

Abstract
Background
Candidate genes associated with idiopathic forms of autism overlap with other disorders including fragile X syndrome. Our laboratory has previously shown reduction in fragile X mental retardation protein (FMRP) and increase in metabotropic glutamate receptor 5 (mGluR5) in cerebellar vermis and superior frontal cortex (BA9) of individuals with autism.
Methods
In the current study we have investigated expression of four targets of FMRP and mGluR5 signaling - homer 1, amyloid beta A4 precursor protein (APP), ras-related C3 botulinum toxin substrate 1 (RAC1), and striatal-enriched protein tyrosine phosphatase (STEP) - in the cerebellar vermis and superior frontal cortex (BA9) via SDS-PAGE and western blotting. Data were analyzed based on stratification with respect to age (children and adolescents vs. adults), anatomic region of the brain (BA9 vs. cerebellar vermis), and impact of medications (children and adolescents on medications (n = 4) vs. total children and adolescents (n = 12); adults on medications (n = 6) vs. total adults (n = 12)).
Results
There were significant increases in RAC1, APP 120 kDa and APP 80 kDa proteins in BA9 of children with autism vs. healthy controls. None of the same proteins were significantly affected in cerebellar vermis of children with autism. In BA9 of adults with autism there were significant increases in RAC1 and STEP 46 kDa and a significant decrease in homer 1 vs. controls. In the vermis of adult subjects with autism, RAC1 was significantly increased while APP 120, STEP 66 kDa, STEP 27 kDa, and homer 1 were significantly decreased when compared with healthy controls. No changes were observed in vermis of children with autism. There was a significant effect of anticonvulsant use on STEP 46 kDa/β-actin and a potential effect on homer 1/NSE, in BA9 of adults with autism. However, no other significant confound effects were observed in this study.
Conclusions
Our findings provide further evidence of abnormalities in FMRP and mGluR5 signaling partners in brains of individuals with autism and open the door to potential targeted treatments which could help ameliorate the symptoms of autism.
Keywords: Autism, RAC1, Homer 1, APP, STEP, BA9, Cerebellar vermis, Children, Adults
Background
Autism is a neurodevelopmental disorder that is characterized by impairments in social reciprocal interaction, communication, and repetitive and stereotyped patterns of behavior, interests, and activities [1]. A number of neuropathologic abnormalities have been characterized in brains from individuals with autism including macrocephaly, volumetric and cellular abnormalities of the frontal cortex, parietal cortex, the limbic structures, and cerebellum, and cortical minicolumnar disorganization [2-4]. Recently, the Centers for Disease Control and Prevention (CDC) reported a prevalence for autism of 11.3 per 1,000 (one in 88) children aged eight years in the United States in 2008 [5]. This represents a 78% increase in incidence over the previously reported finding of 6.4 per 1,000 in 2002 [5].
Beyond core symptoms, people with autism display a number of comorbidities including seizure disorder, intellectual disability, and other cognitive impairments [6]. The presence of seizure disorder has been estimated from 5% to 40% [6]. Epileptiform activity has been shown to cause brief episodes of impaired cognitive function known as transitory cognitive impairment (TCI) [7,8] and may contribute to cognitive deficits in individuals with autism. Due to the heterogeneous nature of autism spectrum disorders, it is not surprising that multiple gene families have been implicated in the pathology of autism [9-11] with 50 gene or gene variants accounting for approximately 30% of autism spectrum cases [11]. Many of these candidate genes overlap with those of other disorders with autistic behavioral deficits including tuberous sclerosis [12], fragile X syndrome (FXS) [13], Rett syndrome [14], and Angelman syndrome [15].
