

Abstract
Most models for tauopathy use a mutated form of the Tau gene, MAPT, that is found in frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17) and that leads to rapid neurofibrillary degeneration (NFD). Use of a wild-type (WT) form of human Tau protein to model the aggregation and associated neurodegenerative processes of Tau in the mouse brain has thus far been unsuccessful. In the present study, we generated an original “sporadic tauopathy-like” model in the rat hippocampus, encoding six Tau isoforms as found in humans, using lentiviral vectors (LVs) for the delivery of a human WT Tau. The overexpression of human WT Tau in pyramidal neurons resulted in NFD, the morphological characteristics and kinetics of which reflected the slow and sporadic neurodegenerative processes observed in sporadic tauopathies, unlike the rapid neurodegenerative processes leading to cell death and ghost tangles triggered by the FTDP-17 mutant Tau P301L. This new model highlights differences in the molecular and cellular mechanisms underlying the pathological processes induced by WT and mutant Tau and suggests that preference should be given to animal models using WT Tau in the quest to understand sporadic tauopathies.
Introduction
The abundant and abnormal accumulation of the hyperphosphorylated microtubule-associated protein Tau is a pathological feature of the neurodegenerative diseases collectively referred to as tauopathies.1 Tau is encoded by a single gene, MAPT, whose mRNA is alternatively spliced to generate the six isoforms expressed in the adult human brain2 as well as in the rat brain.3 In rare tauopathies called FTDP-17 (frontotemporal dementia with Parkinsonism linked to chromosome 17), certain mutations are found in MAPT that are responsible for a neurodegenerative process resulting from the aggregation of mutated Tau. In most tauopathies, non-mutated Tau proteins (wild-type (WT) Tau) aggregate into filaments for ill-defined reasons: changes in post-translational modifications, aberrant proteolysis or the ratio of Tau isoforms.1 From a pathological point of view, Tau proteins accumulate in inclusions of various types in neurons and glia,4 with the most common neuronal lesions being neurofibrillary tangles, in which Tau aggregates into filaments. This neurofibrillary degeneration (NFD) is a slow process that lasts more than 20 years5 and progresses from the pretangle to the ghost tangle stage in tauopathies such as Alzheimer's disease (AD),6 the most common sporadic tauopathy.
WT Tau-associated NFD is poorly represented by the currently available models, most of which are based on the expression of mutated Tau proteins in transgenic animals.7 Only a few models of WT Tau-associated pathology are available. These models exhibit pretangle stages without the development of NFD or cognitive deficits.8,9,10,11 The mouse model engineered by Davies et al. expresses the six human isoforms of Tau in mice in which MAPT has been knocked out. To our knowledge, this is the only transgenic mouse line whose pathological features include the aggregation of hyperphosphorylated Tau to form pathological paired helical filaments.12 A rat transgenic model of tauopathy in which a truncated, rather than full-length, form of WT Tau promotes neurofibrillary tangle formation in the brain is now available.13,14,15
Tau proteins bearing FTDP-17 mutations can directly promote Tau filament formation at different stages of the aggregation pathway.16 Most NFD models that use such mutations have shown that the expression of a mutated gene in the brain leads to a neuropathological phenotype including Tau phosphorylation, aggregation, and neuronal death.7 The P301L and G272V mutations, which increase the rates of both filament nucleation and extension reactions,16 have been extensively used to model neurodegeneration in rodents by gene transfer (transgenesis and viral vectors).17,18,19,20,21,22,23,24,25,26,27
A primary concern with models using mutated/truncated Tau is their relevance, considering that the time-course of Tau aggregation and neuronal death is not comparable to that of classic sporadic tauopathies (e.g., AD, argyrophilic grain disease, corticobasal degeneration, progressive supranuclear palsy, and Pick's disease). Functional differences between the WT and mutated forms of Tau drive differences in the molecular and cellular mechanisms of these models26,28,29 and human tauopathies.16,30,31 For example, in models with FTDP-17 mutations, neurons undergo NFD within a few weeks and rapidly die,21 whereas in most tauopathies, NFD is a very slow process,5 suggesting a difference in the underlying mechanisms of aggregation. Researchers now focus on the development of relevant new models of sporadic tauopathies to study the specific mechanisms regulating their pathology. Transgenic models are useful tools to dissect the pathogenic mechanisms of disease, but in the context of our study, these models suffer from some limitations: (i) despite the use of neuronal promoters, the expression of a transgene driven by a neural promoter is still detectable in large brain regions. Although progress has been made to control gene expression by using ‘tet-off' systems and/or promoters with more restricted patterns of expression, such as the neuropsin promoter, these are also often leaky;32 (ii) most transgenic animals fail to model disease progression steps from pretangles to ghost tangles.
The first step of a pathological process should be initiated after the local delivery, in a specific brain area, of pathological proteins. In this context, the emergence of efficient viral vectors for central nervous system gene transfer offers an approach that bypasses this limitation, and among these vectors, the adeno-associated viral vectors (AAVs)33 and lentiviral vectors (LVs)34 have been previously validated for gene transfer in mouse and primate brains. These vectors are also the most promising candidates for our study, as LVs were recently used to model tauopathies.22,26 Here, we selected LVs rather than AAVs to produce moderate, local, and restricted Tau expression in the pyramidal neurons of CA1, which is a region that is rapidly affected in the most common tauopathy, AD. AAVs could also be used to specifically target neurons; however, our aim was to initiate Tau overexpression in a few neurons and then to observe the aggregation process. When injected into the brain, AAVs show more diffusive infection than LVs, and when overexpressing a WT form of Tau, they induce rapid and dramatic neuronal death.26 The most common pleiotropic envelope protein used in this study is the vesicular stomatitis virus G-protein. This envelope protein meets key criteria: first, it allows for the concentration of the LVs, which is essential for injections into the central nervous system, where only very small volumes can be injected; second, the combination of a vesicular stomatitis virus G-protein envelope with ubiquitous promoters such as phosphoglycerate kinase greatly improves neuronal expression.35
Vesicular stomatitis virus G-protein–pseudotyped LVs were used in the present study to express the four-repeat human WT Tau isoform 2+3−10+ in the hippocampal formation, a key brain region that is prematurely affected by the neurodegenerative processes of AD.36 As expected, a neurodegenerative process mediated by WT Tau is rapidly initiated as soon as 2 months after LV delivery but progresses slowly, thus closely mimicking the steps of sporadic-like tauopathy.
