

Abstract
Engineering CD8+ T cells to deliver interleukin 12 (IL-12) to the tumor site can lead to striking improvements in the ability of adoptively transferred T cells to induce the regression of established murine cancers. We have recently shown that IL-12 triggers an acute inflammatory environment that reverses dysfunctional antigen presentation by myeloid-derived cells within tumors and leads to an increase in the infiltration of adoptively transferred antigen-specific CD8+ T cells. Here, we find that local delivery of IL-12 increased the expression of Fas within tumor-infiltrating macrophages, dendritic cells, and myeloid-derived suppressor cells (MDSC), and that these changes were abrogated in mice deficient in IL-12–receptor signaling. Importantly, upregulation of Fas in host mice played a critical role in the proliferation and antitumor activity of adoptively transferred IL-12–modified CD8+ T cells. We also observed higher percentages of myeloid-derived cell populations within tumors in Fas-deficient mice, indicating that tumor stromal destruction was dependent on the Fas death receptor. Taken together, these results describe the likely requirement for costimulatory reverse signaling through Fasl on T cells that successfully infiltrate tumors, a mechanism triggered by the induction of Fas expression on myeloid-derived cells by IL-12 and the subsequent collapse of the tumor stroma.
Introduction
Malignant cells possess the unique ability to recruit a variety of non-transformed stromal cells to aid in their growth and metastases.1,2 These stromal cells drive carcinogenesis by creating a chronic or low leveled inflammatory environment that maybe critical for immune escape, neo-vascularization, and maintaining the genetic instability and replicative potential of malignant cells.3 Although it is well established that chronic inflammation induces tumor formation, evidence also suggests that under acute conditions, inflammation can orchestrate beneficial cross-talk between innate and acquired immunity against tumors.4,5
We recently described the ability to treat large established melanomas with a single dose of 10,000 CD8+ T cells engineered to secrete a single-chain functional interleukin 12 (IL-12) molecule (IL-12TD cells),5 building on earlier work suggesting that this approach may improve adoptive cell therapies.6 Similar findings have been observed in several different mouse models following the adoptive transfer of IL-12–expressing T cells, providing valuable biological insights.7,8,9,10,11 The mechanisms ascribed to the improvements in antitumor immunity include enhancements in the functionality of engineered T cells and an improved proliferative burst following adoptive transfer.5,10 Furthermore, IL-12 triggers an acute inflammatory environment that reverses stromal cell dysfunction within tumors, enabling them to efficiently cross-present naturally occurring tumor antigens.12,13 This recognition of cross-presented tumor antigens by CD8+ T cells maybe the critical initial step that allows for the arrested migration of T cells within tumors, but additional physiological changes are likely necessary to trigger the cascade of events that leads to the regression of established lesions. Interestingly, we and others witnessed a marked increase in the ability of IL-12–engineered T cells to infiltrate tumors compared with CD8+ T cells not expressing IL-12, but the mechanisms underlying this phenomenon remain to be elucidated.
Here, we describe that the adoptive transfer of tumor-specific CD8+ T cells engineered to secrete IL-12 induces the upregulation of Fas receptors (CD95) on myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells within the tumor stroma, a phenomenon dependent of the ligation of IL-12 receptors on endogenously infiltrating immune cells. Interestingly, IL-12–triggered induction of Fas on host cells delivers a proliferative signal for adoptively transferred, Fas ligand–expressing T cells. These findings suggest that reverse signaling through Fasl on T cells in an inflammatory environment is required for the maintenance of effector memory CD8+ T cells at local sites, although other unknown cross-reactive ligands for the Fas receptor may also contribute to these costimulatory effects. In our previous studies, we witnessed a marked decrease in the number of myeloid-derived cells within tumors immediately before tumor regression. We now find that stromal collapse is prevented in the absence of Fas-receptor expression by endogenous immune cells. These findings highlight the critical Fas–Fasl interactions necessary within the tumor microenvironment to maintain and propagate T-cell–mediated regression of established lesions.
Results
IL-12 increases the expression of Fas and Fasl within tumors
To demonstrate the ability to successfully express the single-chain IL-12 gene into pmel-1 CD8+ T cells (IL-12TD cells), we performed an intracellular stain following retroviral transduction in pmel-1 splenocytes (Figure 1a). Due to the presence of the long-terminal repeat promoter in our retroviral construct, we observed IL-12 expression without the need for restimulation (Figure 1a). In our previous work, we performed a whole transcriptome analysis from tumors 3 and 7 days following the adoptive transfer of 1 × 105 non-transduced (mock) or IL-12TD cells and highlighted the importance of IL-12 to reprogram the tumor microenvironment.12 For this study, we attempted to generate further mechanistic insight by analyzing differentially expressed genes within tumors from mice treated with either mock or IL-12TD cells. Interestingly, after analyzing the robust multichip analysis data, we found a statistically significant increase in the expression of both the Fas receptor (Figure 1b) and Fas ligand (Figure 1c) from tumors in mice treated with IL-12TD cells compared with mock cells at 3 and 7 days following adoptive transfer.
