

The crystal structure of Mast-M1 has been determined at 2.0 Å resolution and provides the first detailed structural description of a TOG-like domain.
Keywords: Dis1-TOG domain, TOG-like domains, microtubules, CLASP, XMAP215/Dis1
Abstract
Mast/Orbit is a nonmotor microtubule-associated protein (MAP) present in Drosophila melanogaster that reportedly binds microtubules at the plus end and is essential for mitosis. Sequence analysis has shown that the N-terminal domain (Mast-M1) resembles TOG domains from the Dis1-TOG family of proteins and stands as a representative of one of the three subclasses of divergent TOG-like domains (TOGL1) that includes human CLASP1. The crystal structure of Mast-M1 has been determined at 2.0 Å resolution and provides the first detailed structural description of any TOG-like domain. The structure confirms that Mast-M1 adopts a similar fold to the previously described Dis1-TOG domains of microtubule-binding proteins. A comparison with three known TOG-domain structures from XMAP215/Dis1 family members exposes significant differences between Mast-M1 and other TOG-domain structures in key residues at the proposed tubulin-binding edge.
1. Introduction
Microtubules are dynamic tubulin polymers that are involved in multiple cellular processes. During mitosis, microtubules are organized at the centrosome to form the mitotic spindle, providing an orientation for cell cleavage as well as a platform for chromosome segregation (Desai & Mitchison, 1997 ▶). Regulation at the centrosome is accomplished by a variety of proteins that interact with microtubules, such as the nonmotor microtubule-associated proteins (MAPs), which aid in the regulation of microtubule dynamics (Al-Bassam & Chang, 2011 ▶). Mast/Orbit (Mast) is a 165 kDa MAP present in Drosophila melanogaster that binds microtubules at the plus end and localizes to centrosomes, spindle microtubules and kinetochores (Inoue et al., 2000 ▶; Lemos et al., 2000 ▶). It is homologous to human CLIP-associated proteins (CLASPS), which also co-localize with CLIPs at the microtubule plus end (Akhmanova et al., 2001 ▶). Data suggest that Mast is involved in maintaining spindle architecture and moving chromosomes along spindle microtubules (Maiato et al., 2002 ▶). Loss-of-function mutations result in bipolar spindles collapsing into monoasters after nuclear envelope breakdown (Inoue et al., 2000 ▶; Lemos et al., 2000 ▶). It is from this phenotypic behavior that Mast derives its name (Inoue et al., 2000 ▶; Lemos et al., 2000 ▶).
Sequence analysis reveals that the N-terminal domain of Mast is 20–25% identical to TOG domains of the Dis1-TOG family of MAPs (Inoue et al., 2000 ▶; Lemos et al., 2000 ▶). TOG domains are typically comprised of six tandem helical repeats and are known to promote microtubule binding (Slep & Vale, 2007 ▶). While isolated TOG domains expressed in vitro often retain little affinity for tubulin, multiple TOG domains appear to act in concert to enhance and promote microtubule assembly and often occur in multiple copies in microtubule plus-end binding proteins such as the XMAP215 family (reviewed by Al-Bassam & Chang, 2011 ▶). Sequence analysis predicts that Mast contains only one identifiable TOG domain at its N-terminal end, but that it embeds two other TOG-like (TOGL) domains that appear to play a similar role (Slep & Vale, 2007 ▶; Al-Bassam & Chang, 2011 ▶).
Previous structural biology has resulted in characterization of the structures of three TOG domains of XMAP215/Dis1 family members representative of diverse eukaryotes, including worms (Caenorhabditis elegans Zyg9 TOG3; PDB entry 2of3; Al-Bassam & Chang, 2011 ▶), flies (D. melanogaster Msps TOG2; PDB entry 2qk2; Slep & Vale, 2007 ▶) and yeast (Saccharomyces cerevisiae Stu2p TOG2; PDB entry 2qk1; Slep & Vale, 2007 ▶). No structures of TOG domains from CLASP family members or of TOG-like domains have previously been reported. Here, we describe the high-resolution crystal structure of the N-terminal TOG domain of D. melanogaster Mast/Orbit and compare it with other TOG-domain structures.