Results
LV-mediated Tau expression in the pyramidal neurons of the rat hippocampus
The ability of our LVs to mediate Tau expression in the pyramidal neurons of the hippocampal CA1 layer was assessed (Figure 1). Phosphate-buffered saline (PBS), LVs encoding enhanced green fluorescent protein (GFP), a WT form of Tau (2+3−10+; hTau46WT) or its mutated counterpart (hTau46P301L), were injected into CA1 of Wistar rats. The well-characterized Tau mutant P301L was also included in this assay as a positive control, as this mutant shows rapid Tau aggregation in viral-mediated and transgenic mouse models.21,27 Fifteen days later, the expression of total Tau was assessed by immunohistochemistry (IHC) using an antibody directed against the N-terminal portion of human Tau (ADx215). We controlled the viral titers of the two batches of LV used in this study such that there was no difference between them (Supplementary Figure S1). Immunohistochemical analyses (Figure 1a–f) indicated that WT and P301L Tau were specifically expressed in the hippocampus at similar levels (Figure 1g, n = 3), and no labeling was detected after PBS (Figure 1a) or LV-enhanced GFP injection (Figure 1d). Tau is mainly found in NeuN-positive cells, i.e., neurons (Figure 1h,j), whereas no colocalization was observed with the astrocytic marker glial fibrillary acidic protein (Figure 1i,k). Moreover, no gliosis was observed under any of the conditions used.
Figure 1.

Lentiviral vectors (LVs) mediate Tau expression in the pyramidal neurons of the rat brain hippocampus. (a–f) Fifteen days post-PBS, LV-eGFP, LV-hTau46WT or LV-hTau46P301L delivery, animals were killed, and their brains were processed for immunohistochemistry using the ADx215 primary antibody. Cresyl violet staining was performed to identify neurons. Bars represent 300 μm (in a,b,d, and e) or 80 μm (in c,f). (g) ADx215 immunoreactivity was quantified using Mercator software and expressed as a percentage of the immunoreactive surface relative to the entire quantified surface. The expression of various forms of Tau (visualized in red and immunodetected using antibodies against the N-terminal portion of Tau (M19G)) was also analyzed by confocal microscopy in 1 μm-thick sections 2 months post-injection of (h) PBS, (i) hTau46WT, (k) eGFP or (l) hTau46P301L. (j,m) Neurons were localized using antibodies against the neuronal marker NeuN (visualized in green). Astrocytes were localized using antibodies against the astrocytic marker GFAP (visualized in green), and Tau was detected as described above. In h–m, nuclei are labeled with DAPI and visualized in blue, and the bars represent 18 μm. Viral titers are available in the Supplementary Figure S1. DAPI, 4′,6-diamidino-2-phenylindole; eGFP, enhanced green fluorescent protein; GFAP, glial fibrillary acidic protein; NS, not significant; PBS, phosphate-buffered saline.
Overexpression of hTau46WT in pyramidal neurons of the hippocampal CA1 layer triggers a slow and progressive “sporadic-like” tauopathy
Immunological tools are available to distinguish among the different biochemical forms of Tau that characterize these pathological stages: the antibody AT8 labels hyperphosphorylated Tau,37 whereas MC1 labels conformational changes38 and AT100 labels aggregated Tau.39 Rats were stereotactically injected with LV-hTau46WT (n = 5), LV-hTau46P301L (n = 5), LV-GFP (n = 3) or PBS (n = 3) into the hippocampus. Immunoreactivities to pTau were studied 2, 4, and 8 months post-injection (p.i.) with various pTau antibodies (AT8, MC1, and AT100) for LV-hTau46WT and LV-hTau46P301L (Figures 2 and 3) and quantified (Figure 4). The absence of pTau immunoreactivity was visualized in control (PBS-injected) rats 2, 4, and 8 months p.i. by staining with the various pTau antibodies (AT8, MC1) (Figure 5a–f). The expression of GFP proteins 8 months after LV-GFP delivery (Figure 5i) did not affect phospho-Tau immunoreactivity (Figure 5g,h). AT8 immunoreactivity progresses in a time-dependent manner for both forms of Tau; however, AT8 immunoreactivity extends from the CA1 to the cortex in rats injected with LV-hTau46WT, whereas it is restricted to the hippocampal formation in rats injected with LV-hTau46P301L, even 8 months p.i. (Figure 6). In LV-hTau46P301L–injected rats, as expected, neuroanatomical modifications appeared rapidly, and tangle-like structures were observed with all antibodies as soon as 4 months p.i. (Figure 2, black arrowheads). Conversely, 2 months following LV-hTau46WT injection, in addition to total Tau immunoreactivity (Figure 1), pTau immunoreactivity (AT8, MC1, AT100) was observed (Figure 3). AT8 and MC1 immunoreactivity was seen in most brains (five out of five and four out of five, respectively). MC1 immunoreactivity was mainly restricted to dystrophic neurites (Figure 3d, black arrows). Finally, a few AT100-immunoreactive neuritic structures were found in one rat brain (Figure 3g). From 4 to 8 months p.i., “tangle-like” structures were observed with AT8 and MC1 antibodies (Figure 3b,c,e,f). AT100-immunoreactive neurites were found in all rat brains (Figure 3h,i, black arrows), and tangle-like neuronal structures were observed only in one rat brain (Figure 7c, upper right panel, black arrowhead).
Figure 2.

Rapid neurodegenerative Tau pathology in CA1 pyramidal neurons of rat brains injected with lentiviral vectors encoding hTau46P301L. Two, 4, and 8 months post-injection, animals were killed, and their brains were processed for immunohistochemistry using the following primary antibodies: (a–c) AT8, (d–f) MC1, and (g–i) AT100. Cresyl violet staining was performed to identify neurons. Black arrowheads indicate pretangles and tangle-like structures; black arrows indicate degenerating neuritis. Bars represent 20 μm.
Figure 3.

Slow and progressive neurodegenerative Tau pathology in CA1 pyramidal neurons of rat brains injected with lentiviral vectors encoding hTau46WT. Two, 4, and 8 months post-injection, animals were killed, and their brains were processed for immunohistochemistry using the following primary antibodies: (a–c) AT8, (d–f) MC1, and (g–i) AT100. Cresyl violet staining was performed to identify neurons. Black arrowheads indicate pretangles and tangle-like structures; black arrows indicate degenerating neurites. Bars represent 20 μm.
Figure 4.

Quantitative analyses of Tau immunoreactivity in the hippocampus. Two, 4, and 8 months post-injection of lentiviral vectorss encoding hTau46WT or hTau46P301L, animals were killed, and their brains were processed for immunohistochemistry using (a) AT8, (b) MC1, and (c) AT100 primary antibodies. Quantitative analyses were then performed using Mercator software, and the results are expressed as antibody immunoreactivity (immunopositive area/whole area analyzed). The P values are shown on the figure.
Figure 5.

Lack of phospho-Tau immunoreactivity in the CA1 pyramidal neurons of rat brains injected with phosphate-buffered saline (PBS) or lentiviral vectors (LVs) encoding green fluorescent protein (GFP). Two, 4, and 8 months post-PBS delivery, animals were killed, and their brains were processed for immunohistochemistry using the following primary antibodies: (a–c) AT8 and (d–f) MC1. Eight months after LV-GFP delivery, the animals were killed, and their brains were processed for immunohistochemistry using the following primary antibodies: (g) AT8 or (h) MC1. (i) GFP expression was controlled by direct fluorescence. Cresyl violet staining was performed to identify neurons. Bars represent 40 μm.
