

Abstract
Posttranslational modification of alpha-dystroglycan (α-DG) by the like-acetylglucosaminyltransferase (LARGE) is required for it to function as an extracellular matrix (ECM) receptor. Mutations in the LARGE gene have been identified in congenital muscular dystrophy patients with brain abnormalities. However, the precise function of LARGE remains unclear. Here we found that LARGE could act as a bifunctional glycosyltransferase, with both xylosyltransferase and glucuronyltransferase activities, which produced repeating units of [–3-xylose–α1,3-glucuronic acid-β1–]. This modification allowed α-DG to bind laminin-G domain–containing ECM ligands.
Protein glycosylation is important in determining cellular localization, modulating structural conformation, and providing recognition sites for other molecules (1). Dystroglycan (DG) is a highly glycosylated basement membrane receptor involved in a variety of physiological processes that maintain skeletal muscle membrane integrity, as well as central nervous system structure and function; it also serves as a receptor for Old World arenaviruses. Composed of a cell surface α subunit and a transmembrane β subunit (2), α-DG acts as a receptor for laminin-G domain–containing extracellular matrix (ECM) proteins such as laminin, agrin, and neurexin (3). α-DG undergoes N-glycosylation, mucin-type O-glycosylation, and O-mannosylation, and a yet-to-be identified modification of a phosphorylated O-mannosyl glycan is believed to be responsible for ligand binding (4). Perturbed glycosylation can lead to reduced ligand binding by α-DG and is a common pathologic feature among several congenital and limb-girdle muscular dystrophies. Mutations in the LARGE gene (5) and several others involved in O-mannosyl glycan synthesis have been identified in these disorders (6). The overexpression of LARGE enhances functional modification of α-DG, as well as laminin binding in cultured cells (7). Moreover, it circumvents defects in the modification of α-DG in cells from patients with several genetically distinct types of congenital muscular dystrophies (8). Although LARGE has been implicated in the postphosphoryl modification pathway that assembles the laminin-binding moiety onto the phosphorylated O-mannose of α-DG, its molecular role and the laminin binding epitope remain to be identified.
We investigated the LARGE-dependent modification on α-DG by performing compositional sugar analysis of the recombinant α-DG produced in LARGE-expressing human embryonic kidney (HEK) 293 cells (fig. S1). Samples were treated with glycosidases to eliminate glycans unrelated to the laminin-binding moiety (4). We detected: (i) known compositional sugars of the α-DG O-mannosyl glycan—specifically N-acetylglucosamine (GlcNAc), galactose (Gal), N-acetylgalactosamine (GalNAc), and mannose (Man) (4); (ii) glucose (Glc), which was likely a contaminant; and (iii) substantial amounts of glucuronic acid (GlcA) and xylose (Xyl) (Fig. 1A). GlcA and Xyl are essential components of heparan sulfate (HS) and chondroitin-dermatan sulfate (CS-DS) glycosaminoglycans (GAGs), whose biosynthesis is initiated by linkage of tetrasaccharide GlcAβ1-3Galβ1-3Galβ1-4Xylβ1- to proteoglycan core proteins. Notably, GAGs are not required for α-DG to bind laminin (9, 10) (fig. S2).
Fig. 1.