There is a wide degree of overlap between behavioral deficits of autism and FXS, the most common inherited form of intellectual disability. Indeed, approximately 25% to 47% of people with FXS display a comorbid diagnosis of autism [16,17]. Similar to those diagnosed with autism, people diagnosed with FXS display learning deficits, delayed language acquisition, impaired motor skills, and repetitive behavior [18]. FXS is caused by mutations in the fragile X mental retardation 1 gene (FMR1) leading ultimately to the loss of fragile X mental retardation protein (FMRP) expression. FMRP binds approximately 4% of all mRNAs expressed in brain [19,20] and acts primarily as a translational repressor. FMRP is expressed in both glia and neurons [21,22] and in neurons is highly localized to the dendrites and spines [23-25].
FMRP acts as a negative regulator of group I metabotropic glutamate signaling, particularly metabotropic glutamate receptor 5 (mGluR5) and it is believed that runaway glutamatergic signaling, particularly in the dendrites, is ultimately responsible for the deficits associated with FXS [26]. An anatomical abnormality of neurons from brains of individuals with FXS, autism, and Fmr1 knockout (KO) mice are dendrites with an overabundance of immature long, thin spines [27-29]. The profusion of dendritic spines could lead to an abnormally large amount of synapse formation and result in the cognitive impairments associated with FXS as well autism. In individuals with autism, greater spine density has been correlated with lower cognitive function [29].
We have previously observed reductions in FMRP in the cerebellar vermis and superior frontal cortex (Brodmann Area 9 (BA9)) of adults with autism and increased expression of mGluR5 in the vermis and BA9 of children with autism [30,31]. These represent the first findings of altered FMRP and mGluR5 in individuals with autism who do not have a comorbid diagnosis of FXS. In the current study, we have expanded upon our initial studies to examine protein expression of four known targets of FMRP and mGluR5 signaling that may play a role in regulating spine density, protein synthesis, and synaptic transmission: homer 1, amyloid beta A4 precursor protein (APP), ras-related C3 botulinum toxin substrate 1 (RAC1), and striatal-enriched protein tyrosine phosphatase (STEP) in the same brain regions. We hypothesized that targets of FMRP would display altered expression, further implicating this signaling pathway in the etiology of autism. Alterations in levels of these targets of FMRP could potentially be corrected therapeutically to ameliorate symptoms of autism.
Methods
Tissue preparation
The Institutional Review Board of the University of Minnesota, School of Medicine approved all experimental procedures for this study. Frozen postmortem blocks of the superior frontal cortex (BA9) and cerebellar vermis were obtained from the NICHD Brain and Tissue Bank for Developmental Disorders, University of Maryland, Baltimore, MD; the Harvard Brain Tissue Resource Center; the Brain Endowment Bank, Miami, Florida; and the Autism Tissue Program. The tissue samples (Table 1) were prepared as described previously [30,31] and each sample included both grey and white matter. None of the controls had a history of neuropsychiatric disorders, seizure disorder, or intellectual disability.
Table 1.