Figure 6.

Spatio-temporal progression of Tau pathology. Two, 4, and 8 months post-injection, animals were killed, and their brains were processed for immunohistochemistry using the AT8 primary antibody. Cresyl violet staining was performed to identify neurons. The injection site is indicated by a black cross. Bars represent 100 μm. The P values in comparison to 2 months post-injection are shown on the histograms.
Figure 7.

From “strings of beads” to ghost tangle neurons in the hippocampal formation. Two (left panels), 4 (middle panels), and 8 months (right panels) post-injection, animals were killed, and their brains were processed for immunohistochemistry with (a) AT8, (b) MC1 or (c) AT100. Cresyl violet staining was performed to identify neurons. Bars are indicated on the photomicrographs. The dendrosomatic localization of Tau can be seen to shift from the neurites, where it aggregates as bead-like structures (black arrows), to the somatic compartment in tangle neurons (black arrowheads).
These observations appear to be specific to rats, as 8 months after the injection of LV-hTau46WT into mouse brains MC1 immunoreactivity was sparse and restricted to neurites, and no AT100-immunoreactive neurons were detected (Supplementary Figures S2 and S3). Together, these results show that the overexpression of hTau46WT in the pyramidal neurons of the rat brain drives a slower and less intense process of Tau aggregation than in the P301L mutant (Figure 4). This slow process is highly similar to the progressive Tau pathology observed in sporadic tauopathies.
From “strings of beads” to ghost tangles in the hippocampal formation
An early pathological hallmark of tauopathies is the abnormal sorting of hyperphosphorylated Tau, which results in its accumulation in the somatic compartment, giving rise to insoluble fibrillar aggregates.40 Small varicosities, which we refer to here as “strings of beads”, are now considered to be early changes in the NFD process.41 We evaluated these time-dependent pathological changes in our model using the AT8 (Figure 7a), MC1 (Figure 7b), and AT100 (Figure 7c) antibodies.
Several neurites with “strings of beads” labeling were observed after the overexpression of WT Tau (Figure 7a,b, upper panels, black arrows). The disappearance of these bead-like structures, mainly present 2 months p.i., seems to be correlated with an increased number of pretangle neurons at 4 (Figure 7a,b, upper middle panels, black arrowheads) and 8 months p.i. (Figure 7, upper right panels, black arrowheads) but also with the dendrosomatic relocalization of Tau. AT100-immunopositive structures were mainly composed of neurites, whereas AT100-positive ghost tangles were observed in only one rat brain (Figure 7c, upper right panel, black arrowhead). The appearance of tangles was concurrent with the shrinkage of CA1/2 in LV-injected rats, as measured between bregma −4.8 and bregma −6.3 (Rat Brain Atlas, Paxinos & Watson, 3rd Edition). Whereas neither PBS- nor LVs-GFP–injected rat brains presented CA1/2 shrinkage, such shrinkage was accelerated in both groups injected with Tau-expressing LVs. This shrinkage appeared to progress more slowly in the hTau46WT-injected rats than in the hTau46P301L-injected rats (Figure 8).
Figure 8.

Neuronal damage in lentiviral vector-injected rat brains. Rats injected with PBS (n = 3), hTau46WT (n = 5, P = 0.06, when compared with the PBS group), hTau46P301L (n = 5, P = 0.05, when compared with the PBS group) or eGFP (n = 3, NS when compared with the PBS group) were killed 8 months post-injection. The thickness of CA1/CA2 was measured in each rat from bregma −4.8 to bregma −6.3 (Rat Brain Atlas Paxinos & Watson, 3rd Edition). eGFP, enhanced green fluorescent protein; NS, not significant; PBS, phosphate-buffered saline.
Taken together, these results clearly show that the overexpression of WT Tau led to the development of NFD via a chronologically defined process composed of distinct steps from strings of beads to neuritic degeneration. The morphological characteristics of this process are similar to those mediated by the aggregation-promoting mutant P301L (Figures 2 and 7), but the kinetics of the process mediated by the P301L mutant are clearly different.
Tau insolubility and Tau aggregates
Differences in the aggregation process triggered by the WT and mutated forms of Tau were confirmed by biochemical fractionation (Figure 9a). Sarkosyl-soluble and sarkosyl-insoluble fractions were prepared from 1 mm-thick brain sections corresponding to the injection site 8 months following LVs injection. AT100 immunoreactivity revealed the presence of aggregated Tau in the sarkosyl-insoluble fraction of hTau46P301L-injected rats but not in that of hTau46WT-injected rats (Figure 9a). As WT Tau immunoreactivity in neurites was found to be weak with this antibody (Figure 3), WT Tau immunoreactivity may have been undetectable in the sarkosyl-insoluble fraction. We overcame this limitation by using electron microscopy. Abnormally phosphorylated Tau was first localized in the section by MC1 immunolabeling, and electron microscopy analyses showed that these structures contain fibrillar material (Figure 9b). These findings suggest that lentivirally expressed WT Tau is capable of forming toxic species in the rat brain 8 months p.i.
Figure 9.
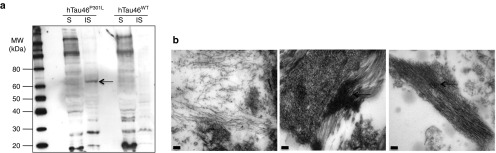
Biochemical and ultrastructural characterization of abnormally phosphorylated Tau species in the rat brain. (a) Rats injected either with hTau46WT or hTau46P301L were killed 8 months post-injection. A 1 mm-thick brain section corresponding to the injection site was fractionated to separate the sarkosyl-soluble (S) and sarkosyl-insoluble (IS) fractions, and AT100 immunoreactivity was analyzed by western blotting. (b) Rats injected with phosphate-buffered saline (left panel), hTau46WT (middle panel) or hTau46P301L (right panel) were killed 8 months post-injection and processed for electron microscopy. Pathological Tau aggregations in the form of fibrils (black arrows) were detected in MC1-positive brain structures in rats injected either with hTau46WT or hTau46P301L. Bars indicate 100 nm. MW, molecular weight.
Discussion
In the present study, we characterized a new model of sporadic tauopathy based on the local expression of the WT form of a four-repeat Tau (hTau46WT) in the adult rat hippocampus. We demonstrate that, in contrast to hTau46P301L, the overexpression of full-length hTau46WT in the hippocampal formation leads to a slow and progressive pathological process. The age-dependent degenerative process initiated by the WT form of Tau is present 2 months post-LV injection. This progressive NFD, which can be characterized by specific antibodies (Figures 2 and 3), is associated with cellular damage (Figure 8), probably resulting from the formation of toxic species in neurons overexpressing WT Tau (Figure 9b). A systematic comparison with a proaggregation control, the P301L Tau mutant, allows us to conclude that early pathological events are delayed with WT Tau; a slower progression of Tau pathology is seen, which may be similar to that of sporadic progressive tauopathies. These findings support the concept that the molecular and cellular mechanisms triggered by the two forms of Tau may be different.
