

Abstract
Summary: MASS is a command-line program to perform meta-analysis of sequencing studies by combining the score statistics from multiple studies. It implements three types of multivariate tests that encompass all commonly used association tests for rare variants. The input files can be generated from the accompanying software SCORE-Seq. This bundle of programs allows analysis of large sequencing studies in a time and memory efficient manner.
Availability and implementation: MASS and SCORE-Seq, including documentations and executables, are available at http://dlin.web.unc.edu/software/.
Contact: lin@bios.unc.edu
1 INTRODUCTION
Meta-analysis of genome-wide association studies (GWAS) has led to the discoveries of common genetic variants for virtually every complex human disease. Recent advances in sequencing technologies have made it possible to extend association studies to rare variants. Because larger sample sizes are required to detect rare variants than common variants (with similar effect sizes), combining evidence from many sources is necessary for sequencing studies. For ethical and logistical reasons, it is strongly preferable to gather summary statistics than collecting original data.
For association testing with rare variants, it is customary to aggregate information across several variant sites within a gene to enrich association signals and to reduce the penalty of multiple testing. The simplest approach is the burden test, which creates a burden score for each subject (by taking a weighted linear combination of the mutation counts within a gene or indicating whether there is any mutation within a gene) (Li and Leal, 2008; Lin and Tang, 2011; Madsen and Browning, 2009; Price et al., 2010). A second approach is the variable threshold (VT) test, which performs a burden test for variants with minor allele frequencies (MAFs) below a certain threshold and minimizes the P-value over the observed MAF thresholds (Lin and Tang, 2011; Price et al., 2010). A third approach is the variance-component test, which is aimed at detecting variants with opposite effects within a gene (Neale et al., 2011; Tzeng and Zhang, 2007; Wu et al., 2011).
After creating a burden score for each subject, one may carry out the burden test by performing a standard regression analysis and combine the Wald statistics of multiple studies through the inverse-variance formula. However, this strategy is problematic for rare variants because the effect estimates may be unstable or even undefined when only a small number of subjects carry any mutation. Indeed, the log odds ratio is undefined if the burden scores are zero for all cases or all controls. We recommend to use score statistics, which are statistically more accurate and numerically more stable than Wald and likelihood ratio statistics, especially for binary traits (Lin and Tang, 2011). Currently, there is no meta-analysis software for combining score statistics. Thus, we developed the Meta-Analysis of Score Statistics (MASS) software, which performs an overall burden test by combining the score statistics of multiple studies. In addition, MASS performs an overall VT test for multiple studies based on score statistics. Finally, MASS performs an overall variance-component test also based on score statistics. The meta-analysis performed by MASS is statistically as efficient as joint analysis of individual-level data. The software is extremely easy to use. Because score statistics are not available in existing software packages, we developed SCORE-Seq, which takes the standard input format of sequencing data and outputs score statistics, which can then be meta-analyzed in MASS.
2 METHODS
Suppose that we are interested in d genetic variables. For the burden and VT tests, the genetic variables pertain to the burden scores; for the variant-component test, the genetic variables pertain to the genotypes of individual variants; for the CMC test (Li and Leal, 2008), the genetic variables contain the genotypes of common variants and the burden scores of rare variants. Suppose that there are K independent studies. For the kth study, we calculate the (multivariate) score statistic 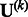 for testing the null hypothesis H0 that none of the d genetic variables have any effect on the trait of interest, and we also calculate the corresponding information matrix
for testing the null hypothesis H0 that none of the d genetic variables have any effect on the trait of interest, and we also calculate the corresponding information matrix 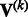 . Note that
. Note that 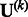 is a
is a 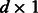 vector and
vector and 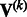 is a
is a 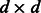 matrix. If a genetic variable is absent in the kth study, then we set the corresponding entries in
matrix. If a genetic variable is absent in the kth study, then we set the corresponding entries in 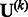 and
and 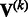 to zero. Given the input of
to zero. Given the input of 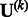 and
and 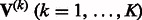 , MASS calculates
, MASS calculates 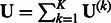 and
and 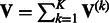 . Under H0, the random vector U is (asymptotically) multivariate normal with mean 0 and covariance matrix V. It can be shown that U is the score statistic for testing H0 from the joint likelihood for the original data of the K studies allowing nuisance parameters (e.g., intercepts and error variances) to be different among the K studies (Lin and Zeng, 2010). Thus, association testing based on U and V is equivalent to the joint analysis of the original data.
. Under H0, the random vector U is (asymptotically) multivariate normal with mean 0 and covariance matrix V. It can be shown that U is the score statistic for testing H0 from the joint likelihood for the original data of the K studies allowing nuisance parameters (e.g., intercepts and error variances) to be different among the K studies (Lin and Zeng, 2010). Thus, association testing based on U and V is equivalent to the joint analysis of the original data.