2. Materials and methods
2.1. Cloning and purification
The first 229 amino acids of Mast (denoted Mast-M1) were amplified by polymerase chain reaction (PCR; reagents purchased from BD Biosciences) using Drosophila QUICK-Clone cDNA purchased from Stratagene. The Mast-M1 construct was generated using the following primers: 1(F), 5′-ATT GCA CAT ATG GCC TAT CGG AAG CCC AGC GAC CTG-3′, and 2(R), 5′-TAG ACC CTC GAG TTT GAC CTG GTC GAA CTT TTG CTC-3′. The PCR products were digested with NdeI and XhoI and inserted into pET-29b vector (Novagen); Escherichia coli BL21(DE3) pLysS cells were transformed with this vector. The bacterial cells were grown at 310 K to an OD of 0.6–0.8 and isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM. Protein production was induced overnight prior to centrifugation at 3000 rev min−1 for 25 min. Following induction, the pellets were frozen at 253 K prior to resuspension in lysis buffer (50 mM HEPES pH 7.5, 100 mM NaCl, 0.5 mM EDTA, 20 mM imidazole pH 7.5). The cell suspension was subjected to a freeze–thaw cycle via freezing at 203 K for at least 30 min and then thawing in a 310 K water bath. The cells were sonicated on ice four times with 30 s bursts followed by 30 s rest intervals prior to the removal of cellular debris via centrifugation at 18 000 rev min−1 for 50 min. The filtered supernatant was applied onto a metal-chelation affinity column loaded with nickel (Pharmacia), which had been equilibrated with 5 mM imidazole pH 7.5, 500 mM NaCl, 20 mM HEPES pH 7.8. The column was washed with the above buffer for five column volumes followed by a five-column-volume wash with 60 mM imidazole. The protein was eluted using a gradient to 1 M imidazole pH 7.5. The fractions containing protein were diluted in half and dialyzed against dialysis buffer (50 mM HEPES pH 7.5, 100 mM NaCl, 5 mM EDTA, 1 mM DTT). The protein was loaded onto an ion-exchange column (Mono Q cation) on an FPLC for further purification. The column was equilibrated in the dialysis buffer above and the protein was eluted using a ten-column-volume gradient from 100 mM to 1 M NaCl. The fractions containing the Mast-M1 construct were dialyzed against the final storage buffer (25 mM HEPES pH 7.5, 50 mM NaCl, 5% glycerol, 5 mM MgCl2, 1 mM DTT).
Selenomethionine-derivative (SeMet) Mast-M1 protein was obtained by growing E. coli BL21(DE3) cells in 250 ml LB medium supplemented with kanamycin overnight at 310 K. The cells were then centrifuged at 4000 rev min−1 for 15 min and the pellet was washed once with distilled sterile water. The pellet was then added to 1 l SeMet minimum medium containing 1 g NH4Cl, 3 g KH2PO4, 6 g Na2HPO4·7H2O and 100 ml of a filter-sterilized solution consisting of 20%(w/v) glucose, 0.3%(w/v) MgSO4, 10 mg Fe2(SO4)3, 10 mg thiamine and filter-sterilized l-SeMet (50 µg ml−1 final concentration). Since this procedure resulted in an OD600 of 0.6–1.0, IPTG was added to a final concentration of 1 mM at the same time as the cells (Guerrero et al., 2001 ▶). The cells were harvested after 12 h of incubation at 298 K with shaking at 180 rev min−1 and were centrifuged at 4000 rev min−1 for 15 min. The cells were resuspended in 50 mM Tris–HCl pH 7.6, 100 mM NaCl, 20 mM imidazole pH 7.8 (buffer A) and lysed by a freeze (193 K)/thaw (310 K) cycle followed by four cycles of 30 s on/off sonication at 40% power. Cell debris was cleared by centrifugation at 18 000 rev min−1 for 50 min and the filtered supernatant was loaded onto a HisTrap FF column (GE Healthcare) pre-equilibrated with buffer A. Nonspecifically bound proteins were removed using buffer A containing 60 mM imidazole, and SeMet Mast-M1 was eluted using a gradient from 60 to 675 mM imidazole. Fractions were checked via SDS–PAGE, pooled and concentrated using an ultracentrifugal filter unit (Millipore) and loaded onto a Sephacryl S100 HR column (GE Healthcare) for buffer exchange into crystallization buffer [25 mM HEPES pH 7.5, 50 mM NaCl, 5 mM MgCl2, 5%(v/v) glycerol, 1 mM DTT].