These findings are in accordance with observations made in various tauopathies: in AD, a tauopathy associated with a non-mutated form of Tau, the NFD process is very slow; conversely, in most FTDP-17, the age of onset is lower, and the progression of pathological changes is faster.42 The mutations found in FTDP-17 mostly occur at the C-terminal region of MAPT that encodes microtubule-binding repeats, resulting in an alteration of the ability of Tau to bind microtubules and rapidly leading to protein aggregation. In our model, the overexpression of full-length hTau46WT in the hippocampal formation induces a slow and progressive pathological process, whereas the expression of the P301L mutation leads to a rapid and acute pathology mediated by Tau aggregation, as previously reported in vitro.16
Recent data from a direct comparison between the effects of WT Tau and P301L Tau on protein phosphorylation, kinase activity, and neuroinflammation also identified different processes.26 The injection of LVs encoding WT Tau and P301L Tau in the rat motor cortex, performed to mimic the advanced stages of human tauopathies, allowed for the development of NFD 4 weeks p.i. LVs expression of both forms has differential effects on the phosphorylation and aggregation of Tau in vivo, but both forms similarly alter kinase and phosphatase activities, and both forms mediate microglial activation and the levels of interleukin-6 and tumor necrosis factor-α in vivo. Here, we confirmed that the phosphorylation state of Tau is altered between both forms and that the conversion of pretangle to ghost tangle neurons is delayed with the WT form of Tau, suggesting that the early pathological events triggered by the two forms are different.
In a previous study, 3 weeks post-AAVs delivery in the ventral mouse hippocampus, the neurotoxicity mediated by the mutant P301L and by the WT Tau was found to be equivalent, with a dramatic and rapid acute neurodegeneration in CA1/2 without Tau aggregation.21 In this study, no correlation between any particular phosphoepitope and neurodegeneration was validated, but two mechanisms might be responsible for the rapid neurodegeneration: neuronal cell cycle re-entry and microgliosis.21 This innovative model could have been clearly informative for tauopathies that are caused by WT Tau 4R, but the acute gene overexpression and rapid-associated neuronal death limit the utility of this model to investigate the slow disease progression associated with some tauopathies, especially AD. The role of microgliosis in the early stages of the pathology has also been suggested in an aged model of rats that received AAVs-Tau 4R in the striatum.43 To our knowledge, these studies and ours are the first to model aggregation mediated by a WT Tau (4R) responsible for sporadic tauopathies. The direct comparison between a WT and a mutated form of Tau in vivo in these models would help us to further investigate the mechanisms leading to sporadic tauopathies.
There are several potential explanations for why attempts to induce a slow or sporadic process of NFD in mouse models have failed for many years. Differences between species may explain the NFD process mediated by the WT form of Tau in our rat model (Supplementary Figure S4). The mouse brain, in contrast to the rat brain,3 does not express the same ratio of 3R/4R isoforms found in humans.44,45 Indeed, a developmental difference exists in the regulation of Tau isoforms expression between human and mouse brains, with a progressive disappearance of the 3R isoforms between postnatal day 6 and postnatal day 90 in the adult mouse brain in contrast to the continued expression of 3R isoforms at this time point in the human brain.45 This hypothesis is supported by the mouse model of Davies et al., where the six isoforms of WT human Tau are expressed against a knockout background1,2 and in a transgenic rat model in which truncated WT Tau is overexpressed.15 In agreement with these findings, a recent study suggested that the deletion of the murine Tau gene increases human Tau aggregation in a transgenic mouse model of mutated human Tau.46 Here, we confirmed that the NFD mediated by WT Tau is initiated in the rat brain but not in the mouse brain (Supplementary Figures S2 and S3). In normal adult brains, 3R and 4R forms of Tau are equimolar, but their ratio undergoes a shift toward 4R forms in several tauopathies.28 This shift may lead to enhanced Tau aggregation and the development of fibrillar Tau pathology.47 In the present study, the overexpression of 4R forms of Tau, which proved to be capable of triggering “sporadic-like” pathological changes, strengthened this shift already present in normal rat brains. With the exception of FTDP-17 mutations in the vicinity of exonic/intronic regions, the mechanisms by which imbalances between the 3R and 4R isoforms cause or contribute to the development of Tau pathology and neurodegeneration remain unknown. Whereas in vitro data show that the coexpression of 3R and 4R isoforms seems to reduce Tau filament assembly and that the 3R Tau isoforms apparently inhibit 4R Tau assembly,48 other studies have shown a role for the 3R isoforms in an AD-like pathology.49 The disruption of the normal equimolar 3R to 4R ratio may be sufficient to drive Tau aggregation, and this shift may require 3R isoforms, even at low levels, that are absent in the adult mouse brain to initiate the 4R nucleation process, thereby leading to enhanced Tau aggregation and the development of Tau fibrillary pathology. Indeed, the tendency of 3R Tau to form oligomers and polymers, in contrast to 4R Tau, was shown in vitro many years ago by Mandelkow's group.50 The cysteine (C322) present in 3R Tau allows for the formation of intermolecular bridges, whereas the two cysteines (C291, C322) in 4R Tau mainly drive the formation of intramolecular bridges. These differences in the ability to form inter- or intra-disulfide bridges may explain why 3R isoforms are required to induce NFD.
In this article, we provide proof-of-principle for the use of lentiviral technology to induce, in the rat hippocampus, pyramidal aggregation that is mediated by a WT 4R form of Tau. This model offers an opportunity to obtain further insights into the specific molecular and cellular mechanisms implicated in sporadic tauopathies.
Materials and Methods
Antibodies. The following antibodies were used for IHC and biochemical assays at the dilutions indicated: monoclonal antibody (mAb) AT8, which recognizes the phosphoserine 202 and phosphothreonine 205 residues of Tau (MN1020; Thermo Scientific, Illkirch, France; 1/400 for IHC); mAb AT100, which recognizes the phosphothreonine 212 and phosphoserine 214 residues of Tau (MN1060; Thermo Scientific; 1/400 for IHC and 1/1,000 for biochemical assays); polyclonal rabbit Ab M19G,51,52 which recognizes the amino terminal region of Tau (raised in-house; 1/1,000 for IHC); polyclonal rabbit Ab Tau C-Ter,52 which recognizes the carboxyl terminal region of Tau (raised in-house; 1/400 for ICC); a mAb-recognizing neuron-specific nuclear protein or NeuN (MAB377; Millipore, Molsheim, France; 1/200 for IHC); and a mAb-recognizing glial fibrillary acidic protein (G3893; Sigma, Lyon, France; 1/100 for IHC). The mAb MC1 was a generous gift from Dr Peter Davis (1/100 for IHC). The mAb ADx215 (1/1,000 for IHC) is a new human-specific mAb raised against the GTYGLGDRK human sequence.