After calculating U and V, MASS can perform three types of multivariate tests, which encompass all commonly used rare-variant tests.
1. Quadratic statistic:
Under H0, the test statistic Q is distributed as 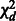 . If d = 1 and the genetic variable pertains to a specific burden score (based on a MAF threshold or the Madsen–Browning weighting), then Q is a burden test with 1 degree of freedom. If the genetic variables consist of the genotypes of common SNPs and the burden score of rare variants, then Q is the CMC test.
. If d = 1 and the genetic variable pertains to a specific burden score (based on a MAF threshold or the Madsen–Browning weighting), then Q is a burden test with 1 degree of freedom. If the genetic variables consist of the genotypes of common SNPs and the burden score of rare variants, then Q is the CMC test.
2. Maximum statistic:
where Uj is the jth component of U, and Vj is the jth diagonal element of V. The P-value of 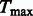 is determined by the multivariate normal distribution of U (Lin and Tang, 2011). If the d genetic variables are the burden scores at d MAF thresholds, then
is determined by the multivariate normal distribution of U (Lin and Tang, 2011). If the d genetic variables are the burden scores at d MAF thresholds, then 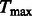 yields the VT test. If the genetic variables pertain to different types of burden scores, such as T1, T5, and Madsen–Browning, then
yields the VT test. If the genetic variables pertain to different types of burden scores, such as T1, T5, and Madsen–Browning, then 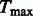 can be used to adjust for multiple testing with those burden tests.
can be used to adjust for multiple testing with those burden tests.
3. Weighted quadratic statistic:
where W is a weight matrix. The null distribution of Qw is determined by 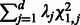 , where
, where 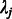 is the jth eigenvalue of
is the jth eigenvalue of 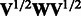 , and
, and  are independent
are independent 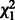 random variables. If the genetic variables are the genotypes of individual SNPs, then Qw becomes the SKAT or C-alpha test. For SKAT, W is a diagonal matrix that depends on the MAFs through a beta function; for C-alpha, W is an identity matrix.
random variables. If the genetic variables are the genotypes of individual SNPs, then Qw becomes the SKAT or C-alpha test. For SKAT, W is a diagonal matrix that depends on the MAFs through a beta function; for C-alpha, W is an identity matrix.
3 RESULTS
MASS is a freely available C program that runs on Unix and Linux systems. The basic command line is MASS [-method method][-sfile script][-ofile outfile][options]. The option -method selects one of the three test statistics: quadratic, maximum or weighted quadratic. The option -sfile specifies a script file that describes the input files from multiple studies. For the weighted quadratic statistic, the option -weight can be used to specify a file with a weight for each component of U. MASS can filter out genetic variables based on minor allele counts (MACs). Full documentation is available at http://dlin.web.unc.edu/software/.
The summary statistics for individual studies can be obtained from SCORE-Seq, which inputs the sequencing data with a quantitative or binary trait and outputs the score statistics and information matrices for all commonly used rare-variant tests. The basic command line is SCORE-Seq [-pfile phenofile] [-gfile genofile] [-mfile mapfile] [-ofile outfile] [-vtlog vtlog] [-snplog snplog] [options]. There are three input files: phenofile contains the phenotype and covariates; genofile contains the genotypes; mapfile provides the gene–SNP mapping and SNP annotation. The output files outfile, vtlog and snplog contain the score statistics and information matrices for the burden test, VT and SKAT. In the output files, each row corresponds to a component of the score statistic and each column of the score statistic is followed by the corresponding information matrix. The SCORE-Seq output files for different studies can be input directly into MASS.
We recently applied MASS to the NHLBI Exome Sequencing Project. The meta-analysis involved 11 studies and 15 404 genes, with an average of 7 genetic variables per test. In three of the studies, subjects were selected for sequencing because they had extreme values of a quantitative trait. Thus, we developed a special program called SCORE-SeqTDS to perform quantitative trait analysis under trait-dependent sampling. We obtained the summary statistics from SCORE-Seq or SCORE-SeqTDS, dependent on the study design. The total size of the input files for MASS was 172 MB. We ran the three types of tests on an IBM HS22 machine: the quadratic statistic took 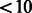 seconds and 1 MB memory; the maximum statistic took
seconds and 1 MB memory; the maximum statistic took 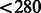 seconds and 33 MB memory; and the weighted quadratic statistic took
seconds and 33 MB memory; and the weighted quadratic statistic took 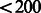 seconds and 33 MB memory.
seconds and 33 MB memory.
Figure 1 is a flow chart for performing the VT test. The top panel shows the SCORE-Seq input and output files for the first two studies. The first gene, ABHD8, has three MAF thresholds. Thus, the score vector for this gene is provided in three rows, and the information matrix is a 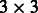 matrix, whose upper diagonal elements are redundant and thus set to zero. The 11 output files generated by SCORE-Seq and SCORE-SeqTDS are input into MASS via the script file shown in the bottom panel of Figure 1. The output file of MASS provides the maximum test statistic and P-value for each gene.
matrix, whose upper diagonal elements are redundant and thus set to zero. The 11 output files generated by SCORE-Seq and SCORE-SeqTDS are input into MASS via the script file shown in the bottom panel of Figure 1. The output file of MASS provides the maximum test statistic and P-value for each gene.
