

Abstract
Myocardial infarction (MI) is the leading cause of death worldwide. MicroRNAs regulate the expression of their target genes, thus mediating a plethora of pathophysiological functions. Recently, miRNA-24 emerged as an important but controversial miRNA involved in post-MI responses. Here, we aimed at clarifying the effect of adenovirus-mediate intra-myocardial delivery of a decoy for miRNA-24 in a mouse MI model and to investigate the impact of miRNA-24 inhibition on angiogenesis and cardiovascular apoptosis. After MI induction, miRNA-24 expression was lower in the peri-infarct tissue and its resident cardiomyocytes and fibroblasts; while it increased in endothelial cells (ECs). Local adenovirus-mediated miRNA-24 decoy delivery increased angiogenesis and blood perfusion in the peri-infarct myocardium, reduced infarct size, induced fibroblast apopotosis and overall improved cardiac function. Notwithstanding these beneficial effects, miRNA-24 decoy increased cardiomyocytes apoptosis. In vitro, miRNA-24 inhibition enhanced ECs survival, proliferation and networking in capillary-like tubes and induced cardiomyocyte and fibroblast apoptosis. Finally, we identified eNOS as a novel direct target of miR-24 in human cultured ECs and in vivo. Our findings suggest that miRNA-24 inhibition exerts distinct biological effects on ECs, cardiomyocytes and fibroblasts. The overall result of post-infarction local miRNA-24 inhibition appears to be therapeutic.
Introduction
Ischemic heart disease is the leading cause of death worldwide.1 Myocardial infarction (MI) occurs when a coronary artery is occluded, leading to insufficient oxygen supply to the myocardium and resulting in death of cardiomyocytes and non-myocyte cells, including fibroblasts and endothelial cells (ECs).2,3 This is accompanied by hypertrophic growth of cardiomyocytes and deposition of interstitial fibrosis in the non-infarcted tissues, which collectively and ultimately results in maladaptive left ventricular (LV) remodeling and heart failure.4 Conventional revascularization procedures aimed at reopening the obstructed artery, such as thrombolysis, angioplasty, and bypass surgery have improved the post-MI survival rate. However, these techniques cannot be applied to many patients and are prone to failure. Regenerative medicine represents a new hope for overcoming the limitation of conventional treatments for ischemic heart disease. Although traditional revascularization approaches target large vessels, therapeutic angiogenesis research seeks to improve the microcirculation by stimulating new capillary and collateral arterial vessel formation.5 Angiogenesis gene therapy based on the delivery of single growth factors has shown promises in small animal models of acute MI, but has so far failed to conclusively demonstrate therapeutic efficacy in double blinded, randomized clinical trials on large patient populations.6,7 This may be accounted by a series of factors, including the chosen growth factors, delivery strategy, and patient selections and should be addressed at translational level.
Recently, microRNAs (miRs) have come into focus of cardiovascular and angiogenesis research.8 In their mature form, miRs are small (21 to 25 nucleotides) endogenous non-coding RNAs that modulate gene expression, mainly by binding to the 3′ untranslated region (3′-UTR) of their targeted messenger RNAs (mRNAs), thus resulting in mRNA degradation or translational repression.9 So far, in excess of 1,000 miRs have been described in mammals and it is believed that >60% of protein coding genes are under the control of miRs.10 Each miR regulates the expression of several target genes, with the possibility to modulate several molecular pathways. Hence, it is possible that some miRs could contribute into post-ischemic vascular repair and cardiac regeneration at multiple levels. In line with this hypothesis, an increasing number of studies show that miR expression is altered in the infarcted heart and that miRs are involved in the regulation of post-MI cardiac angiogenesis,11,12 apoptosis,13,14,15 and fibrosis.16,17,18,19 Of particular interest is miR-24, which has been recently implicated in post-MI responses. However, two leading groups have reported contrasting results on the expressional regulation and actions of this miR in the mouse acute MI setting.12,14 Fiedler et al. found that cardiac ECs express miR-24 under basal conditions and that endothelial miR-24 levels are increased after MI. They additionally showed that silencing of miR-24 by systemic delivery of a modified antisense oligonucleotide (antagomir) improves post-MI reparative angiogenesis and preserves cardiac function in mice.12 By contrast, Qian et al. could not detect miR-24 in cardiac ECs and observed a MI-induced downregulation of miR-24 in the peri-infarct area that correlates with increased cardiomyocyte apoptosis.14 Most important, in striking contrast with Fielder et al., Qian et al. found that one intra-myocardial injection of precursor miR-24 (pre-miR-24) was enough to improve post-MI cardiac function in mice. The two studies attributed the cardiac effects of miR-24 to different target genes, namely the pro-angiogenic PAK4 (p21 protein-Cdc42/Rac-activated kinase 4) and GATA2 (globin transcription factor binding protein 2) in Fiedler et al. and the pro-apoptotic BIM (BCL2-like 11 apoptosis facilitator) in Qian et al.12,14 In support to the latter study, a report showed that ex-vivo miR-24 enrichment improves the therapeutic potential of cardiac progenitor cells upon their transplantation in a rodent MI model.20 More recently, lentivirus-mediated cardiac miR-24 overexpression has been shown to attenuate cardiac fibrosis and improve cardiac function after MI in mice.21
Given the aforementioned controversies present in the literature and the importance of defining miR-24 functions before moving to translational studies, we aimed at clarifying the cellular expression of miR-24 in the post-MI setting, the miR-24 role in angiogenesis and cardiac apoptosis, and the effect of local miR-24 inhibition by using an adenoviral vector carrying a decoy sequence for miR-24 (Ad.decoymiR-24; control: Ad.Null) in the same mouse MI model used in refs. 12,14,21.
Results
Expression of cardiac miR-24 in the post-MI setting
To evaluate the expression of miR-24 after MI, different cardiac regions were isolated at 1 and 3 days after surgery to induce MI or sham-operation. Whereas miR-24 levels were similar in the right ventricle (RV) and septum (S) of infarcted and sham-operated mice, miR-24 expression was downregulated in the peri-infarct area as compared with either the LV of sham-operated mice or the LV remote zone isolated from infarcted hearts (Figure 1a and Supplementary Figure S1a). The mRNA relative expression of the miR-24 target genes BIM and GATA2 was similar in all cardiac regions (Supplementary Figure S1b,c) while PAK4 was upregulated by MI at 1 and 3 days after MI (Supplementary Figure S1d). Next, to better understand miR-24 expressional changes at the cellular level, we extracted ECs, cardiomyocytes and fibroblasts from infarcted and sham-operated hearts. Myocardial CD146+ ECs were magnetic sorted from the peri-infarct and remote areas of the LV at 1 and 3 days after MI. ECs prepared from the LV of sham-operated mice were used as reference. As shown in Supplementary Figure S2a–c, 84.5 ± 3.9% of CD146+ cells magnetic sorted from the mouse LV coexpressed the CD31 endothelial marker, thus confirming that our protocol was highly enriching for ECs. As shown in Figure 1b, in comparison with ECs taken from the sham-operated heart, miR-24 expression was significantly increased in CD146+ ECs isolated from the peri-infarct zone but not from the remote area, suggesting that myocardial ischemia increases miR-24 expression in ECs. In addition, miR-24 expression was evaluated in cardiomyocytes and fibroblasts isolated from the infarcted or sham-operated LV. Supplementary Figure S1d,e, respectively, present the characterization of the cardiomyocytes (stained with an antibody for α-sarcomeric actin) and fibroblasts (stained with an antibody for vimentin) prepared from the mouse heart. In agreement with Qian et al.,14 in comparison with cardiomyocytes from sham-operated heart, miR-24 was downregulated in cardiomyocytes extracted from the LV peri-infarct, but not the remote zone (Figure 1c). Finally, miR-24 expression was also reduced in fibroblasts isolated from the LV peri-infarct in comparisons with cells from either sham-LV or LV remote area (Figure 1d).
