25–26 (350 and 400 mmol/l, respectively). Lysosomal enzyme activity of the fractions was determined as described above.
Statistical analysis. Serum antibody titers to vector or human HexA protein were analyzed for significance by Wilcoxon two-sample test and survival data was created using the LIFETEST procedure and log-rank Test with SAS software (SAS, Cary, NC). Significance of HexA activity and HexA:HexB ratios was determined with a two-tailed t-test.
SUPPLEMENTARY MATERIAL Table S1. AAV vectors for treatment of SD and normal cats. Table S2. In vitro functionality of AAVrh8 vectors expressing feline HexA or HexB cDNAs. Table S3. Correlation of Hex activity with neurons containing eosinophilic, botryoid inclusions. Table S4. Primers used in the current study. Video S1. Untreated SD cat at 4 months of age. Video S2. AAV-treated SD cat at 9.8 months of age.
Acknowledgments
This work was funded by National Institutes of Health grant U01 NS064096, the National Tay-Sachs and Allied Diseases Association, the Cure Tay-Sachs Foundation, Cambridge in America, and the Scott-Ritchey Research Center. S.H.C. is an employee and stockholder of Genzyme Corporation. R.C.B. is an employee of Genzyme Corporation. T.M.C. and M.B.C.-G. received an unrestricted grant for gene therapy research from the Genzyme Corporation. The other authors declared no conflict of interest.
Supplementary Material
References
- Gravel RA, Clarke JTR, Kaback MM, Mahuran D, Sandhoff K, Suzuki K.1995The GM2 gangliosidoses. Scriver CR, Beaudet AL, Sly WS.Valle D.eds). The Metabolic and Molecular Bases of Inherited Disease7th edn., vol. 2McGraw-Hill, Inc.New York; pp. 2839–2879. [Google Scholar]
- Tay W. Symmetrical changes in the region of the yellow spot in each eye of an infant. Tran Ophthalmol Soc UK. 1881;1:155–157. [Google Scholar]
- Arfi A, Bourgoin C, Basso L, Emiliani C, Tancini B, Chigorno V, et al. Bicistronic lentiviral vector corrects beta-hexosaminidase deficiency in transduced and cross-corrected human Sandhoff fibroblasts. Neurobiol Dis. 2005;20:583–593. doi: 10.1016/j.nbd.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Guidotti JE, Mignon A, Haase G, Caillaud C, McDonell N, Kahn A, et al. Adenoviral gene therapy of the Tay-Sachs disease in hexosaminidase A-deficient knock-out mice. Hum Mol Genet. 1999;8:831–838. doi: 10.1093/hmg/8.5.831. [DOI] [PubMed] [Google Scholar]
- Itakura T, Kuroki A, Ishibashi Y, Tsuji D, Kawashita E, Higashine Y, et al. Inefficiency in GM2 ganglioside elimination by human lysosomal beta-hexosaminidase beta-subunit gene transfer to fibroblastic cell line derived from Sandhoff disease model mice. Biol Pharm Bull. 2006;29:1564–1569. doi: 10.1248/bpb.29.1564. [DOI] [PubMed] [Google Scholar]
- Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- Cachón-González MB, Wang SZ, Lynch A, Ziegler R, Cheng SH, Cox TM. Effective gene therapy in an authentic model of Tay-Sachs-related diseases. Proc Natl Acad Sci USA. 2006;103:10373–10378. doi: 10.1073/pnas.0603765103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachón-González MB, Wang SZ, McNair R, Bradley J, Lunn D, Ziegler R, et al. Gene transfer corrects acute GM2 gangliosidosis–potential therapeutic contribution of perivascular enzyme flow. Mol Ther. 2012;20:1489–1500. doi: 10.1038/mt.2012.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cork LC, Munnell JF, Lorenz MD, Murphy JV, Baker HJ, Rattazzi MC. GM2 ganglioside lysosomal storage disease in cats with beta-hexosaminidase deficiency. Science. 1977;196:1014–1017. doi: 10.1126/science.404709. [DOI] [PubMed] [Google Scholar]
