

Abstract
Many primary cancers including chronic lymphocytic leukemia (CLL) are resistant to vesicular stomatitis virus (VSV)-induced oncolysis due to overexpression of the antiapoptotic and antiautophagic members of the B-cell lymphoma-2 (BCL-2) family. In the present study, we investigated the mechanisms of CLL cell death induced as a consequence of VSV infection in the presence of BCL-2 inhibitors, obatoclax, and ABT-737 in primary ex vivo CLL patient samples. Microarray analysis of primary CD19+ CD5+ CLL cells treated with obatoclax and VSV revealed changes in expression of genes regulating apoptosis, the mechanistic target of rapamycin (mTOR) pathway, and cellular metabolism. A combined therapeutic effect was observed for VSV and BCL-2 inhibitors in cells from untreated patients and from patients unresponsive to standard of care therapy. In addition, combination treatment induced several markers of autophagy—LC3-II accumulation, p62 degradation, and staining of autophagic vacuoles. Inhibition of early stage autophagy using 3-methyladenine (3-MA) led to increased apoptosis in CLL samples. Mechanistically, a combination of BCL-2 inhibitors and VSV disrupted inhibitory interactions of Beclin-1 with BCL-2 and myeloid cell leukemia-1 (MCL-1), thus biasing cells toward autophagy. We propose a mechanism in which changes in cellular metabolism, coupled with pharmacologic disruption of the BCL-2–Beclin-1 interactions, facilitate induction of apoptosis and autophagy to mediate the cytolytic effect of VSV.
Introduction
Chronic lymphocytic leukemia (CLL) is the most common form of leukemia in the Western hemisphere,1,2 a clonal malignancy characterized by peripheral blood lymphocytosis as a result of defective apoptosis signaling1 and the abnormal accumulation of CD5+ monoclonal B lymphocytes.1,3 Overexpression of antiapoptotic regulators of the B-cell lymphoma-2 (BCL-2) family contribute to resistance to programmed cell death, drug resistance, disease progression, and poor clinical outcome in CLL patients in response to conventional therapies.4 The BCL-2 family is divided into three groups: (i) antiapoptotic (BCL-2, myeloid cell leukemia-1 (MCL-1), and BCL-XL,), (ii) proapoptotic (BAX and BAK), and (iii) BH3-only (NOXA, PUMA, BID, BIM, BAD, BIK, and BMF) proteins.5,6,7 In addition to their contribution to apoptosis, the BCL-2 family is involved in regulation of autophagy, a cellular process characterized by the sequestration of cytoplasmic material into vacuoles for bulk degradation.8,9,10 Beclin-1 is a BH3-only protein, a central autophagy regulator and a haploinsufficient tumor suppressor that is inhibited by antiapoptotic BCL-2 and BCL2-XL proteins; this interaction blocks autophagy progression in cancer cells9,10,11 and serves as a regulatory point of cross-talk between the apoptotic and autophagic pathways.12,13,14 Several other proteins also negatively regulate autophagy, including mechanistic target of rapamycin (mTOR), a Ser/Thr protein kinase involved in growth, proliferation, and cell cycle progression.15
Oncolytic virotherapy is currently being tested with promising results in phase I–III clinical trials.16,17,18,19,20 Vesicular stomatitis virus (VSV) has emerged as a prototypical oncolytic virus that induces direct tumor cell lysis, is sensitive to type I interferon (IFN) induction and cellular antiviral responses,21,22,23,24 and activates intrinsic and extrinsic apoptotic signaling.25,26,27 VSV used in this study has a M51R substitution in the viral matrix (M) protein that was shown to enhance the safety profile of the virus; the attenuated mutant is a potent inducer of the IFN response in healthy cells.21,23
BCL-2 inhibitors (BH3 mimetics) represent a new class of anticancer therapeutics that display promising results in preclinical and clinical studies when used as single agents or in combination with conventional cancer therapies.28,29,30 Functionally, this class of inhibitors competes with BH3-only proapoptotic proteins for binding to antiapoptotic BCL-2 proteins.31,32 Obatoclax (GX15-070) is a pan BCL-2 inhibitor and a synthetic derivative of prodiginine.33,34 ABT-737 and its orally active analogue ABT-263 (navitoclax) are BAD-like mimetics; the ABT compounds target the majority of the antiapoptotic BCL-2 proteins but have low affinity for MCL-1.31,35 Obatoclax and ABT-263 are currently in multiple phase I/II clinical trials for the treatment of various solid and hematological malignancies including CLL, non-Hodgkin's lymphoma, and lung cancer.36,37,38
We previously reported that obatoclax displaced prosurvival interactions, whereas VSV infection both stimulated expression of the BH3-only NOXA in an IRF-3–dependent manner and the formation of a NOXA-BAX proapoptotic complex.25 With a growing interest in pharmacological disruption of antiapoptotic and antiautophagic interactions in CLL treatment, we expanded our previous studies to also examine a novel, more specific BCL-2 inhibitor ABT-737, performed microarray analysis of both leukemic and non-leukemic cells exposed to VSV + obatoclax, and examined contribution of an alternative cell death pathway (autophagy) to CLL cell death. Gene expression profiling of CLL patient samples exposed to the therapies was performed to gain insight into mechanisms that are responsible for the combined therapeutic effects. Combination therapy in primary CLL cells ex vivo selectively targeted CD19+ CD5+ leukemic cells from both untreated patients and patients resistant to standard of care therapy. Mechanistically, VSV and ABT-737 treatment induced both apoptosis and autophagy in CLL cells by disrupting the inhibitory interactions of Beclin-1 with BCL-2 and MCL-1, thus biasing cells toward autophagy and apoptotic cell death.
Results
Molecular and clinical features of the CLL patients
Several clinical prognostic and biological characteristics1,3,39 of CLL patients included in this study are summarized in Table 1. Elevated ZAP70 and CD38 expression, p53 mutation, chromosomal abnormalities (fluorescence in situ hybridization), and unmutated IgVH all have prognostic significance and have been associated with an unfavorable clinical course and resistance to traditional therapy. Patients examined in the study were grouped as: (i) previously untreated; (ii) non-responders/refractory; or (iii) responders to chemotherapy and/or antibody-based therapies. Given that CLL is characterized by the progressive accumulation of small mature CD5+ B lymphocytes,1 we measured by flow cytometry, the CD19 and CD5 B cell subsets in CLL patients and healthy donors as listed in Table 1. The percentage of leukemic CD19+ CD5+ B-cells was dramatically higher in untreated (68.0%) and refractory (57.8%) representative CLL patients, compared with responders (1.4%) or healthy controls (1.8%) (Figure 1a–d). The total percentage of cells with CD19+ CD5+ and CD19+ CD5− marker expression for all the CLL patients and healthy donors included in this study are listed in Figure 1e.