Figure 1.

Myocardial infarct-induced microRNA-24 expressional changes in the mouse myocardium and its resident cells. At 1 and 3 days after surgery to myocardial infarct (MI) or sham-operation in mice, microRNA-24 (miR-24) relative expression was evaluated in (a) different cardiac regions and in (b) CD146+ endothelial cells (ECs), (c) cardiomyocytes and (d) fibroblasts isolated from sham-operated or infarcted (peri-infract area and remote area) left ventricles. Data are expressed as mean ± SEM. N = 3–4/group for cardiac regions analyses. For cellular expressional analyses, each preparation was obtained pooling cells from two hearts (N = 3–5 pools/group). Small nuclear RNA U6 (snU6) was used for normalization and data are reported to the control group by the 2-ΔΔCt formula. *P < 0.05 versus sham-operated LV; †P < 0.05 versus LV remote area; §P < 0.05 versus cells of sham-operated LV; ‡P < 0.05 versus cells of LV remote area. LV, left ventricle; Rem, LV remote area; RV, right ventricle; Peri, LV peri-infarct area; S, septum.
miR-24 in angiogenesis and EC apoptosis
Fiedler et al. reported that blocking miR-24 increases the angiogenic potential of human umbilical vein ECs (HUVECs).12 We have confirmed the data. Supplementary Figure S3a show the expected changes in miR-24 expression after transfection of HUVECs with synthetic miR-24 precursor (pre-miR-24), an anti-miR-24 inhibitor (anti-miR-24) or a negative control (scramble). Supplementary Figure S4a–d show Matrigel assays performed with HUVECs after miR-24 inhibition. Next, we extended the investigation to human microvascular ECs (HMVECs) because they better reflect the EC populations contributing into neoangiogenesis. Efficiency of HMVECs transfection by either pre-miR-24 or anti-miR-24 was confirmed by Taqman RT-PCR (Supplementary Figure S3b). As shown in Figure 2a–c, in comparison with the negative control, pre-miR-24 impaired the formation of cellular networks from HMVECs seeded on Matrigel, whereas anti-miR-24 induced the opposite effect. In addition, pre-miR-24 inhibited HMVEC proliferation (Figure 2d) and increased HMVECs apoptosis (Figure 2e). Fiedler et al. reported PAK4 and GATA2 as miR-24 direct target genes and regulators of the pro-angiogenic effects mediated by miR-24 inhibition in HUVECs.12 As shown in Figure 2f, we confirm the data in HMVECs, where pre-miR-24 reduces and anti-miR-24 increase the expression of both target genes. In addition, the pro-apoptotic BIM was increased by anti-miR-24 in HMVECs. This is at contrast with the knowledge that BIM is a direct target gene of miR-24.14 Nonetheless, this is in line with the observed anti-apoptotic action exerted by miR-24 inhibition in ECs (Figure 2e).
Figure 2.

miR-24 modulates HMVEC networking on Matrigel, proliferation and apoptosis and regulates the expression of target genes. (a) Photomicrograph show the endothelial network formation on Matrigel (scale bar 20 µm) of HMVECs transfected with pre-miR-24, anti-miR-24, or negative control (scramble). Bar graphs show (b) total length of tube-like structures and (c) percentage of area covered by connected tubular structures. Bar graphs show (d) BrdU incorporation and (e) Caspase activity assay of transfected HMVECs. Bar graphs show the expression of (f) the miR-24 target genes GATA2, PAK4 and BIM on HMVECs after transfection with pre-miR-24, anti-miR-24 or scramble and of (g) GATA2 and (h) PAK4 after infection with Ad.decoymiR-24 or Ad.Null (control). (j) GATA2 and (k) PAK4 relative expression was additionally evaluated in CD146+ ECs isolated from the total LV (for sham-operated mice) or the LV peri-infarct myocardium at 3 days after surgery and gene transfer. Data are expressed as mean ± SEM. Experiments in HMVECs were performed in triplicate and repeated 3 times. Each preparation of CD146+ cells was obtained pooling cells from two hearts (N = 3–4 pools/group). Three days in advance, the hearts received surgery and were infected with either Ad.decoymiR-24 or Ad.Null. Expressional analyses employed snU6 (for miRNA analyses) and 18S ribosomal RNA (RNA 18S) (for mRNA analyses) for normalization and data are reported to the control group by the 2-ΔΔCt formula. RLU: Relative Luminescent Units. *P < 0.05 and **P < 0.01 versus Scramble; †P < 0.05 and ††P < 0.01 versus pre-miR-24; §§P < 0.01 versus Ad.Null in HMVECs; ‡P < 0.05 versus ECs from Sham/Ad.Null hearts; #P < 0.05 versus ECs from MI/Ad.Null hearts.
As additional tool to inhibit miR-24, we have prepared a decoy for miR-24 using a similar method already published in ref. 22 for miR-503 inhibition. Supplementary Figure S4e shows the miR-24 decoy structure. The decoy was then inserted in an adenoviral vector (Ad.decoymiR-24). The capacity of the decoy to bind to miR-24, thus reducing EFGP signal was validated in vitro (Supplementary Figure S4f). In HMVECs Ad.decoymiR-24 upregulated GATA2 and PAK4 mRNA expression and increases angiogenesis in comparison with Ad.Null (Figure 2g,h Supplementary Figure S4c,d, respectively). The effect of in vivo miR-24 inhibition on GATA2, PAK4 and BIM was additionally evaluated in CD146+ ECs isolated from the LV at 3 and 14 days after MI and Ad.decoymiR-24 or Ad.Null. As shown in Figure 2j, GATA2 expression was unchanged. However, Ad.decoymiR-24 increased PAK4 mRNA relative expression in cardiac ECs at 3 days after MI (Figure 2k), thus validating PAK4 as miR-24 target in our in vivo model. At the same time point, Ad.decoymiR-24 decreased miR-24 levels in cardiac ECs (Supplementary Figure S5a). At 14 days, this effect was lost (data not shown), thus suggesting that the inhibitory effect of Ad.decoymiR-24 are no longer effective on ECs at this time point. Of note, BIM expression in CD146+ ECs was not affected by miR-24 inhibition (data not shown).
Identification of eNOS as a miR-24 direct target gene
To investigate whether additional factors besides PAK4 and GATA2 could be involved in the pro-angiogenic effects triggered by miR-24 inhibition, we searched for predicted target of miR-24 which may favor EC homeostasis and angiogenesis. Both mouse vascular endothelial growth factor (vegf) and human VEGF were predicted as direct targets of miR-24 by six (DIANAmT, miRanda, miRWalk, PICTAR5, RNA22, and TargetScan) out of the nine searched bioinformatic platforms. Human VEGF was additionally indicated by miRwalk as a validated target gene of miR-24.23 Moreover, human endothelial nitric oxide synthases (eNOS, NOS3) was predicted as target gene of miR-24 by TargetScan (Figure 3a). A luciferase reporter gene assay confirmed miR-24 binding to wild type eNOS 3′-UTR, while the miR-24 binding was impaired by mutation in the eNOS-3′-UTR (Figure 3b), thus validating eNOS as direct target gene of miR-24. Moreover, by aligning human miR-24 sequence to mouse eNOS 3′-UTR sequence using RNAhybrid software, we additionally verified that the binding site for miR-24 on eNOS 3′-UTR is conserved in mouse and human (Supplementary Figure S6).
Figure 3.