- Baker HJ, Reynolds GD, Walkley SU, Cox NR, Baker GH. The gangliosidoses: comparative features and research applications. Vet Pathol. 1979;16:635–649. doi: 10.1177/030098587901600602. [DOI] [PubMed] [Google Scholar]
- Martin DR, Krum BK, Varadarajan GS, Hathcock TL, Smith BF, Baker HJ. An inversion of 25 base pairs causes feline GM2 gangliosidosis variant. Exp Neurol. 2004;187:30–37. doi: 10.1016/j.expneurol.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Rattazzi MC, Appel AM, Baker HJ. Enzyme replacement in feline GM2 gangliosidosis: catabolic effects of human beta-hexosaminidase A. Prog Clin Biol Res. 1982;94:213–220. [PubMed] [Google Scholar]
- Rattazzi MC, Appel AM, Baker HJ, Nester JA.1981Towards enzyme replacement in GM2 gangliosidosis: inhibition of hepatic uptake and induction of CNS uptake of human beta-hexosaminidase in the cat Callahan JW, Lowden JA.eds). Lysosomes and Lysosomal Storage Diseases Raven Press: New York, NY; 213–220. [Google Scholar]
- Walkley SU, Baker HJ, Rattazzi MC, Haskins ME, Wu JY. Neuroaxonal dystrophy in neuronal storage disorders: evidence for major GABAergic neuron involvement. J Neurol Sci. 1991;104:1–8. doi: 10.1016/0022-510x(91)90208-o. [DOI] [PubMed] [Google Scholar]
- Wood PA, McBride MR, Baker HJ, Christian ST. Fluorescence polarization analysis, lipid composition, and Na+, K+-ATPase kinetics of synaptosomal membranes in feline GM1 and GM2 gangliosidosis. J Neurochem. 1985;44:947–956. doi: 10.1111/j.1471-4159.1985.tb12909.x. [DOI] [PubMed] [Google Scholar]
- Walkley SU, Baker HJ, Rattazzi MC. Initiation and growth of ectopic neurites and meganeurites during postnatal cortical development in ganglioside storage disease. Brain Res Dev Brain Res. 1990;51:167–178. doi: 10.1016/0165-3806(90)90273-2. [DOI] [PubMed] [Google Scholar]
- Walkley SU, Wurzelmann S, Rattazzi MC, Baker HJ. Distribution of ectopic neurite growth and other geometrical distortions of CNS neurons in feline GM2 gangliosidosis. Brain Res. 1990;510:63–73. doi: 10.1016/0006-8993(90)90728-t. [DOI] [PubMed] [Google Scholar]
- Baek RC, Martin DR, Cox NR, Seyfried TN. Comparative analysis of brain lipids in mice, cats, and humans with Sandhoff disease. Lipids. 2009;44:197–205. doi: 10.1007/s11745-008-3268-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passini MA, Bu J, Fidler JA, Ziegler RJ, Foley JW, Dodge JC, et al. Combination brain and systemic injections of AAV provide maximal functional and survival benefits in the Niemann-Pick mouse. Proc Natl Acad Sci USA. 2007;104:9505–9510. doi: 10.1073/pnas.0703509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs F, Snoeys J, Feng Y, Van Craeyveld E, Lievens J, Armentano D, et al. Direct comparison of hepatocyte-specific expression cassettes following adenoviral and nonviral hydrodynamic gene transfer. Gene Ther. 2008;15:594–603. doi: 10.1038/sj.gt.3303096. [DOI] [PubMed] [Google Scholar]
- Cox NR, Ewald SJ, Morrison NE, Gentry AS, Schuler M, Baker HJ. Thymic alterations in feline GM1 gangliosidosis. Vet Immunol Immunopathol. 1998;63:335–353. doi: 10.1016/s0165-2427(98)00113-5. [DOI] [PubMed] [Google Scholar]
- Zhou J, Cox NR, Ewald SJ, Morrison NE, Basker HJ. Evaluation of GM1 ganglioside-mediated apoptosis in feline thymocytes. Vet Immunol Immunopathol. 1998;66:25–42. doi: 10.1016/s0165-2427(98)00180-9. [DOI] [PubMed] [Google Scholar]