Table 1. Patient characteristics.
Figure 1.

Distribution of lymphocyte subsets in chronic lymphocytic leukemia (CLL) patients. Flow cytometry analysis of CD5+ B cells and CD5− B cells, gated on lymphocytes from peripheral blood in (a) a healthy donor and (b) responder, (c) untreated and (d) refractory (non-responder), and CLL patients. (e) Percent marker expression for all patients. Blue bars indicate CD19+ CD5+ cells; black bars indicate CD19+ CD5− cells (mean ± SD). FSC, forward scatter; SSC, side scatter.
Combination treatment selectively kills CLL cells ex vivo and improves survival of CLL xenograft model
The leukemic CD19+ CD5+ subpopulation of peripheral blood mononuclear cells (PBMCs) from untreated (Figure 2a) and non-responder/refractory (Figure 2b) CLL patients were examined for cell viability following treatment with a low, sub-optimal dose of ABT-737 (5 nmol/l) or by VSV (10 multiplicity of infection) infection. Treatment with VSV + ABT-737 decreased cell viability to 25% (untreated) and 33% (non-responder) by 48 hours while only minimally reducing the viability of non-leukemic, CD19+ CD5− cells from the same patient to 83% (untreated) and 89% (non-responder), demonstrating the capacity of the combination treatment to specifically kill leukemic cells. Next, we sought to investigate the antitumor efficacy of VSV + ABT-737 combination therapy in a murine A20 lymphoma model, which is partially resistant to VSV treatment.25 Tumors grew aggressively in untreated mice with an average tumor volume of >2,000 mm3 measured 10 days post-treatment initiation (Figure 2c). Single agent treatments, ABT-737 or VSV, decreased tumor volume by 32 and 43%, respectively. A statistically significant reduction in tumor size (P ≤ 0.05) was observed for the combination-treated animals compared with single agents or the vehicle. The effect of VSV + ABT-737 on tumor growth translated to increased animal survival (Figure 2d) and a statistically significant increase in median survival time from 10 days (VSV– or ABT-737–treated groups) to 24 days (VSV + ABT-737–treated mice) was observed (P ≤ 0.05).
Figure 2.

ABT-737 + vesicular stomatitis virus (VSV) therapy selectively reduces viability of chronic lymphocytic leukemia (CLL) cells ex vivo and in vivo. MTT assay showing sensitivity of CD19+ CD5+ cells of (a) untreated (n = 3) and (b) non-responder (n = 3) CLL patients to VSV + ABT-737 combination treatment in comparison to CD19+ CD5− cells (mean ± SD). White bars indicate control cells; light gray bars indicate ABT-737 treatment; dark gray bars indicate VSV infection, and black bars indicate VSV + ABT-737 treatment. (c) Tumor volumes were calculated as ½ (length × width)2 and values are expressed as the mean ± SD of tumor volume (n = 7). (d) Animals were evaluated for signs of stress such as infection, dehydration, weight loss, and limb paralysis. Mice were killed when tumor volumes exceed 2,000 mm3. *P ≤ 0.05 comparing tumor size between the single and combination treatment groups. MTT, 3-(4,5-dimethylthiazol)-2,5-diphenyl tetrazolium; NT, non-treated.
Gene expression profiles in CLL cells
We performed analysis of transcriptional profiles of the leukemic CD19+ CD5+ population exposed to obatoclax, VSV or the combination and compared them to untreated CLL cells as a tool to investigate the biological responses of CLL cells to either single agents or the combination. Comparisons of expression profiles between VSV versus control (vehicle-treated) cells, obatoclax versus control cells, and VSV + obatoclax versus control cells were generated and differentially regulated pathways were evaluated using ingenuity systems pathway analysis (IPA) (Figure 3a). Expression profile comparisons revealed changes in several pathways, including changes in mTOR and apoptotic signaling pathways, which were the only differentially regulated pathways common for the three comparisons. Expression profile analysis of individual genes (Figure 3b) of the apoptotic pathway revealed decreases in antiapoptotic BIRC6 (obatoclax- and combination-treated cells), and increases in both proapoptotic BID and the inhibitor of apoptosis (IAP)-binding protein, DIABLO (VSV- and combination-treated cells). Changes in transcription of genes belonging to mTOR pathway (Figure 3c) revealed downregulation in mRNA levels of PRKAB2 only in CLL cells exposed to obatoclax + VSV combination. The protein encoded by this gene is a regulatory subunit of the AMP-activated protein kinase, an enzyme that monitors cellular energy status.40
Figure 3.

Analysis of gene expression profiles in chronic lymphocytic leukemia (CLL) cells. (a) For each cluster, the differentially regulated pathways are indicated, and were compared with non-treated cells. Genes that belong to multiple Clusters of Orthologous Groups (COG) were placed into each assigned COG category. (b,c) The heatmaps show statistically significant differentially regulated genes for (b) apoptosis and (c) mTOR pathways. The genes shown in the heatmaps are derived from the following comparisons (left to right): VSV to control, obatoclax to control, and VSV + obatoclax to control and were selected based on the following criteria: absolute fold change >1.3 and nominal P value <0.05. Missing values that did not pass these criteria are shown in black. HGF, hepatocyte growth factor; mTOR, mechanistic target of rapamycin; NGF, nerve growth factor; NHEJ, non-homologous end joining; PDGF, platelet-derived growth factor; VSV, vesicular stomatitis virus.
