

Abstract
Glioblastoma (GBM) comprises 51% of all gliomas and is the most malignant form of brain tumors with a median survival of 18–21 months. Standard-of-care treatment includes maximal surgical resection of the tumor mass in combination with radiation and chemotherapy. However, as the poor survival rate indicates, these treatments have not been effective in preventing disease progression. Cellular immunotherapy is currently being explored as therapeutic approach to treat malignant brain tumors. In this review, we discuss advances in active, passive, and vaccine-based immunotherapeutic strategies for gliomas both at the bench and in the clinic.
Introduction
Gliomas account for about 60% of all primary central nervous system tumors. Glioblastoma (GBM), which comprises 51.2% of all gliomas, is the most malignant form with a 2-year survival rate of 40% and a median survival of 18–21 months.1,2 The current standard-of-care includes surgical debulking of the tumor mass followed by radiation and chemotherapy using temozolomide.3 However, as indicated by the poor survival rate, these treatments have not been effective in preventing disease progression. Complete surgical resection of a tumor mass is nearly impossible due to the location and invasive properties of malignant gliomas. High doses of radiation therapy cannot be delivered due to potential damage to the normal brain. Likewise, chemotherapeutics often cannot penetrate the blood–brain barrier efficiently; and the resistances that gliomas are known to develop further compound the issues involved with these treatments.4,5,6,7,8
Cellular therapy is based on the idea of introducing a specific cell type into a particular tissue to treat the disease. Its earliest applications can be dated back to the 1950's where it was used in the bone marrow transplantation field.9,10 Currently, a broader spectrum for the application of cellular therapy is being pursued. Different cell types are used in replacement therapies to take over the function of diseased cells in the target organ. This is exhibited in cases of diabetes where insulin-producing cells can be injected to replace the malfunctioning diseased cells in the pancreas.11,12 Tissue engineering, in which ex vivo whole organs are recreated out of cells, is in early phases of development but holds a tremendous potential for the future.13 One example that has reached the clinic is the seeding of artificial skin, grown from collagen scaffolds, with the patient's own epidermal skin cells.14 This technique is Food and Drug Administration-approved and has been shown to drastically improve the life of patients with burn injuries. While both replacement therapy and tissue engineering focus on the use of cells for their inherent function (e.g., myoblasts for the generation of muscle tissue), other research is focusing on the application of cells for tasks outside of their pre-programmed function. For instance, stem cells and immune cells can be used for immunotherapy and as carriers of therapeutic genes or prodrugs that get activated at a specific location in the body. This cell-based therapy provides a new and interesting strategy for the treatment of cancers including brain tumors. Cellular immunotherapy in particular has the potential to both specifically target brain tumor cells, thereby limiting brain damage, and to establish a long-term antitumor response by stimulating the immune system. Thus, cellular immunotherapy is being explored as a new alternative therapy for gliomas. Currently, a wide range of strategies are being investigated at the bench, with a slow but steady portion of this research getting transitioned to the clinic. In this review, we cover recent advances in the field of immuno-cellular therapy for malignant gliomas both in the early experimental phase, as well as in the clinical setting.
Experimental Immuno-Cell Therapy
Over the last decade, extensive studies have been performed evaluating the use of modified immune cells as a potential therapeutic approach for gliomas. In vivo glioma xenografts of intracranial or subcutaneously implanted cells, as well as spontaneously induced gliomas, are widely used and commonly accepted models that depict an accurate and reproducible tumor environment in rodents. Histopathological changes such as pseudopalisade necrosis, glomeruloid vascular hyperplasia, and infiltrating cells mimic those found in human gliomas.15 In this section, we discuss experimental approaches using a variety of cells to boost the immune system and to establish a potent immune response against malignant brain tumors.
Overview of immuno-cell therapy
Cellular immunotherapy is based on the use of cells from the innate and adaptive immune system to elicit an antitumor response. Either passive or active immunotherapy can be pursued. Passive immunotherapy involves the ex vivo activation of immune cells, which are subsequently injected back into the patient to attack the tumor directly. Often, these cells are not only activated, but also genetically modified to have enhanced antitumor properties. This approach has proven to be successful, but has the limitation of lacking prolonged or continuous antitumor response. Recent studies are focused towards active immunotherapy or a combination of both. Active immunotherapy relies on the activation of the endogenous immune system either by vaccines or ex vivo-activated cells. This approach elicits long-term antitumor effects, which not only enhance the likelihood of the tumor being eradicated, but also decrease the risk of tumor recurrence. T cells, dendritic cells (DCs), and macrophages are the cells of choice for this therapeutic strategy (Table 1; Box 1 and Box 2).
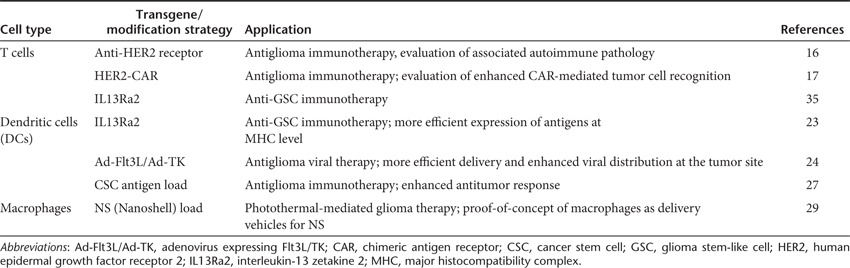
Table 1. Overview of experimental immuno-cell therapy against gliomas.
Box 1 Cells used for immune-cellular therapy.

Box 2 Cell isolation and preparation for immunotherapy.