eNOS is a direct target of miR-24. (a) Prediction of human eNOS as miR-24 target gene by TargetScan. (b) Luciferase assay for eNOS 3′-UTR. HEK293 cells were cotransfected with pre-miR-24 (miR-24) or scramble oligonucleotides (control) and 3′-UTR-eNOS plasmid, expressing wild type or mutated (mut) sequences at the putative miR-24 target site. Luciferase activity was measure at 48 hours from transfection. Bar graphs show (c) eNOS mRNA relative expression and (d) representative western blots bands and relative protein quantification for eNOS on HMVECs transfected with pre-miR-24, anti-miR-24, or scramble. Tubulin was used as loading control. (d) Bar graphs show eNOS mRNA relative expression on HMVECs infected with Ad.decoymiR-24 or Ad.Null. Experiments on cultured ECs were performed in triplicate and repeated two times. (f) eNOS mRNA relative expression and (g) eNOS activity assay on CD146+ ECs isolated at 3 days after surgery and gene transfer as described in legend to Figure 2. N = 3–4 pools/group. (h) Representative western blot bands and relative quantification of eNOS protein expression in mouse myocardium at 3 days after MI and gene transfer. N = 4–5 mice/group. eNOS mRNA analyses employed RNA 18S for normalization and data are reported to the control group by the 2-ΔΔCt formula. All data are expressed as mean ± SEM. *P < 0.05 versus premiR-24; †P < 0.05 versus scramble; §P < 0.05 and §§P < 0.01 versus Scramble; ‡‡P < 0.01 versus pre-miR-24; MI/Ad.Null; ##P < 0.01 versus Ad.Null; ##P < 0.01 and #P < 0.05 versus Sham/Ad.Null; ¥P < 0.05 versus MI/Ad.Null.
Next, we performed eNOS and VEGF-A western blots analyses on HMVECs pretreated with pre-miR-24, anti-miR-24 or scramble negative control. As shown in Figure 3c,d, in comparison with negative control, pre-miR-24 reduced eNOS mRNA and protein expression, whereas anti-miR-24 showed the opposite effects. By contrast, VEGF-A protein level were not changed by either pre-miR-24 or anti-miR-24 (data not shown). In addition, eNOS relative expression was upregulated by Ad.decoymiR-24 in HMVECs (Figure 2e). Next, the effect of in vivo miR-24 inhibition were evaluated in CD146+ ECs isolated at 3 days after surgery from the peri-infarct myocardium treated with either Ad.decoymiR-24 or Ad.Null or the sham-operated LV treated with Ad.Null. CD146+ ECs eNOS mRNA relative expression was similar in all groups (Figure 3f). However, eNOS activity was increased in CD146+ ECs isolated at 3 days after MI and Ad.decoymiR-24 (P < 0.05 versus MI/Ad.Null, Figure 3g). In addition, western blot analyses on the peri-infarct tissue at 3 days after MI showed that eNOS expression was increased after Ad.decoymiR-24 (Figure 3h), thus further suggesting eNOS to be targeted by miR-24 in vitro and in vivo. Similarly to what observed in HMVECs, VEGF-A protein level in the peri-infarcted heart was unchanged by miR-24 inhibition (data not shown).
miR-24 in cardiomyocyte and fibroblast apoptosis
To confirm the pro-apoptotic effect of miR-24 inhibition described by Qian et al. in cardiomyocytes,14 the murine cardiomyocyte cell line HL1 was transfected with Ad.decoymiR-24 or control Ad.Null and subjected to two apoptosis assays. Both Caspase-GLO assay and Cell Death Detection ELISA showed increased apoptosis in HL1 cells after Ad.decoymiR-24 (Figure 4a,b, respectively), thus confirming that miR-24 exerts pro-survival actions in cardiomyocytes. In addition, in line with Qian et al.,14 BIM mRNA relative expression in HL1 increased by twofold following Ad.decoymiR-24 (Figure 4c). In vitro experiments on adult mouse cardiomyocytes confirmed that Ad.decoymiR-24 induces cardiomyocyte apoptosis and upregulates BIM mRNA level (Figure 4d,e, respectively). These data confirm with a different approach the anti-apoptotic effect of miR-24 in cardiomyocytes14 and the possible involvement of BIM inhibition in this response. Ad.decoymiR-24 additionally upregulated eNOS RNA level in cultured cardiomyocytes (Figure 4f). However, this was not sufficient to promote cell survival, probably because of the prevalent action of BIM or other possible pro-apoptotic miR-24 target genes in the cardiomyocyte. Next, we examined BIM changes induced by MI and in vivo Ad.decoymiR-24 in cardiomyocytes. At 14 days after MI, as compared with MI/Ad.Null, Ad.decoymiR-24 increased BIM relative expression in cardiomyocytes (Figure 4g). At the same time point, Ad.decoymiR-24 decreased miR-24 levels in cardiomyocytes (Supplementary Figure S5b), thus suggesting that Ad.decoymiR-24-mediated miR-24 inhibition is still effective on cardiomyocytes at this time point. Of note, in line with Fiedler et al., GATA2 and PAK4 were not detected in cardiomyocytes (data not shown).
Figure 4.

miR-24 inhibition induces apoptosis and increases BIM expression in cardiac myocytes. Apoptosis of cultured murine HL1 cardiomyocytes treated with either Ad.Null or Ad.decoymir-24 was measured by (a) Caspase-GLO activity assay and (b) Cell Death Detection ELISA. (c) BIM relative expression in HL1 cells infected with Ad.Null or Ad.decoymiR-24. (d) Caspase-GLO activity assay and mRNA relative expression of (e) BIM and (f) eNOS on adult cardiomyocytes after in vitro infection with Ad.Null or Ad.decoymiR-24. Experiments were performed in duplicate and repeated 3 times. (g) BIM mRNA relative expression on cardiomyocytes isolated at 3 and 14 days after surgery and gene transfer (N = 3–5 pools/group). Data are expressed as mean ± SEM. BIM and eNOS mRNA analyses employed RNA 18S for normalization and data are reported to the control group by the 2-ΔΔCt formula. RLU: Relative Luminescent Units; OD: optical density. *P < 0.05 versus Ad.Null; ††P < 0.01 and †P < 0.05 versus Sham/Ad.Null; §P < 0.05 versus MI/Ad.Null.
The effect of miR-24 inhibition on cardiac fibroblasts has been also taken into account. Fibroblasts were transfected with anti-miR-24 or scramble control. Efficacy of transfection in regulating miR-24 expression was validated by PCR (Supplementary Figure S3c). Next, fibroblasts were prompted in Caspase activity assay, which showed their increased apoptosis following miR-24 inhibition (Figure 5a). Moreover, anti-miR-24 led to a significant upregulation of BIM, but did not affect the other studied target genes (Figure 5b). Similar results were obtained by Ad.decoymiR-24-induced miR-24 inhibition (data not shown). Finally, target gene analyses in cardiac fibroblasts isolated from the LV at 3 days after MI and adenovirus infection showed that Ad.decoymiR-24 induced upregulation of BIM in comparison with Ad.Null (Figure 5c), whereas no significant effect on GATA2 and eNOS were observed (data not shown) and PAK4 was not detected.
Figure 5.
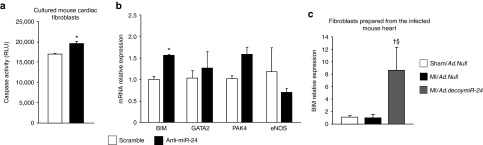
miR-24 inhibition induces apoptosis and increases BIM expression in cardiac fibroblasts. Bar graph shows (a) apoptosis (evaluated by Caspase-GLO activity assay) and (b) miR-24 target genes on cultured cardiac fibroblasts after transfection with anti-miR-24 or scramble (control). (c) RT-PCR for BIM assessed on cardiac fibroblasts isolated from mouse LV at 3 days after MI and gene transfer. Data are expressed as mean ± SEM. N = 2 samples/group for transfection experiments. Fibroblasts isolated form infected LV were combined in pools of 2 samples (N = 3–4 pools/group). mRNA analyses employed RNA 18S for normalization and data are reported to the control group by the 2-ΔΔCt formula. *P < 0.05 versus Scramble; †P < 0.05 versus Sham/Ad.Null; §P < 0.05 versus MI/Ad.Null.