2.2. Crystallization and data collection
Following buffer exchange, SeMet Mast-M1 was concentrated to 9.3 mg ml−1; the concentration was measured using a Nanodrop 1000 spectrophotometer (Thermo Scientific). Crystals of SeMet Mast-M1 were obtained from 12.5% PEG 4000, 0.1 M bis-tris pH 6.5, 0.1 M NaCl, 20%(v/v) glycerol by the hanging-drop method with a 1:1 ratio of protein to precipitant at 292 K. Long rectangular crystals appeared within 3 d and grew fully within a week to final dimensions of 0.50 × 0.060 × 0.060 mm.
Crystals were picked out from the drop and quickly flash-cooled in liquid nitrogen. Diffraction data were collected remotely on beamline 4.2.2 at the Advanced Light Source (ALS), Berkeley, California, USA, which was equipped with a NOIR-1 MBC detector, using a beam size of 0.85 × 0.12 mm. Data for MAD phasing were collected at three wavelengths from a single crystal at 100 K. On a second occasion at ALS, an SeMet Mast-M1 crystal was found to diffract to beyond the resolution of the initial crystals; thus, this crystal was utilized for the final model determination. The program d*TREK (Pflugrath, 1999 ▶) was utilized to index, integrate and scale the data. Data-collection and refinement statistics can be found in Table 1 ▶.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Crystal 1 | ||||
|---|---|---|---|---|
| Data set | Crystal 2 | Peak | Inflection | Remote |
| Data collection | ||||
| Wavelength () | 1.0000 | 0.9788 | 0.9791 | 1.0000 |
| Temperature (K) | 100 | 100 | 100 | 100 |
| Unit-cell parameters () | a = 36.2, b = 97.6, c = 149.4 | a = 36.4, b = 97.7, c = 149.3 | a = 36.5, b = 97.6, c = 149.2 | a = 36.1, b = 97.7, c = 149.1 |
| Space group | P212121 | P212121 | P212121 | P212121 |
| Resolution () | 50.002.00 (2.072.00) | 46.422.40 (2.492.40) | 46.382.40 (2.492.40) | 49.692.30 (2.382.30) |
| Total/unique reflections | 437740/37049 | 107909/39900 | 99339/39328 | 124166/44988 |
| Mean I/(I) | 28.9 (2.3) | 6.8 (1.9) | 6.4 (1.7) | 6.5 (2.1) |
| R merge (%)† | 5.9 (35.9) | 7.8 (34.3) | 8.4 (35.8) | 9.0 (35.7) |
| Completeness (%) | 97.7 (84.1) | 99.3 (98.0) | 97.7 (96.5) | 99.3 (99.8) |
| Multiplicity | 5.8 (4.4) | 2.70 (2.71) | 2.53 (2.53) | 2.76 (2.71) |
| V M (3Da1) | 2.40 | |||
| Solvent content (%) | 48.72 | |||
| Refinement | ||||
| R work/R free (%) | 18.8/22.9 | |||
| No. of subunits in asymmetric unit | 2 | |||
| No. of non-H protein atoms | 3676 | |||
| No. of water molecules | 211 | |||
| No. of glycerol molecules | 2 | |||
| R.m.s.d., bonds () | 0.007 | |||
| R.m.s.d., angles () | 1.014 | |||
| Mean B factor (2) | 45.7 | |||
| Ramachandran analysis (%) | ||||
| Favored regions | 98.5 | |||
| Allowed regions | 1.5 | |||
| Outliers | 0 | |||
| PDB code | 4g3a | |||
R
merge = 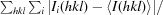
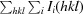 , where I(hkl) is the intensity of reflection hkl,
, where I(hkl) is the intensity of reflection hkl, 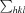 is the sum over all reflections and
is the sum over all reflections and 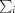 is the sum over i measurements of reflection hkl.
is the sum over i measurements of reflection hkl.