Figure 1.

Adoptive transfer of IL-12–engineered CD8+ T cells into C56BL/6 mice bearing established subcutaneous B16 tumors induces an increase in Fas receptor (CD95) and Fasl (CD95L) expression within whole tumor samples. (a) Representative intracellular flow cytometry plot for expression of IL-12 in pmel-1 CD8+ T cells transduced with a retroviral vector encoding the single-chain IL-12A sequence linked to IL-12B with a (Gly4Ser)3 flexible linker. (b) Robust multichip analysis (RMA) in log base 2 format for expression of Fas in B16 tumors 3 and 7 days following treatment with either mock or IL-12–transduced pmel-1 CD8+ T cells into sublethally irradiated C56BL/6 mice bearing established tumors. (c) RMA in log base 2 format for expression of Fasl from tumors of mice treated similarly to b. All data are expressed as a mean ± SEM and RMA analysis was obtained from previously published whole genome transcriptome analysis of four independent tumor samples. *P < 0.05, compared with no treatment control tumors, **P < 0.05, compared with B16 tumors from mice treated with non-transduced CD8+ T cells (mock). NT, no treatment.
The majority of cells expressing the IL-12Rβ2 within tumors are MDSC and dendritic cells
Solid tumors harbor a complex mixture of endogenous cells that supports the growth of mutated neoplastic cells. To visualize the contribution of host-derived, non-transformed cells within an established and growing tumor, we implanted subcutaneous B16 tumors on albino C57BL/6 dsRed × CD11c-YFP mice (all endogenous cells express the red-fluorescent protein) and noted a dense network of endogenous cells (red) infiltrating the tumor mass directly below the collagen stained (blue) tumor capsule (Figure 2a). Given the heterogeneity of cells within tumors, we sought to determine the specific lineage of cells capable of responding to IL-12 within the tumor mass. We generated single cell suspensions of 1 week established B16 subcutaneous tumors and determined that the majority of cells that expressed the IL-12Rβ2 and capable of functionally responding to IL-12 were Gr-1Hi MDSCs and CD11cHi dendritic cells (Figure 2b). Thus, myeloid-derived cells within tumors possess the intrinsic capability to be functionally reprogrammed by IL-12.
Figure 2.

The majority of cells that express the IL-12Rβ2 within tumors are MDSC and dendritic cells. (a) Static images from a 2 photon microscope of an 8-day–established subcutaneous B16 tumor (blue, collagen; red, endogenous cells). (b) Representative flow cytometric plots for IL-12Rβ2 expression in single cell tumor suspensions from established subcutaneous B16 tumors. Cells staining positive for IL-12Rβ2 were further analyzed for expression of Gr-1, CD11c, and F4/80. All plots gated on PI− live cells. Data are representative of three independent experiments.
MDSC, macrophages, and dendritic cells in the tumor stroma upregulate Fas expression in response to IL-12
We next attempted to decipher the cell populations responsible for the increased expression of Fas within tumors following treatment with IL-12TD cells. Our previous studies demonstrated the importance for IL-12 to induce the cross-presentation of natural tumor antigens, allowing transferred CD8+ T cells to form an immunologic synapse with antigen-presenting cells (APC) within tumors. We therefore hypothesized that these myeloid-derived APCs, possessing a functional IL-12Rβ2, may increase their expression of Fas in response to the secreted IL-12. To measure the direct effects of IL-12, either mock-transduced or IL-12–transduced pmel-1 CD8+ T cells were in vitro cocultured for 48 hours with a single cell tumor suspension created from 7-day–established subcutaneous B16 melanomas. Following coculture with IL-12TD cells, there indeed was an increase in the expression of Fas within all myeloid-derived subpopulations in comparison with cocultures with mock-transduced cells (Figure 3a,b).
Figure 3.

MDSC, macrophages, and dendritic cells residing within tumors upregulate the expression of Fas in response to IL-12 secretion by transferred CD8+ T cells. (a) Flow cytometric analysis of the expression of Fas in CD11b+ Gr1Hi MDSC, CD11b+ F4/80Hi macrophages, and CD11b+ CD11cHi dendritic cells following a 48 hour in vitro coculture of single cell suspensions from 1 week established B16 tumors with mock-transduced or IL-12–transduced pmel-1 CD8+ T cells. (b) Quantification of the percentage of Fas-positive cells within the different subpopulations of myeloid cells from a. All data are expressed as a mean ± SEM and representative of two independent experiments. *P < 0.05 compared with coculture with mock-transduced cells. (c) Representative flow cytometry plot for the in vivo expression of Fas in tumor infiltrating CD11b+ Gr1Hi/Mid MDSC, CD11b+ F4/80Hi macrophages, and CD11b+ CD11cHi dendritic cells in wild-type (WT) or IL-12Rβ2−/− mice bearing established B16 tumors following treatment with IL-12TD CD8+ T cells. All plots gated on PI−, CD11b+ cells. (d) Quantification of the percentage of Fas-positive cells in vivo within the different subpopulations of myeloid cells from c. All data are expressed as a mean ± SEM and representative of two independent experiments. *P < 0.05 compared with non-treated tumors established on WT hosts. **P < 0.05 compared with IL-12–expressing T cells transferred into tumor-bearing IL-12Rβ2−/− mice. NT, no treatment.