T cell-based immunotherapy
T cells are often used as cellular vehicles for the treatment of different types of cancers typically resistant to conventional therapy. These cells can be easily modified with tumor-specific antigens to target the tumor. An interesting strategy in T cell-based immunotherapy is the use of chimeric antigen receptors (CARs). CARs combine the antigen-binding domain of antibodies with the ζ chain of T cell receptors, creating an increased binding between the T cell and the tumor antigen. In a proof-of-concept study, Wang et al.16 showed that CD8+ T cells can become tumor-specific when directed towards a tumor epitope such as the human epidermal growth factor receptor 2 (HER2), overexpressed in tumors versus normal tissues. Genetically engineered T cells expressing an anti-HER2 chimeric receptor were intravenously transferred in combination with lymphoablation and interleukin-2 (IL-2). The autoimmune effect had no significant toxicity on normal mammary and brain tissues expressing HER2, whereas an antitumor effect could be demonstrated, indicating that the modified T cells were specific to the tumor. Nakazawa et al.17 similarly combined HER2 and CARs to modify a T cell population, creating a functional HER2-CAR. Although HER2 is overexpressed by many different tumors, including malignant gliomas, the expression is often too low to be recognized by T cells. Intratumoral injection of CARs demonstrated efficient killing of tumor cells in vivo, including the CD133-positive glioma stem-like cells (GSC; also known as tumor-initiating cells resistant to chemo- and radiotherapy), and resulted in increased survival rate.18 The challenge of this therapeutic strategy is to establish stable expression of the activated antigen while achieving enough expansion with nonspecific activated T cells (known to be intolerant to transfection) as well as a persistent antitumor effect. To overcome these limitations, the same group used Epstein-Barr virus (EBV) to stimulate T cells (EBV-cytotoxic T lymphocytes (CTLs)), a method known to elicit an enhanced immune response.19 EBV-CTLs outperformed the activated T cells in both expansion and antitumor persistence.20,21,22 Further, the nonviral piggybac (PB) transposon system was evaluated for HER2-CAR gene delivery to T cells. While this model has been successfully applied in mouse primary cells, human cell lines, and inducible pluripotent stem cells, PB has not been evaluated for in vivo immunotherapeutic models. No preferential integration near or into oncogenes was observed, as typically is the case with retrovirus and lentivirus-based transduction methods. PB gene transfer was highly effective and the transduced cells could be maintained for >100 days in culture, while retaining stable transgene expression and T cell properties. Since transposons are less expensive and easier to produce than viral vectors, this method provides an interesting new approach for gene delivery.
DC and macrophage-based therapy
DCs are the most potent antigen-presenting cells of the human body, sensitizing T cells to all acquired antigens. In contrast to activation of T cells ex vivo, activation and stimulation of DCs induces a long-term immune response and is therefore considered active immunotherapy. DCs can be loaded with tumor antigens ex vivo, which can subsequently activate the endogenous immune system upon injection. Although this technique is safe, clinical efficiency still needs to be improved to achieve high expression of the antitumor antigen at the major histocompatibility complex (MHC) on the DC surface, and to expand the subgroup of these cells that primes naive T cells. Saka et al.23 designed a DC vaccine-based strategy aimed at targeting the IL-13 zetakine (IL13Ra2), which is overexpressed in gliomas. Since proper expression of this antitumor antigen at the DC MHC was problematic, a late endosomal/lysosomal sorting signal was added to the IL13Ra2 plasmid. DCs were transduced with this plasmid and injected intraperitoneally in glioma-bearing mice on days 3 and 10 of post-tumor implantation. A significant increase in the number of CD4+ and CD8+ T lymphocytes in the IL13Ra2-DC–treated tumor environment was observed, resulting in an increased survival rate.23 In a model in which gene therapy is effective but DC vaccination is not effective, Mineharu et al.24 demonstrated that combining in situ Ad-Flt3L/Ad-TK–mediated gene therapy (an Food and Drug Administration-approved adenoviral vector expressing either fms-like tyrosine kinase 3 ligand or thymidine kinase) with DC vaccination increased therapeutic efficacy and antitumor immunity as compared with in situ Ad-Flt3L/Ad-TK–mediated gene therapy alone. Ad-Flt3L and Ad-TK were intratumorally injected, followed by systemic administration of the prodrug ganciclovir. Flt3L causes DCs to migrate, differentiate, and expand within the tumor microenvironment of mice and rats. Thymidine kinase at the tumor site converts ganciclovir into a highly toxic phosphorylated drug causing the death of dividing tumor cells. Further, tumors treated with ganciclovir released high mobility group 1 protein, which serves as an adjuvant of the innate immune system by stimulating toll-like receptor 2 in signaling bone marrow-derived DCs thus confirming previous studies.25,26 The DCs were conditioned ex vivo with Fl3t and IL-6 to achieve enhanced proliferation and antitumor effects.24
As an alternative of loading DCs with regular tumor antigens, Xu et al.27 explored the use of cancer stem cell (CSC) antigens as a source for DC antiglioma vaccination. CSCs are thought to play an important role in the onset, progression, and recurrence of malignant gliomas and are known to express high levels of MHCs and tumor-associated antigens. A sufficient T cell response against CSCs and an increase in survival of mice bearing 9L gliosarcoma CSC tumors were observed as compared to DCs loaded with daughter or conventionally cultured 9L cells after intradermal injection of the vaccine. Albeit conventional loading and CSC antigen loading of DCs require further comparison in DC maturation and memory T cell generation in vivo, the authors speculate that CSC antigens might indeed be more suited for DC loading as compared with conventional tumor antigens. A clinical trial evaluating DC vaccines using CSCs is being considered and will start shortly.27
Another antigen-presenting cell used for immuno-cell therapy are macrophages. The advantage of these cells is their ability to easily travel across the blood–brain barrier, which often remains a great limitation for effective brain tumor therapy. Tumor-associated macrophages are often observed in the glioma microenvironment, and intravenously injected macrophages target the brain tumor site.28 In a recent study, Baek et al.,29 used macrophages loaded with gold-coated nanoshells for the treatment of human multicellular glioma spheroids. This in vitro model has similar characteristics in both resistance to radio- and chemotherapy, as well as growth and metabolic rates of glioma tumors in vivo, while simulating the tumor before vascularization. Nanoshells are spherical nanoparticles consisting of a dielectric core (called the silica), and an outer layer coated with a thin metallic shell (often made of gold) that converts the absorbed light to heat with great efficiency. Nanoshells are relatively small and can thereby easily be taken up by macrophages. In this study, using the glioma spheroid model, the authors compared macrophages loaded with empty nanoshells to macrophages with gold-coated nanoshells and found that the latter were able to inhibit tumor growth by photothermal therapy, whereas no response was observed with the empty control group.