Xylosylation is required for functional modification of α-DG. (A) Compositional sugar analysis by gas chromatography-MS with trimethylsilyl derivatization using α-DG produced by LARGE-overexpressing HEK293 cells digested with peptide: N-glycosidase F, O-glycosidase, and neuraminidase. Peaks shown in red represent Xyl, those in blue represent GlcA, and numbers indicate 1, Man; 2, Gal; 3, Glc; 4, GlcNAc; 5, GalNAc; and 6, inositol (internal control). Some of the detected Gal was derived from the Jacalin elution buffer we used for the purification. (B) Flow cytometry of WT or UXS1-deficient (pgsI-208) CHO cells surface stained with antibodies against the laminin-binding epitope of α-DG (IIH6), the α-DG core protein (CORE), or heparan sulfate (HS). Dashed line, secondary antibody alone. (C) Functional modification of α-DG in cells overexpressing UXS1. (Left) Immunoblotting or laminin overlay (OL) assays of glycoproteins. Mr, relative molecular mass. Asterisk indicates nonfunctional α-DG that appeared as a sharp band because of hypomodification by CORE staining. (Right) Flow cytometry for surface staining with HS or IIH6. (D) Immunoblotting for IIH6 and β-DG–specific antibody reactivity in WT and pgsI-208 cells with or without overexpression of LARGE. (E) Immunoblotting for IIH6, CORE, or β-DG–specific antibody reactivity in LARGE-expressing WT cells treated with or without Xyl-α-pNP.
We tested whether xylosylation unrelated to GAG formation is involved in functional modification of α-DG by assessing uridine 5′-diphosphate (UDP)–xylose synthase 1 (UXS1)–deficient Chinese hamster ovary (CHO) cells (line pgsI-208), which are deficient in GAG synthesis because they lack UDP-Xyl (11). Immunoblotting with IIH6 (an antibody that recognizes the laminin-binding form of α-DG), laminin overlay of wheat germ agglutinin–enriched proteins (glycoproteins), and flow cytometric analysis revealed that these cells were defective in synthesis of the functional modification of α-DG (Fig. 1, B and C). Ectopic expression of UXS1 in pgsI-208 cells rescued HS-GAG synthesis, as well as the IIH6 reactivity and laminin-binding ability of α-DG (Fig. 1C). In contrast, overexpression of LARGE in these cells did not overcome the defect (Fig. 1D). Thus, xylosylation may be required for formation of the acceptor substrate on which LARGE acts, and/or LARGE itself may be involved in the transfer of Xyl to α-DG. Xylosides with hydrophobic moieties that help penetrate lipid bilayers are widely used to inhibit certain glycosyltransferases acting on Xyl, because they serve as artificial acceptors (12). Given that one of the two domains of LARGE belongs to glycosyltransferase family 8 (GT8), whose members generate products with an α-linked glycosidic bond (13), we tested α-D-xyloside for the ability to inhibit functional modification of α-DG. Indeed, treatment of LARGE-expressing CHO cells with p-nitrophenyl-α-D-xyloside (Xyl-α-pNP) reduced α-DG glycosylation (Fig. 1E). Thus xyloside may compete against endogenous α-DG bearing an α-linked Xyl for LARGE-dependent modification.
Next, we examined the enzymatic activity of LARGE with respect to Xyl-α-pNP as the acceptor, using a secreted form of LARGE that lacks its transmembrane domain (LARGEdTM). Purified LARGEdTM (fig. S3) was incubated with Xyl-α-pNP and each donor substrate, and the reaction products were separated by high-performance liquid chromatography (HPLC). A unique peak was detected only when UDP-GlcA was used as the donor (Fig. 2A for UDP-GlcA and fig. S4 for other donors). This activity was specific to α-linked Xyl (fig. S5). We purified the product (fig. S6) and analyzed it by mass spectrometry (MS). The MS/MS fragmentation pattern of a parent ion at mass/charge ratio, m/z, of 446.25 [M-H]− demonstrated that GlcA was attached to Xyl-α-pNP (Fig. 2B and fig. S7). Because LARGE showed glucuronyltransferase (GlcA-T) activity toward α-linked Xyl and has two glycosyltransferase-like domains, we hypothesized that it could also act on a nonreducing terminal GlcA. When 4-methylumbelliferyl-β-D-glucuronide (GlcA-β-MU) was used as an acceptor, a unique peak was observed only with UDP-Xyl (Fig. 2C for UDP-Xyl and fig. S8 for other donors); the attachment of Xyl to GlcA-β-MU was confirmed by MS (Fig. 2D and fig. S9).
Fig. 2.