Cell culture. Cells of the human embryonic kidney cell line HEK293T were cultivated in Dulbecco's modified Eagle's medium with 10% fetal bovine serum and 1% penicillin/streptomycin. The cells were maintained in a humidified incubator with 5% CO2. All cell lines were passaged twice a week.
Plasmid construction. The packaging construct used was pCMVΔR8.92. To further decrease the risk of recombination and the generation of replication-competent retroviruses, the Rev gene was inserted into the pRSV-Rev plasmid. Viral particles were pseudotyped using the VSV-G encoded by the pMD.2G plasmid described previously.53 cDNAs encoding the isoform 2+3−10+ of human WT Tau (hTau46WT), the P301L mutant (hTau46P301L) or GFP were first cloned into the Gateway Entry pCR8/GW/TOPO vector (Invitrogen, St Aubin, France) using TOPO TA cloning methodology, as follows. The Gateway LR Clonase (Invitrogen) was used to catalyze the in vitro recombination between the Gateway Entry pCR8/GW/TOPO vector (containing the Tau cDNA flanked by attL sites) and the lentiviral destination vector (SIN-cPPT-PGK-Gateway-WPRE, containing homologous attR sites).
Production and assay of recombinant LVs. LVs were amplified as previously described.54 HEK293T cells (4 × 106) were plated on 10-cm plates and transfected the following day with 13 μg of hTau cDNA, 13 μg of pCMVΔR8.92, 3 μg of pRSV-Rev, and 3.75 μg of pMD.2G using the calcium phosphate DNA precipitation procedure. Four to six hours later, the medium was removed and replaced by fresh medium. Forty-eight hours later, the supernatant was collected and filtered. High-titer stocks were obtained by two successive ultracentrifugation steps at 19,000 rpm (SW 32Ti and SW 60Ti rotors; Beckman Coulter, Villepinte, France) at 4 °C. The pellet was resuspended in PBS with 1% bovine serum albumin and stored frozen at −80 °C until use.
Viral titer. Viral concentrations were determined using independent assays as follows: (i) The physical titer was quantified using ELISA for the HIV-1 p24 antigen (Gentaur BVBA, Paris, France). The p24 protein is a lentiviral capsid protein that is commonly used in ELISA assays to determine the physical titer of lentiviral batches per milliliter. The ability of LVs to induce the expression of Tau in vitro was tested by infecting 5 × 105 HEK293T cells with increasing amounts of viral vectors (200 and 400 ng of p24). Forty-eight hours later, cells were processed by western blotting (data not shown). (ii) The functional titer was determined in a limiting dilution assay to detect Tau expression by immunofluorescence. HEK293T cells (3 × 105 per well) were seeded onto six-well plates and transduced with increasing amounts (4,260 × 10−5 to 1 × 10−5 ng of p24 per cell) of LVs encoding hTau46WT or hTau46P301L in serum-free medium. Six hours later, complete medium was added to the well, and the cells were incubated for 48 hours at 37 °C. The cells were rinsed in PBS, fixed with 4% paraformaldehyde for 20 minutes at room temperature and rinsed twice with 50 mmol/l of NH4Cl. Subsequently, the cells were permeabilized with Triton X-100 (0.2%, 10 minutes at room temperature) and rinsed twice with 50 mmol/l of NH4Cl. The cells were then incubated with a polyclonal rabbit antibody (Tau C-Ter) that recognizes the carboxyl terminal region of Tau at 4 °C overnight, and labeling was completed the following day using a secondary goat anti-rabbit IgG conjugated to Alexa Fluor 488 (1/400; Invitrogen) for 45 minutes at room temperature. The cells were mounted with Vectashield medium containing DAPI (Vector Laboratories, Burlingame, CA). Confocal microscopy was carried out using a Zeiss LSM 710 inverted confocal microscope (Carl Zeiss, Le Pecq, France). For each optical section, double fluorescence images were obtained. The signal was treated by line averaging to integrate the signal collected over two or four lines to reduce noise. The confocal pinhole was adjusted to allow for a minimum depth of field. A focal series was collected for each specimen. The focal step between each section and the next was generally 1 μm. (iii) The functional titer was determined in a limiting dilution assay to detect the viral genome using specific primers. HEK293T cells were transduced as described in (ii), and DNA was extracted using the “Nucleospin Tissue” kit as recommended by the manufacturer (Macherey-Nagel, Hoerdt, France). DNA purity and concentrations were ascertained using a NanoDrop 1000 system (Thermo Scientific). Viral cDNAs were then amplified from 200 ng of total DNA using oligonucleotides (1 μmol/l) specific to the viral element WPRE (woodchuck-hepatitis post-transcriptional regulatory element; forward: 5′-GTCCTTTCCATGGCTGCTC-3′ and reverse: 5′-CCGAAGGGACGTAGCAGA-3′) in the presence of dNTPs (200 nmol/l) and 1 unit of DNA polymerase in its buffer (DyNAzyme EXT; Finnzymes, a part of Thermo Scientific, Illkirch, France). The PCR products were subjected to electrophoresis on a 2% agarose gel with 1 μg/ml of ethidium bromide.
Animals. Two months old male Wistar rats (Rattus norvegicus, 300 g) and 3 months old male C57BL/6 mice (Mus musculus) were included in this study. The animals were purchased from Janvier Laboratories (St Berthevin, France) and maintained in compliance with institutional protocols (Comité d'éthique en expérimentation animale du Nord Pas-de-Calais, n° 0508003). The animals were housed in a temperature-controlled room and maintained on a 12-hour day/night cycle with food and water ad libitum. All experiments on animals were performed in compliance with, and following the approval of, the local Animal Resources Committee (CEEA 342012), standards for the care and use of laboratory animals, and French and European Community rules.
Stereotaxic injection. Intracerebral injections of PBS or viral particles into the brains of anesthetized (ketamine 100 mg/kg, xylazine 10 mg/kg intraperitoneally) 2 months old Wistar rats or 3 months old C57BL/6 mice were performed using standard stereotactic procedures at the following coordinates relative to the bregma: posterior: −5.3 mm; lateral: ±6.2 mm; depth: −7 and −6.2 mm. Injections were performed bilaterally. The standard injection procedure consisted of the delivery of 400 ng of p24 using a 10 μl glass syringe with a fixed needle (Hamilton; Dutscher, Brumath, France). After injection at a rate of 0.25 μl per minute, the needle was left in place for 1 minute. A similar injection was then performed after 5 minutes at the second depth.
Animals were anesthetized (chloral hydrate 8%) and transcardially perfused with cold PBS followed by 4% paraformaldehyde for 20 minutes. The brains were removed rapidly, fixed overnight in 4% paraformaldehyde, placed in 20% sucrose for a week and then frozen and stored until use. Free-floating coronal cryostat sections (40 μm) were used for IHC.