Fig. 1.

Pipeline for performing the VT test in the NHLBI Exome Sequencing Project
4 DISCUSSION
Protection of human subjects and other study policies make it difficult to share individual-level data, even in well-organized consortia. Thus, meta-analysis is strongly preferable to joint analysis. The MASS software enables one to perform meta-analysis of score statistics for sequencing studies, which is statistically as efficient as and indeed numerically equivalent to joint analysis of individual-level data. This software can also be used to combine results from other types of genetic studies as well as non-genetic studies.
In meta-analysis of sequencing data, the participating studies should use the same annotation file so that the summary statistics are generated in a consistent manner across studies. This does not mean that the variants have to be the same among studies. If a genetic variable is missing in one study, then zero can be used as a placeholder in the summary statistics and MASS will combine all available information.
The calculation of the burden scores requires specification of the MAFs. The MAFs may be estimated separately in each study or jointly across all studies; they may also be determined from an external source. We recommend that the same MAFs be used by all participating studies. In this way, the same variants are included in the calculations of the burden scores among the studies, and the MAF thresholds for the VT test are consistent across the studies.
For studies that use different exome capturing kits or studies in which some have exome sequencing while others have exome chip data, the input variants are different. If the genetic variable pertains to the genotype of a variant and that variant is not measured in the kth study, then we simply set the corresponding entries in 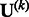 and
and 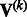 to zero, so that MASS will combine all available data. If the genetic variable pertains to a burden score, then a variant that is absent in a study will not contribute to the calculation of the burden score for that study. The score statistics from such studies can still be combined in MASS, although the results need to be interpreted with extra care.
to zero, so that MASS will combine all available data. If the genetic variable pertains to a burden score, then a variant that is absent in a study will not contribute to the calculation of the burden score for that study. The score statistics from such studies can still be combined in MASS, although the results need to be interpreted with extra care.
If the burden score is a (weighted) linear combination of the genotype values (e.g. the total number of mutations or a weighted sum of the mutation counts), then the score statistics for the burden and VT tests are (weighted) linear combinations of the score statistic for testing the null hypothesis that the genotypes of individual variants are not associated with the trait of interest. In that case, it would be sufficient to input only the score vector and information matrix for individual variants because they can be used to create the score statistics and information matrices for the burden, VT and SKAT tests. We did not take this approach because it requires the burden scores to be calculated under the additive mode of inheritance. Indeed, it has become a common practice to define the burden score as the presence or absence of any mutation within a gene rather than the total number or weighted linear combination of the mutations. SCORE-Seq allows burden scores to be calculated under the additive, dominant or recessive mode of inheritance. By asking the user to input the score statistics and information matrices for each specific test, MASS can accommodate any mode of inheritance.
MASS adopts the fixed-effect model, which assumes that the genetic effects are the same among the participating studies. This approach will have reasonable power as long as the effects are in the same direction across studies. An alternative approach is the random-effect model, under which the effects in different studies follow a normal distribution. The random-effect model tends to be less powerful than the fixed-effect model even when the effects are heterogeneous and thus has rarely been used in genetic association studies.
ACKNOWLEDGEMENTS
We thank the Associate Editor and three referees for reviewing our work and providing helpful comments.
Funding: National Institutes of Health awards R01CA082659, P01CA142538 and R37GM047845.
Conflict of Interest: none declared.
REFERENCES
- Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am. J. Hum. Genet. 2008;83:311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DY, Tang ZZ. A general framework for detecting disease associations with rare variants in sequencing studies. Am. J. Hum. Genet. 2011;89:354–367. doi: 10.1016/j.ajhg.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DY, Zeng D. On the relative efficiency of using summary statistics versus individual level data in meta-analysis. Biometrika. 2010;97:321–332. doi: 10.1093/biomet/asq006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen BE, Browning SR. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5:e1000384. doi: 10.1371/journal.pgen.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, et al. Testing for an unusual distribution of rare variants. PLoS Genet. 2011;7:e1001322. doi: 10.1371/journal.pgen.1001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, et al. Pooled association tests for rare variants in exon-resequencing studies. Am. J. Hum. Genet. 2010;86:832–838. doi: 10.1016/j.ajhg.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng JY, Zhang D. Haplotype-based association analysis via variance component score test. Am. J. Hum. Genet. 2007;81:939–963. doi: 10.1086/521558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MC, et al. Rare variant association testing for sequencing data using the sequence kernel association test (SKAT) Am. J. Hum. Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]