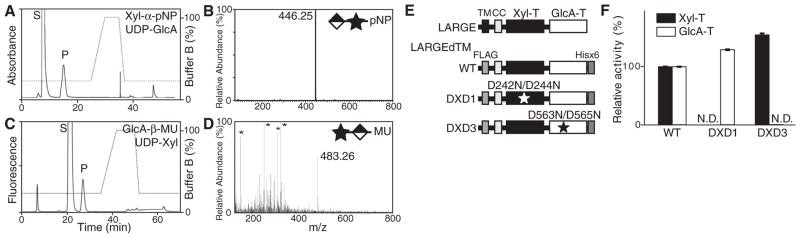
LARGE is a bifunctional glycosyltransferase that has Xyl-T and GlcA-T activities. (A) HPLC elution profile from the amide column GlycoSep N of the products obtained from the reaction of LARGEdTM with Xyl-α-pNP and UDP-GlcA. S, unreacted substrate. P, product. (B) Linear trap quadrupole (LTQ)–MS analysis of the product peak detected in (A). Stars and diamonds indicate Xyl and GlcA, respectively. MS/MS fragmentation pattern (fig. S7) confirmed that the ion with m/z of 446.3 [M-H]− is Xyl-α-pNP with an added GlcA. (C) HPLC elution profile from GlycoSep N of the products obtained from the reaction of LARGEdTM with GlcA-β-MU and UDP-Xyl. (D) As in the legend to (B), for the product isolated from the reaction analyzed in (C). The MS/MS fragmentation pattern (fig. S9) confirmed that the ion with m/z of 483.3 [M-H]− is GlcA-β-MU with an added Xyl. Asterisks indicate background ions. (E) Schematic representation of the WT and DXD mutants of LARGEdTM constructs used in the assay. The locations of the mutations in the DXD motifs are symbolized by stars. (F) GlcA-T and Xyl-T activities of WT and mutant LARGEdTMs. Relative activity (%) with respect to WT, and the standard deviation in triplicate experiments, are shown. TM, transmembrane domain. CC, coiled-coil domain. N.D., not detected.
Because mutations in the LARGE Asp-X-Asp (DXD; X, any amino acid) motifs (Fig. 2E), which are highly conserved across glycosyltransferases, lead to failed functional modification of α-DG (14), we generated constructs with such mutations for further testing of LARGE glycosyltransferase activity. In the case of the DXD1 mutant in which Asn replaces Asp at two sites (D242N/D244N), GlcA-Tactivity was comparable to that in wild type (WT), whereas Xyl-T activity was undetectable (Fig. 2F). In the DXD3 mutant D563N/D565N, in contrast, Xyl-T activity, but not GlcA-T activity, was present (Fig. 2F). Thus, LARGE is a bifunctional glycosyltransferase composed of two distinct catalytic domains, Xyl-T and GlcA-T.
Negatively charged oligosaccharides likely contribute to binding between α-DG and laminin (15), and anionic sugars distinct from sialic acid are involved (16). Thus, we tested whether LARGE could add Xyl and GlcA to the acceptor on α-DG, by performing the LARGEdTM reaction using both donors. Multiple products were generated for both acceptors (Xyl-α-pNP and GlcA-β-MU) (Fig. 3, A and B). We next asked whether this enzymatic activity could confer ligand-binding ability to α-DG in vitro, using skeletal muscle glycoproteins from the Largemyd mouse [in which a mutation in the LARGE gene causes defects in α-DG glycosylation (17)] as the acceptor substrate. Functional modification was robust only when both UDP-GlcA and UDP-Xyl were present (Fig. 3C). Thus, LARGE can assemble a polysaccharide with ligand-binding activity onto the immature glycan of the Largemyd α-DG.
Fig. 3.