Effect of miR-24 inhibition on microvascular density and blood flow in the infarcted myocardium
We next sought to evaluate the effect of local miR-24 inhibition by Ad.decoymiR-24 in the mouse MI model. At 14 days after MI induction, the numbers of capillaries (Figure 6a,d) and of small (diameter <50 µm) arterioles (Figure 6b,d) in the peri-infarct area were significantly increased by Ad.decoymiR-24 in comparison with Ad.Null. Importantly, the increase in microvascular density was associated with improved myocardial blood perfusion (revealed by using fluorescent microspheres) in mice given Ad.decoymiR-24 (Figure 6c).
Figure 6.

miR-24 inhibition increases vessels density and improves blood perfusion in the infarcted myocardium. Bar graphs show the (a) capillary and (b) small arteriole (<50 µm in lumen) densities in the peri-infarct region and the (c) LV blood flow at 14 days after surgery. (d) Representative microphotographs showing the peri-infarct area of Ad.Null and Ad.decoymiR-24 injected hearts after staining with the endothelial marker Isolectin-B4 (in red) and with a fluorescent antibody for the mural cell marker α-SMA (in green and pointed by arrows, for arterioles). Scale bars: 20 µm. Data are expressed as mean ± SEM. (N = 6–7 mice/group for histological analyses, 8 mice/group for evaluation of LV perfusion). *P < 0.05 and **P < 0.01 versus Sham/Ad.Null; †P < 0.05 versus MI/Ad.Null.
Effects of miR-24 inhibition on cardiovascular apoptosis in the infarcted myocardium
Apoptosis of ECs, cardiomyocytes, and fibroblasts is reportedly increased after MI.24,25,26 Here, we evaluated whether Ad.decoymiR-24 impacts on apoptosis in the peri-infarct area at 14 days after surgery. By in situ TUNEL assay, we found that Ad.decoymiR-24 increased by 2.2-folds the percentage of apoptotic cardiomyocyte nuclei (11.6 ± 0.7 versus 5.3 ± 1.5 in mice treated with Ad.Null; P < 0.05) (Figure 7a,d). By contrast, no effect of Ad.decoymiR-24 on EC survival was observed at this time point (Figure 7b,e). In addition, Ad.decoymiR-24 increased the percentage of apoptotic fibroblasts in the peri-infarct area (Figure 7c,f).
Figure 7.

miR-24 inhibition induces apoptosis of cardiomyocytes and fibroblasts but not ECs in the peri-infarct myocardium. Quantification of TUNEL positive (a) apoptotic cardiomyocyte nuclei, (b) ECs, and (c) fibroblasts following TUNEL assay in paraffin sections from peri-infarct region at 14 days after MI. (d) Representative pictures after staining for TUNEL (red) and α-sarcomeric actin (green) showing apoptotic cardiomyocytes (in purple and pointed by white arrows). (e) Representative pictures of apoptotic ECs (in purple and pointed by red arrows) evaluated after staining for TUNEL (in red) and isolectin-B4 (in green). Nuclei are depicted in blue (DAPI). (f ) Representative pictures of apoptotic fibroblasts (in purple and pointed by red arrows) evaluated after staining for TUNEL (in red) and vimentin (in green). Asterisks indicate the infarct area. Scale bars: 20 µm. Data are expressed as mean ± SEM. (N = 5 mice/group). **P < 0.01 and *P < 0.05 versus MI/Ad.Null.
Effects of miR-24 inhibition on cardiac function after MI
The infarcted heart undergoes functional and dimensional changes.27 Occlusion of a coronary artery leads to enlargement of the LV chamber volume and reduction of the myocardial contractile performance.25,27 To date, positive effects on cardiac function have been described after either inhibition12 or over expression14 of the miR-24 in the setting of acute MI in mice. In the present study, cardiac function was measured by echocardiography and Millar tip-catheter at 14 days after MI. As expected, in comparison with sham-operated mice, worsening of LV function was observed in MI mice for all analyzed parameters (Figure 8a–f). However, Ad.decoymiR-24 improved ejection fraction (EF) and fractional shortening (FS) (Figure 8a,b, respectively) in comparison with Ad.Null. In addition, dP/dtmax (an index of ventricular contractile performance) was improved by miR-24 inhibition (Figure 8c), whereas no significant changes were observed for dP/dtmin (Figure 8d). Moreover, despite ventricular dilatation being evident in the MI mice, Ad.decoymiR-24 led to a small but significant reduction of both the LV internal diameter (Figure 8e) and LV chamber volume (Figure 8f) compared with Ad.Null, thus suggesting that miR-24 inhibition positively impacts on post-MI LV remodeling and function. This set of data is in agreement with what reported by Fiedler et al. using antagomir for miR-24.12 In addition, TTC staining performed at 14 days after MI revealed that Ad.decoymiR-24 reduced infarct size (Figure 8g), thus further supporting the cardiac functional improvement observed after MI and miR-24 inhibition.
Figure 8.

Local Ad.decoy-mediated inhibition of miR-24 improves cardiac function after MI. Bar graphs showing (a) LV ejection fraction, (b) fractional shortening, (c) dP/dtmax, (d) dP/dtmin, (e) end-systolic LV internal diameter (LVID), and (f) end-systolic LV chamber volume evaluated at 14 days after MI and gene transfer. a, b, e, and f were measured by echocardiography; c and d were assessed by Millar tip-catheter. (g) Bar graphs and representative pictures show the percent of infarct area evaluated in MI hearts at 14 days after surgery after TTC staining. Data are expressed as mean ± SEM. (N = 12 mice/group for functional analyses and five mice/groups for infarct size). *P < 0.05 and **P < 0.01 versus Sham/Ad.Null; †P < 0.05 versus MI/Ad.Null.
Discussion
Recent studies have implicated miRs in a substantial variety of biological processes and diseases. Some miRs and several hundred miR-regulated target genes have been shown to participate in cardiovascular development28,29,30,31 as well as cardiovascular diseases, including MI.12,14
To better understand the role of miRs in physiology, diseases and as therapeutics, molecular tools allowing for miR forced expression or inhibition are essential. Therapeutic delivery of “good” miRs by using either adenoviral (Ad)32 or adeno-associated viral (AAV) vectors33 proved to be adequate at modulating miR expression and showed therapeutic promises in animal MI models. On the other side, inhibition of pathogenetic miRs by modified antisense oligonucleotides (antagomirs),12,18 miR sponges34 or miRs decoys22,32 has also showed therapeutic efficacy. Antagomirs are still the predominant tools employed for systemic miR inhibition.35 Notwithstanding, we believe that the use or improved viral vectors for either induction or repression of miRs can be useful, especially to confer a local, longer-lasting and robust expression of the therapeutic construct after a single administration dose.