To further evaluate differentially regulated genes and pathways, we analyzed differences in expression profiles of genes belonging to other top 10 statistically significant pathways based on IPA that are unique to each treatment. Exposure of cells to VSV alone (Figure 3a) induced upregulation of numerous genes that belong to oxidative phosphorylation (45/159 or 28.3%) and mitochondrial dysfunction (38/174 or 21.8%) including ATPases, ATP synthases, NADH dehydrogenases, and cytochrome c oxidases. Interestingly, the data above indicate that the most prominent effects of VSV infection in CLL cells are changes in mitochondrial function and oxidative phosphorylation. In cells exposed to obatoclax (Figure 3a) we observed changes in mRNA levels of several proteins that are involved in regulation of transcription (estrogen receptor signaling), mostly downregulation of complex subunit moderators and RNA polymerases. Moreover, a subset of downregulated molecules clustered to B cell receptor signaling pathway, belong to a prosurvival mitogen-activated protein kinase pathway. Together, these data indicate that the treatment of CLL cells with obatoclax induces downregulation of prosurvival signaling. Transcriptional profiling in cells exposed to VSV + obatoclax revealed differences in the expression of a subset of genes that belong to receptor signaling and metabolic regulation, revealing downregulation of several mitogen-activated protein kinases, inositol metabolizing proteins, and several acyl-CoA dehydrogenase family members. Overall, these data indicate that in CD19+ CD5+ cells, changes in expression of genes that are involved in mTOR and apoptotic pathways are observed in all three treatment groups (VSV, obatoclax or the combination) and are likely the most prominent biological response to therapies, although pathways that regulate metabolism, protein synthesis, and prosurvival signaling also contribute to such responses. We did not observe dramatic changes in IFN antiviral response genes, which is likely due to defective IFN signaling frequently observed in cancer cells and specifically in primary CLL.41,42,43
Combination treatment activates markers of autophagy
Next, we examined the markers of autophagy in CLL cells exposed to Bcl-2 inhibitor and VSV. The combined therapeutic effect observed in cells treated with VSV + ABT-737 (75% cytotoxicity) was partially reversed by the late stage autophagy-lysosomal inhibitor chloroquine (Figure 4a), without a significant change in viral replication (Figure 4b), suggesting a role of autophagy in CLL cell death induced with VSV + ABT-737. We next evaluated several markers of autophagy, including protein levels of mTOR and phosphorylation of FOX03A in CLL cells exposed to either single or combination therapies. Two hallmarks of autophagy were also measured–conversion of LC3-I (19 kDa) to LC3-II (17 kDa) and p62 degradation (Figure 4c). At 24 hours post-infection, the levels of LC3-II increased fivefold in VSV + obatoclax-treated cells when compared with non-treated cells and p62 decreased to undetectable levels, indicating the formation of autophagosomes and an increase in protein flux, respectively. Autophagy is regulated by a variety of cell signaling pathways including mTOR.44 Functionally, activated mTOR suppresses the initiation of the autophagic pathway. A threefold decrease in mTOR protein levels was observed in VSV + obatoclax-treated cells (Figure 4c). FOX03A, a transcriptional regulator implicated in tumorigenesis, stimulates autophagy when activated by dephosphorylation.45,46 FOX03A phosphorylation at serine residue 253 was reduced by threefold in the presence of VSV + obatoclax (Figure 4c). Similar to obatoclax + VSV combination treatment, LC3-I was proteolytically cleaved yielding LC3-II (twofold increase) and p62 was completely degraded in cells exposed to ABT-737 + VSV (Figure 4d). mTOR levels were reduced by threefold and serine 253 dephosphorylation of FOXO3A diminished by twofold with VSV + ABT-737 treatment (Figure 4d). Collectively, these results demonstrate the induction of autophagy in CLL cells treated with VSV + BCL-2 inhibitor.
Figure 4.
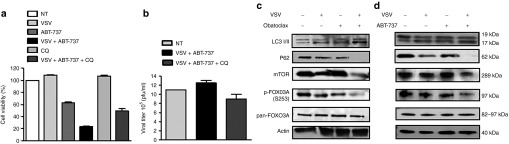
BCL-2 inhibitor + vesicular stomatitis virus (VSV) combination induces markers of autophagy in chronic lymphocytic leukemia (CLL) cells. (a) MTT assays showing sensitivity of CLL cells of a CLL patient to VSV (10 multiplicity of infection) and ABT-737 (5 nmol/l) combination treatment with or without 30 minutes pre-treatment with autophagy inhibitor chloroquine (CQ). Treatments are indicated above bars. (b) Viral titer was examined by plaque assay (mean ± SD). Light gray bars indicate VSV infection, black bars indicate VSV + ABT-737 treatment, and dark gray bars indicate VSV + ABT-737 + CQ treatment. (c,d) Twenty-four hours post-treatment, CLL cells were lysed, and LC3-II accrual and p62 degradation were analyzed by immunoblotting. LC3-I (19 kDa), LC3-II (17 kDa), p62 (62 kDa), mTOR (289 kDa), phospho-FOX03A (S243) (97 kDa), total FOX03A (82–97 kDA), and actin (40 kDa). mTOR, mechanistic target of rapamycin; NT, non-treated; pfu, plaque-forming unit.
It is well known that overexpression of prosurvival BCL-2 family members inhibits the induction of apoptosis and autophagy in cancer cells.11 Immunoblot analysis showed that BCL-2 was overexpressed in PBMCs isolated from a CLL patient, compared with a healthy volunteer; BCL-2 and MCL-1 were also overexpressed in human B-cell non-Hodgkin's lymphoma Karpas-422 cell line (Figure 5a). Karpas-422 cells display similar characteristics to primary CLL cells and are refractory to VSV oncolysis.25,26 It has been suggested that disruption of protein–protein interactions between antiapoptotic BCL-2 and proautophagic Beclin-1 could promote autophagic cell death.9,10,11 Co-immunoprecipitation experiments revealed that BCL-2/Beclin-1 and MCL-1/Beclin-1 heterodimer complexes were constitutively present in untreated Karpas-422 cells (Figure 5b, lane 1). VSV or obatoclax alone had minimal effect on heterodimer formation (Figure 5b, lanes 2, 3), but VSV + obatoclax almost completely abrogates BCL-2 and MCL-1 interactions with Beclin-1 (Figure 5b, lane 4). Collectively, these results suggest obatoclax disrupts BCL-2–MCL-1/Beclin-1 interactions, but only in the presence of VSV. Next, we immunoprecipitated BCL-2 and measured Beclin-1 displacement in cells treated with ABT-737 + VSV. Such treatment neutralized BCL-2/Beclin-1 inhibitory interactions allowing the release of Beclin-1 (Figure 5c, lane 4). These results suggest that ABT-737 in combination with VSV disrupt BCL-2/Beclin-1 interactions which likely facilitates induction of both apoptosis and autophagy. Based on the data above, both inhibitors, obatoclax and ABT-737, utilize a comparable mechanism to potentiate VSV-mediated oncolysis in CLL cells.