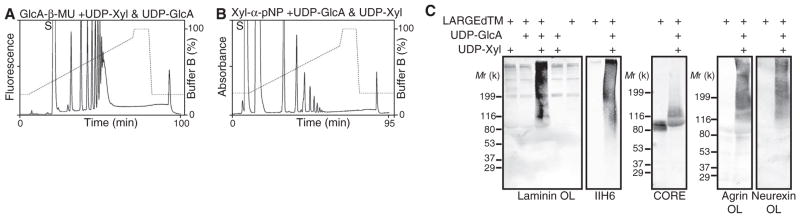
Polymerizing activity of LARGE confers ligand-binding ability on α-DG. HPLC profile of the reaction products of LARGEdTM generated from GlcA-β-MU (A) and Xyl-α-pNP (B) in the presence of both donors, UDP-Xyl and UDP-GlcA, on the amide column. S, unreacted substrate. (C) Glycoproteins extracted from Largemyd mouse skeletal muscle were incubated with LARGEdTM, with or without UDP-GlcA and UDP-Xyl, and analyzed by immunoblotting with IIH6 or the CORE antibody, or by overlay assays (OLs) using laminin-G domain–containing ECM ligands (laminin, agrin, or neurexin).
To identify the precise glycan structure that LARGE generates, we performed a large-scale enzymatic reaction with GlcA-β-MU, separated the products by gel filtration (Fig. 4A), and further purified those peaks (fig. S10). MS analysis of the product peaks ranging from a degree of polymerization (dp) of 3 to 6 showed m/z values corresponding to those of the products expected to be sequentially modified by GlcA and Xyl (Fig. 4B). Products dp2 to dp6 were further analyzed by nuclear magnetic resonance (NMR) to determine how the sugars are linked (fig. S11 to S15). Representative 13C/1H heteronuclear multiple quantum coherence spectroscopy (HMQC), total correlation spectroscopy (TOCSY), and rotating-frame Overhauser effect spectroscopy (ROESY) spectra for dp5 are shown in Fig. 4, C to E, respectively. The majority of the spin systems of the sugar residues can be traced and assigned using different TOCSY mixing times together with double-quantum filtered correlated spectroscopy (COSY) spectra (Fig. 4D and fig. S11). GlcA residues c and e were linked via β1-3 linkages to Xyl residues b and d, respectively (Fig. 4E); and Xyl residues b and d were connected to GlcA residues a and c via α1-3 linkages, respectively (Fig. 4E). The structures of products dp2 to dp6 are shown in Fig. 4F; complete NMR assignments are listed in table S1. The fact that the glycosidic linkages were preserved among all tested products indicates that LARGE is a polymerizing enzyme with UDP-GlcA:αXyl β1,3-GlcA-T and UDP-Xyl: β GlcA α1,3-Xyl-T activities.
Fig. 4.

MS and NMR analyses of products obtained from in vitro LARGE enzymatic reaction products, using GlcA-β-MU as the acceptor and UDP-GlcA and UDP-Xyl as the donors. (A) Separation profile of polymeric oligosaccharide by Superdex Peptide 10/300. S, unreacted substrate. (B) Matrix-assisted laser desorption–ionization tandem MS analysis of products dp3 to dp6. (C) HMQC spectrum of dp5 at 15°C. Assigned cross peaks are labeled with a letter representing the subunit [as designated in (F)], and a number representing the position on that subunit. The cross peak derived from sample impurities is marked by an asterisk. (D) TOCSY spectrum of dp5, collected with a mixing time of 120 ms at 15°C. (E) ROESY spectrum of dp5, collected with a mixing time of 300 ms at 15°C for the assignment of interglycosidic linkages (indicated by blue circles). The first letter in each label refers to the sugar subunit and the second to the hydrogen position of that subunit. (F) Structures of the polymeric oligosaccharides produced by the LARGE enzymatic reaction in vitro, with the sugar subunits labeled a to f.