Recently, miR-24 has emerged as an important but controversial miR involved in post-MI recovery.12,14,21 Hence, we considered worth to further investigating on the cardiovascular actions and possible therapeutic potential of miR-24 expression modulation. miR-24 level has been previously shown to be increased in ECs of the mouse heart peri-infarct zone at early time points after MI.12 However, Qian et al. disputed the presence of miR-24 expression in mouse myocardial ECs.14 This is quite surprising, as miR-24 has been described to be highly expressed in ECs isolated from several tissues.36 In line with these observations, we found that miR-24 is expressed in HUVECs, HVMECs and mouse myocardial CD146+ ECs and it is upregulated in CD146+ cells of the peri-infarct myocardium. This is in agreement with Fiedler et al.12 On the other hand, similarly to what has been shown by Qian et al.14 and Wang et al.,21 we found that miR-24 is downregulated in the peri-infarct tissue as compared with both sham-operated LV and the LV remote zone isolated from infarcted hearts. Moreover, our data show that MI reduces miR-24 levels in both cardiomyocytes and fibroblasts.
To understand the impact of miR-24 on the angiogenic process, in vitro studies on ECs were performed. The pro-angiogenic role of miR-24 has been already elucidated by Fiedler et al. during zebrafish development and using HUVECs.12 Here, to better represent the EC contributing to adult neoangiogenesis we additionally employed adult HMVECs. Similarly to what previously observed in HUVECs,12 synthetic miR-24 precursor (pre-miR-24) impaired HMVEC networks formation on Matrigel, whereas miR-24 inhibition by anti-miR-24 improved HMVEC networking capacity.
Many pro- and anti-angiogenic factors are involved in the angiogenesis response to physiological or pathological stimuli. Among them, nitric oxide released by eNOS is recognized as an essential factor which induces vasodilation and modulates post-ischemic angiogenesis.37 In the present study, we have newly identified eNOS as a target of miR-24 in cultured ECs as well as in the infarcted heart. In fact, miR-24 inhibition increased eNOS protein expression in both HMVECs and the mouse peri-infarct tissue as well as eNOS activity in CD146+ ECs isolated from the peri-infarct myocardium. eNOS protein upregulation induced by miR-24 inhibition might contribute to the increased myocardial capillary and arteriole densities together with the improved LV perfusion observed at 14 days after MI. Thus far, the pro-angiogenic effect of miR-24 inhibition has been described to be mediated by PAK4 and GATA2.12 The finding that eNOS is another target of miR-24 considerably advances our understanding on the mechanisms used by miR-24 to control angiogenesis, including in the setting of tissue ischemia. However, as each miR can regulate the expression of multiple target genes, we cannot exclude that more factors able to modulate vascular biology could additionally contribute to the miR-24 anti-angiogenic actions.
Fiedler et al. used the systemic delivery of an antagomir to inhibit miR-24 in mice with MI.12 By contrast, in our study, in vivo miR-24 inhibition was obtained by using an Ad carrying a decoy sequence. This tool was prepared consistently with what published by Care et al., who previously worked on miR-133.32 Ad.decoymiR-24 was directly injected in the mouse peri-infarct myocardium soon after coronary artery ligation. Our mouse MI model is comparable with that used in refs. 12,14,21. Here, using Ad-mediated local delivery of miR-24 decoy, we confirmed that miR-24 inhibition promotes the formation of new capillaries and arterioles in the peri-infarct myocardium. In addition, the increment in LV perfusion observed after miR-24 inhibition suggests the functionality of the newly formed microvessels to supply the ischemic region of the myocardium.
Finally, Qian et al. provided evidence of an anti-apoptotic effect of miR-24 in cardiomyocytes.14 We confirmed this data in vitro as well as in the MI setting. Notwithstanding, in apparent contrast with Qian et al.14 and Wang et al.21 who showed therapeutic advantages of overexpressing miR-24 in the mouse infarcted heart,14 our study shows an overall positive effect of local miR-24 inhibition in post-MI LV remodeling and cardiac function. Our in vitro data confirm the observation previously reported by either Fiedler in ECs or Qian in cardiomyocytes and these findings are additionally validated by our in vivo data. In addition, we newly report that miR-24 inhibition induces fibroblast apoptosis, possibly via BIM, which is upregulated in cardiac fibroblast after either anti-miR-24 or Ad.decoymiR-24 (versus the respective controls). This pro-apoptotic effect on fibroblasts might support our finding of reduced infarct size in the heart of Ad.decoymiR-24 injected mice.
We believe that, in our experimental model, Ad.decoymiR-24-mediated inhibition of miR-24 has an initial predominant effect on ECs rather than on cardiomyocytes, thus leading to a potent pro-angiogenic response which is possibly accompanied by a control of fibroblast expansion at 2 weeks after MI. However, we cannot exclude that a prolonged inhibition of miR-24 followed by extended apoptosis of cardiomyocytes might have a detrimental effect on infarct size and cardiac function.
In conclusion, we have provided strong evidence of the pro-angiogenic and overall therapeutic effects of miR-24 inhibition in the setting of acute MI and revealed for the first time that eNOS is a target of miR-24.
Materials and Methods
MI protocol in mice. The in vivo experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals prepared by the Institute of Laboratory Animal Resources and with the prior approval of the UK Home Office. MI or sham-operation was induced in anaesthetized CD1 male mice, as previously described.25 Briefly, for MI induction, mice were anesthetized by intraperitoneal injection of Avertin (880 mmol/kg i.p., Sigma, Poole, UK), intubated and artificially ventilated using a Minivent mouse ventilator (Harvard Apparatus, Kent, UK). Under a surgical microscope, an incision was made at the level of the left 5th intercostal space and MI was induced by permanent ligation of the proximal left anterior descending coronary artery (LAD) by using a 7.0 Mersilene suture (Ethicon, Somerville, NJ).
Isolation of ECs, cardiomyocytes and fibroblasts from the mouse LV. To investigate miR-24 mRNA relative expression in different cardiac cells after MI, LVs were harvested at 1, 3 and 14 days after MI or sham-operation. Briefly, the peri-infarct area was separated from the remote area and pools of 2 samples were rinsed and digested with collagenase II (Worthington, Lakewood, NJ) plus DNase I (Sigma) using gentleMACS Dissociator (Miltenyi Biotec, Bisley, UK), following manufacturer's instructions. Then, ECs were immunomagnetic sorted using a CD146 antibody (clone ME-9F1) (Miltenyi Biotech).22,38 To verify EC enrichment following CD146+ separation, single cell suspensions were incubated with FITC-conjugated CD146 and APC-conjugated CD31 antibodies (or the respective isotype for negative control). Unstained and single stained controls were performed to define positivity. Fluorescence was analyzed in a Canto II flow cytometer using the Diva software (BD Biosciences, Oxford, UK). Separation of cardiomyocytes was obtained by sedimentation at room temperature for 10 minutes. Cardiomyocyte preparations were validated by staining with a fluorescent antibody for α-sarcomeric actin (Sigma). Fibroblasts were isolated form the total LV of adult healthy mice (for in vitro experiments) as well as from the peri-infarct area and the sham-operated MI. To extract fibroblasts, LVs were harvested, minced and digested with liberase (Sigma) for 30 minutes at 37 °C. Cells were plated for 2 hours and non-adherent cells were then separated from adherent fibroblasts. Fibroblast preparations were validated by staining with a fluorescent antibody for vimentin (AbCam, Cambridge, UK).