Figure 5.

Disruption of Beclin-1/BCL-2 and Beclin-1/MCL-1 interactions. (a) Antiapoptotic BCL-2, MCL-1, and BCL-XL as well as Beclin-1 levels were analyzed by immunoblotting in peripheral blood mononuclear cells isolated from a healthy donor, a chronic lymphocytic leukemia (CLL) patient, and in Karpas-422 cell line. Karpas-422 cells were treated with (b) obatoclax and vesicular stomatitis virus (VSV) or (c) ABT-737 and VSV for 24 hours. Cells were lysed in 1% CHAPS lysis buffer and BCL-2 and MCL-1 were immunoprecipitated followed by immunoblotting for Beclin-1. IB, immunoblotting; IP, immunoprecipitation.
Combination treatment induces Atg5-mediated cell death and autophagosome formation
To confirm the role of autophagy in VSV + ABT-737–mediated cell killing, mouse embryonic fibroblasts (MEFs) derived from wild-type (WT) and autophagy-deficient Atg5 knockout (KO) mice were treated with VSV and/or ABT-737 and cytotoxicity was measured by 3-(4,5-dimethylthiazol)-2,5-diphenyl tetrazolium (MTT) assay. Both WT and KO cell lines displayed complete resistance to ABT-737 (5 nmol/l) treatment after 24 hours (Figure 6a). VSV-mediated lysis is markedly decreased in Atg5 KO MEFs in comparison to WT cells. Cell viability decreased from 100 to 24% with VSV infection (0.01 multiplicity of infection) in WT MEFs compared with 67% in Atg5 KO cells 24 hours post-infection. Combining ABT-737 with VSV did not further decrease cell viability in WT MEFs compared with VSV alone (Figure 6a) (22% VSV alone and 21% VSV ± ABT-737); slightly better therapeutic effect for the combination was observed in the KO cell line. Viral titer was determined by plaque assay and a one log decrease was observed in Atg5 KO MEFs compared with Atg5 WT MEFs (Figure 6b). Thus, viral replication is augmented in the WT cell line (Figure 6b,c) and VSV oncolysis appears to be in part dependent on autophagy. Immunoblot studies revealed that LC3-II did not accumulate in Atg5−/− cells but was evident in Atg5+/+ cells with combination treatment (Figure 6c, top panel). These results were validated by immunofluorescence studies using green fluorescent protein (GFP)-LC3 as a label for autophagosome formation. Combination VSV + ABT-737 induced both GFP-LC3–positive autophagic vesicles and punctate foci in Atg5 WT MEFs (Figure 6d). As expected, vesicle formation and foci were not detected in Atg5 KO MEFs. Altogether these results imply that VSV + ABT-737–induced cell death is in part dependent on autophagy.
Figure 6.

VSV + ABT-737 combination treatment induces Atg5-mediated cell death. (a) Cell viability analysis was performed on wild-type (WT) mouse embryonic fibroblasts (MEFs) and Atg5 knockout (KO) MEFs treated for 24 hours with VSV (0.1 multiplicity of infection (MOI)) and ABT-737 (5 nmol/l) alone and in combination. Cells were treated or not with CQ (10 μmol/l) for 30 minutes before VSV + ABT-737 therapy. White bars indicate non-treated (NT) cells; light gray bars indicate VSV infection; dark gray bars indicate ABT-737 treatment; black bars indicate VSV + ABT-737 treatment; light blue bars indicate CQ treatment, and dark blue bars indicate VSV + ABT-737 + CQ treatment. (b) WT Atg5 KO MEFs were infected with VSV or ABT-737 alone and in combination followed by inhibition with CQ. Twenty-four hours after infection, the cells and culture supernatants were recovered, and the levels of viral titer were examined by the plaque assay. Viral titers were significantly lower in Atg5 KO MEFs. Light gray bars indicate VSV infection; black bars indicate VSV + ABT-737 treatment, and dark blue bars indicate VSV + ABT-737 + CQ treatment. (c) Western blot measured processing of LC3 and VSV replication in lysates after 24 hours of VSV infection (0.1 MOI). LC3-I (19 kDa) and LC3-II (17 kDa). Viron G, glycoprotein; M, matrix; N, nucleocapsid. (d) WT (top panel) and KO (bottom panel) Atg5 MEFs were transfected with LC3-GFP plasmid, treated with VSV ± ABT-737 and analyzed by immunofluorescence. 3-MA, 3-methyladenine (autophagy inhibitor). Green spots (foci) indicate LC3-GFP localized at autophagosomes. CQ, chloroquine; GFP, green fluorescent protein; pfu, plaque-forming unit; VSV, vesicular stomatitis virus.
Upregulation of apoptotic and autophagic markers in Karpas-422 cells exposed to combination treatment
Karpas-422 cells were examined for autophagy and apoptosis induction by Cyto-ID staining (selective label of autophagic vacuoles), p62 expression, and annexin V staining by flow cytometry analysis. Following VSV + ABT-737 combination, Cyto-ID+ cells increased from 13% in non-treated samples to 29% (Figure 7a, top panel). The corresponding p62 levels were reduced significantly from 93% in non-treated cells to 33% in cells treated with the combination (Figure 7a, middle panel), thus confirming stimulation of the autophagic pathway. Combination treatment also dramatically increased annexin V-positive staining from 7 to 62% in Karpas-422 cells (Figure 7a, bottom panel). The Pearson's correlation test revealed a strong association (R2 = 0.8698) between Cyto-ID staining and p62 expression (Figure 7b). We subsequently identified Cyto-ID+ and p62− cells as autophagic. To examine the relationship between the apoptotic and autophagic pathways, we measured early and late apoptosis markers, annexin V, and cleaved caspase 3, respectively, and mitochondrial membrane potential (ΔΨ) using 3,3′-dihexyloxacarbocyanine iodide (DIOC6(3)) in autophagic cells (Cyto-ID+ and p62−). (Figure 7c); DIOC6(3) is used to label living cells. As shown in Figure 7c, 5% of autophagic Karpas-422 cells stained positive for annexin V with ABT-737 single agent treatment and 2% with VSV infection; an increase to 40% was detected in the combination-treated cells. Similarly, cleaved caspase 3 staining increased from 5 and 1% to 46% in autophagic Karpas-422 cells. Loss of mitochondrial membrane potential (DIOC6(3) low) increased from 2% in non-treated cells to 44% with VSV + ABT-737 treatment in these cells (Figure 7c). Pre-treatment for 1 hour with the pan-caspase inhibitor zVAD-FMK (100 μmol/l) inhibited apoptosis in autophagic combination-treated cells (Figure 7c) reducing annexin V, cleaved caspase 3, and DIOC6(3) low levels from 40 to 4%, 46 to 1%, and 44 to 8%, respectively. Moreover, chemical inhibition of the autophagic pathway increased activation of the apoptotic pathway; when 3-methyladenine (3-MA) (10 mmol/l) was administered to combination-treated apoptotic cells, markers increased from 40 to 67% (annexin V), from 46 to 73% (caspase 3), and from 44 to 75% (DIOC6(3) low) (Figure 7c). These findings confirm that both apoptotic and autophagic pathways contribute to CLL cell death.