2.3. Structure determination
Crystals of underivatized (native) Mast-M1 were first obtained and diffraction data were collected from a single crystal; however, molecular replacement using protein models from the Dis1-TOG family [PDB entries 2of3 (Al-Bassam et al., 2007 ▶), 2qk1 (Slep & Vale, 2007 ▶) and 2qk2 (Slep & Vale, 2007 ▶)] gave no solutions and SeMet-derivative crystals were therefore prepared. These SeMet-derivative crystals were used for both phasing and structure refinement. A MAD data set collected from a single crystal to 2.40 Å resolution was used in the AutoSol module of PHENIX (Adams et al., 2002 ▶), where ten out of 16 Se sites were found, resulting in a figure of merit of 0.29. The AutoBuild module (Adams et al., 2002 ▶) of PHENIX was employed to trace 80% of the two Mast-M1 molecules in the crystallographic asymmetric unit into the initial electron-density map. The complete structure resulted from subsequent rounds of model building using Coot (Emsley & Cowtan, 2004 ▶) and refinement using REFMAC (Murshudov et al., 2011 ▶) and phenix.refine (Afonine et al., 2005 ▶).
The final model was further improved by refinement against 2.0 Å resolution diffraction data obtained from an improved SeMet Mast-M1 crystal using phenix.refine (Afonine et al., 2005 ▶). Refinement statistics are summarized in Table 1 ▶.
2.4. Computational methods
Crystallographic computations that are not explicitly named above were carried out using the CCP4 program suite (Winn et al., 2011 ▶). Macromolecular structure illustrations were prepared with PyMOL (DeLano, 2010 ▶). Structure-based alignment of multiple TOG-domain structures was performed using secondary-structure matching in PDBeFold (http://www.ebi.ac.uk/msd-srv/ssm/; Krissinel & Henrick, 2004 ▶). The conformational substructure searching and substructure overlays needed in the preparation of Fig. 2 were performed using the search services of the DrugSite server (https:/drugsite.msi.umn.edu/; Finzel et al., 2011 ▶).
2.5. αβ-Tubulin gel-filtration binding assay
A gel-filtration binding assay was used to assess the binding of Mast-M1 to αβ-tubulin. Purified Mast-M1 and pure αβ-tubulin (Cytoskeleton Inc.) were dialyzed against 25 mM Tris pH 7.5, 200 mM NaCl, 1 mM MgCl2, 1 mM EGTA buffer prior to use. Each protein was loaded separately onto a Sephacryl S200 HR column (GE Healthcare) at a concentration of 5 µM. To assay binding, Mast-M1 and αβ-tubulin were mixed at concentrations of 5 µM each and incubated for 30 min at 277 K. The mixture was then loaded onto the same Sephacryl column as mentioned above. For all of the runs, 3 ml fractions were collected and analyzed by SDS–PAGE electrophoresis.
2.6. Taxol-stabilized microtubule co-sedimentation assay
Microtubule binding of the Mast-M1 domain was performed using the Microtubule Binding Protein Spin-Down Assay kit from Cytoskeleton Inc. Mast-M1 was dialyzed against 80 mM HEPES pH 7.5, 25 mM NaCl, 5 mM MgCl2 buffer. Pure αβ-tubulin was reconstituted in 80 mM PIPES pH 7.0, 2 mM MgCl2, 0.5 mM EGTA buffer (tubulin buffer) supplemented with 1 mM GTP. Microtubules were assembled from pure αβ-tubulin by adding 80 mM PIPES pH 7.0, 1 mM MgCl2, 1 mM EGTA, 60% glycerol buffer and were incubated for 20 min at 308 K. Tubulin buffer enhanced with 20 µM Taxol (paclitaxel) was added to stabilize the microtubules.
Microtubules (4 µM) were mixed with the proteins Mast-M1 (4 and 2 µM), BSA (2.3 µM) and MAP (0.5 µM) in a total volume of 50 µl. Each solution was laid on top of a 60% glycerol cushion and spun at 100 000g for 40 min at 298 K. The resulting pellet and supernatant of each mixture was loaded onto an SDS–PAGE gel for analysis of microtubule binding.
2.7. Data deposition
The atomic coordinates of SeMet Mast-M1 have been deposited in the Protein Data Bank under accession code 4g3a.