Immuno-Cell Therapy in the Clinic
Despite an abundance of experimental research, only a small number of clinical trials are currently in progress focusing on the safety, efficacy, and feasibility of immuno-cellular therapeutic approaches in a phase I/II setting. While experimental therapies show a wide variety of strategic approaches, the clinic reflects a somewhat more conservative approach with DC vaccines and modified T lymphocytes dominating the picture (Table 2). Recently, two clinical trials showed the potential of immuno-cell therapy for the treatment of cancer. A study led by Professor Carl June at the University of Pennsylvania showed the success of adoptive CAR T cell therapy in treating chronic lymphocytic leukemia (CLL), where two out of three patients saw complete remission after CAR-CD19 therapy and had remained so for >1 year after treatment.30,31 Another trial led by Drs Renier Brentjens and Michel Sadelain at the Memorial Sloan Kettering Cancer Institute showed that the same CAR strategy can successfully treat patients with CLL and B cell acute lymphoblastic leukemia forms of blood cancer.32,33 These successful trials lend support to the significance of immuno-cellular therapeutic strategies for treating different tumors such as gliomas.
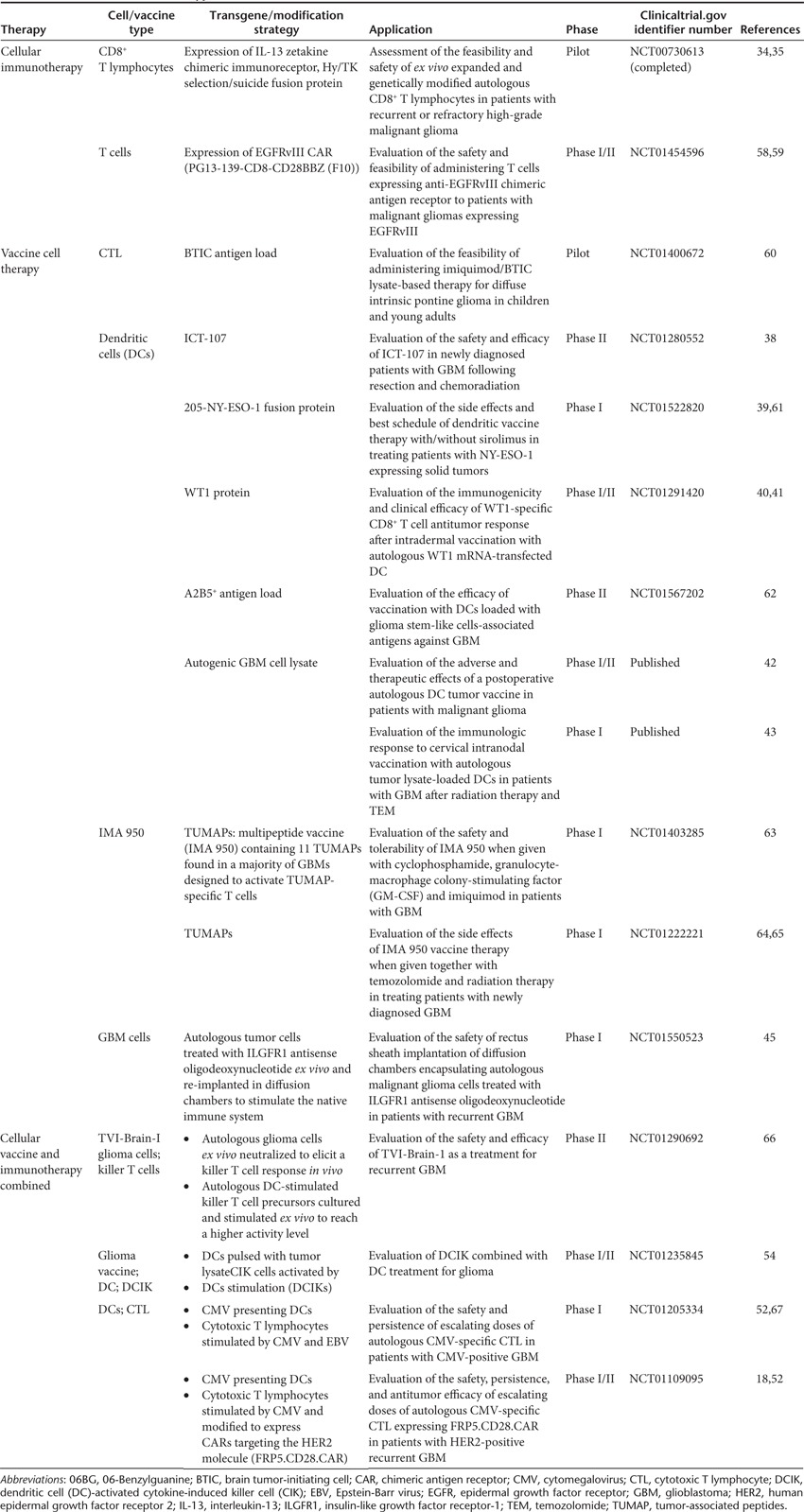
Table 2. Current immuno-cell therapy in clinical trials.
T cell immunotherapy in the clinic
It is well known that malignant gliomas evade provocation of an immune response. However, immune cells could be of tremendous value in the fight against brain tumors. These cells can provide a very efficient elimination mechanism of the tumor bulk and metastatic/invasive cells without the need to impair patient quality-of-life, as is the case with chemo- and radiotherapeutic paradigms. Many of the current strategies are exploring methods to overcome the lack of an immune response to glioma cells by artificially stimulating the immune system by either passive or active immunization. The use of T cells is one of the most popular strategies in the clinic, and is often used in combination with DC vaccines. Two clinical trials focused on ex vivo stimulation of T cells to boost a passive immune response are currently in progress.
In one trial, by Forsman et al. at the City of Hope Medical Center, autologous peripheral blood mononuclear cells are collected and genetically modified to express the membrane-tethered IL-13 cytokine chimeric T cell receptor targeting the IL-13 receptor α2 (IL13Rα2) present in over 80% of malignant gliomas. This IL-13 zetakine has an E13Y mutation, which enhances its specificity for the IL13Rα2 receptor by >50-fold, as compared with the normal IL-13 receptor expressed by healthy brain tissue.34,35 In addition to the IL-13 zetakine, the CTL were further modified to express the thymidine kinase suicide gene (Hy/TK) under the control of the constitutively active cytomegalovirus (CMV) promoter incase immediate ablation of CTL activity is required. Repeated CTL infusion was performed over 2 weeks (three times/week), followed by an injection every 3 weeks in the absence of disease progression and signs of autoimmunity. Recently, an experimental study from the same group was published discussing the use of the IL-13 zetakine in an orthotopic mouse tumor model.34,35 The authors showed that IL13Rα2 is expressed by both GSCs and the more differentiated tumor cell population, and that IL13Rα2 zetakine therapy ablates the tumor-initiating activity of IL13Rα2-positive GSCs. At time of writing, the pilot study had been completed though the results had not been published yet.