Figure 7.

ABT-737 + VSV treatment induces markers of autophagy. (a) Karpas-422 cells were infected with VSV (10 multiplicity of infection) and ABT-737 (150 nmol/l) alone or in combination; 18 hours after infection, the cells and culture supernatants were recovered and stained with Cyto-ID, p62, or annexin V antibody. (b) Pearson correlation was used to quantify the degree of association between p62 and Cyto-ID. (c) Annexin V, cleaved caspase 3, and DIOC6(3) low levels were monitored by flow cytometry. White bars indicate annexin V; dark gray bars indicate cleaved caspase, and black bars indicate DIOC6(3) low staining. zVAD (apoptosis inhibitor); 3-MA, 3-methyladenine (autophagy inhibitor); NT, non-treated. All experiments were performed in triplicate. VSV, vesicular stomatitis virus.
Discussion
CLL accounts for about one-third of all leukemias, with an estimated 16,060 new cases and 4,580 deaths in the United States alone in 2012 and no curative therapies are available.1,3,47,48,49 These statistics underline the unmet need for new alternative strategies to target CLL and improve survival. In the present manuscript, we demonstrate that the combination of VSV and BCL-2 inhibitors: (i) induce cell death in leukemic CLL cells from patients that had not responded to standard of care therapies; (ii) induce changes in expression of genes involved in the regulation of mTOR, apoptotic, and metabolic pathways; (iii) trigger both autophagy and apoptosis in CLL cells; (iv) enhance Atg5-dependent and Beclin-1–mediated cell killing; and (v) degrade mTOR and decrease FOX03A phosphorylation.
The ABT-737 + VSV combination therapy was also evaluated in a murine model of lymphoma (Figure 2a). Although modest effects were observed with single agent–ABT-737 or VSV–treatments, therapeutic benefit and prolonged survival was observed in combination-treated animals without overt toxicity, indicating that such therapeutic combination selectively kills tumor cells in vivo. Microarray analysis was performed to gain insight into the mechanism(s) responsible for therapeutic efficacy and selectivity of VSV + BCL-2 inhibitor combination. Changes in transcription profiles and clustering of such changes into pathways revealed differential regulation of mTOR, apoptotic, and metabolic pathways as well as in genes involved in protein translation, oxidative stress, and prosurvival pathways. Activation of IFN-responsive antiviral pathways were not observed in our array analysis; such pathways are significantly diminished or aberrant in many cancers including in CLL42 which could explain the lack of antiviral genes observed in the microarray following VSV infection. Interestingly, only a handful of genes in CD19+ CD5− (non-leukemic) cell population exposed to VSV, obatoclax or the combination had altered gene expression, indicating that healthy cells do not activate similar stress-response mechanisms in response to such therapies (data not shown).
BCL-2 has a dual function to regulate both apoptosis and autophagy.10,17,18,19 Antiapoptotic BCL-2 members have been shown to inhibit Beclin-1–induced autophagy. Executioner caspases can cleave Beclin-1 and Atg5 inactivating the autophagic pathway.50 Conversely Beclin-1 can inhibit activation of caspases and BID protein. Consistent with these conclusions, we observed a strong association between apoptosis and autophagy: inhibition of apoptosis with zVAD completely abrogated VSV + ABT-737–induced apoptosis in autophagic (Cyto-ID+, p62−) Karpas-422 cells (Figure 7c), and inhibition of autophagy with 3-MA enhanced the apoptotic effect of the combination-induced apoptosis. Unfortunately, little is known about the role of autophagy in VSV-induced lysis of cancer cells. Here, we demonstrate a major role of autophagy in VSV-mediated CLL cell death. Because of its dual role in inhibiting both apoptosis and autophagy, BCL-2 represents an attractive target for development of therapies that will stimulate both pathways.
We propose a mechanism in which CLL cells exposed to VSV undergo metabolic and oxidative stress that leads to changes in protein translation and cellular metabolism. Such changes alone do not activate autophagy, probably because of the tight association between Beclin-1 and BCL-2. However, upon addition of BCL-2 inhibitors, VSV-induced cellular stress initiates the autophagic pathway, in part because Beclin-1 is available to participate in the scaffolding and initiation of autophagosome formation. These data are further corroborated by co-immunoprecipitation studies demonstrating that inhibition of the autophagic pathway in CLL is due to overexpression of prosurvival BCL-2 family members. Disruption of the BCL-2/Beclin-1 complex indicates that autophagy is inhibited in CLL cells and that activation of the autophagic pathway by combination treatment promotes tumor killing. In conclusion, we show for the first time that oncolytic VSV induces cell death in CLL cells via activation of both autophagic and apoptotic signaling and demonstrate a mechanism of cell death that is in part dependent on autophagy. Due to the dose-limiting cytotoxity of BCL-2 agonists as single agents, low-dose combination with VSV provides an alternative strategy that may have a greater safety profile for the treatment of CLL and other malignancies that could be applied in clinics.