- Zhou J, Shao H, Cox NR, Baker HJ, Ewald SJ. Gangliosides enhance apoptosis of thymocytes. Cell Immunol. 1998;183:90–98. doi: 10.1006/cimm.1998.1247. [DOI] [PubMed] [Google Scholar]
- Kanzaki S, Yamaguchi A, Yamaguchi K, Kojima Y, Suzuki K, Koumitsu N, et al. Thymic alterations in GM2 gangliosidoses model mice. PLoS ONE. 2010;5 8:e12105. doi: 10.1371/journal.pone.0012105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka K, Tsuji D, Taki T, Itoh K. Thymic involution and corticosterone level in Sandhoff disease model mice: new aspects the pathogenesis of GM2 gangliosidosis. J Inherit Metab Dis. 2011;34:1061–1068. doi: 10.1007/s10545-011-9316-6. [DOI] [PubMed] [Google Scholar]
- Ciron C, Cressant A, Roux F, Raoul S, Cherel Y, Hantraye P, et al. Human alpha-iduronidase gene transfer mediated by adeno-associated virus types 1, 2, and 5 in the brain of nonhuman primates: vector diffusion and biodistribution. Hum Gene Ther. 2009;20:350–360. doi: 10.1089/hum.2008.155. [DOI] [PubMed] [Google Scholar]
- Sondhi D, Hackett NR, Peterson DA, Stratton J, Baad M, Travis KM, et al. Enhanced survival of the LINCL mouse following CLN2 gene transfer using the rh.10 rhesus macaque-derived adeno-associated virus vector. Mol Ther. 2007;15:481–491. doi: 10.1038/sj.mt.6300049. [DOI] [PubMed] [Google Scholar]
- Cabrera-Salazar MA, Roskelley EM, Bu J, Hodges BL, Yew N, Dodge JC, et al. Timing of therapeutic intervention determines functional and survival outcomes in a mouse model of late infantile batten disease. Mol Ther. 2007;15:1782–1788. doi: 10.1038/sj.mt.6300249. [DOI] [PubMed] [Google Scholar]
- Vite CH, McGowan JC, Niogi SN, Passini MA, Drobatz KJ, Haskins ME, et al. Effective gene therapy for an inherited CNS disease in a large animal model. Ann Neurol. 2005;57:355–364. doi: 10.1002/ana.20392. [DOI] [PubMed] [Google Scholar]
- Mays LE, Wilson JM. The complex and evolving story of T cell activation to AAV vector-encoded transgene products. Mol Ther. 2011;19:16–27. doi: 10.1038/mt.2010.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RL, Dayton RD, Leidenheimer NJ, Jansen K, Golde TE, Zweig RM. Efficient neuronal gene transfer with AAV8 leads to neurotoxic levels of tau or green fluorescent proteins. Mol Ther. 2006;13:517–527. doi: 10.1016/j.ymthe.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgievska B, Kirik D, Björklund A. Aberrant sprouting and downregulation of tyrosine hydroxylase in lesioned nigrostriatal dopamine neurons induced by long-lasting overexpression of glial cell line derived neurotrophic factor in the striatum by lentiviral gene transfer. Exp Neurol. 2002;177:461–474. doi: 10.1006/exnr.2002.8006. [DOI] [PubMed] [Google Scholar]
- Shapiro BE, Logigian EL, Kolodny EH, Pastores GM. Late-onset Tay-Sachs disease: the spectrum of peripheral neuropathy in 30 affected patients. Muscle Nerve. 2008;38:1012–1015. doi: 10.1002/mus.21061. [DOI] [PubMed] [Google Scholar]
- Muldoon LL, Neuwelt EA, Pagel MA, Weiss DL. Characterization of the molecular defect in a feline model for type II GM2-gangliosidosis (Sandhoff disease). Am J Pathol. 1994;144:1109–1118. [PMC free article] [PubMed] [Google Scholar]