Demographic data for individuals with autism and controls
| Case | Dx | Sex | Age | PMI (hrs) | Ethnicity | Medication history | Cause of death | Seizure | ID | Side of brain | Brain region |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UMB-4670 |
Control |
M |
4 |
17 |
Caucasian |
None |
Commotio cordis |
No |
No |
Unknown |
BA9, Vermis |
| UMB-4898 |
Control |
M |
7 |
12 |
Caucasian |
Methylphenidate; clonidine |
Drowning |
No |
No |
Unknown |
Vermis |
| UMB-1674 |
Control |
M |
8 |
36 |
Caucasian |
None |
Drowning |
No |
No |
Unknown |
BA9, Vermis |
| UMB-4787 |
Control |
M |
12 |
15 |
African American |
Montelukast; albuteral; prednisone, loratadine |
Asthma |
No |
No |
Unknown |
BA9, Vermis |
| UMB-1823 |
Control |
M |
15 |
18 |
Caucasian |
None |
MVA |
No |
No |
Unknown |
BA9, Vermis |
| AN17425 |
Control |
M |
16 |
26.16 |
Unknown |
None |
Heart attack |
No |
No |
Left |
BA9 |
| AN15105 |
Control |
M |
18 |
19.83 |
Unknown |
None |
Unknown |
No |
No |
Unknown |
BA9, Vermis |
| AN03217 |
Control |
M |
19 |
18.58 |
Caucasian |
None |
Pneumonia |
No |
No |
Left |
BA9 |
| UMB-1846 |
Control |
F |
20 |
9 |
Caucasian |
None |
MVA |
No |
No |
Unknown |
BA9, Vermis |
| AN07176 |
Control |
M |
21 |
29.91 |
Unknown |
None |
MVA |
No |
No |
Left |
BA9 |
| AN14368 |
Control |
M |
22 |
24.2 |
Unknown |
None |
Unknown |
No |
No |
Left |
BA9 |
| AN19760 |
Control |
M |
28 |
23.25 |
Unknown |
None |
Unknown |
No |
No |
Left |
BA9 |
| AN15566 |
Control |
F |
32 |
28.92 |
Unknown |
None |
Unknown |
No |
No |
Left |
BA9, Vermis |
| UMB-1169 |
Control |
M |
33 |
27 |
African American |
Metoclopramide; loratadine |
Dilated cardiomyopathy (morbid obesity) |
No |
No |
Unknown |
BA9, Vermis |
| UMB-1376 |
Control |
M |
37 |
12 |
African American |
None |
ASCVD |
No |
No |
Unknown |
BA9, Vermis |
| AN15151 |
Control |
M |
41 |
30.4 |
Unknown |
None |
Heart attack |
No |
No |
Unknown |
BA9, Vermis |
| AN19440 |
Control |
F |
50 |
20.25 |
Unknown |
None |
Heart attack |
No |
No |
Unknown |
BA9 |
| AN12240 |
Control |
M |
51 |
4.75 |
Caucasian |
None |
Heart attack |
No |
No |
Right |
BA9 |
| AN13295 |
Control |
M |
56 |
22.12 |
Unknown |
None |
Unknown |
No |
No |
Right |
BA9 |
| AN08873 |
Autism |
M |
5 |
25.5 |
Caucasian |
None |
Drowning |
No |
No |
Left |
BA9 |
| AN13872 |
Autism |
F |
5 |
32.73 |
Asian |
None |
Drowning |
No |
No |
Left |
BA9, Vermis |
| UMB-1349 |
Autism |
M |
5 |
39 |
Caucasian |
None |
Drowning |
No |
No |
Unknown |
Vermis |
| UMB-1174 |
Autism |
F |
7 |
14 |
Caucasian |
None |
Seizure disorder |
Yes |
No |
Unknown |
BA9 |
| UMB-4231 |
Autism |
M |
8 |
12 |
African American |
Olanzapine; galantamine |
Drowning |
No |
Yes |
Unknown |
BA9, Vermis |
| AN19511 |
Autism |
M |
8 |
22.16 |
Caucasian |
None |
Cancer |
Yes |
No |
Left |
BA9, Vermis |
| UMB-4721 |
Autism |
M |
8 |
16 |
African American |
None |
Drowning |
No |
No |
Unknown |
Vermis |
| AN16641 |
Autism |
M |
9 |
27 |
Caucasian |
Carbamazepine; methylphenidate; clonidine |
Seizure disorder |