3. Results and discussion
3.1. Structure of the N-terminal domain of MAST
The Mast-M1 structure embodies a fold similar to that of the Dis1-TOG family of proteins, possessing six HEAT-like repeats (A, B, C, D, E and F; Andrade & Bork, 1995 ▶) that arrange in a parallel manner, giving the characteristic flat paddle-like domain. HEAT-like repeats are characterized by a pair of antiparallel α-helices of 10–15 residues connected by a loop forming a helical hairpin (Fig. 1 ▶ a). One helix is frequently kinked. Many HEAT-repeat-containing proteins form long superhelical structures that possess an extraordinary right-handed curvature (Andrade et al., 2001 ▶). Mast-M1 and other TOG domains maintain a flat structure by interjecting a left-handed twist between centralized repeats; in Mast-M1, this twist exists between repeats C and D (Fig. 1 ▶ b).
Figure 1.

(a) Domain organization of Mast. (b) Structure of Mast-M1 displaying each HEAT-like repeat, where helix 1 is behind helix 2 and the helices are colored in pairs. (c) Top view of Mast-M1 obtained by rotating (b) 90° around the x axis and 90° around the y axis. (d) Structure-based alignment of the sequences of Mast-M1 with TOG domains from S. cerevisiae (Stu2p), D. melanogaster (Msps) and C. elegans (Zyg9). Capitalization signifies the presence of structural correspondence. Blue residues are 100% conserved, green residues are 100% conserved in size and/or charge and red residues are only 75% conserved in size and/or charge. The secondary structure at the top of the sequence is colored as in (b). Amino acids in the central turn of each HEAT-like repeat are identified with boxes labeled TA–TE.
The final structure of Mast-M1 contains two protein molecules in the asymmetric unit; monomer A includes residues 5–231 and monomer B includes residues 2–231. In molecule B, four extra residues from the C-terminal His tag added to facilitate protein isolation extend helix αF2 unnaturally. Otherwise, the two molecules are quite similar. Superposition of all common backbone atoms in the two monomers results in a root-mean-square difference in position of 0.87 Å. The largest differences are the result of a twist in the orientation of helices αB1 and αB2, culminating in an ∼5 Å slide in residues 61–64 that comprise the turn that connects them. A close contact between these residues in monomer B with αD2 of monomer A that arises from crystal packing may necessitate this conformational difference. This change illustrates the conformational malleability of this protein fold, in particular how it can adapt in response to intermolecular interactions. In the discussion below, we confine our comments and analysis to the conformation represented by monomer A.
Only one of the HEAT-like repeats in Mast-M1 is matched by the HEAT sequence profile (Pfam02985; Marchler-Bauer et al., 2013 ▶). This segment embeds αE1–αE2 (residues 170–198). Nevertheless, there is exceptional conservation in the detail of the conformation of several of the repeats, particularly in and around the central turn (identified as Tx in Fig. 1 ▶ d, where x refers to the repeat position A–E). This is important because it is the face of TOG domains dominated by these internal repeat turns that has consistently been implicated by mutation studies as being important for tubulin binding (Slep & Vale, 2007 ▶; Al-Bassam et al., 2007 ▶; Al-Bassam & Chang, 2011 ▶). A typical consensus turn geometry is represented by Mast-M1 turn αD1–αD2 (Mast1-M1 TD; Fig. 2 ▶ a), where residues are numbered with respect to a canonical position within a consensus 35-residue HEAT-like repeat beginning ten residues from the end of the first of the tandem helices. At residue 11 the helix begins to unwind into a more extended coil of exactly six residues that turns into the second helix beginning at residue 17. While the conformation of repeats varies wildly outside of this turn (Fig. 2 ▶ b), the turn backbone geometry is preserved intact in most repeats. The same turn geometry is conserved in Mast-M1 TB and TC, S. cerevisiae Stu2p TA, D. melanogaster Msps TA, C. elegans Zyg9 TA and TC and in turns TD and TE of all four TOG domains.
Figure 2.