Cells and cell culture. HUVECs and HMVECs (Lonza, Verviers, Belgium) were grown in EGM-2 (EBM-2 added with growth factors and other supplements, Lonza) with 10% fetal bovine serum and used at P2 to P5. HL1 cardiomyocytes (a gift from Prof William Claycomb, Louisiana State University Medical Center, New Orleans, LA) were cultured in Claycomb medium as originally described.39
Isolation and culture of cardiomyocytes from adult mice. The mouse heart was excised and cannulated via the aorta onto a modified Langendorff apparatus. All solutions perfusing the heart were gassed with 100% O2 and heated to 37 °C. Perfusion was performed retrogradely with a myocyte isolation buffer (containing 113 mmol/l NaCl, 4.7 mmol/l KCl, 1.2 mmol/l MgSO4, 0.6 mmol/l NaH2PO4, 0.6 mmol/l KH2PO4, 10 mmol/l HEPES, 10 mmol/l 2, 3 –BDM, 1.6 mmol/l NaHCO3, 20 mmol/l glucose, and 30 mmol/l taurine; pH 7.4) followed by an enzyme solution (myocyte isolation buffer containing 1 mg/ml collagenase type 1 (Worthington), 0.05 mg/ml protease (Sigma) and 0.015 mg/ml DNase I grade 2 (Roche, Indianapolis, IN). After 2 minutes of perfusion the enzyme solution was supplemented with 50 µmol/l CaCl2 and the heart was perfused for another 15–20 minutes. The ventricular tissue was then collected and gently disaggregated to obtain single cells suspension. The cells were pelleted and resuspended in minimum essential medium (with 2.5% fetal bovine serum) for 1 hour before being infected with either Ad.Null or Ad.decoymiR-24 for 48 hours (in minimum essential medium without fetal bovine serum). Cardiomyocyte preparations were validated by staining with a fluorescent antibody for α-sarcomeric actin (Sigma).
Preparation of decoymiR-24 adenoviral vector. For Ad.decoymiR-24 generation, tandem sequences complementary to miR-24 separated by a 18 bp unrelated spacer (decoymiR-24 sequence: 5′-CTGTTCCTGCTGAACTGAGCCAagagaacttagagaacttCTGTTCCTGCTGAACTGAGCCA-3′) were synthesized as oligonucleotides (Invitrogen) and inserted into XhoI/XbaI multicloning site in a TW3′-UTR vector40 (Supplementary Figure S4e). After sequencing, the CMV-eGFP decoymiR-24 fragment was subcloned into an adenoviral vector, pDC515 (Microbix Biosystems, Mississauga, Ontario, Canada), for adenovirus production. The vector produces a stable eGFP transgene under CMV promoter. This design, using multiple copies of complementary targets, was intended to optimize the transgene repression in the presence of the miRNA.22,40 Replication-deficient adenoviruses were generated by site-specific FLP-mediated recombination of the cotransfected shuttle and genomic plasmids in 293 cells. Viral stocks were amplified, CsCl banded, and titrated as previously described.41 Cells were plated 24 hours before infection, incubated with fresh media containing the required multiplicity of infection (MOI) per cell of virus, left for 6 hours, washed, and maintained until harvesting.
Pre-miR-24 and Anti-miR-24 transfection of HUVECs, HMVECs, and mouse cardiac fibroblasts. A miR-24 precursor (pre-miR-24, AM17100; Ambion-Life Technologies, Paisley, UK) and a miR-24 inhibitor oligonucleotide (anti-miR-24, AM17000; Ambion) and a scrambled oligonucleotide (Cy3 dye-labeled anti-miR Negative Control, AM17011; Ambion) was transfected into HUVECs, HMVECs or mouse cardiac fibroblasts using Lipofectamine 2000 Reagent (Invitrogen, Carlsbad, CA).
Evaluation of apoptosis in cultured cells. HUVECs and HMVECs (1.5 × 105/well) were seeded in six-well plates and treated with pre-miR-24, anti-miR-24 or negative control (a scramble miR sequence) (all at 50 nmol/l/l) for 5 hours then kept in Opti-MEM for 48 hours before being trypsinized and plated in a flat-bottomed 96-well plates (4 × 103/well). Caspase-3 activity was then measured using a luminescent cell death detection kit (Caspase-GLO assay; Promega, Southampton, UK). HL1 cells (2 × 105/well) were seeded in flat-bottomed six-well plates and infected with Ad.Null or Ad.decoymiR-24 (100 MOI each) for 24 hours, then trypsinized and plated in a 96-well plates (5 × 103/well) to perform Caspase-GLO assay or Cell Death Detection ELISA assay (Roche) following manufacturer's instructions. Adult mouse cardiomyocytes (2 × 103/well) or cardiac fibroblasts (5 × 103/well) were seeded in a 96-well plates and infected with Ad.Null or Ad.decoymiR-24 (100 MOI each) for 48 hours before performing Caspase-GLO assay. Additional cardiac fibroblasts were transfected with anti-miR-24 or scramble (control) before undergo Caspase-GLO assay. For Caspase-GLO assay, the luminometer reading were taken at 30 minutes after adding the Caspase-GLO reagent.
Evaluation of tube-like structures by Matrigel assay. HUVECs and HMVECs (1.5 × 104/well) were seeded in six-well plates and treated with pre-miR-24, anti-miR-24 or scramble control for 48 hours, then trypsinized and plated in a flat-bottomed 96-well plates (8 × 103/well) coated with growth factors-enriched Matrigel (Matrigel Matrix, Basement Membrane, BD). Endothelial network formation was quantified at 5 hours in randomly captured microscopic fields (magnification 40×) by counting the length of cellular network and by measuring the % of area covered by connected vascular-like structures.
BrdU incorporation assay. BrdU proliferation assay was performed on HUVECs and HMVECs transfected with pre-miR-24, anti-miR-24, or scramble control. At 48 hours after transfection, ECs (4 × 103 cells/well) were seeded on 96-well plates. The medium was then replaced with a complete medium added with BrdU (10 μmol/l) for 24 hours. BrdU incorporation was measured by the BrdU ELISA assay kit (Roche).
Bioinformatic prediction of miR-24 target genes. MiRwalk (http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk/index.html) was searched for predicted target genes of miR-24 These analyses were performed allowing miRwalk to search in 9 bioinfomatic platforms, namely DIANAmT, miRanda, miRDB, miRWalk, RNAhybrid, PICTAR5, PITA, RNA22, and Targetscan.
Luciferase assay. To investigate whether miR-24 directly regulates eNOS expression, portions of the 3′-UTR of these potentials target genes were inserted downstream of a luciferase open reading frame (pLUC). An eNOS 3′-UTR vector was purchased from Origene. For controls, we prepared similar vectors in which five nucleotide mutations were inserted in the 3′-UTR sequences (eNOS 3′-UTR: position 119–125) complementary to the miR-24 “seed” sequences (primers used for mutation are: forward AAGTGCTGCGATTATAGACGatttaatCTGCACCTGG, reverse CGTCTATAATCGCAGCACTTTGGGAGGCCGA).
HPLC purified oligonucleotides (Sigma) were used for mutagenesis, performed with Pfu enzyme following the in vitro mutagenesis kit protocol (Invitrogen). The different luciferase constructs were transfected into HEK293 cells together with pre-miR-24 a scrambled oligonucleotide sequence (control). Cells were cultured for 48 hours and assayed with the Dual-Luciferase Reporter Assay System (Promega). Values are normalized using Renilla expression level.
In vivo nucleic acid therapy. Immediately after LAD ligation, Ad.decoymi-R24 (108 p.f.u. in 15 µl) or an empty vector (Ad.Null) was injected in the peri-infarct myocardium. Sham-operated mice given Ad.Null in the LV were used as reference.
Measurement of cardiac dimensions, function and myocardial perfusion. Cardiac dimensions and function were evaluated in anaesthetized mice (Avertin) at basal (before the induction of MI) and at 14 days after MI by using a High-Resolution Echocardiography System (Visual Sonics, Toronto, Ontario, Canada). The following parameters were analyzed: LV ejection fraction (EF, %), LV fractional shortening (FS, %), LV chamber volume (µl), LV internal diameter (mm). In addition, LV function was measured at 14 days after MI with a miniaturized 1.4F transducer Millar tip-catheter. Peak systolic LV pressure (mmHg), maximal rate of LV pressure rise (dP/dtmax, mmHg/s) and minimal rate of LVP fall (dP/dtmin, mmHg/s) were digitally recorded and analyzed with a dedicated software (PowerLab/4SP, ADInstrument).