Rosenberg et al., at the National Institutes of Health Clinical Center, took on a similar approach by genetically modifying peripheral blood lymphocytes to express the anti-EGFRvIII chimeric antigen receptor. As in the case for IL13Rα2, the mutant EGFRvIII receptor is overexpressed in 30–70% of glioblastomas, whereas no expression is seen in the normal brain.36 After ex vivo preparation, the autologous-modified cells would be intravenously injected and safety, feasibility, and progression-free interval would be monitored.
Vaccine therapy in the clinic
With nine clinical trials either in progress or recently completed, vaccine therapy is the most popular clinical immuno-cellular therapeutic approach for malignant gliomas. Vaccine therapy is based on active immunization of the body against glioma, resulting in a permanent and sustained attack on the tumor by the immune system. In five out of the nine trials, autologous DCs are used to stimulate the patient immune system to evoke an antitumor immune response. DCs are the most potent antigen-presenting cells with the capability of presenting antigenic material not only by the MHC II pathway (stimulating CD4+ T lymphocytes), but also by MHC I pathway (stimulating a CD8+ lymphocyte response) through a process called “cross presenting” which results in a diversification of the immune response.37,38 ImmunoCellular Therapeutics (Woodland Hills, CA) recently initiated a Phase II study using the immunotherapeutic vaccine ICT-107 composed of synthetically purified antitumor antigens corresponding to epitopes found on GBM cells. Autologous DCs are ex vivo pulsed with ICT-107 and injected intradermally upon completion of tumor removal and after 6 weeks of temozolomide therapy. An earlier Phase I study demonstrated safety and efficacy of this therapeutic strategy. Earlier this year Odumsi et al., at the Roswell Park Cancer Institute, initiated a large Phase I study evaluating the safety and feasibility of a new vaccine aimed at NY-ESO-1 expressing solid tumors in combination with the mTOR inhibitor sirolimus.39 Autologous DCs are ex vivo pulsed with the 205-NY-ESO-1 fusion protein and intranodally injected. The investigators hope that this strategy will elicit a stronger immune response yielding to enhanced tumor killing. At the same time, Berneman et al., at the University Hospital (Antwerp, Belgium), are evaluating immunogenicity and efficacy of intradermal vaccination with autologous DCs genetically modified to express WT1 protein which is overexpressed in a variety of solid tumors. A previous Phase I study in patients with acute myeloid leukemia demonstrated the vaccine is well tolerated and elicits a CD8+ T lymphocyte response.40,41 In China, Zhou et al., at the Huashan Hospital, initiated a Phase II study evaluating the overall survival (OS) of patients with primary and/or secondary GBM after treatment with autologous DCs loaded with autogenic GSCs (A2B5+). A study investigating the adverse and therapeutic effect of a postoperative DC-derived tumor vaccine was recently published by Chang et al.,42 in the Journal of Clinical Neuroscience, reporting an increase in the median survival to 525 days and a 5-year survival rate to 18.8% as compared with the historical control group (380 days and 0%). Patients underwent surgery to debulk the tumor mass and the vaccine was prepared using cells from the surgical specimen. Autologous DCs were administered with 10 injections over the course of 6 months. The authors report that 47% of the enrolled patients developed a transient elevation in both alanine aminotransferase and aspartate aminotransferase levels, which correlated with the vaccination schedule and high doses of the DC vaccine. At lower levels of DC vaccine, no increase in serum alanine aminotransferase/aspartate aminotransferase was observed, suggesting a safe upper limit of 2 × 107 DCs/dose.
Another recently completed study by Fadul et al.43 was reported in the Journal of Immunotherapy which focused on the immune response, progression-free survival, and OS of GBM patients treated with an intranodal autologous tumor lysate DC vaccination. CTL tumor-specific activation was measured and correlated with both progression-free survival and OS. All patients survived past 6 months post-diagnosis and a progression-free survival of 9.5 months was reported. Median OS was 28 months, which is significantly higher than the OS of 18–21 months in GBM patients receiving standard therapy.2
As an alternative to the standard DC approach, Andrews et al., at the Thomas Jefferson University (Philadelphia, PA), initiated a pilot study evaluating the possibility of stimulating the DC population in vivo. In vivo stimulation is thought to be more effective and is expected to elicit a stronger and longer immune response as compared with ex vivo stimulation.44 Diffusion chambers containing autologous tumor cells treated ex vivo with insulin-like growth factor receptor-1 antisense oligodeoxynucleotide were re-implanted in the rectus sheet to stimulate the native immune system. Loss of insulin-like growth factor receptor-1 is expected to result in apoptosis with subsequent release of tumor antigens containing exosomes (microvesicles), which will allow the diffusion chamber to act as a slow-release antigen depot.45 Since a wound containing a foreign body is created upon implantation of the diffusion chamber, high levels of DCs are expected to be present in the immediate surroundings which enhances the efficacy of antitumor activation of the immune system.
A pilot study by Moertel et al., at the Masonic Cancer Center (University of Minnesota) developed a cell-based cancer vaccine composed of glioma stem-like associated antigens found in the brain tumor-initiating cell line GBM6.46,47 Upon administration, the brain tumor-initiating cell vaccine is thought to stimulate an antitumor CTL response against both GSCs and the more proliferated tumor bulk. Since GSCs have the ability of self-renewal and seem to drive tumor growth and initiation, elimination of this specific group of glioma cells would be of tremendous benefit. Vaccine administration will start following radiation therapy and will be given every 2 weeks for 4 weeks in combination with the drug imiquimod, which acts as an immune response modifier.
Two separate groups are conducting a Phase I study to test the safety and feasibility of IMA 950, which is a therapeutic multipeptide vaccine containing 11 tumor-associated peptides found in a majority of GBMs designed to activate tumor-associated peptide-specific T cells. Rumpling et al., (Cancer Research UK) are testing the vaccine in combination with granulocyte-macrophage colony-stimulating factor, radiation, and chemotherapy (temozolomide) for patients with newly diagnosed gliomas.48 Sul et al. (Immatics Biotechnologies (Tuebingen, Germany) in collaboration with the National Cancer Institute) follow a similar approach to test IMA 950 with granulocyte-macrophage colony-stimulating factor and locally applied imiquimod 20 minutes after each vaccination. Patients will further be treated with one dose of cyclophosphamide before the first vaccination.