- Hoover DM, Lubkowski J. DNAWorks: an automated method for designing oligonucleotides for PCR-based gene synthesis. Nucleic Acids Res. 2002;30:e43. doi: 10.1093/nar/30.10.e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broekman ML, Baek RC, Comer LA, Fernandez JL, Seyfried TN, Sena-Esteves M. Complete correction of enzymatic deficiency and neurochemistry in the GM1-gangliosidosis mouse brain by neonatal adeno-associated virus-mediated gene delivery. Mol Ther. 2007;15:30–37. doi: 10.1038/sj.mt.6300004. [DOI] [PubMed] [Google Scholar]
- Matalon R, Surendran S, Rady PL, Quast MJ, Campbell GA, Matalon KM, et al. Adeno-associated virus-mediated aspartoacylase gene transfer to the brain of knockout mouse for canavan disease. Mol Ther. 2003;7 5 Pt 1:580–587. doi: 10.1016/s1525-0016(03)00066-2. [DOI] [PubMed] [Google Scholar]
- Broekman ML, Comer LA, Hyman BT, Sena-Esteves M. Adeno-associated virus vectors serotyped with AAV8 capsid are more efficient than AAV-1 or -2 serotypes for widespread gene delivery to the neonatal mouse brain. Neuroscience. 2006;138:501–510. doi: 10.1016/j.neuroscience.2005.11.057. [DOI] [PubMed] [Google Scholar]
- Baek RC, Kasperzyk JL, Platt FM, Seyfried TN. N-butyldeoxygalactonojirimycin reduces brain ganglioside and GM2 content in neonatal Sandhoff disease mice. Neurochem Int. 2008;52:1125–1133. doi: 10.1016/j.neuint.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Seyfried TN, Yu RK, Miyazawa N. Differential cellular enrichment of gangliosides in the mouse cerebellum: analysis using neurological mutants. J Neurochem. 1982;38:551–559. doi: 10.1111/j.1471-4159.1982.tb08662.x. [DOI] [PubMed] [Google Scholar]
- Kasperzyk JL, El-Abbadi MM, Hauser EC, D'Azzo A, Platt FM, Seyfried TN. N-butyldeoxygalactonojirimycin reduces neonatal brain ganglioside content in a mouse model of GM1 gangliosidosis. J Neurochem. 2004;89:645–653. doi: 10.1046/j.1471-4159.2004.02381.x. [DOI] [PubMed] [Google Scholar]
- Macala LJ, Yu RK, Ando S. Analysis of brain lipids by high performance thin-layer chromatography and densitometry. J Lipid Res. 1983;24:1243–1250. [PubMed] [Google Scholar]
- Seyfried TN, Glaser GH, Yu RK. Cerebral, cerebellar, and brain stem gangliosides in mice susceptible to audiogenic seizures. J Neurochem. 1978;31:21–27. doi: 10.1111/j.1471-4159.1978.tb12428.x. [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Kasperzyk JL, d'Azzo A, Platt FM, Alroy J, Seyfried TN. Substrate reduction reduces gangliosides in postnatal cerebrum-brainstem and cerebellum in GM1 gangliosidosis mice. J Lipid Res. 2005;46:744–751. doi: 10.1194/jlr.M400411-JLR200. [DOI] [PubMed] [Google Scholar]
- Ando S, Chang NC, Yu RK. High-performance thin-layer chromatography and densitometric determination of brain ganglioside compositions of several species. Anal Biochem. 1978;89:437–450. doi: 10.1016/0003-2697(78)90373-1. [DOI] [PubMed] [Google Scholar]
- Martin DR, Rigat BA, Foureman P, Varadarajan GS, Hwang M, Krum BK, et al. Molecular consequences of the pathogenic mutation in feline GM1 gangliosidosis. Mol Genet Metab. 2008;94:212–221. doi: 10.1016/j.ymgme.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury AM, Morrison NE, Hwang M, Cox NR, Baker HJ, Martin DR. Neurodegenerative lysosomal storage disease in European Burmese cats with hexosaminidase beta-subunit deficiency. Mol Genet Metab. 2009;97:53–59. doi: 10.1016/j.ymgme.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Lacorazza HD, Jendoubi M. In situ assessment of beta-hexosaminidase activity. BioTechniques. 1995;19:434–440. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.