Yes |
Yes |
Left |
BA9 |
| AN16115 |
Autism |
F |
11 |
12.88 |
Caucasian |
Carbamazepine; lamotrigine; dextroamphetamine; topiramate |
Seizure/drowning |
Yes |
Yes |
Right |
Vermis |
| UMB-4899 |
Autism |
M |
14 |
9 |
Caucasian |
None |
Drowning |
No |
No |
Unknown |
BA9, Vermis |
| AN17138 |
Autism |
M |
16 |
24 |
Asian |
Fexofenadine; buspirone; topiramate |
Seizure disorder |
Yes |
No |
Left |
BA9, Vermis |
| AN01570 |
Autism |
F |
18 |
6.75 |
Caucasian |
None |
Seizure disorder |
Yes |
No |
Right |
BA9 |
| AN00764 |
Autism |
M |
20 |
23.66 |
Caucasian |
Erythromycin gel; minocycline |
MVA |
No |
Yes |
Left |
BA9 |
| UMB-1638 |
Autism |
F |
20 |
50 |
Caucasian |
None |
Seizure disorder |
Yes |
Yes |
Unknown |
BA9 |
| AN09730 |
Autism |
M |
22 |
25 |
Caucasian |
Aripiprazole; lamotrigine; zonisamide |
Aspiration |
Yes |
No |
Left |
BA9, Vermis |
| AN08166 |
Autism |
M |
29 |
43.25 |
Caucasian |
Fexofenadine; ziprasidone HCl; carbamazepine |
Seizure (suspected) |
Yes |
No |
Left |
BA9, Vermis |
| AN12457 |
Autism |
F |
29 |
17.83 |
Caucasian |
Fluvoxamine |
Seizure disorder |
Yes |
No |
Left |
BA9, Vermis |
| AN11989 |
Autism |
M |
30 |
16.06 |
Caucasian |
None |
Heart failure (congestive) |
No |
No |
Left |
BA9 |
| AN08792 |
Autism |
M |
30 |
20.33 |
Caucasian |
Cisapride; clorazepate; sodium valproate; phenytoin; folic acid; primidone; phenobarbital; omeprazole; metoclopramide |
Gastrointestinal bleeding |
Yes |
No |
Left |
BA9, Vermis |
| UMB-5027 |
Autism |
M |
37 |
26 |
African American |
None |
Obstruction of bowel due to adhesion |
No |
No |
Unknown |
BA9, Vermis |
| AN06420 |
Autism |
M |
39 |
13.95 |
Caucasian |
None |
Cardiac tamponade |
No |
No |
Left |
BA9, Vermis |
| AN14613 |
Autism |
M |
39 |
22.75 |
Caucasian |
None |
Sudden unexpected death |
Yes |
No |
Unknown |
BA9 |
| AN17777 |
Autism |
F |
49 |
16.33 |
Caucasian |
Wafarin; venlafaxine; erythromycin; lansoprazole; risperidone; metformin; gabapentin; propranolol; levothyroxine |
Pulmonary arrest |
No |
Yes |
Left |
BA9, Vermis |
| AN01093 |
Autism |
M |
56 |
19.48 |
Caucasian |
Benztropine mesylate; haloperidol; lithium; chlorpromazine; alprazolam |
Anoxic encephalopathy |
Yes |
No |
Right |
BA9, Vermis |
|
Children |
Control |
Autistic |
Change |
P-value |
d |
|
|||||
| Age ± SD (years) |
11.4 ± 5.22 |
9.5 ± 4.38 |
↓16.6% |
ns |
nd |
|
|||||
| PMI ± SD (years) |
20.6 ± 8.1 |
20.1 ± 9.94 |
↓2.4% |
ns |
nd |
|
|||||
| Gender |
7M |
8M:4F |
nd |
nd |
nd |
|
|||||
|
Adults |
Control |
Autistic |
Change |
P-value |
d |
|
|||||
| Age ± SD (years) |
34.2 ± 13 |
33.3 ± 11.2 |
↓2.6% |
ns |
nd |
|
|||||
| PMI ± SD (years) |
20.9 ± 8.41 |
24.6 ± 11.1 |
↑17.7% |
ns |
nd |
|
|||||
| Gender | 9M:3F | 9M:3F | nd | nd | nd | ||||||
ASCVD, Arteriosclerotic cardiovascular disease; Dx, Diagnosis; Hrs, Hours; PMI, Postmortem interval; M, Male; F, Female; EtOH, Alcohol; ID, Intellectual disability; MVA, Motor vehicle accident; nd, Not determined; ns, Not significant; d, Cohen’s test statistic.