Consensus geometry of central turns in TOG-domain HEAT-like repeats. (a) A representative HEAT-like repeat of the conserved turn geometry. Shown is the backbone and secondary structure of Mast-M1 residues 125–159 (turn D; TD) renumbered to reflect a position within a canonical HEAT-like repeat. Ribbon residues colored red (canonical residues 11–23) served as a target for substructure searching using distance geometry matching (Finzel et al., 2011 ▶). (b) 14 matching substructures from known TOG-domain structures including Mast-M1 TB, TC, TD and TE, Stu2p TA, TD and TE, Msps TA, TD and TE and Zyg9 TA, TC, TD and TE. Some repeats exhibit very different geometry outside of the turn (shown as a Cα trace). (c) The sequence logo (Schneider & Stephens, 1990 ▶) arising from alignment of these 14 similar substructures.
The sequences accommodated within this framework are diverse (Fig. 2 ▶ c), but typically include an arginine at position 21, where the guanidinium makes hydrogen bonds to the backbone carbonyl O atoms of turn residues 15 and 16. A histidine or lysine can fulfill a similar role. A second key amino acid occupies position 15. The side chain of this residue hydrogen-bonds to the backbone amide NH of residue 17 and makes hydrophobic interactions with the side chain of residue 20 (most often a valine). In the HEAT-repeat profile, aspartic acid is strongly preferred at the position corresponding to position 15 (Andrade et al., 2001 ▶), but in TOG domains the aspartic acid is often replaced by asparagine, serine, histidine or glutamic acid. It is notable that Stu2p TB and TC, Msps TB and TC, Zyg9 TB and Mast-M1 TA all lack the arginine (or alternate) at position 21 and all adopt a different turn geometry. Only Mast-M1 TB adopts this geometry in the absence of this arginine and in this case Arg98 from TC crosses over to make similar interactions that stabilize the turn.
Mast-M1 has previously been identified as a TOG domain (Slep & Vale, 2007 ▶). A structure-based alignment with other published dis1-TOG domain structures confirms the overall similarity (Fig. 1 ▶ d). Using aligned sequences, Mast-M1 appears to be more like Zyg9 TOG3 (28% identity) than Msps TOG2 (22%) or Stu2p TOG2 (16%), but it is even more closely related to the N-terminal TOG-like domain of human CLASP1 (43% identity). CLASP1 TOGL1 is one of three TOG-like domains subclassified in the phylogenetic analysis of Al-Bassam & Chang (2011 ▶). The Mast-M1 domain structure that is reported here is the first TOG-like domain to be structurally characterized. An alignment of the human CLASP1 TOGL1 sequence is also included in Fig. 1 ▶(d).
There are significant differences between Mast-M1 and the known TOG-domain structures in key residues conserved at the proposed tubulin-binding edge dominated by intra-repeat turns TA–TE (Al-Bassam & Chang, 2011 ▶). These turns collect specific charged amino acids at specific locations that are thought to create an electrostatic surface that promotes tubulin binding (Al-Bassam et al., 2007 ▶). Turn TA conserves the features of the comparable turns in the TOG5 and TOGL1 subgroups, but differs from the conserved x-W-x-x-R-x of the other TOG domains. Turn TB is unique to Mast-M1, but turns TC, TD and TE all resemble TOGL1 domains. Based on these important similarities, we would classify Mast-M1 as a TOGL1 domain.
3.2. Microtubule binding of Mast-M1
Although it has been reported that CLASP1 TOGL1 domains form stable complexes with soluble tubulin (Al-Bassam & Chang, 2011 ▶), we wanted to determine whether Mast-M1 could do so. We have tested Mast-M1 to assess its binding to soluble tubulin and/or tubulin incorporated into a microtubule lattice by using gel-filtration binding and microtubule co-sedimentation assays, respectively. The results obtained with a gel-filtration binding assay are shown in Fig. 3 ▶(b). Tubulin and Mast-M1 were individually run through the size-exclusion column in order to establish their elution times (red line, tubulin; blue line, Mast-M1). After incubating both proteins for 30 min, the sample was passed through the column and the eluted graph is displayed as a green line. If Mast-M1 bound tubulin, there would be a shift of the tubulin peak towards earlier elution; the absence of such a shift (peak of the green line at 107 min) confirms that tubulin binding does not occur. These results were also confirmed by running the eluted fractions of each trial on an SDS–PAGE gel, demonstrating that no Mast-M1 protein was present in the fractions containing eluted tubulin (data not shown).