Vaccine and cellular therapy combined
Two clinical trials, using a combined approach of vaccines and immunotherapy, are being performed by Ahmed et al. at the Baylor College of Medicine. In the first trial, autologous CTLs are stimulated ex vivo with human β-herpes CMV presenting DCs. CMV-specific antigens can be detected in 70–90% of malignant glioma cells, but not in the normal brain.49,50 The CMV-specific CTLs are then cultured in the presence of EBV-infected cells to elicit a stronger immune response upon intravenous administration.19 The second trial furthers this through the genetic modification of the CMV-specific CTLs to express the CAR targeting HER2, which is associated with 70% of GBM malignancies.51 Both trials are still in their initial Phase I stage; however, a recently published pilot study by the same group evaluated the use of CMV-specific T cells and demonstrated that autologous T cells could successfully be activated and expanded, are able to recognize the CMV antigens pp65 and IEI, and are capable of killing CMV-infected autologous GBM cells.52 Concurrently, Wood et al. (TVAX Biomedical, Lenexa, KS) are testing in a Phase II trial (supported by positive safety and efficacy studies in a Phase I trial) a brain cancer vaccine called TVI-Brain I that consists of neutralized autologous tumor cells. An immune response of killer T cells is expected upon vaccination, yielding highly effective antitumor activities. Yao et al., at Quindao University, used a somewhat similar approach in a Phase I/II trial combining intranodal DC vaccination with subsequent ex vivo expansion of activated T cells in patients with recurrent glioma. The study is specifically aimed at a group of T cells, named cytokine-induced killer cells, which are known to express a very potent antitumor activity.53,54 These cells will be selected by expression of the cell markers CD3 and CD56.
Limitations and Future Prospects of Immuno-Cell Therapy
Although a wide range of potential targets and immuno-cellular therapeutic strategies have been investigated experimentally, only the most successful ones are transitioned to the clinic. The translation from bench to bedside remains a difficult path, with the DC vaccine strategy being the most successful example. DC therapy has been proven safe with some therapeutic success; however, no breakthrough has been achieved using this therapeutic strategy for gliomas. The clinical outcome did not reflect the expected results on the bench, showing perhaps a limitation in the existing glioma models. Vaccination is mostly given before tumor implantation for DC therapy to be effective in animal models. This, of course, is impossible in human patients. While many pathophysiological similarities between the rodent glioma models and the human tumors can be observed, many models are performed in immunocompromised mice. Therefore, tumor-associated immunosuppression and immune-modulating events are not likely to be reflected accurately and their usefulness as models for evaluating immuno-cellular therapy might be limited. Furthermore, tumor xenografts will not mimic the process of tumorigenesis de novo, resulting in a slightly different tumor microenvironment. The use of rodents with intact immune systems, and the development of genetically induced glioma models, could help optimize preclinical studies and lead to a more predictable transition to the clinic.
Another difficulty in assessing the efficacy and success of DC vaccination in the clinic is the relatively low number of glioma patients per trial group, which often leads to a weak statistical significance. Further, it is difficult to compare study outcomes from different trials because inclusion criteria and injection route differs from one group to another. This can have a substantial effect on patient survival. The use of corticosteroids and other co-medication, as often seen in malignant glioma patients such as GBM, impairs objective assessment even further as efficacy of treatment might be limited, side effects might get masked, and differentiation of immune cells are halted. In the case of vaccination, improvements have only been seen when compared with historical controls, which are improper controls to use for glioma studies. When compared with standard-of-care, no clinically significant benefits have been reported. Furthermore, caution has to be exerted when interpreting effects on immune function following vaccination. Brain inflammation has never been detected inmost, if not all, clinical trials of vaccination. Although this is usually incorrectly interpreted as the vaccines being safe, the absence of any adverse effects in hundreds of immunized patients most likely speaks to the vaccination being ineffective. Thus, at this stage, we remain unable to differentiate the absence of side effects as a result of non-effective vaccination or actual safety. To date, only a single study combining gene/vaccine therapy in dogs showed physiologically effective immune activation associated with brain inflammation which resulted in clinical benefits.55 This study is the only objective description supporting the idea that, under the right conditions, it is possible to stimulate a systemic immune response that can attack the brain and brain tumors.
To underline some of these problems, and to get a true understanding of the working mechanism and antitumor effect of immuno-cellular therapies, the development of adequate imaging tools is of the utmost importance. The ability to track immune cells and to determine their fate, tropism, migration, interaction with surroundings, and mechanism of action will answer important questions regarding safety and efficacy. Several imaging tools are currently available in the preclinical setting such as bioluminescence and fluorescence. However, these techniques are not yet translatable for use in humans due to several concerns including substrate toxicity and sensitivity. Labeling of stem cells with ferumoxide, which allows them to be tracked in vivo by magnetic resonance imaging, has been successfully reported to monitor real-time migration and distribution of these cells at the tumor site.56 Similar approaches might be translated to the clinic to track immune cells, however, additional studies are required to fine tune this technique and increase its sensitivity to make it suitable for human use. While new imaging tools are a necessity to further develop the immuno-cell therapy field, another issue that needs to be addressed is the availability and efficacy of the cells themselves. High passage number of effector cells in vitro, in order to reach adequate levels, could lead to differentiation and change of phenotype, limiting their therapeutic potential. New techniques that allow rapid growth and expansion of these cells while maintaining their characteristics will be of extreme importance for the cellular immunotherapy field (Box 2). Similar problems can be seen in the clinic where a lack of in vivo expansion and inability to maintain high expression levels over a sufficient period of time could limit treatment efficacy. This may result not only in unsuccessful clinical trials, but also in the abandonment of a potentially successful strategy. The success of the CAR-CD19 adoptive T cell therapy study for CLL and acute lymphoblastic leukemia shows that, once the immune cells are manipulated, extensive in vivo expansion and high levels of gene expression could be maintained over time, therefore, immunotherapy can indeed be an effective strategy in the battle against cancer. In order to stimulate cell survival and proliferation, a 4-1 BB costimulatory domain was added to the CAR construct, resulting in >1,000-fold higher proliferation rate of T cells once injected in vivo, with each T cell killing ~1,000 CLL cells. Three out of three CLL patients showed clinical activity lasting for over 6 months, with two out of three patients reaching complete remission.30,31 Kloss et al. demonstrated a similar successful approach in a prostate cancer model using a chimeric costimulator receptor together with CARs, with increased selectivity of the modified T cells for prostate cancer cells.57 Although still at the experimental level, this strategy may greatly increase efficacy and safety of T cell adaptive immunotherapy. Both approaches could easily be adapted to T cell glioma therapy (similar to the Nakazawa17 and Wang16 studies), potentially in combination with EBV-CTL.