Myocardial blood flow (ml/minute/g of tissue) was measured 14 days after MI or sham-operation by using fluorescent microspheres (Invitrogen) injected into the LV cavity, as previously described.25 Briefly, anesthetized mice (Avertin) were intubated and a polyethylene catheter (PE10) was inserted into the right carotid artery and connected to a syringe pump for collection of reference blood. Microspheres (0.02 μm in diameter) were then injected into the left ventricle cavity (200 μl of total volume). Reference blood was collected at a rate of 0.15 ml/minute. Mice were killed 2 minute later, the heart was removed and the LV was separated, weighed, and minced before being digested in 10 ml of 2 mol/l ethanolic KOH (Sigma) at 60 °C for 48 hours. Finally, microspheres were collected and fluorescence intensity was determined by a fluorometer. Blood flow was additionally measured in the kidneys to confirm the homogenous distribution of microspheres into the bloodstream.
Histological analyses. Mice were perfusion/fixed at 14 days after surgery and hearts collected and embedded in paraffin. Heart sections including the middle part of the infarct, the peri-infarct zone (which contains the borderline area around the MI), and the remote zone (classified as myocardial tissue unaffected by the MI) were used. Capillary and arteriole densities were measured in transverse sections (5 µm thickness) of the LV peri-infarct zone. Density of arterioles was evaluated after staining for α-smooth muscle actin (α-SMA, for marking vascular smooth muscle cells) and with isolectin-B4 (EC marker). Capillary density was evaluated in the same sections by counting the number of isolectin B4-positive and α-SMA-negative microvessels (40× magnification). Apoptosis of ECs and cardiomyocytes was determined by transferase-mediated dUTP nick-end labeling (TUNEL) assay (40× magnification) (in situ cell death detection kit Fluorescein; Roche). TUNEL assay was performed in combination with isolectin-B4 (to determine apoptotic ECs), α-sarcomeric actin (to distinguish apoptotic cardiomyocytes) or vimentin (to recognize apoptotic fibroblasts).
Determination of myocardial infarct size. Infarct size at 2 weeks after MI was determined by triphenyltetrazolium chloride (Sigma) staining. Briefly, the hearts were collected and sliced transversely to yield five slices. Slices were incubated in 1% triphenyltetrazolium chloride for 10 minutes at 37 °C and fixed in 10% formalin. The area of infarction in both sides was determined by an imager analyzer, summed for each hearts and infarct size was expressed as a percentage of the LV.
Determination of protein and gene expression in the mouse heart. For RT-PCR analyses, cardiac regions (LV, RV, and septum of sham-operated mice and LV peri-infarct, LV remote area, RV and septum of MI mice) were isolated at 1, and 3 days after surgery. Protein expressional analyses were performed in LV peri-infarct zone or LV of sham-operated mice at 3 days after surgery. Briefly, mice were anaesthetized (Avertin), the heart removed and the required cardiac regions separated and immediately snap-frozen in liquid nitrogen for subsequent extraction of either mRNA or protein. Total RNA was extracted using the miRNeasy mini kit (Qiagen, Crawley, UK) and RNA concentration measured with the NanoDrop spectrophotometer. Real time quantification to measure miR-24 was performed with the TaqMan miRNA reverse transcription kit and miRNA Assay (Applied Biosystems, Carlsbad, CA). miR-24 expression was normalized to the U6 small nucleolar RNA (snRU6) (Applied Biosystem). Primers identification numbers for miRs were as follows: miR-24: Assay ID 000402; snRU6: Assay ID 001973 (Applied Biosystem). Relative miR-24 expression data reported to the control group by the 2-ΔΔCt formula. For mRNA analysis, single-strand complementary DNA was synthetized from 500 ng of total RNA. Quantitative reverse transcription PCR (RT-PCR) was used to measure miR-24 target genes. 18S ribosomal RNA was used for normalization. The primers used for mRNA expressional analyses are indicated in Supplementary Table S1. Relative target gene mRNA expression data reported to the control group by the 2-ΔΔCt formula.
Proteins were extracted from either mouse LV samples using RIPA buffer and western blot analyses were performed loading the equivalent of 50 µg of total proteins resolved by SDS-PAGE and transferred to a nitrocellulose membrane (Amersham Bioscience, Piscataway, NJ). Primary antibodies were: total eNOS (Santa Cruz Biotechnology, Santa Cruz, CA) and tubulin (loading control; Research Diagnostics, Flanders, NJ). Secondary antibody was anti-rabbit horseradish peroxidase conjugate (Amersham Bioscience).
Determination of protein and gene expression in cells. Total RNA was extracted from HMVECs, HL1 and CD146+ ECs, adult mouse cardiomyocytes and fibroblasts (isolated as described above) using the miRNeasy mini kit (Qiagen, Crawley, UK) and RNA concentration was measured with the NanoDrop spectrophotometer. Real time quantification to measure miR-24 was performed with the TaqMan miRNA reverse transcription kit and miRNA Assay. miR-24 expression was normalized as described above. For mRNA analysis, single-strand complementary DNA was synthetized from 500 ng of total RNA. Methods for mRNA analyses and quantification are described above. Protein extraction and western blot analyses on cultured ECs were performed as described above.
eNOS activity assay. eNOS-derived nitric oxide production was evaluated in CD146+ ECs isolated from the peri-infarct LV at 3 days after infection with Ad.Null or Ad.decoymiR-24. After isolation, cells were loaded with 10 µmol/l of the rhodamine-based fluorescent chromophore DAR4M-AM (Sigma) in Dulbecco's modified Eagle's medium (without phenol red) for 30 minutes at 37 °C. Nitric oxide production was evaluated by using a fluorometer.
Statistical analyses. Values are presented as mean ± SEM. Statistical significance was evaluated through the use of an unpaired t-test for comparisons between two groups. For comparison among more than two groups, ANOVA was used, followed by an unpaired t-test. Analyses were performed using the SigmaStat 3.1 software (Sigma). A P value of <0.05 was interpreted to denote statistical significance.
SUPPLEMENTARY MATERIAL Figure S1. microRNA-24 (miR-24) and target genes expression in the mouse peri-infarct myocardium. Figure S2. Characterization of cells isolated form the mouse LV. Figure S3. miR-24 relative expression in cultured cells after transfection. Figure S4. Proangiogenic effect miR-24 inhibition in HUVECs and presentation of the decoymiR-24. Figure S5. miR-24 expression on cells isolated form the mouse LV after MI and Ad.decoy-mediated miR-24 inhibition. Figure S6. Alignment of human mature miR-24 (red sequence) on mouse eNOS 3′-UTR (green sequence) using RNAhybrid software. Table S1. Mouse and human PCR primer sequences.
Acknowledgments
The study was supported by a grant from Diabetes UK and the British Heart Foundation (BHF). I.F. is supported by a PhD studentship bursary from the University of Sassari Italy; A.C. is a BHF Intermediate Research Fellow; and C.E. is a BHF Senior Research Fellow. We thank Carlotta Reni and Hua Lin (both form the University of Bristol) for technical assistance and acknowledge the Bristol National Institute Health Research (NIHR) Biomedical Research Unit in Cardiovascular Medicine, in which our laboratories are embedded. The authors declare no conflict of interest.