To demonstrate these changes in vivo, we adoptively transferred 1 × 105 IL-12TD cells into sublethally irradiated wild type (WT) or IL-12Rβ2−/− mice and analyzed the expression of Fas on different bone-marrow–derived stromal cells within the tumor microenvironment 7 days following adoptive transfer. Flow cytometric analysis of tumor-infiltrating cells revealed an increase in the percentage of Fas-expressing cells within the CD11b+ Gr-1Mid/CD11b+ Gr-1Hi MDSC, CD11b+ F4/80Hi macrophages, and CD11b+ CD11cHi dendritic cell populations (Figure 3c). We also witnessed similar changes 3 days following the adoptive transfer of IL-12–expressing CD8+ T cells (Supplementary Figure S1). Interestingly, the increased expression of Fas within the tumor stroma was significantly abrogated in IL-12Rβ2−/− mice, indicating the direct importance for IL-12 receptor ligation on endogenous immune cells to induce the upregulation of Fas (Figure 3c). To quantify these findings, we examined samples from separate experiments and indeed found a statistically significant increase in both the percentage of Fas-positive cells (Figure 3d) and the mean fluorescence intensity (Supplementary Figure S2) of Fas expression within the tumor-infiltrating MDSC, macrophage and dendritic cell populations from mice treated with the IL-12TD compared with mock-transduced cells. These results indicate that IL-12 secreted by adoptively transferred CD8+ T cells induces the expression of Fas on cross-presenting myeloid-derived cells within the tumor microenvironment.
IL-12–engineered T cells within tumors express Fas ligand
We next hypothesized that the increased expression of Fas within tumors may have an effect on transferred CD8+ T cells forming an immunologic synapse with Fas-expressing APC. We therefore looked for the expression of Fasl on adoptively transferred T cells within tumors. We harvested tumors 7 days following the transfer of 1 × 105 thy1.1+ IL-12–expressing pmel-1 CD8+ T cells or mock-transduced thy1.1+ pmel-1 CD8+ T cells into sublethally irradiated WT mice bearing subcutaneous B16 tumors and found that thy1.1+ CD8+ T cells within tumors did indeed express Fasl (Figure 4a,b). Interestingly, thy1.1− CD8+ T cells within tumors did not express Fasl, indicating that only the adoptively transferred CD8+ T cells and not the endogenous CD8+ T-cell fraction expressed measurable levels of surface Fasl expression. However, there did not appear to be an increase in Fasl expression in IL-12TD cells as adoptively transferred mock-transduced pmel-1 CD8+ T cells also expressed Fasl to a similar degree (Supplementary Figure S3). Thus, there exists the possibility that reverse signaling through Fasl on adoptively transferred cells plays a costimulatory role for effector memory T cells at local sites of acute-inflammation, a physiological event that is critically dependent on the increased expression of Fas by IL-12.
Figure 4.
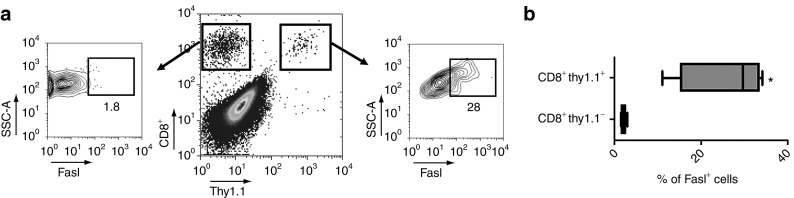
Adoptively transferred IL-12–engineered CD8+ T cells express Fasl within the tumor microenvironment. (a) Representative flow cytometry plot for the expression of Fasl on adoptively transferred T cells (CD8+ thy1.1+) from single cell tumor suspensions 7 days following treatment with 105 IL-12–engineered T cells into B16 tumor–bearing mice. (b) Quantification of the percentage of Fasl+ T cells within the CD8+ thy1.1+ transferred and CD8+ thy1.1− endogenous populations. All cells gated on live PI− cells. All data are expressed as a mean ± SEM and representative of two independent experiments.
Tumor-stromal cell expression of Fas is required for the infiltration and proliferation of IL-12–expressing T cells
On the basis of our experiments showing an increase in the expression of Fas in APCs within tumors combined with the expression of Fasl on transferred CD8+ T cells, we hypothesized that these interactions may play a role in the ability of transferred CD8+ T cells to infiltrate and proliferate at the tumor site. We therefore harvested tumors 7 days following the adoptive transfer of 1 × 105 IL-12–expressing thy1.1+ pmel-1 CD8+ T cells into either WT or Faslpr mice and examined the infiltration of transferred T cells within tumors. Strikingly, we observed minimal infiltration of adoptively transferred IL-12–expressing thy1.1+ pmel-1 CD8+ T cells in mice deficient in Fas-receptor signaling (Figure 5a; left panel). These results were in stark contrast to the substantial infiltration of transferred T cells observed in WT mice. Quantification through flow cytometry–based assays showed a statistically significant increase in the infiltration of transferred T cells within B16 tumors of WT mice compared with Faslpr mice (Figure 5a; right panel). Thus, IL-12–induced expression of Fas on APCs within tumors likely delivers a costimulatory reverse signal to Fasl-expressing antigen-specific T cells forming an immunologic synapse within the tumor microenvironment.