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and western blot analysis
Tissue samples from the cerebellar vermis (vermal lobule unknown) (in adults, n = 5 controls and 8 autistic adults; in children, n = 6 controls and 8 autistic children) and BA9 (in adults, n = 12 controls and 12 autistic adults; in children, n = 6 controls and 9 autistic children) were prepared. Children were defined as those younger than 13 years of age; adolescents were defined as those between the ages of 13 years to 18 years; and adults were defined as those 19 years of age or older. For this study, children and adolescents were grouped together. Samples were mixed with denaturing SDS sample buffer and denatured by heating at 100°C for 5 minutes. SDS-PAGE gels were prepared using standard Laemmli solutions. For experiments involving RAC1 or STEP, 12% resolving gels were used; 10% resolving gels were used for experiments involving homer 1, neuronal specific enolase (NSE), or β-actin, and for experiments involving APP, 6% resolving gels were used. In all cases, 5% stacking gels were used. Thirty μg of protein per lane was loaded onto the gel and electrophoresed for 15 minutes at 75 V followed by 55 minutes at 150 V at room temperature (RT). We minimized interblot variability by including samples from subjects of each group (control and autism) on each gel. Samples were run in duplicate. Proteins were electroblotted onto nitrocellulose membranes for 2 hrs at 300 mAmp at 4°C as previously described [30,31]. Blots were blocked with 0.2% I-Block (Tropix, Bedford, MA, USA) in PBS with 0.3% Tween 20 for one hour at RT followed by an overnight incubation in primary antibodies at 4°C. The primary antibodies used were: anti-RAC1 (1:500; BD Transduction, San Jose, CA, USA), anti-APP (1:250; Abcam Inc., Cambridge, MA, USA), anti-homer 1 (1:500; Abnova, Taipei, Taiwan), anti-STEP (1:200; Abgent, San Diego, CA, USA), anti-NSE (1:2,000; Abcam Inc.), and anti-β-actin (1:5,000; Sigma Aldrich, St. Louis, MO, USA). Blots were washed for 30 minutes and incubated in secondary antibodies for one hour at RT as previously described [30,31]. Secondary antibodies were goat anti-rabbit IgG (A9169, Sigma Aldrich, 1:80,000) or goat anti-mouse IgG (A9044, Sigma Aldrich, 1:80,000). Blots were washed twice in PBS supplemented with Tween-20 (PBST) and bands were visualized using the ECL plus detection system (GE Healthcare, Little Chalfont, Buckinghamshire, UK) and exposed to CL-Xposure film (Thermo Scientific, Rockford, IL, USA). The molecular weights of approximately 120 kDa and 88 kDa (APP); 66 kDa, 61 kDa, 46 kDa, 33 kDa, and 27 kDa (STEP); 46 kDa (NSE); 45 kDa (homer 1); 42 kDa (β-actin); and 21 kDa (RAC1) immunoreactive bands were quantified with background subtraction using a RioRad densitometer and BioRad MultiAnalyst software (Bio-Rad, Hercules, CA, USA). Results obtained were based on two to four independent experiments.
Statistical analysis
Statistical analysis of protein data was performed as previously described [30,32-34]. The independent samples t-test was used to compare people with autism to healthy controls. Separate analyses were conducted for children and adults. Analysis of covariance was used to estimate the effect of covariates on group comparisons. Effect sizes were calculated using Cohen’s d statistic, with values >0.8 considered a large effect [35]. For analysis of confounders, significance was set at P <0.06.