Supplementary Material
References
- Rosamond W, Flegal K, Friday G, Furie K, Go A, Greenlund K, American Heart Association Statistics Committee and Stroke Statistics Subcommittee et al. Heart disease and stroke statistics–2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007;115:e69–171. doi: 10.1161/CIRCULATIONAHA.106.179918. [DOI] [PubMed] [Google Scholar]
- Olivetti G, Quaini F, Sala R, Lagrasta C, Corradi D, Bonacina E, et al. Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart. J Mol Cell Cardiol. 1996;28:2005–2016. doi: 10.1006/jmcc.1996.0193. [DOI] [PubMed] [Google Scholar]
- Anversa P, Cheng W, Liu Y, Leri A, Redaelli G, Kajstura J. Apoptosis and myocardial infarction. Basic Res Cardiol. 1998;93 Suppl 3:8–12. doi: 10.1007/s003950050195. [DOI] [PubMed] [Google Scholar]
- Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000;101:2981–2988. doi: 10.1161/01.cir.101.25.2981. [DOI] [PubMed] [Google Scholar]
- Krichavsky MZ, Losordo DW. Prevention and recovery of hibernating myocardium by microvascular repair. Circulation. 2011;124:998–1000. doi: 10.1161/CIRCULATIONAHA.111.047746. [DOI] [PubMed] [Google Scholar]
- Henry TD, Annex BH, McKendall GR, Azrin MA, Lopez JJ, Giordano FJ, VIVA Investigators et al. The VIVA trial: Vascular endothelial growth factor in Ischemia for Vascular Angiogenesis. Circulation. 2003;107:1359–1365. doi: 10.1161/01.cir.0000061911.47710.8a. [DOI] [PubMed] [Google Scholar]
- Stewart DJ, Kutryk MJ, Fitchett D, Freeman M, Camack N, Su Y, NORTHERN Trial Investigators et al. VEGF gene therapy fails to improve perfusion of ischemic myocardium in patients with advanced coronary disease: results of the NORTHERN trial. Mol Ther. 2009;17:1109–1115. doi: 10.1038/mt.2009.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporali A, Emanueli C. MicroRNA regulation in angiogenesis. Vascul Pharmacol. 2011;55:79–86. doi: 10.1016/j.vph.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710–1713. doi: 10.1126/science.1174381. [DOI] [PubMed] [Google Scholar]
- Fiedler J, Jazbutyte V, Kirchmaier BC, Gupta SK, Lorenzen J, Hartmann D, et al. MicroRNA-24 regulates vascularity after myocardial infarction. Circulation. 2011;124:720–730. doi: 10.1161/CIRCULATIONAHA.111.039008. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhang X, Ren XP, Chen J, Liu H, Yang J, et al. MicroRNA-494 targeting both proapoptotic and antiapoptotic proteins protects against ischemia/reperfusion-induced cardiac injury. Circulation. 2010;122:1308–1318. doi: 10.1161/CIRCULATIONAHA.110.964684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Van Laake LW, Huang Y, Liu S, Wendland MF, Srivastava D. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J Exp Med. 2011;208:549–560. doi: 10.1084/jem.20101547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JX, Jiao JQ, Li Q, Long B, Wang K, Liu JP, et al. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat Med. 2011;17:71–78. doi: 10.1038/nm.2282. [DOI] [PubMed] [Google Scholar]
- Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G, et al. MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ Res. 2010;106:166–175. doi: 10.1161/CIRCRESAHA.109.202176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Huang M, Li Z, Jia F, Ghosh Z, Lijkwan MA, et al. MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation. 2010;122 11 Suppl:S124–S131. doi: 10.1161/CIRCULATIONAHA.109.928424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- Hu S, Huang M, Nguyen PK, Gong Y, Li Z, Jia F, et al. Novel microRNA prosurvival cocktail for improving engraftment and function of cardiac progenitor cell transplantation. Circulation. 2011;124 11 Suppl:S27–S34. doi: 10.1161/CIRCULATIONAHA.111.017954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Huang W, Xu R, Nie Y, Cao X, Meng J, et al. MicroRNA-24 regulates cardiac fibrosis after myocardial infarction. J Cell Mol Med. 2012;16:2150–2160. doi: 10.1111/j.1582-4934.2012.01523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporali A, Meloni M, Völlenkle C, Bonci D, Sala-Newby GB, Addis R, et al. Deregulation of microRNA-503 contributes to diabetes mellitus-induced impairment of endothelial function and reparative angiogenesis after limb ischemia. Circulation. 2011;123:282–291. doi: 10.1161/CIRCULATIONAHA.110.952325. [DOI] [PubMed] [Google Scholar]
- Kiriakidou M, Nelson PT, Kouranov A, Fitziev P, Bouyioukos C, Mourelatos Z, et al. A combined computational-experimental approach predicts human microRNA targets. Genes Dev. 2004;18:1165–1178. doi: 10.1101/gad.1184704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill C, Mestril R, Samali A. Losing heart: the role of apoptosis in heart disease–a novel therapeutic target. FASEB J. 2002;16:135–146. doi: 10.1096/fj.01-0629com. [DOI] [PubMed] [Google Scholar]
- Meloni M, Caporali A, Graiani G, Lagrasta C, Katare R, Van Linthout S, et al. Nerve growth factor promotes cardiac repair following myocardial infarction. Circ Res. 2010;106:1275–1284. doi: 10.1161/CIRCRESAHA.109.210088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Shen YT, Gaussin V, Heyndrickx GR, Bartunek J, Resuello RR, et al. Apoptosis predominates in nonmyocytes in heart failure. Am J Physiol Heart Circ Physiol. 2009;297:H785–H791. doi: 10.1152/ajpheart.00310.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutgens E, Daemen MJ, de Muinck ED, Debets J, Leenders P, Smits JF. Chronic myocardial infarction in the mouse: cardiac structural and functional changes. Cardiovasc Res. 1999;41:586–593. doi: 10.1016/s0008-6363(98)00216-8. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, et al. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436:214–220. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
- Morton SU, Scherz PJ, Cordes KR, Ivey KN, Stainier DY, Srivastava D. microRNA-138 modulates cardiac patterning during embryonic development. Proc Natl Acad Sci USA. 2008;105:17830–17835. doi: 10.1073/pnas.0804673105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordes KR, Srivastava D. MicroRNA regulation of cardiovascular development. Circ Res. 2009;104:724–732. doi: 10.1161/CIRCRESAHA.108.192872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13:613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S, et al. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492:376–381. doi: 10.1038/nature11739. [DOI] [PubMed] [Google Scholar]
- Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721–726. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij E, Purcell AL, Levin AA. Developing microRNA therapeutics. Circ Res. 2012;110:496–507. doi: 10.1161/CIRCRESAHA.111.247916. [DOI] [PubMed] [Google Scholar]
- Larsson E, Fredlund Fuchs P, Heldin J, Barkefors I, Bondjers C, Genové G, et al. Discovery of microvascular miRNAs using public gene expression data: miR-145 is expressed in pericytes and is a regulator of Fli1. Genome Med. 2009;1:108. doi: 10.1186/gm108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, et al. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest. 1998;101:2567–2578. doi: 10.1172/JCI1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrage A, Loddenkemper C, Erben U, Lauer U, Hausdorf G, Jungblut PR, et al. Murine CD146 is widely expressed on endothelial cells and is recognized by the monoclonal antibody ME-9F1. Histochem Cell Biol. 2008;129:441–451. doi: 10.1007/s00418-008-0379-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claycomb WC, Lanson NA, Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, et al. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA. 1998;95:2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci D, Coppola V, Musumeci M, Addario A, Giuffrida R, Memeo L, et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med. 2008;14:1271–1277. doi: 10.1038/nm.1880. [DOI] [PubMed] [Google Scholar]
- Sala-Newby GB, Freeman NV, Curto MA, Newby AC. Metabolic and functional consequences of cytosolic 5'-nucleotidase-IA overexpression in neonatal rat cardiomyocytes. Am J Physiol Heart Circ Physiol. 2003;285:H991–H998. doi: 10.1152/ajpheart.00053.2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.