Several studies are exploring different strategies to deliver immune cells to the tumor. While many choose a direct injection route, others are exploring intranodal, intradermal, and systemic injections in an attempt to enhance the delivery success. Direct comparison of these delivery strategies should be performed to reach the optimal injection route for effective glioma therapy. Other research groups argue that ex vivo cell manipulation is time consuming and may result in cellular differentiation and an increased risk of infection. Thus, the focus should not be on “how to deliver the manipulated cells”, but on “how to manipulate the cells in vivo”. The studies being performed by Andrews et al., at the Thomas Jefferson University, will shed new light on these possibilities.
Finally, when discussing treatment efficacy and success of new clinical strategies, it is important to bear in mind the current prognosis and treatment options available for glioma patients. While the results of the CAR-CD19 trial showed that two out of the three CLL patients are in remission for over a year, which is extraordinary, one must realize that with a median survival of 8–10 years, the CLL population is not comparable to glioma patients. We advocate that in a patient population where the 2-year survival rate is only 40%, and in the past 25 years the median survival rate has only increased by 3 months, our expectation on efficacy should be as equally moderate.1,2 Furthermore, a gain of months rather than years should be valued as well as the decrease in side effects and/or increase in patient's well-being. The aim of trials should therefore not only be directed towards increased survival, but also for better quality-of-life. It is the hope that optimization of some of the strategies discussed here would increase both potential goals and expectations. For now, it is difficult to conclude what role and effect immuno-cellular therapy has on malignant gliomas. If some of the discussed issues can be addressed, and current clinical trials show promising results, this therapeutic strategy has potentially a tremendous value in the search for a cure for tumors as heterogeneous as GBM while complementing current standard therapy.
Acknowledgments
Dr Tannous is supported by grants from the National Institutes of Health, the National Institute of Neurological Disorders and Stroke (1R01NS064983), and the National Cancer Institute (1R01CA166077). M.H.D. is supported by a Fulbright scholarship, the Saal van Zwanenberg Foundation, VSB fonds, Dr. Hendrik Muller Vaderlandsch Fonds, the Dutch Cancer Foundation (KWF Kankerbestrijding), the Hersenstichting brain fund as well as the Jo Keur (Leiden hospital). M.S.S.B. was supported by a Fulbright scholarship, the Huygens Scholarship Program, VSB Fonds, and the Saal van Zwanenberg Foundation. The authors thank Grant Lewandrowski for editorial assistance. The authors declared no conflict of interest.
References
- Johnson DR, Ma DJ, Buckner JC, Hammack JE. Conditional probability of long-term survival in glioblastoma: a population-based analysis. Cancer. 2012;118:5608–5613. doi: 10.1002/cncr.27590. [DOI] [PubMed] [Google Scholar]
- Grossman SA, Ye X, Piantadosi S, Desideri S, Nabors LB, Rosenfeld M, NABTT CNS Consortium et al. Survival of patients with newly diagnosed glioblastoma treated with radiation and temozolomide in research studies in the United States. Clin Cancer Res. 2010;16:2443–2449. doi: 10.1158/1078-0432.CCR-09-3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- Albesiano E, Han JE, Lim M. Mechanisms of local immunoresistance in glioma. Neurosurg Clin N Am. 2010;21:17–29. doi: 10.1016/j.nec.2009.08.008. [DOI] [PubMed] [Google Scholar]
- Haar CP, Hebbar P, Wallace GC, 4th, Das A, Vandergrift WA, 3rd, Smith JA, et al. Drug resistance in glioblastoma: a mini review. Neurochem Res. 2012;37:1192–1200. doi: 10.1007/s11064-011-0701-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro-oncology. 2010;12:1113–1125. doi: 10.1093/neuonc/noq082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R, Pica A, Mirimanoff RO, Michielin O. [A practical guide for the management of gliomas]. Bull Cancer. 2007;94:817–822. [PubMed] [Google Scholar]
- Stupp R, Roila F, ESMO Guidelines Working Group Malignant glioma: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2009;20 suppl. 4:126–128. doi: 10.1093/annonc/mdp151. [DOI] [PubMed] [Google Scholar]
- Gale RP. Bone marrow transplantation in acute leukemia: current status and future directions. Haematol Blood Transfus. 1979;23:71–78. doi: 10.1007/978-3-642-67057-2_8. [DOI] [PubMed] [Google Scholar]
- Santos GW, Elfenbein GJ, Tutschka PJ. Bone marrow transplantation–present status. Transplant Proc. 1979;11:182–188. [PubMed] [Google Scholar]
- Joglekar MV, Hardikar AA. Isolation, expansion, and characterization of human islet-derived progenitor cells. Methods Mol Biol. 2012;879:351–366. doi: 10.1007/978-1-61779-815-3_21. [DOI] [PubMed] [Google Scholar]
- Efrat S, Russ HA. Making ß cells from adult tissues. Trends Endocrinol Metab. 2012;23:278–285. doi: 10.1016/j.tem.2012.03.005. [DOI] [PubMed] [Google Scholar]
- Vacanti JP. Tissue engineering and the road to whole organs. Br J Surg. 2012;99:451–453. doi: 10.1002/bjs.7819. [DOI] [PubMed] [Google Scholar]
- Wain RA, Shah SH, Senarath-Yapa K, Laitung JK. Dermal substitutes do well on dura: comparison of split skin grafting +/- artificial dermis for reconstruction of full-thickness calvarial defects. Clin Plast Surg. 2012;39:65–67. doi: 10.1016/j.cps.2011.09.010. [DOI] [PubMed] [Google Scholar]
- Jones TS, Holland EC. Animal models for glioma drug discovery. Expert Opin Drug Discov. 2011;6:1271–1283. doi: 10.1517/17460441.2011.632628. [DOI] [PubMed] [Google Scholar]