Figure 5.

IL-12–engineered CD8+ T cells fail to infiltrate tumors and do not cause stromal collapse in mice deficient in Fas-receptor signaling. (a) Wild-type (WT) or Faslpr mice bearing 10-day–established B16 subcutaneous tumors were treated with 105 IL-12–engineered CD8+ T cells following sublethal irradiation. Seven days following treatment, tumors were harvested and examined by flow cytometry for infiltration of adoptively transferred CD8+ thy1.1+ T cells. Representative flow cytometry plots in the left panel are gated on live PI−, CD8+ T cells with the quantification of data in the right panel. All data are expressed as a mean ± SEM and representative of two independent experiments. *P < 0.05 compared with treatment in WT mice. (b) Tumors and spleens from mice treated similarly as a were harvested and examined for the presence of CD11b+ myeloid-derived cells and CD3+ lymphocytes. The left panel represents flow cytometry plots for CD11b+ cells in B16 tumors and the right panel is a representative plot for CD11b+ and CD3+ cells within spleens. All plots gated on live PI− cells and are representative of two independent experiments. (c) Quantification of the percentage of CD11b+ and CD3+ cells from b. All data are expressed as a mean ± SEM and representative of two independent experiments. *P < 0.05 compared with treatment in WT mice.
IL-12–expressing T cells induce tumor-stromal collapse through Fas-receptor signaling
We next wanted to investigate the effects of signaling through the Fas receptor on APCs within the tumor. Our previous studies showed a significant decrease in the percentage of CD11b+ stromal cells within tumors of mice treated with IL-12–expressing T cells.12 These changes occurred just before regression of established lesions (10–14 days following transfer). We therefore hypothesized that signaling through the Fas receptor may contribute to the active loss of these stromal populations within tumors. We adoptively transferred IL-12–engineered T cells into sublethally irradiated B16 tumor–bearing WT or Faslpr mice and measured the percentage of CD11b+ cells within tumors and spleens 10 days following transfer. We observed a marked increase in the percentage of CD11b+ cells within tumors in Faslpr mice compared with WT mice (Figure 5b; left panel). To examine whether these differences were a generalized phenomenon or localized specifically to the tumor, we harvested spleens from the same mice that had tumors analyzed and measured the percentage of CD11b+ myeloid-derived cells and CD3+ T cells. In contrast to tumors, we did not observe higher percentages of CD11b+ cells within spleens of Faslpr mice compared with WT mice treated with IL-12–engineered T cells (Figure 5b; right panel). Thus, in locations where tumor antigens are present and an immunologic synapse with antigen-specific CD8+ T cells is theoretically possible, we observed a decrease in the percentage of CD11b+ myeloid-derived stromal cells in WT mice compared with mice deficient in Fas-receptor signaling. These changes were quantified and found to be statistically significant (Figure 5c). Thus, T-cell delivery of IL-12 within the tumor environment upregulates Fas expression in myeloid-derived cells within tumors, leading to the increased local proliferation of Fasl-expressing T cells in combination with the likely delivery of apoptotic signals to Fas receptor expressing stromal cells.
In light of the well-known lymphoproliferative effects in Faslpr mice, we also compared the percentages of T cells within spleens of WT and Faslpr mice. We noted only minor differences (<15%) in the percentage of T cells in Faslpr mice compared with WT mice, likely due to experimentation with young mice (6–7 weeks old) and the use of sublethal lymphodepleting irradiation (5 Gy) before all our cell transfer experiments (Figure 5b; right panel).
Fas–Fasl cross-talk is required for IL-12–engineered T cells to mediate antitumor immunity
We next sought to assess the functional significance of Fas–Fasl signaling in vivo and measured antitumor responses following the adoptive transfer of 1 × 105 IL-12–expressing pmel-1 CD8+ T cells into sublethally irradiated WT or Faslpr mice bearing B16 tumors established for 10 days. We observed a significant impairment in the antitumor immunity of IL-12–engineered T cells in mice deficient in Fas-receptor signaling compared with WT mice (Figure 6a). Furthermore, we observed a significant decrease in survival in Faslpr mice compared with WT mice treated with the IL-12TD cells (Figure 6b). Taken together, these findings highlight the critical need for IL-12–induced Fas–Fasl interactions within the tumor microenvironment to maintain and propagate T-cell–mediated regression of established lesions.
Figure 6.