Figure 3.

(a) SDS–PAGE gel with results from the microtubule co-sedimentation assay. The ladder is labeled in kDa. Lanes 2–5 are supernatant samples; lanes 6–9 are pellet samples. In the following, the amount of protein loaded into each gel lane is given in parentheses. Lane 1, Mast-M1 (5.0 mg); lanes 2 and 6, MAPF incubated with microtubules (1.6 mg MAPF + 1.0 mg microtubules); lanes 3 and 7, Mast-M1 not incubated with microtubules (0.5 mg); lanes 4 and 8, Mast-M1 incubated with microtubules (0.5 mg Mast-M1 + 1.0 mg microtubules); lanes 5 and 9, Mast-M1 incubated with microtubules (0.2 mg Mast-M1 + 1.0 mg microtubules). (b) Gel-filtration graph of tubulin (red line), Mast-M1 (blue line) and Mast-M1 incubated with tubulin (green line).
In the microtubule co-sedimentation assay, the microtubules will precipitate below the glycerol cushion, leaving the supernatant empty or almost empty of protein. If a protein binds to microtubules, it will co-precipitate with the microtubules; if not, it will stay on top of the glycerol cushion and will appear in the supernatant. The precipitated and supernatant samples were inspected by SDS–PAGE. Fig. 3 ▶(a) displays the SDS–PAGE gel of Mast-M1 exposed to microtubules. Lanes 4 and 5 demonstrate that the supernatant samples of Mast-M1 (4 and 2 µM, respectively) mixed with microtubules (4 µM). The Mast-M1 band is visible above the 21.5 kDa ladder band. (The small band above 45 kDa results from contamination with microtubules owing to difficulty in extracting the layers from the glycerol cushion.) Lane 3 presents Mast-M1 without exposure to microtubules at the same concentration as in lane 4; lane 7 is the complementary pellet sample. Lanes 8 and 9 contain precipitated samples of Mast-M1 exposed to microtubules, complementary to lanes 4 and 5, respectively. As can be observed, no significant amount of Mast-M1 was precipitated with microtubules. Lanes 2 and 6 represent a positive control created using a mixture of microtubule-associated proteins provided as part of the test kit (MAPF). These proteins are known to bind microtubules and demonstrate co-precipitation with microtubules (lane 6, band above 200 kDa.). There is no visible MAPF band for the supernatant sample (lane 2), just minor microtubule contamination, indicating that MAPF binds to microtubules. Comparing lane 2 with lanes 4 and 5 and lane 6 with lanes 8 and 9 leads to the conclusion that Mast-M1 is not binding to microtubules.
D. melanogaster tubulin is not commercially available; the tubulin used in the above experiments was isolated from rabbit. The possibility exists that Mast-M1 might bind native D. melanogaster microtubules with higher affinity. It should be noted, however, that tubulin sequences are very highly conserved; D. melanogaster, human, rabbit and bovine tubulin are all 97% identical. In other microtubulin-binding proteins, multiple TOG-like domains are required for microtubulin binding (Al-Bassam & Chang, 2011 ▶). Our data do not definitively show that Mast-M1 does not contribute to microtubulin binding, only that it is insufficient to manifest detectable binding on its own.
4. Conclusions
Mast-M1 adopts a flat paddle-like fold similar to that found in the Dis1-TOG domains of other published structures. The structure is the first representative of a class of TOG-like domains from CLIP-associated proteins and their homologs that are integral and essential structural features in proteins required for regulating microtubule dynamics during cell division. It is likely that variation in the sequence of loops central to each HEAT-like repeat work in concert with other TOG-like domains in these proteins to regulate the kinetics of microtubule assembly and collapse. The conformation of these loops is the same in the four central HEAT-like repeats of Mast-M1 and these dominate the proposed tubulin-binding edge. D. melanogaster Mast/Orbit possesses 57% sequence similarity to human CLIP-associating protein (CLASP; Galjart, 2005 ▶), and the N-terminal CLASP TOGL1 domain shares 43% sequence identity with the D. melanogaster Mast-M1 structure reported here. This report provides the first detailed characterization of a TOGL domain.