Materials and Methods
Patients and PBMC isolation. PBMCs were obtained from healthy individuals and CLL patients at the Robert & Carol Weissman Cancer Center at Martin Memorial Health Systems (Stuart, FL), the Vaccine & Gene Therapy Institute of Florida (Port St. Lucie, FL), and the Jewish General Hospital (Montreal, Quebec, Canada) following written informed consent, in agreement with the Vaccine & Gene Therapy Institute of Florida, Jewish General Hospital, and McGill University Research Ethics Committee. The absolute lymphocyte counts were typical of CLL patients in general. PBMCs were isolated as previously described.25,26 PBMCs were cultured in RPMI 1640 supplemented with 15% heat-inactivated fetal bovine serum (Wisent, St-Bruno, Quebec, Canada) and 100 U/ml penicillin–streptomycin. PBMCs were cultured at 37 °C in a humidified, 5% CO2 incubator.
Cell lines. The human B-lymphoma cell line Karpas-422 was purchased from the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany) and the A20 mouse B-lymphoma cell line was purchased from ATCC (Manassas, VA). Atg5 WT (Atg5+/+) and Atg5 KO (Atg5−/−) MEFs were obtained from Dr Nathalie Grandvaux, CRCHUM-Centre Hospitalier de l'Université de Montréal, Montréal, Quebec, Canada, with the permission of Dr Noboru Mizushima, Tokyo Medical and Dental University, Tokyo, Japan, who generated the original Atg5 WT and KO MEFs. For all experiments early passage primary MEFs were used. Karpas-422 and A20 cell lines were grown in RPMI 1640 medium (Wisent) supplemented with 10% fetal calf serum, penicillin, and streptomycin. Atg5 WT and KO MEFs were cultured in Dulbecco's modified Eagle's medium medium (Wisent) supplemented with 10% fetal calf serum, penicillin, and streptomycin. All cell lines were maintained at 37 °C and 5% CO2.
Virus production, quantification, and infection. Construction of VSV was previously described.21 Virus stock was grown in Vero cells (purchased from ATCC, Bethesda, MD), concentrated from cell-free supernatants by centrifugation (15,000 rpm/4 °C/90 minutes) and titrated in duplicate by standard plaque assay as previously described.26 Primary PBMC isolates and Karpas-422 cells were infected with VSV at a multiplicity of infection of 10 plaque-forming units/cell for 1 hour in serum-free media at 37 °C. The cells were then incubated with complete medium at 37 °C for the indicated times.
Viability assay. Cell viability was assessed by MTT dye absorbance according to the manufacturer's instructions (Chemicon, Billerica, MA) as previously described.25 For drug combination studies, cells were incubated with or without obatoclax (100 nmol/l) or ABT-737 (150 nmol/l, Karpas-422, 5 nmol/l PBMCs) and infected or not with VSV (10 multiplicity of infection) as indicated. Plates were incubated at 37 °C, 5% CO2 and cells were analyzed every 24 hours for 3 days. Each experimental condition was performed in triplicate. Cell viability was also analyzed by annexin V, cleaved caspase 3, and DIOC6(3) staining by flow cytometry analysis.
Protein extraction and western blot analysis. Cells were washed twice with ice-cold phosphate-buffered saline, and proteins were extracted as described previously.25,26 Membranes were blocked for 1 hour in 5% non-fat dried milk in TBST (Tris-buffered saline + 0.5% Tween-20). Followed by incubation with any of the following primary antibodies: mTOR, FOX03A-S256, and Beclin-1 (Cell Signaling Technologies, Danvers, MA; 1:2,000); β-actin (Millipore); LC3 (Novus Biologicals, Oakville, ON); p62 (Santa Cruz Biotechnology, Santa Cruz, CA; 1:2,000); BCL-2 (Santa Cruz Biotechnology; 1:2,000); BAX (Santa Cruz Biotechnology; 1 μg/ml); and NOXA (Calbiochem, San Diego, CA; 1 μg/ml). For LC3 protein extraction, cell pellets were lysed in ice-cold buffer containing 20 mmol/l Tris, 400 mmol/l NaCl, 1 mmol/l EDTA, 1% Triton-X, 30 mmol/l NaF, 40 mmol/l β-glycerophosphate, 10 mmol/l Na3VO4, 0.1 mmol/l PMSF, 1 mmol/l DTT, and protease inhibitor cocktail (Sigma-Aldrich, St Louis, MO) in 1/1,000 dilution. Extracts were vortexed for 30 minutes to 1 hour at 4 °C and centrifuged at 10,000g for 10 minutes (4 °C); supernatants were stored at −80 °C. Protein concentration was determined with Bio-Rad protein assay reagent (Bio-Rad, Hercules, CA). Protein extracts were resolved using mini-PROTEAN TGX precast gels (Bio-Rad) and transferred to polyvinylidene difluoride membrane (Bio-Rad).
In vivo murine lymphoma model. This study was approved by the local animal care and institutional animal ethics committee of Jewish General Hospital and McGill University. A total of seven animals per treatment group were used for this study, 4–6 weeks old female nu/nu mice (Charles River Laboratories, Pointe Claire, Quebec, Canada) were injected subcutaneously with 1 × 106 A20 cells in a 100-μl volume into the hind flanks. Tumor volumes were measured and calculated as ½ (length × width2). Once tumors were palpable, animals were randomly assigned to treatment groups and received 10 intraperitoneal injections of ABT-737 (75 mg/day/kg). ABT-737 was dissolved in 30% propylene glycol, 5% Tween 80, 65% D5W (5% dextrose in water), pH 4–5. At days 2 and 5 following ABT-737 injection, VSV was inoculated intratumorally at 5 × 107 plaque-forming units of virus each. Animals were evaluated for signs of stress such as infection, dehydration, weight loss (>20%), and limb paralysis.
Co-immunoprecipitation of BCL-2 family proteins. Co-immunoprecipitation studies were performed as previously described.25 Membranes were blocked for 1 hour in 5% non-fat dried milk in TBST followed by incubation with the following primary antibodies: BCL-2, MCL-1 (Santa Cruz Biotechnology; 1:2,000), and Beclin-1 (Cell Signaling Technologies; 1:2,000).
Autophagy analysis by flow cytometry. Autophagy was also analyzed by Cyto-ID and p62 staining by flow cytomtery. Cyto-ID was used according to the manufacturer's instructions (Enzo Life Sciences, Farmingdale, NY). p62 (Santa Cruz Biotechnology) was labeled with Alexa Fluor 647 using the Zenon Alexa Fluor 647 Mouse IgG1 Labeling Kit (Invitrogen, Carlsbad, CA).