- Wang LX, Westwood JA, Moeller M, Duong CP, Wei WZ, Malaterre J, et al. Tumor ablation by gene-modified T cells in the absence of autoimmunity. Cancer Res. 2010;70:9591–9598. doi: 10.1158/0008-5472.CAN-10-2884. [DOI] [PubMed] [Google Scholar]
- Nakazawa Y, Huye LE, Salsman VS, Leen AM, Ahmed N, Rollins L, et al. PiggyBac-mediated cancer immunotherapy using EBV-specific cytotoxic T-cells expressing HER2-specific chimeric antigen receptor. Mol Ther. 2011;19:2133–2143. doi: 10.1038/mt.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16:474–485. doi: 10.1158/1078-0432.CCR-09-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintarelli C, Savoldo B, Dotti G. Gene therapy to improve function of T cells for adoptive immunotherapy. Methods Mol Biol. 2010;651:119–130. doi: 10.1007/978-1-60761-786-0_8. [DOI] [PubMed] [Google Scholar]
- Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woltjen K, Hämäläinen R, Kibschull M, Mileikovsky M, Nagy A. Transgene-free production of pluripotent stem cells using piggyBac transposons. Methods Mol Biol. 2011;767:87–103. doi: 10.1007/978-1-61779-201-4_7. [DOI] [PubMed] [Google Scholar]
- Woltjen K, Michael IP, Mohseni P, Desai R, Mileikovsky M, Hämäläinen R, et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458:766–770. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka M, Amano T, Kajiwara K, Yoshikawa K, Ideguchi M, Nomura S, et al. Vaccine therapy with dendritic cells transfected with Il13ra2 mRNA for glioma in mice. J Neurosurg. 2010;113:270–279. doi: 10.3171/2009.9.JNS09708. [DOI] [PubMed] [Google Scholar]
- Mineharu Y, King GD, Muhammad AK, Bannykh S, Kroeger KM, Liu C, et al. Engineering the brain tumor microenvironment enhances the efficacy of dendritic cell vaccination: implications for clinical trial design. Clin Cancer Res. 2011;17:4705–4718. doi: 10.1158/1078-0432.CCR-11-0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali S, King GD, Curtin JF, Candolfi M, Xiong W, Liu C, et al. Combined immunostimulation and conditional cytotoxic gene therapy provide long-term survival in a large glioma model. Cancer Res. 2005;65:7194–7204. doi: 10.1158/0008-5472.CAN-04-3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009;6:e10. doi: 10.1371/journal.pmed.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Liu G, Yuan X, Xu M, Wang H, Ji J, et al. Antigen-specific T-cell response from dendritic cell vaccination using cancer stem-like cell-associated antigens. Stem Cells. 2009;27:1734–1740. doi: 10.1002/stem.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinonaga M, Chang CC, Suzuki N, Sato M, Kuwabara T. Immunohistological evaluation of macrophage infiltrates in brain tumors. Correlation with peritumoral edema. J Neurosurg. 1988;68:259–265. doi: 10.3171/jns.1988.68.2.0259. [DOI] [PubMed] [Google Scholar]
- Baek SK, Makkouk AR, Krasieva T, Sun CH, Madsen SJ, Hirschberg H. Photothermal treatment of glioma; an in vitro study of macrophage-mediated delivery of gold nanoshells. J Neurooncol. 2011;104:439–448. doi: 10.1007/s11060-010-0511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3 95:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, La Perle K, et al. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2007;13 18 Pt 1:5426–5435. doi: 10.1158/1078-0432.CCR-07-0674. [DOI] [PubMed] [Google Scholar]
- Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004;64:9160–9166. doi: 10.1158/0008-5472.CAN-04-0454. [DOI] [PubMed] [Google Scholar]
- Brown CE, Starr R, Aguilar B, Shami A, Martinez C, D'Apuzzo M, et al. Stem-like tumor initiating cells isolated from IL13Ralpha2-expressing gliomas are targeted and killed by IL13-zetakine redirected T cells. Clin Cancer Res. 2012;18:2199–2209. doi: 10.1158/1078-0432.CCR-11-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatanpaa KJ, Burma S, Zhao D, Habib AA. Epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia. 2010;12:675–684. doi: 10.1593/neo.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Regnault A, Kleijmeer M, Ricciardi-Castagnoli P, Amigorena S. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat Cell Biol. 1999;1:362–368. doi: 10.1038/14058. [DOI] [PubMed] [Google Scholar]
- Kim W, Liau LM. Dendritic cell vaccines for brain tumors. Neurosurg Clin N Am. 2010;21:139–157. doi: 10.1016/j.nec.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji T, Matsuzaki J, Kelly MP, Ramakrishna V, Vitale L, He LZ, et al. Antibody-targeted NY-ESO-1 to mannose receptor or DEC-205 in vitro elicits dual human CD8+ and CD4+ T cell responses with broad antigen specificity. J Immunol. 2011;186:1218–1227. doi: 10.4049/jimmunol.1000808. [DOI] [PubMed] [Google Scholar]
- Van Tendeloo VF, Van de Velde A, Van Driessche A, Cools N, Anguille S, Ladell K, et al. Induction of complete and molecular remissions in acute myeloid leukemia by Wilms' tumor 1 antigen-targeted dendritic cell vaccination. Proc Natl Acad Sci USA. 2010;107:13824–13829. doi: 10.1073/pnas.1008051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Driessche A, Van de Velde AL, Nijs G, Braeckman T, Stein B, De Vries JM, et al. Clinical-grade manufacturing of autologous mature mRNA-electroporated dendritic cells and safety testing in acute myeloid leukemia patients in a phase I dose-escalation clinical trial. Cytotherapy. 2009;11:653–668. doi: 10.1080/14653240902960411. [DOI] [PubMed] [Google Scholar]
- Chang CN, Huang YC, Yang DM, Kikuta K, Wei KJ, Kubota T, et al. A phase I/II clinical trial investigating the adverse and therapeutic effects of a postoperative autologous dendritic cell tumor vaccine in patients with malignant glioma. J Clin Neurosci. 2011;18:1048–1054. doi: 10.1016/j.jocn.2010.11.034. [DOI] [PubMed] [Google Scholar]
- Fadul CE, Fisher JL, Hampton TH, Lallana EC, Li Z, Gui J, et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J Immunother. 2011;34:382–389. doi: 10.1097/CJI.0b013e318215e300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol. 2007;7:790–802. doi: 10.1038/nri2173. [DOI] [PubMed] [Google Scholar]