IL-12–induced Fas expression on endogenous stromal cells is critical for the antitumor function of adoptively transferred CD8+ T cells. (a) Antitumor immunity of 105 IL-12–engineered pmel-1 CD8+ T cells transferred into sublethally irradiated wild-type (WT) or Faslpr mice bearing subcutaneous B16 tumors established for 10 days. (b) Kaplan–Meier survival curves for mice treated in a. All data are expressed as a mean ± SEM and are representative of two independent experiments. *P < 0.05, Wilcoxon rank sum test compared with no treatment control, **P < 0.05 compared with IL-12 CD8+ T cells into Faslpr host mice. NT, no treatment.
Discussion
A budding body of evidence now suggests that the immunological components of a tumor mass help form a framework that enables for the sustained growth of cancers.2,14 This stromal network includes a variety of cells of hematopoietic origin from both innate and acquired immunity.1,15,16,17,18 Evidence now suggests that tumors can harbor myeloid-derived cells that phenotypically resemble APCs. However, these immunologic constituents of a tumor mass are often not capable of functionally activating CD4+ and CD8+ T lymphocytes.19,20,21 Several studies now show that ablating or reprogramming the intrinsic properties of stromal cells can enhance cancer therapies and provides a rationale for using these agents in a multimodality approach.22,23,24
The death receptor Fas is expressed in a variety of mammalian tissues and cells and has been reported to be functionally active in various myeloid lineage cells.24,25 Although classically viewed as a receptor that induces apoptosis, recent studies are starting to highlight alternative functions for this intriguing molecule.26,27 The natural ligand for Fas, appropriately named Fas ligand, is also expressed by a variety of cells including activated lymphocytes. A well-studied facet of Fasl is the ability to deliver a death signal through the Fas receptor on target cells, but compelling studies also show the importance of reverse signaling through Fas ligand and the costimulatory effects of this molecule.28,29
IL-12 is a hallmark inflammatory cytokine capable of eliciting potent immune responses and directly augments the functionality of multiple end effectors such as CD4+ T, CD8+ T, natural killer (NK), and NKT cells.30,31,32,33,34,35,36 The antitumor effects of IL-12 are thought to be well known. IL-12 enhances the ability of CD8+ T, NK, and NKT cells to kill tumor targets, and recent studies point to the ability of IL-12 to improve the functionality of APCs within the tumor stroma.5,12 The link between IL-12 and Fas–Fasl signaling for tumor destruction is well described, but the mechanism that has classically been described is the induction of Fas directly on tumor cells and the delivery of a death signal by Fasl on a variety of cells such as lymphocytes, NK, and tumor cells.37,38,39
In this study, we demonstrate that T-cell delivery of IL-12 to the tumor microenvironment increased the expression of Fas on MDSC, macrophages, and dendritic cells within tumors. We also show that the IL-12–expressing CD8+ T cells infiltrating tumors express Fas ligand. Our previous studies highlighted the importance for major histocompatibility complex class I on host APC within tumors, indicating the need for cross-presentation of natural tumor antigens and the formation of an immunologic synapse between transferred CD8+ T cells and APCs within the tumor microenvironment. The arrested migration of T cells with cross-presenting APCs likely enables for further beneficial cross-talk in an inflammatory local environment.
Here, we show that in the absence of Fas-receptor signaling on host immune cells, adoptively transferred IL-12–engineered CD8+ T cells do not engraft well and fail to infiltrate tumors to the same degree as in mice with an intact Fas receptor. The increase in Fas expression on endogenous myeloid-derived cells by IL-12 is likely the critical biological change because Fasl is stably expressed on transferred T cells and not upregulated by IL-12. These findings suggest that reverse signaling through Fasl on transferred T cells, induced by the increased expression of Fas on APCs, maintains populations of effector memory cells at local sites of inflammation, although we cannot rule out the possibility that the Fas receptor triggers a proliferative signal through reverse signaling on other cross-reactive T-cell surface ligands.
Another importance facet of the Fas–Fasl communication is the delivery of apoptotic signals through the Fas receptor on APCs forming the tumor stroma. In our previous studies, we demonstrated the ability of transferred IL-12–expressing CD8+ T cells to induce a significant decrease in the number of Ly-6CHi monocyte-derived myeloid cells, dendritic cells, and macrophages, a phenomenon occurring just before the collapse of large vascularized lesions. We did not see this decrease in stromal cells following treatment with IL-12–engineered T cells into mice deficient in Fas signaling, indicating that the mechanism of stromal elimination induced by T cells likely involves signaling through the Fas receptor on APCs within the tumor microenvironment.
Importantly, the communication between Fas on APCs and Fasl on T cells was critical for the antitumor efficacy of adoptively transferred IL-12–expressing CD8+ T cells. Following a lymphodepleting 5Gy irradiation regimen to help normalize the number of endogenously circulating lymphocytes between WT and Faslpr mice, we witnessed a marked decrease in the ability of IL-12–engineered CD8+ T cells to induce tumor regression in mice lacking Fas receptors. Taken together, these findings highlight the critical Fas–Fasl interactions necessary within the tumor microenvironment to maintain and propagate T-cell–mediated regression of established lesions. Future studies using different established tumor models such as lung, colorectal, renal cell, sarcomas, or lymphomas will need to be conducted to show that Fas–Fasl cross-talk within tumors is important in other tumor histologies besides melanoma.