Immunofluorescence. Atg5 WT and KO MEFs grown on glass coverslips were transfected with constructs encoding GFP and LC3-GFP using JETPRIME (Polyplus-transfection, Illkirch, France) according to the manufacturer's instructions. Following transfection, cells were treated or not with ABT-737 and infected with VSV. Six hours post-infection, cover slips were washed two times in phosphate-buffered saline and fixed in phosphate-buffered saline containing 4% formaldehyde for 12 minutes. Cover slips were then washed three times in phosphate-buffered saline and mounted on glass slides with Shandon Immuno-Mount (Thermo Scientific, Ottawa, Ontario, Canada), and left to dry overnight at room temperature in the dark.
Statistical analysis. Graphics and statistical analysis were executed using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA). Differences among the treatment groups were analyzed by paired t-test. The P values <0.05 were considered statistically significant. Average values were expressed as mean ± SD.
Microarray analyses. CLL cells from both non-responders and non-treated patients were treated with VSV, obatoclax or combination and total RNA was isolated from samples (n = 3 per treatment group) using RNeasy Micro Kits (Qiagen, Valencia, CA). The quantity and quality of the RNA was confirmed using a NanoDrop 2000c (Thermo Fisher, Montreal, Quebec, Canada) and an Experion Electrophoresis System. Samples (50 ng) were then amplified using Illumina TotalPrep RNA amplification kits (Ambion, Austin, TX). The microarray analysis was conducted using 750 ng of biotinylated complementary RNA hybridized to HumanHT-12_V4 BeadChips (Illumina, San Diego, CA) at 58 °C for 20 hours. The arrays were scanned using Illumina's iSCAN and quantified using Genome Studio (Illumina). The analysis of the GenomeStudio output data was conducted using the R and Bioconductor software packages. Quantile normalization was applied, followed by a log 2 transformation. The LIMMA package was used to fit a linear model to each probe and perform (moderated) t-tests or F-tests on the groups being compared. To control the expected proportions of false positives, the false discovery rate for each unadjusted P value was calculated using the Benjamini and Hochberg method implemented in LIMMA. Multidimensional scaling was used as a dimensionality reduction method in R to generate plots for the evaluation of similarities or dissimilarities between datasets. Ingenuity Pathway Analysis software (IPA; Ingenuity Systems, Redwood City, CA) was used to annotate genes and rank canonical pathways. Canonical pathway analysis identified the pathways from the IPA library of pathways that were most significant to the data set. Molecules from the data set that met the cut-off of 1.3 and were associated with a canonical pathway in the Ingenuity Knowledge Base were considered for the analysis. The significance of the association between the data set and the canonical pathway was measured by determining the ratio between the number of molecules from the data set that map to the pathway divided by the total number of molecules that map to the canonical pathway is displayed. Fisher's exact test was used to calculate a P value determining the probability that the association between the genes in the dataset and the canonical pathway is explained by chance alone.
Acknowledgments
We thank GeminX for the generous gift of Obatoclax. We also thank AbbVie Laboratories for providing the ABT-737 compound. This work was supported by grants from the Canadian Institutes of Health Research (MOP 42562). Terry Fox Foundation with the support of the Canadian Cancer Society, and the Department of Defense grant LC110658 to J.H. The authors declared no conflict of interest.
References
- Shanshal M, Haddad RY. Chronic lymphocytic leukemia. Dis Mon. 2012;58:153–167. doi: 10.1016/j.disamonth.2012.01.009. [DOI] [PubMed] [Google Scholar]
- Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med. 2005;352:804–815. doi: 10.1056/NEJMra041720. [DOI] [PubMed] [Google Scholar]
- Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, International Workshop on Chronic Lymphocytic Leukemia et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111:5446–5456. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggins AG, Pepper CJ. The role of Bcl-2 family proteins in chronic lymphocytic leukaemia. Leuk Res. 2010;34:837–842. doi: 10.1016/j.leukres.2010.03.011. [DOI] [PubMed] [Google Scholar]
- García-Sáez AJ. The secrets of the Bcl-2 family. Cell Death Differ. 2012;19:1733–1740. doi: 10.1038/cdd.2012.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci. 2009;122 Pt 4:437–441. doi: 10.1242/jcs.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4:600–606. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattingre S, Levine B. Bcl-2 inhibition of autophagy: a new route to cancer. Cancer Res. 2006;66:2885–2888. doi: 10.1158/0008-5472.CAN-05-4412. [DOI] [PubMed] [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Sinha S, Levine B. The autophagy effector Beclin 1: a novel BH3-only protein. Oncogene. 2008;27 suppl. 1:S137–S148. doi: 10.1038/onc.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordy C, He YW. The crosstalk between autophagy and apoptosis: where does this lead. Protein Cell. 2012;3:17–27. doi: 10.1007/s13238-011-1127-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou W, Han J, Lu C, Goldstein LA, Rabinowich H. Autophagic degradation of active caspase-8: a crosstalk mechanism between autophagy and apoptosis. Autophagy. 2010;6:891–900. doi: 10.4161/auto.6.7.13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, Yang Y, Xing D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2011;278:403–413. doi: 10.1111/j.1742-4658.2010.07965.x. [DOI] [PubMed] [Google Scholar]
- Townsend KN, Hughson LR, Schlie K, Poon VI, Westerback A, Lum JJ. Autophagy inhibition in cancer therapy: metabolic considerations for antitumor immunity. Immunol Rev. 2012;249:176–194. doi: 10.1111/j.1600-065X.2012.01141.x. [DOI] [PubMed] [Google Scholar]
- Eager RM, Nemunaitis J. Clinical development directions in oncolytic viral therapy. Cancer Gene Ther. 2011;18:305–317. doi: 10.1038/cgt.2011.7. [DOI] [PubMed] [Google Scholar]