Our experimental evidence confirms that the Mast-M1 TOGL domain alone is insufficient to support the binding of microtubules in vitro and that it does not bind αβ-tubulin. It has been demonstrated that the N-terminal Dis1-TOG domain of CLASP binds β-actin in vivo (Tsvetkov et al., 2007 ▶). An evaluation of the binding of actin by Mast-M1 lies beyond the scope of this report.
Supplementary Material
PDB reference: Mast-M1, 4g3a
Acknowledgments
The authors thank Todd Geders for help with data collection and processing, Can Ergenekan and the facilities of the University of Minnesota Supercomputing Institute for access to software and Jay Nix at beamline 4.2.2 at ALS for access to the beam and help with data collection. Grant funding was provided by MN Department of Employment and Economic Development #SPAP-06-0014-P-FY07.
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Afonine, P. V., Grosse-Kunstleve, R. W. & Adams, P. D. (2005). Acta Cryst. D61, 850–855. [DOI] [PMC free article] [PubMed]
- Akhmanova, A., Hoogenraad, C. C., Drabek, K., Stepanova, T., Dortland, B., Verkerk, T., Vermeulen, W., Burgering, B. M., De Zeeuw, C. I., Grosveld, F. & Galjart, N. (2001). Cell, 104, 923–935. [DOI] [PubMed]
- Al-Bassam, J. & Chang, F. (2011). Trends Cell Biol. 21, 604–614. [DOI] [PMC free article] [PubMed]
- Al-Bassam, J., Larsen, N. A., Hyman, A. A. & Harrison, S. C. (2007). Structure, 15, 355–362. [DOI] [PubMed]
- Andrade, M. A. & Bork, P. (1995). Nature Genet. 11, 115–116. [DOI] [PubMed]
- Andrade, M. A., Petosa, C., O’Donoghue, S. I., Müller, C. W. & Bork, P. (2001). J. Mol. Biol. 309, 1–18. [DOI] [PubMed]
- DeLano, W. L. (2010). PyMOL http://www.pymol.org.
- Desai, A. & Mitchison, T. J. (1997). Annu. Rev. Cell Dev. Biol. 13, 83–117. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Finzel, B. C., Akavaram, R., Ragipindi, A., Van Voorst, J. R., Cahn, M., Davis, M. E., Pokross, M. E., Sheriff, S. & Baldwin, E. T. (2011). J. Chem. Inf. Model. 51, 1931–1941. [DOI] [PubMed]
- Galjart, N. (2005). Nature Rev. Mol. Cell Biol. 6, 487–498. [DOI] [PubMed]
- Guerrero, S. A., Hecht, H.-J., Hofmann, B., Biebl, H. & Singh, M. (2001). Appl. Microbiol. Biotechnol. 56, 718–723. [DOI] [PubMed]
- Inoue, Y. H., do Carmo Avides, M., Shiraki, M., Deak, P., Yamaguchi, M., Nishimoto, Y., Matsukage, A. & Glover, D. M. (2000). J. Cell Biol. 149, 153–166. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Lemos, C. L., Sampaio, P., Maiato, H., Costa, M., Omel’yanchuk, L. V., Liberal, V. & Sunkel, C. E. (2000). EMBO J. 19, 3668–3682. [DOI] [PMC free article] [PubMed]
- Maiato, H., Sampaio, P., Lemos, C. L., Findlay, J., Carmena, M., Earnshaw, W. C. & Sunkel, C. E. (2002). J. Cell Biol. 157, 749–760. [DOI] [PMC free article] [PubMed]
- Marchler-Bauer, A. et al. (2013). Nucleic Acids Res. 41, D348–D352. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Pflugrath, J. W. (1999). Acta Cryst. D55, 1718–1725. [DOI] [PubMed]
- Schneider, T. D. & Stephens, R. M. (1990). Nucleic Acids Res. 18, 6097–6100. [DOI] [PMC free article] [PubMed]
- Slep, K. C. & Vale, R. D. (2007). Mol. Cell, 27, 976–991. [DOI] [PMC free article] [PubMed]
- Tsvetkov, A. S., Samsonov, A., Akhmanova, A., Galjart, N. & Popov, S. V. (2007). Cell Motil. Cytoskeleton, 64, 519–530. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Mast-M1, 4g3a