- Andrews DW, Resnicoff M, Flanders AE, Kenyon L, Curtis M, Merli G, et al. Results of a pilot study involving the use of an antisense oligodeoxynucleotide directed against the insulin-like growth factor type I receptor in malignant astrocytomas. J Clin Oncol. 2001;19:2189–2200. doi: 10.1200/JCO.2001.19.8.2189. [DOI] [PubMed] [Google Scholar]
- Hu Y, Fu L. Targeting cancer stem cells: a new therapy to cure cancer patients. Am J Cancer Res. 2012;2:340–356. [PMC free article] [PubMed] [Google Scholar]
- Selvan SR, Carbonell DJ, Fowler AW, Beatty AR, Ravindranath MH, Dillman RO. Establishment of stable cell lines for personalized melanoma cell vaccine. Melanoma Res. 2010;20:280–292. doi: 10.1097/CMR.0b013e3283390696. [DOI] [PubMed] [Google Scholar]
- Zhan Y, Xu Y, Lew AM. The regulation of the development and function of dendritic cell subsets by GM-CSF: more than a hematopoietic growth factor. Mol Immunol. 2012;52:30–37. doi: 10.1016/j.molimm.2012.04.009. [DOI] [PubMed] [Google Scholar]
- Dziurzynski K, Chang SM, Heimberger AB, Kalejta RF, McGregor Dallas SR, Smit M, HCMV and Gliomas Symposium et al. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro-oncology. 2012;14:246–255. doi: 10.1093/neuonc/nor227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas KG, Bao L, Bruggeman R, Dunham K, Specht C. The detection of CMV pp65 and IE1 in glioblastoma multiforme. J Neurooncol. 2011;103:231–238. doi: 10.1007/s11060-010-0383-6. [DOI] [PubMed] [Google Scholar]
- Potti A, Forseen SE, Koka VK, Pervez H, Koch M, Fraiman G, et al. Determination of HER-2/neu overexpression and clinical predictors of survival in a cohort of 347 patients with primary malignant brain tumors. Cancer Invest. 2004;22:537–544. doi: 10.1081/cnv-200026523. [DOI] [PubMed] [Google Scholar]
- Ghazi A, Ashoori A, Hanley PJ, Brawley VS, Shaffer DR, Kew Y, et al. Generation of polyclonal CMV-specific T cells for the adoptive immunotherapy of glioblastoma. J Immunother. 2012;35:159–168. doi: 10.1097/CJI.0b013e318247642f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesiano G, Todorovic M, Gammaitoni L, Leuci V, Giraudo Diego L, Carnevale-Schianca F, et al. Cytokine-induced killer (CIK) cells as feasible and effective adoptive immunotherapy for the treatment of solid tumors. Expert Opin Biol Ther. 2012;12:673–684. doi: 10.1517/14712598.2012.675323. [DOI] [PubMed] [Google Scholar]
- Li H, Wang C, Yu J, Cao S, Wei F, Zhang W, et al. Dendritic cell-activated cytokine-induced killer cells enhance the anti-tumor effect of chemotherapy on non-small cell lung cancer in patients after surgery. Cytotherapy. 2009;11:1076–1083. doi: 10.3109/14653240903121252. [DOI] [PubMed] [Google Scholar]
- Pluhar GE, Grogan PT, Seiler C, Goulart M, Santacruz KS, Carlson C, et al. Anti-tumor immune response correlates with neurological symptoms in a dog with spontaneous astrocytoma treated by gene and vaccine therapy. Vaccine. 2010;28:3371–3378. doi: 10.1016/j.vaccine.2010.02.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thu MS, Najbauer J, Kendall SE, Harutyunyan I, Sangalang N, Gutova M, et al. Iron labeling and pre-clinical MRI visualization of therapeutic human neural stem cells in a murine glioma model. PLoS ONE. 2009;4:e7218. doi: 10.1371/journal.pone.0007218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroncek DF, Berger C, Cheever MA, Childs RW, Dudley ME, Flynn P, et al. New directions in cellular therapy of cancer: a summary of the summit on cellular therapy for cancer. J Transl Med. 2012;10:48. doi: 10.1186/1479-5876-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Finocchiaro G, Pellegatta S. Immunotherapy for glioma: getting closer to the clinical arena. Curr Opin Neurol. 2011;24:641–647. doi: 10.1097/WCO.0b013e32834cbb17. [DOI] [PubMed] [Google Scholar]
- Wadle A, Mischo A, Strahl S, Nishikawa H, Held G, Neumann F, et al. NY-ESO-1 protein glycosylated by yeast induces enhanced immune responses. Yeast. 2010;27:919–931. doi: 10.1002/yea.1796. [DOI] [PubMed] [Google Scholar]
- Hua W, Yao Y, Chu Y, Zhong P, Sheng X, Xiao B, et al. The CD133+ tumor stem-like cell-associated antigen may elicit highly intense immune responses against human malignant glioma. J Neurooncol. 2011;105:149–157. doi: 10.1007/s11060-011-0572-y. [DOI] [PubMed] [Google Scholar]
- Sul J, Fine HA. Malignant gliomas: new translational therapies. Mt Sinai J Med. 2010;77:655–666. doi: 10.1002/msj.20223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brada M, Stenning S, Gabe R, Thompson LC, Levy D, Rampling R, et al. Temozolomide versus procarbazine, lomustine, and vincristine in recurrent high-grade glioma. J Clin Oncol. 2010;28:4601–4608. doi: 10.1200/JCO.2009.27.1932. [DOI] [PubMed] [Google Scholar]
- Gorlia T, Stupp R, Brandes AA, Rampling RR, Fumoleau P, Dittrich C, et al. New prognostic factors and calculators for outcome prediction in patients with recurrent glioblastoma: a pooled analysis of EORTC Brain Tumour Group phase I and II clinical trials. Eur J Cancer. 2012;48:1176–1184. doi: 10.1016/j.ejca.2012.02.004. [DOI] [PubMed] [Google Scholar]
- Sloan AE, Dansey R, Zamorano L, Barger G, Hamm C, Diaz F, et al. Adoptive immunotherapy in patients with recurrent malignant glioma: preliminary results of using autologous whole-tumor vaccine plus granulocyte-macrophage colony-stimulating factor and adoptive transfer of anti-CD3-activated lymphocytes. Neurosurg Focus. 2000;9:e9. doi: 10.3171/foc.2000.9.6.10. [DOI] [PubMed] [Google Scholar]
- Hoa N, Ge L, Kuznetsov Y, McPherson A, Cornforth AN, Pham JT, et al. Glioma cells display complex cell surface topographies that resist the actions of cytolytic effector lymphocytes. J Immunol. 2010;185:4793–4803. doi: 10.4049/jimmunol.1001526. [DOI] [PubMed] [Google Scholar]