- Rowan K. Oncolytic viruses move forward in clinical trials. J Natl Cancer Inst. 2010;102:590–595. doi: 10.1093/jnci/djq165. [DOI] [PubMed] [Google Scholar]
- Breitbach CJ, Burke J, Jonker D, Stephenson J, Haas AR, Chow LQ, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477:99–102. doi: 10.1038/nature10358. [DOI] [PubMed] [Google Scholar]
- Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly OG, Errington-Mais F, Prestwich R, Harrington K, Pandha H, Vile R, et al. Recent clinical experience with oncolytic viruses. Curr Pharm Biotechnol. 2012;13:1834–1841. doi: 10.2174/138920112800958904. [DOI] [PubMed] [Google Scholar]
- Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–275. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- Lichty BD, Stojdl DF, Taylor RA, Miller L, Frenkel I, Atkins H, et al. Vesicular stomatitis virus: a potential therapeutic virus for the treatment of hematologic malignancy. Hum Gene Ther. 2004;15:821–831. doi: 10.1089/hum.2004.15.821. [DOI] [PubMed] [Google Scholar]
- Barber GN. Vesicular stomatitis virus as an oncolytic vector. Viral Immunol. 2004;17:516–527. doi: 10.1089/vim.2004.17.516. [DOI] [PubMed] [Google Scholar]
- Lichty BD, Power AT, Stojdl DF, Bell JC. Vesicular stomatitis virus: re-inventing the bullet. Trends Mol Med. 2004;10:210–216. doi: 10.1016/j.molmed.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Samuel S, Tumilasci VF, Oliere S, Nguyên TL, Shamy A, Bell J, et al. VSV oncolysis in combination with the BCL-2 inhibitor obatoclax overcomes apoptosis resistance in chronic lymphocytic leukemia. Mol Ther. 2010;18:2094–2103. doi: 10.1038/mt.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumilasci VF, Olière S, Nguyên TL, Shamy A, Bell J, Hiscott J. Targeting the apoptotic pathway with BCL-2 inhibitors sensitizes primary chronic lymphocytic leukemia cells to vesicular stomatitis virus-induced oncolysis. J Virol. 2008;82:8487–8499. doi: 10.1128/JVI.00851-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaddy DF, Lyles DS. Oncolytic vesicular stomatitis virus induces apoptosis via signaling through PKR, Fas, and Daxx. J Virol. 2007;81:2792–2804. doi: 10.1128/JVI.01760-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason KD, Khaw SL, Rayeroux KC, Chew E, Lee EF, Fairlie WD, et al. The BH3 mimetic compound, ABT-737, synergizes with a range of cytotoxic chemotherapy agents in chronic lymphocytic leukemia. Leukemia. 2009;23:2034–2041. doi: 10.1038/leu.2009.151. [DOI] [PubMed] [Google Scholar]
- High LM, Szymanska B, Wilczynska-Kalak U, Barber N, O'Brien R, Khaw SL, et al. The Bcl-2 homology domain 3 mimetic ABT-737 targets the apoptotic machinery in acute lymphoblastic leukemia resulting in synergistic in vitro and in vivo interactions with established drugs. Mol Pharmacol. 2010;77:483–494. doi: 10.1124/mol.109.060780. [DOI] [PubMed] [Google Scholar]
- Campàs C, Cosialls AM, Barragán M, Iglesias-Serret D, Santidrián AF, Coll-Mulet L, et al. Bcl-2 inhibitors induce apoptosis in chronic lymphocytic leukemia cells. Exp Hematol. 2006;34:1663–1669. doi: 10.1016/j.exphem.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Bajwa N, Liao C, Nikolovska-Coleska Z. Inhibitors of the anti-apoptotic Bcl-2 proteins: a patent review. Expert Opin Ther Pat. 2012;22:37–55. doi: 10.1517/13543776.2012.644274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR, et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci USA. 2007;104:19512–19517. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey R, Chander R, Sainis KB. Prodigiosins as anti cancer agents: living upto their name. Curr Pharm Des. 2009;15:732–741. doi: 10.2174/138161209787582192. [DOI] [PubMed] [Google Scholar]
- Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. 2011;29:909–916. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik PK, Rudin CM, Brown A, Rizvi NA, Takebe N, Travis W, et al. A phase I study of obatoclax mesylate, a Bcl-2 antagonist, plus topotecan in solid tumor malignancies. Cancer Chemother Pharmacol. 2010;66:1079–1085. doi: 10.1007/s00280-010-1265-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30:488–496. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer P, Hallek M. Prognostic factors in chronic lymphocytic leukemia-what do we need to know. Nat Rev Clin Oncol. 2011;8:38–47. doi: 10.1038/nrclinonc.2010.167. [DOI] [PubMed] [Google Scholar]
- Zaha VG, Young LH. AMP-activated protein kinase regulation and biological actions in the heart. Circ Res. 2012;111:800–814. doi: 10.1161/CIRCRESAHA.111.255505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- Tomic J, Lichty B, Spaner DE. Aberrant interferon-signaling is associated with aggressive chronic lymphocytic leukemia. Blood. 2011;117:2668–2680. doi: 10.1182/blood-2010-05-285999. [DOI] [PubMed] [Google Scholar]
- Wong LH, Krauer KG, Hatzinisiriou I, Estcourt MJ, Hersey P, Tam ND, et al. Interferon-resistant human melanoma cells are deficient in ISGF3 components, STAT1, STAT2, and p48-ISGF3gamma. J Biol Chem. 1997;272:28779–28785. doi: 10.1074/jbc.272.45.28779. [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiacchiera F, Simone C. Inhibition of p38alpha unveils an AMPK-FoxO3A axis linking autophagy to cancer-specific metabolism. Autophagy. 2009;5:1030–1033. doi: 10.4161/auto.5.7.9252. [DOI] [PubMed] [Google Scholar]
- Chiacchiera F, Simone C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle. 2010;9:1091–1096. doi: 10.4161/cc.9.6.11035. [DOI] [PubMed] [Google Scholar]
- Hallek M, Kuhn-Hallek I, Emmerich B. Prognostic factors in chronic lymphocytic leukemia. Leukemia. 1997;11 suppl. 2:S4–13. [PubMed] [Google Scholar]
- Hallek M, German CLL Study Group Prognostic factors in chronic lymphocytic leukemia. Ann Oncol. 2008;19 suppl. 4:iv51–iv53. doi: 10.1093/annonc/mdn196. [DOI] [PubMed] [Google Scholar]
- Eichhorst B, Dreyling M, Robak T, Montserrat E, Hallek M, ESMO Guidelines Working Group Chronic lymphocytic leukemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2011;22 suppl. 6:vi50–vi54. doi: 10.1093/annonc/mdr377. [DOI] [PubMed] [Google Scholar]
- Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]