In conclusion, we highlight what we believe is a critical mechanism for the ability of IL-12–expressing CD8+ T cells to induce tumor regression. IL-12 within the tumor microenvironment enables for the recruitment and maintenance of a population of activated CD8+ T cells that induce stromal collapse through the induction of Fas–Fasl signaling. This concept is currently being translated in human clinical trials and in the future, engineering tumor-specific T cells with IL-12 may enable adoptive immunotherapies to reach their full therapeutic potential against cancers of various histologies.5,7,8,10,12,40,41,42
Materials and Methods
Mice and tumor lines. Female thy1.1+ pmel-1 T-cell receptor transgenic (Tg) mice were generated in our laboratory and made available at http://www.jax.org. Female C57BL/6 mice, C57BL/6 CD90.1 (thy1.1+), C57BL/6 Il12rb2−/−, and C57BL/6 Faslpr mice were obtained from Jackson Laboratories (Bar Harbor, ME). C57BL/6 Albino dsRed (RFP transgene; Jackson Laboratories) mice were crossed with CD11c-YFP C57BL/6 mice to generate mice for two photon microscopic images. Tumor lines used for in vivo experiments (B16, an H-2b+/gp100+ murine melanoma) were maintained in culture through previously described methodologies.43
Retroviral production and transduction. Platinum Eco 293–based cells (Cell Bio Labs, San Diego, CA) were plated on poly-D-lysine coated 100 mm plates (BD Biosciences, San Jose, CA) and transfected with 6 µg of pCL-Eco helper plasmid (Imgenex, San Diego, CA) and 9.3 µg of the MSGV-1 IL-12 vector with lipofectamine 2000 (Invitrogen, Carlsbad, CA) overnight in antibiotic-free complete media. Viral supernatants were harvested 36–48 hours after transfection. Retroviral transductions: Pmel-1 splenocytes were cultured in the presence of 1 µmol/l hgp10025–33 and complete media containing 60 International Units (IU)/ml of recombinant human (rh) IL-2 (Chiron, Emeryville, CA). For transduction of C57BL/6 splenocytes, 1 µg/ml of soluble anti-CD3 (BD Biosciences) and 1 µg/ml of soluble anti-CD28 (BD Biosciences) were used to stimulate bulk splenocytes. After 2 days, splenocytes were collected and resuspended in retroviral supernatant with 60 IU/ml rhIL-2 and 10 µg/ml protamine sulfate (Abraxis Pharmaceutical Products, Schaumburg, IL), and spun at 1,000 g at 32 °C for 90 minutes in 24-well plates. Cultured cells were adoptively transferred 3–5 days after transduction (>90% CD8+ T cell).43
Adoptive cell transfer. Six- to seven-week-old mice of different strains (n = 5 for all groups) were injected subcutaneously with 5 × 105 B16 melanoma cells. After 10–14 days, they were irradiated with 5 Gy total body irradiation and given by tail vein 1 × 105 IL-12–transduced pmel-1 CD8+ T cells (IL-12) cells or 1 × 105 pmel-1 CD8+ T cells undergoing the full transduction protocol without the addition of viral supernatant (mock). Tumors were measured using digital calipers and the tumor area was calculated as the product of perpendicular diameter by investigators in a blinded manner. All experiments were performed independently at least twice with similar results and all tumor curve data are shown as mean ± SEM.
Flow cytometry–based analysis. Following adoptive transfer, tumor samples were harvested, digested with 200 U/ml collagenase IV (Invitrogen) and 100 µg/ml DNAse I (Roche, San Francisco, CA), and evaluated by flow cytometry for CD3, CD90.1 (thy1.1), CD11b, F4/80, Gr-1, CD11c, CD95, and CD95L (BD Biosciences). Transferred pmel-thy1.1+ cells were analyzed by flow cytometry for thy1.1 and CD8α expression.
Two photon microscopy of established tumors. C57BL/6 Albino dsRed × CD11c-YFP mice were anesthetized with isoflurane in 30% O2/70% air, with body temperatures maintained at 37 °C via a temperature-controlled environmental chamber and heating pads. Tumors were surgically exposed and imaged through a 20× water immersion lens (N.A. 1.0) using a Zeiss 710 system (Multiphoton Confocal Microscope Zeiss 710) tuned to 920 nm. Imaging planes were collected at 4-μm z-intervals to yield xyz data sets for processing and analysis by Imaris (BitPlane, South Windsor, CT). Collagen second harmonic signals were detected with a 495DCLP mirror in combination with a 447/60 BP filter, yellow fluorescent protein signals were detected with a 565 DCXR mirror in combination with a 530/11 BP filter, and dSRed signals were detected with a 565DCXR filter in combination with a 630/90 BP filter.