IHC. Brain sections (three or five rats and three mice per group) were washed with PBS-Triton (0.2%) and treated for 30 minutes with 0.3% H2O2, and nonspecific binding was blocked with goat serum (1/100 in PBS; Vector Laboratories) for 60 minutes. The sections were then incubated with the primary antibody in PBS-Triton 0.2% overnight at 4 °C. After several washes, labeling was amplified using a biotinylated anti-mouse IgG (1/400 in PBS-Triton 0.2%; Vector Laboratories) for 60 minutes, followed by the ABC kit (1/400 in PBS; Vector Laboratories), and labeling was completed using 0.5 mg/ml DAB (Vector Laboratories) in 50 mmol/l Tris-HCl, pH 7.6, containing 0.075% H2O2. Brain sections were mounted on gelatin-coated slides, stained for 1 minute in a 0.5% cresyl violet solution, washed with 2% acetic acid, dehydrated through a graded series of alcohol and toluene, and then mounted with Vectamount (Vector Laboratories) for microscopic analysis using a Leica DMRB microscope coupled with a Leica DC300 camera (Leica Microsystems, Nanterre, France).
Image analysis. Tissue sections were quantitatively analyzed using stereology software (Mercator image analysis system; Explora Nova, La Rochelle, France). The software uses hue, saturation, and intensity to distinguish objects (cellular bodies) in the image field. Thresholds were established using accurately identified objects on a standard set of slides, and these segmentation thresholds remained constant throughout the analysis session (the region of interest was the whole hippocampus). The Mercator software calculates the measurement parameters for the entire field of view at 500X magnification. For quantitative image analysis of IHC, we selected all brain sections from bregma −4.8 to −6.3 (10–15 sections were analyzed per animal). The area immunoreactive to the antibodies (ADx215, AT8, MC1 or AT100) was normalized to the whole area analyzed. For CA1/2 thickness measurements, quantification was performed based on cresyl violet coloration in all brain sections from bregma −4.8 to −6.3. In each individual section, the entire pyramidal layer was drawn using Mercator software (Explora Nova) to calculate the average thickness.
Immunofluorescence. Brain sections (three or five rats per group) were washed with PBS-Triton 0.2%, and nonspecific binding was blocked through incubation with goat serum (1/100 in PBS; Vector Laboratories) for 60 minutes. The sections were then incubated with the first primary antibody in PBS-Triton 0.2% overnight at 4 °C. After several washes, the sections were incubated with the primary antibody against the second target in PBS-Triton 0.2% overnight at 4 °C. Sections were then incubated with the two Alexa Fluor-conjugated secondary antibodies (1/1,000 in PBS) for 60 minutes at room temperature. The sections were mounted with Vectashield containing DAPI to label the nuclei (Vector Laboratories). Confocal microscopy was performed on a Zeiss LSM 710 inverted confocal microscope as indicated above.
Extraction and characterization of sarkosyl-insoluble Tau. For immunoblot analyses, 1-mm thick brain sections (three rats per group) were dissected using an acrylic coronal brain matrix (World Precision Instruments, Sarasota, FL). The section containing the injection site was weighed, and 150 mg of tissue was homogenized in 1 ml of Tris buffer, pH 7.4, containing 0.32 mol/l sucrose and 1% N-lauroylsarcosine (sarkosyl) using a Potter Teflon-glass homogenizer (40 strokes). The homogenate was sonicated and spun at 100,000g for 1 hour at 4 °C. The pellet was resuspended in 800 μl of Tris/sucrose/sarkosyl buffer, sonicated and spun a second time at 100,000g for 1 hour at 4 °C. The second pellet, containing sarkosyl-insoluble Tau protein, was resuspended in 30 μl of Tris/sucrose buffer. For western blot analysis, NuPAGE LDS and Sample Reducing Agent (Invitrogen) were added to samples before denaturation at 100 °C for 5 minutes. Then, proteins were loaded onto a 4–12% Bis-Tris NuPAGE Novex gel (Invitrogen) and transferred to a 0.45 μm nitrocellulose membrane. The membrane was incubated with the AT100 antibody, and signals were visualized by chemiluminescence (ECL; GE Healthcare Life Sciences, Velizy-Villacoublay, France) using Image Reader LAS-3000 (Fujifilm, Bois d'Arcy, France).
RNA extraction from rat brain and viral genome analysis by reverse transcription-PCR. Wistar rats (three rats per group) were anesthetized with 8% chloride hydrate. The brains were dissected to isolate the hippocampus, and total RNA was extracted using the MIRVana PARis kit (Invitrogen) as recommended by the manufacturer. RNA purity and concentration were determined using a NanoDrop 1000 (Thermo Scientific). RNA (1 μg) was denatured for 10 minutes at 68 °C, and cDNA was generated by reverse transcription with 200 nmol/l of dNTPs, 1 ng/μl of random primers, 1 ng/μl of oligo-dT, 5 mmol/l of dithiothreitol, 2 units/μl of RNase Out, and 10 units/μl of M-MLV reverse transcriptase. Viral cDNAs were then amplified using oligonucleotides specific to Tau exons (exons 2+3+, 2+3−, 2−3−: forward 5′-ATGGCTGAACCCCGCCAGGAGT-3′, reverse: 5′-TTGCTTACGCCGGCCACTCGAG-3′ exon 10+, 10−: forward 5′-CATGCCAGACCTAAAGAACGTCAGG-3′, reverse 5′-TCACAAACCCTGCTTGGCCA-3′). PCR was performed using 2 μl of the previously obtained reverse transcription products using forward and reverse primers (1 μmol/l), dNTPs (1 μmol/l), and 0.02 units/μl of DNA polymerase in its buffer (DyNAzyme EXT; Finnzymes). The PCR products were subjected to electrophoresis on a 2% agarose gel with 1 μg/ml of ethidium bromide.
Electron microscopy. Brain sections from LV-injected rats (three rats per group) were processed 8 months p.i. for MC1 immunostaining as explained previously, with the following exception, post-fixation in 0.1% of glutaraldehyde was carried out for 1 hour before H2O2 treatment. Slides were then post-fixed for 15 minutes in phosphate buffer (0.1 mol/l) containing 1% osmium tetroxide. After rinsing in PBS, sections were dehydrated through a graded ethanol series. The slides were then treated with araldite/propylene oxide for 20 minutes at 56 °C, and the polymerization process continued overnight at 56 °C. MC1-positive brain areas were then cut into ultra-thin sections that were placed onto nickel grids and floated on a drop of staining solution (2% uranyl acetate) for 1 minute. After air-drying, the sections were observed under a transmission electron microscope (EM902; Zeiss).
Statistical analysis. Data are presented as the means (± SEM) of experiments performed in triplicate and are representative of the results obtained from three independent experiments that produced similar results. Statistical analysis was performed using the Mann–Whitney test.