Here, we have identified the enzymatic function of LARGE, which produces a polysaccharide with repeating units of [-3Xyl-α1,3GlcAβ1-]. We believe that LARGE synthesizes the polymer on the O-mannosyl glycan of α-DG in vivo, but can possibly act on other glycans and/or other protein substrates when the enzyme concentration is artificially high. α-DG binds the LG4 and 5 domains of laminin alpha chains, and it is suggested that three basic patches contribute to these interactions (18). The LARGE-synthesized, negatively charged glycan on α-DG likely binds to laminin through electrostatic associations with these basic patches, a notion supported by the fact that one patch, 2719RKR contributes to binding with heparin, which contains acidic sugars GlcA and iduronic acid. The glycan we have observed resembles heparin-HS– and CS-DS–GAGs, linear polysaccharides that consist of repeating disaccharides and are synthesized by copolymerases with dual glycosyltransferase activities. Our findings may extend the types of GAG that provide a platform to retain molecules onto cell surface and/or in the ECM.
Supplementary Material
Acknowledgments
We thank all members of the Campbell laboratory for fruitful discussions and D. Venzke and Z. Zhu for technical assistance, J. D. Esko for providing pgsI-208 cells, and the University of Iowa Proteomics Facility and Flow Cytometry Facility, Iowa State University Hybridoma Facility, and the Glycotechnology Core Resource (UCSD) for their services. This work was supported in part by a Paul D. Wellstone Muscular Dystrophy Cooperative Research Center grant (1U54NS053672). K.P.C. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
References and Notes
- 1.Varki A, et al., editors. Essentials of Glycobiology. 2. Cold Spring Harbor; New York: 2009. [PubMed] [Google Scholar]
- 2.Ibraghimov-Beskrovnaya O, et al. Nature. 1992;355:696. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- 3.Barresi R, Campbell KP. J Cell Sci. 2006;119:199. doi: 10.1242/jcs.02814. [DOI] [PubMed] [Google Scholar]
- 4.Yoshida-Moriguchi T, et al. Science. 2010;327:88. doi: 10.1126/science.1180512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Longman C, et al. Hum Mol Genet. 2003;12:2853. doi: 10.1093/hmg/ddg307. [DOI] [PubMed] [Google Scholar]
- 6.Godfrey C, Foley AR, Clement E, Muntoni F. Curr Opin Genet Dev. 2011;21:278. doi: 10.1016/j.gde.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 7.Kanagawa M, et al. Cell. 2004;117:953. doi: 10.1016/j.cell.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 8.Barresi R, et al. Nat Med. 2004;10:696. doi: 10.1038/nm1059. [DOI] [PubMed] [Google Scholar]
- 9.Ervasti JM, Campbell KP. J Cell Biol. 1993;122:809. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamada H, et al. J Neurochem. 1996;66:1518. doi: 10.1046/j.1471-4159.1996.66041518.x. [DOI] [PubMed] [Google Scholar]
- 11.Bakker H, et al. J Biol Chem. 2009;284:2576. doi: 10.1074/jbc.M804394200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwartz NB, Galligani L, Ho PL, Dorfman A. Proc Natl Acad Sci USA. 1974;71:4047. doi: 10.1073/pnas.71.10.4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coutinho PM, Deleury E, Davies GJ, Henrissat B. J Mol Biol. 2003;328:307. doi: 10.1016/s0022-2836(03)00307-3. [DOI] [PubMed] [Google Scholar]
- 14.Brockington M, et al. Hum Mol Genet. 2005;14:657. doi: 10.1093/hmg/ddi062. [DOI] [PubMed] [Google Scholar]
- 15.Hohenester E, Tisi D, Talts JF, Timpl R. Mol Cell. 1999;4:783. doi: 10.1016/s1097-2765(00)80388-3. [DOI] [PubMed] [Google Scholar]
- 16.Combs AC, Ervasti JM. Biochem J. 2005;390:303. doi: 10.1042/BJ20050375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grewal PK, Holzfeind PJ, Bittner RE, Hewitt JE. Nat Genet. 2001;28:151. doi: 10.1038/88865. [DOI] [PubMed] [Google Scholar]
- 18.Harrison D, et al. J Biol Chem. 2007;282:11573. doi: 10.1074/jbc.M610657200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.