Statistical analysis. Tumor growth slopes were compared using Wilcoxon rank sum test. One-way analysis of variance and two-tailed Student's t-tests were used to test for significant differences in enumeration assays. P < 0.05 was considered significant.
Study approval. All experiments were conducted with the approval of the National Cancer Institute Animal Use and Care Committee.
SUPPLEMENTARY MATERIAL Figure S1. Quantification of the percentage of Fas-positive cells within the CD11b+ GrlHi cells, CD11b+ GrlMid cells, CD11b+ F4/80Hi macrophages, or CD11b+ CD11cHi dendritic cells within tumors from sublethally irradiated WT mice bearing established B16 tumors 3 days following treatment with mock or IL-12–expressing CD8+ T cells. Figure S2. The mean fluorescence intensity of Fas (CD95) in tumor infiltrating CD11b+ GrlHi/Mid cells, CD11b+ F4/80Hi macrophages, and CD11b+ CD11cHi dendritic cells in sublethally irradiated WT or IL-12Rβ2−/− mice bearing established B16 tumors 7 days following treatment with IL-12–expressing CD8+ T cells. Figure S3. Representative flow cytometry plot for the expression of Fasl on adoptively transferred T cells (CD8+ thy1.1+) frome single cell tumor suspensions 7 days following treatment with 105 mock-transduced T cells into B16 tumor bearing mice.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The authors declare no conflict of interest.
Supplementary Material
References
- Bronte V. Myeloid-derived suppressor cells in inflammation: uncovering cell subsets with enhanced immunosuppressive functions. Eur J Immunol. 2009;39:2670–2672. doi: 10.1002/eji.200939892. [DOI] [PubMed] [Google Scholar]
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkar SP, Restifo NP. Cellular constituents of immune escape within the tumor microenvironment. Cancer Res. 2012;72:3125–3130. doi: 10.1158/0008-5472.CAN-11-4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria S, Pikarsky E, Karin M, Coussens LM, Chen YC, El-Omar EM, et al. Cancer and inflammation: promise for biologic therapy. J Immunother. 2010;33:335–351. doi: 10.1097/CJI.0b013e3181d32e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkar SP, Muranski P, Kaiser A, Boni A, Sanchez-Perez L, Yu Z, et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 2010;70:6725–6734. doi: 10.1158/0008-5472.CAN-10-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner HJ, Bollard CM, Vigouroux S, Huls MH, Anderson R, Prentice HG, et al. A strategy for treatment of Epstein-Barr virus-positive Hodgkin's disease by targeting interleukin 12 to the tumor environment using tumor antigen-specific T cells. Cancer Gene Ther. 2004;11:81–91. doi: 10.1038/sj.cgt.7700664. [DOI] [PubMed] [Google Scholar]
- Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, et al. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res. 2012;18:1672–1683. doi: 10.1158/1078-0432.CCR-11-3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71:5697–5706. doi: 10.1158/0008-5472.CAN-11-0103. [DOI] [PubMed] [Google Scholar]
- Kerkar SP, Restifo NP. The power and pitfalls of IL-12. Blood. 2012;119:4096–4097. doi: 10.1182/blood-2012-03-415018. [DOI] [PubMed] [Google Scholar]
- Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–4141. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP, et al. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther. 2011;19:751–759. doi: 10.1038/mt.2010.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest. 2011;121:4746–4757. doi: 10.1172/JCI58814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Bose A, Komita H, Taylor JL, Kawabe M, Chi N, et al. Intratumoral IL-12 gene therapy results in the crosspriming of Tc1 cells reactive against tumor-associated stromal antigens. Mol Ther. 2011;19:805–814. doi: 10.1038/mt.2010.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz JB, Muller AJ, Prendergast GC. Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunol Rev. 2008;222:206–221. doi: 10.1111/j.1600-065X.2008.00610.x. [DOI] [PubMed] [Google Scholar]
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiotto MT, Rowley DA, Schreiber H. Bystander elimination of antigen loss variants in established tumors. Nat Med. 2004;10:294–298. doi: 10.1038/nm999. [DOI] [PubMed] [Google Scholar]
- Zhang B, Bowerman NA, Salama JK, Schmidt H, Spiotto MT, Schietinger A, et al. Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J Exp Med. 2007;204:49–55. doi: 10.1084/jem.20062056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375–379. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- Motz GT, Coukos G. The parallel lives of angiogenesis and immunosuppression: cancer and other tales. Nat Rev Immunol. 2011;11:702–711. doi: 10.1038/nri3064. [DOI] [PubMed] [Google Scholar]
- Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J, et al. Immune resistance orchestrated by the tumor microenvironment. Immunol Rev. 2006;213:131–145. doi: 10.1111/j.1600-065X.2006.00442.x. [DOI] [PubMed] [Google Scholar]
- Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330:827–830. doi: 10.1126/science.1195300. [DOI] [PubMed] [Google Scholar]
- Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 2005;65:3437–3446. doi: 10.1158/0008-5472.CAN-04-4262. [DOI] [PubMed] [Google Scholar]
- Listopad JJ, Kammertoens T, Anders K, Silkenstedt B, Willimsky G, Schmidt K, et al. Fas expression by tumor stroma is required for cancer eradication. Proc Natl Acad Sci USA. 2013;110:2276–2281. doi: 10.1073/pnas.1218295110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RM, Chan FK, Chun HJ, Lenardo MJ. The multifaceted role of Fas signaling in immune cell homeostasis and autoimmunity. Nat Immunol. 2000;1:469–474. doi: 10.1038/82712. [DOI] [PubMed] [Google Scholar]
- Liu K, Caldwell SA, Abrams SI. Immune selection and emergence of aggressive tumor variants as negative consequences of Fas-mediated cytotoxicity and altered IFN-gamma-regulated gene expression. Cancer Res. 2005;65:4376–4388. doi: 10.1158/0008-5472.CAN-04-4269. [DOI] [PubMed] [Google Scholar]
- Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boursalian TE, Fink PJ. Mutation in fas ligand impairs maturation of thymocytes bearing moderate affinity T cell receptors. J Exp Med. 2003;198:349–360. doi: 10.1084/jem.20030220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Ames KT, Suzuki I, Fink PJ. The cytoplasmic domain of Fas ligand costimulates TCR signals. J Immunol. 2006;177:1481–1491. doi: 10.4049/jimmunol.177.3.1481. [DOI] [PubMed] [Google Scholar]
- Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- O'Garra A, Murphy KM. From IL-10 to IL-12: how pathogens and their products stimulate APCs to induce T(H)1 development. Nat Immunol. 2009;10:929–932. doi: 10.1038/ni0909-929. [DOI] [PubMed] [Google Scholar]
- Colombo MP, Trinchieri G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002;13:155–168. doi: 10.1016/s1359-6101(01)00032-6. [DOI] [PubMed] [Google Scholar]
- Zitvogel L, Lotze MT. Role of interleukin-12 (IL12) as an anti-tumour agent: experimental biology and clinical application. Res Immunol. 1995;146:628–638. doi: 10.1016/0923-2494(96)83041-0. [DOI] [PubMed] [Google Scholar]
- Weiss JM, Subleski JJ, Wigginton JM, Wiltrout RH. Immunotherapy of cancer by IL-12-based cytokine combinations. Expert Opin Biol Ther. 2007;7:1705–1721. doi: 10.1517/14712598.7.11.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj N, Seder RA, Reddy A, Feldman MV. IL-12 in conjunction with dendritic cells enhances antiviral CD8+ CTL responses in vitro. J Clin Invest. 1996;98:715–722. doi: 10.1172/JCI118843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinke DJ, 2nd, Cheng N, Chambers E. Quantifying crosstalk among interferon-gamma, interleukin-12, and tumor necrosis factor signaling pathways within a TH1 cell model. Sci Signal. 2012;5:ra32. doi: 10.1126/scisignal.2002657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne M, Rimoldi D, Schröter M, Romero P, Schreier M, French LE, et al. Melanoma cell expression of Fas(Apo-1/CD95) ligand: implications for tumor immune escape. Science. 1996;274:1363–1366. doi: 10.1126/science.274.5291.1363. [DOI] [PubMed] [Google Scholar]
- Zeytun A, Hassuneh M, Nagarkatti M, Nagarkatti PS. Fas-Fas ligand-based interactions between tumor cells and tumor-specific cytotoxic T lymphocytes: a lethal two-way street. Blood. 1997;90:1952–1959. [PubMed] [Google Scholar]
- Wigginton JM, Park JW, Gruys ME, Young HA, Jorcyk CL, Back TC, et al. Complete regression of established spontaneous mammary carcinoma and the therapeutic prevention of genetically programmed neoplastic transition by IL-12/pulse IL-2: induction of local T cell infiltration, Fas/Fas ligand gene expression, and mammary epithelial apoptosis. J Immunol. 2001;166:1156–1168. doi: 10.4049/jimmunol.166.2.1156. [DOI] [PubMed] [Google Scholar]
- Hwu P, Rosenberg SA. The use of gene-modified tumor-infiltrating lymphocytes for cancer therapy. Ann N Y Acad Sci. 1994;716:188–97; discussion 197. doi: 10.1111/j.1749-6632.1994.tb21712.x. [DOI] [PubMed] [Google Scholar]
- June C, Rosenberg SA, Sadelain M, Weber JS. T-cell therapy at the threshold. Nat Biotechnol. 2012;30:611–614. doi: 10.1038/nbt.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner MK. Will T-cell therapy for cancer ever be a standard of care. Cancer Gene Ther. 2012;19:818–821. doi: 10.1038/cgt.2012.74. [DOI] [PubMed] [Google Scholar]
- Kerkar SP, Sanchez-Perez L, Yang S, Borman ZA, Muranski P, Ji Y, et al. Genetic engineering of murine CD8+ and CD4+ T cells for preclinical adoptive immunotherapy studies. J Immunother. 2011;34:343–352. doi: 10.1097/CJI.0b013e3182187600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.