SUPPLEMENTARY MATERIAL Figure S1. Equivalent viral titers of LVs-hTau46WT and LVs-hTau46P301L. Figure S2. hTau46WT did not drive Tau aggregation in mouse brains. Figure S3. Tau is expressed at a similar level in rat and mouse brains after lentiviral delivery. Figure S4. The Wistar rat brain hippocampus expresses six Tau isoforms, as found in the human brain.
Acknowledgments
This work was developed and supported through the LABEX (excellence laboratory, Invest for the Future) and DISTALZ programs (Development of Innovative Strategies for a Transdisciplinary approach to ALZheimer's disease) programs. It was also supported by the Inserm, CNRS, IMPRT (Institut de Médecine Prédictive et de RechercheThérapeutique, Lille), University of Lille 2, Région Nord/Pas-de-Calais, FEDER and DN2M (VICTAUR), and grants from ANR-08-MNPS-002/AMYTOXTAU and the European Community, including MEMOSAD (FP7 contract 200611) and FRC (“Fondation pour la Recherche sur le Cerveau”). We are also grateful to the IMPRT for access to core facilities (electron microscopy, Cécile Allet and confocal microscopy, Meryem Tardivel). We thank KU Leuven-LRD (Leuven, Belgium) for providing the ADx215 antibody for academic research purposes and Eugeen Vanmechelen (Ghent, Belgium) for advice. The authors declared no conflict of interest.
Supplementary Material
References
- Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- Hanes J, Zilka N, Bartkova M, Caletkova M, Dobrota D, Novak M. Rat tau proteome consists of six tau isoforms: implication for animal models of human tauopathies. J Neurochem. 2009;108:1167–1176. doi: 10.1111/j.1471-4159.2009.05869.x. [DOI] [PubMed] [Google Scholar]
- Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, Consortium for Frontotemporal Lobar Degeneration et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K. Alzheimer's disease: pathogenesis and prevention. Alzheimers Dement. 2012;8:227–233. doi: 10.1016/j.jalz.2012.01.011. [DOI] [PubMed] [Google Scholar]
- Augustinack JC, Schneider A, Mandelkow EM, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol. 2002;103:26–35. doi: 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- Götz J, Ittner LM. Animal models of Alzheimer's disease and frontotemporal dementia. Nat Rev Neurosci. 2008;9:532–544. doi: 10.1038/nrn2420. [DOI] [PubMed] [Google Scholar]
- Götz J, Probst A, Spillantini MG, Schäfer T, Jakes R, Bürki K, et al. Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J. 1995;14:1304–1313. doi: 10.1002/j.1460-2075.1995.tb07116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst A, Götz J, Wiederhold KH, Tolnay M, Mistl C, Jaton AL, et al. Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol. 2000;99:469–481. doi: 10.1007/s004010051148. [DOI] [PubMed] [Google Scholar]
- Brion JP, Tremp G, Octave JN. Transgenic expression of the shortest human tau affects its compartmentalization and its phosphorylation as in the pretangle stage of Alzheimer's disease. Am J Pathol. 1999;154:255–270. doi: 10.1016/S0002-9440(10)65272-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, et al. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–762. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- Zilka N, Filipcik P, Koson P, Fialova L, Skrabana R, Zilkova M, et al. Truncated tau from sporadic Alzheimer's disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett. 2006;580:3582–3588. doi: 10.1016/j.febslet.2006.05.029. [DOI] [PubMed] [Google Scholar]
- Zilka N, Korenova M, Novak M. Misfolded tau protein and disease modifying pathways in transgenic rodent models of human tauopathies. Acta Neuropathol. 2009;118:71–86. doi: 10.1007/s00401-009-0499-y. [DOI] [PubMed] [Google Scholar]
- Filipcik P, Zilka N, Bugos O, Kucerak J, Koson P, Novak P, et al. First transgenic rat model developing progressive cortical neurofibrillary tangles. Neurobiol Aging. 2012;33:1448–1456. doi: 10.1016/j.neurobiolaging.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Chang E, Kim S, Yin H, Nagaraja HN, Kuret J. Pathogenic missense MAPT mutations differentially modulate tau aggregation propensity at nucleation and extension steps. J Neurochem. 2008;107:1113–1123. doi: 10.1111/j.1471-4159.2008.05692.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Bugiani O, Murrell JR, Giaccone G, Hasegawa M, Ghigo G, Tabaton M, et al. Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in tau. J Neuropathol Exp Neurol. 1999;58:667–677. doi: 10.1097/00005072-199906000-00011. [DOI] [PubMed] [Google Scholar]
- Schindowski K, Bretteville A, Leroy K, Bégard S, Brion JP, Hamdane M, et al. Alzheimer's disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am J Pathol. 2006;169:599–616. doi: 10.2353/ajpath.2006.060002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy K, Bretteville A, Schindowski K, Gilissen E, Authelet M, De Decker R, et al. Early axonopathy preceding neurofibrillary tangles in mutant tau transgenic mice. Am J Pathol. 2007;171:976–992. doi: 10.2353/ajpath.2007.070345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski T, Dewachter I, Lechat B, Croes S, Termont A, Demedts D, et al. AAV-tau mediates pyramidal neurodegeneration by cell-cycle re-entry without neurofibrillary tangle formation in wild-type mice. PLoS ONE. 2009;4:e7280. doi: 10.1371/journal.pone.0007280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osinde M, Clavaguera F, May-Nass R, Tolnay M, Dev KK. Lentivirus Tau (P301S) expression in adult amyloid precursor protein (APP)-transgenic mice leads to tangle formation. Neuropathol Appl Neurobiol. 2008;34:523–531. doi: 10.1111/j.1365-2990.2008.00936.x. [DOI] [PubMed] [Google Scholar]
- Ramirez JJ, Poulton WE, Knelson E, Barton C, King MA, Klein RL. Focal expression of mutated tau in entorhinal cortex neurons of rats impairs spatial working memory. Behav Brain Res. 2011;216:332–340. doi: 10.1016/j.bbr.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustroph ML, King MA, Klein RL, Ramirez JJ. Adult-onset focal expression of mutated human tau in the hippocampus impairs spatial working memory of rats. Behav Brain Res. 2012;233:141–148. doi: 10.1016/j.bbr.2012.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayton RD, Wang DB, Cain CD, Schrott LM, Ramirez JJ, King MA, et al. Frontotemporal lobar degeneration-related proteins induce only subtle memory-related deficits when bilaterally overexpressed in the dorsal hippocampus. Exp Neurol. 2012;233:807–814. doi: 10.1016/j.expneurol.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal PJ, Dumanis SB, Herman AM, Rebeck GW, Moussa CE. Wild type and P301L mutant Tau promote neuro-inflammation and a-Synuclein accumulation in lentiviral gene delivery models. Mol Cell Neurosci. 2012;49:44–53. doi: 10.1016/j.mcn.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RL, Dayton RD, Tatom JB, Diaczynsky CG, Salvatore MF. Tau expression levels from various adeno-associated virus vector serotypes produce graded neurodegenerative disease states. Eur J Neurosci. 2008;27:1615–1625. doi: 10.1111/j.1460-9568.2008.06161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- Dayanandan R, Van Slegtenhorst M, Mack TG, Ko L, Yen SH, Leroy K, et al. Mutations in tau reduce its microtubule binding properties in intact cells and affect its phosphorylation. FEBS Lett. 1999;446:228–232. doi: 10.1016/s0014-5793(99)00222-7. [DOI] [PubMed] [Google Scholar]
- DeTure M, Ko LW, Easson C, Yen SH. Tau assembly in inducible transfectants expressing wild-type or FTDP-17 tau. Am J Pathol. 2002;161:1711–1722. doi: 10.1016/S0002-9440(10)64448-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretteville A, Ando K, Ghestem A, Loyens A, Bégard S, Beauvillain JC, et al. Two-dimensional electrophoresis of tau mutants reveals specific phosphorylation pattern likely linked to early tau conformational changes. PLoS ONE. 2009;4:e4843. doi: 10.1371/journal.pone.0004843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, et al. Trans-synaptic spread of tau pathology in vivo. PLoS ONE. 2012;7:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peel AL, Klein RL. Adeno-associated virus vectors: activity and applications in the CNS. J Neurosci Methods. 2000;98:95–104. doi: 10.1016/s0165-0270(00)00183-7. [DOI] [PubMed] [Google Scholar]
- Lundberg C, Björklund T, Carlsson T, Jakobsson J, Hantraye P, Déglon N, et al. Applications of lentiviral vectors for biology and gene therapy of neurological disorders. Curr Gene Ther. 2008;8:461–473. doi: 10.2174/156652308786847996. [DOI] [PubMed] [Google Scholar]
- Jakobsson J, Ericson C, Jansson M, Björk E, Lundberg C. Targeted transgene expression in rat brain using lentiviral vectors. J Neurosci Res. 2003;73:876–885. doi: 10.1002/jnr.10719. [DOI] [PubMed] [Google Scholar]
- Lace G, Savva GM, Forster G, de Silva R, Brayne C, Matthews FE, MRC-CFAS et al. Hippocampal tau pathology is related to neuroanatomical connections: an ageing population-based study. Brain. 2009;132 Pt 5:1324–1334. doi: 10.1093/brain/awp059. [DOI] [PubMed] [Google Scholar]
- Mercken M, Vandermeeren M, Lübke U, Six J, Boons J, Van de Voorde A, et al. Monoclonal antibodies with selective specificity for Alzheimer Tau are directed against phosphatase-sensitive epitopes. Acta Neuropathol. 1992;84:265–272. doi: 10.1007/BF00227819. [DOI] [PubMed] [Google Scholar]
- Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J Biol Chem. 2008;283:32066–32076. doi: 10.1074/jbc.M805300200. [DOI] [PubMed] [Google Scholar]
- Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, et al. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci. 2002;22:9340–9351. doi: 10.1523/JNEUROSCI.22-21-09340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegener. 2009;4:13. doi: 10.1186/1750-1326-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K. The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol. 2011;121:171–181. doi: 10.1007/s00401-010-0789-4. [DOI] [PubMed] [Google Scholar]
- van Swieten JC, Rosso SM, Heutink P.2000MAPT-related disorders. Pagon RA, Bird TD, Dolan CR, Stephens K.andAdam MP.eds). GeneReviews™ [Internet] University of Washington: Seattle, WA [Google Scholar]
- Klein RL, Dayton RD, Diaczynsky CG, Wang DB. Pronounced microgliosis and neurodegeneration in aged rats after tau gene transfer. Neurobiol Aging. 2010;31:2091–2102. doi: 10.1016/j.neurobiolaging.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuma H, Arawaka S, Mori H. Isoforms changes of tau protein during development in various species. Brain Res Dev Brain Res. 2003;142:121–127. doi: 10.1016/s0165-3806(03)00056-7. [DOI] [PubMed] [Google Scholar]
- McMillan P, Korvatska E, Poorkaj P, Evstafjeva Z, Robinson L, Greenup L, et al. Tau isoform regulation is region- and cell-specific in mouse brain. J Comp Neurol. 2008;511:788–803. doi: 10.1002/cne.21867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando K, Leroy K, Héraud C, Yilmaz Z, Authelet M, Suain V, et al. Accelerated human mutant tau aggregation by knocking out murine tau in a transgenic mouse model. Am J Pathol. 2011;178:803–816. doi: 10.1016/j.ajpath.2010.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad C, Zhu J, Conrad C, Schoenfeld D, Fang Z, Ingelsson M, et al. Single molecule profiling of tau gene expression in Alzheimer's disease. J Neurochem. 2007;103:1228–1236. doi: 10.1111/j.1471-4159.2007.04857.x. [DOI] [PubMed] [Google Scholar]
- Adams SJ, DeTure MA, McBride M, Dickson DW, Petrucelli L. Three repeat isoforms of tau inhibit assembly of four repeat tau filaments. PLoS ONE. 2010;5:e10810. doi: 10.1371/journal.pone.0010810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HJ, Park SS, Mok JO, Lee TK, Park CS, Park SA. Increased expression of three-repeat isoforms of tau contributes to tau pathology in a rat model of chronic type 2 diabetes. Exp Neurol. 2011;228:232–241. doi: 10.1016/j.expneurol.2011.01.012. [DOI] [PubMed] [Google Scholar]
- Schweers O, Mandelkow EV, Biernat J, Mandelkow E. Oxidation of cystein-322 in the repeat domain of microtubule-associated protein tau controls the in vitro assembly of paired helical filament. Proc Natl Acad Sci USA. 1995;92:8463–8467. doi: 10.1073/pnas.92.18.8463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sautiere PE, Caillet-Boudin ML, Wattez A, Delacourte A. Detection of Alzheimer-type tau proteins in okadaic acid-treated SKNSH-SY5Y neuroblastoma cells. Neurodegeneration. 1994;3:53–60. [Google Scholar]
- Sergeant N, Sablonnière B, Schraen-Maschke S, Ghestem A, Maurage CA, Wattez A, et al. Dysregulation of human brain microtubule-associated tau mRNA maturation in myotonic dystrophy type 1. Hum Mol Genet. 2001;10:2143–2155. doi: 10.1093/hmg/10.19.2143. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Bloch J, Ma SY, Chu Y, Palfi S, Roitberg BZ, et al. Lentiviral gene transfer to the nonhuman primate brain. Exp Neurol. 1999;160:1–16. doi: 10.1006/exnr.1999.7178. [DOI] [PubMed] [Google Scholar]
- Hottinger AF, Azzouz M, Déglon N, Aebischer P, Zurn AD. Complete and long-term rescue of lesioned adult motoneurons by lentiviral-mediated expression of glial cell line-derived neurotrophic factor in the facial nucleus. J Neurosci. 2000;20:5587–5593. doi: 10.1523/JNEUROSCI.20-15-05587.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.