Results
All western blotting results were normalized against β-actin and NSE and are expressed as ratios to β-actin and NSE. In the cerebellar vermis of adults with autism we observed significantly increased expression of the RAC1/β-actin ratio (P <0.025, d = 1.62) and the RAC1/NSE ratio (P <0.016, d = 1.73) (Figures 1 and 2, Table 2). We also observed a significant reduction in expression of the APP 120 kDa/β-actin ratio (P <0.018, d = −2.04) and the APP 120 kDa/NSE ratio (P <0.012, d = −2.40) (Figures 1 and 2, Table 2). Ratios for STEP 66 kDa/β-actin (P <0.023, d = −1.51), STEP 66 kDa/NSE (P <0.018, d = −1.59), STEP 33 kDa/β-actin (P <0.024, d = −1.63) and STEP 33 kDa/NSE (P <0.020, d = −1.68) were significantly reduced in the cerebellar vermis of adults with autism (Figures 1 and 3, Table 2). STEP 27 kDa/NSE was also significantly reduced in the cerebellar vermis of subjects with autism (P <0.038, d = −1.40) (Table 2). We did not observe alterations in levels of homer 1 or APP 88 kDa in the cerebellar vermis of adults with autism and there were no significant changes for any of the proteins in the cerebellar vermis of children with autism (Figures 1, 2, 3, Table 2).
Figure 1.
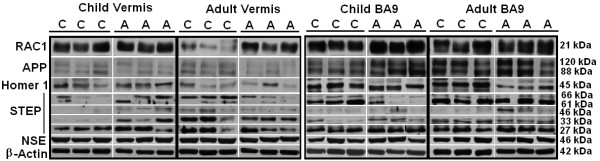
Representative samples of RAC1, APP, Homer 1, STEP, NSE, and β-actin from the cerebellar vermis and BA9 from controls and people with autism. C, controls; A, people with autism; RAC1, Ras-related C3 botulinum toxin substrate 1; APP, amyloid beta A4 precursor protein; STEP, striatal-enriched protein tyrosine phosphatase; NSE, neuronal specific enolase; BA9, Brodmann Area 9.
Figure 2.

Expression of RAC1, APP, and homer 1 in the cerebellar vermis of subjects with autism and controls. (A) Mean RAC1/β-actin, APP 120 kDa/β-actin, APP 88/β-actin, and homer 1/β-actin ratios for controls (gray histogram bars) and people with autism (black histogram bars) are shown for the cerebellar vermis. (B) Mean RAC1/NSE, APP 120 kDa/NSE, APP 88/NSE, and homer 1/NSE ratios for controls (gray histogram bars) and people with autism (black histogram bars) are shown for the cerebellar vermis. Error bars express standard error of the mean. *P <0.05. RAC1, Ras-related C3 botulinum toxin substrate 1; amyloid beta A4 precursor protein; NSE, neuronal specific enolase.
Table 2.
Western blotting results for RAC1, homer 1, APP, STEP, NSE, and β-actin and their ratios in the cerebellar vermisa
| Control | Autistic | Change | P-value | Cohen’s d | |
|---|---|---|---|---|---|
|
Adults |
|
|
|
|
|
| RAC1/β-actin |
0.315 ± 0.206 |
1.3 ± 0.714 |
313%↑ |
0.025b |
1.62b |
| Homer 1/β-actin |
0.237 ± 0.303 |
0.147 ± 0.147 |
38%↓ |
ns |
0.42 |
| APP 120 kDa/β-actin |
0.091 ± 0.029 |
0.050 ± 0.016 |
45%↓ |
0.018b |
−2.04b |
| APP 88 kDa/β-actin |
0.08 ± 0.06 |
0.03 ± 0.02 |
63%↓ |
ns |
1.26 |
| STEP 66 kDa/β-actin |
0.136 ± 0.116 |
0.025 ± 0.029 |
82%↓ |
0.023b |
−1.51b |
| STEP 61 kDa/β-actin |
0.015 ± 0.028 |
0.003 ± 0.004 |
80%↓ |
ns |
−0.67 |
| STEP 46 kDa/β-actin |
0.03 ± 0.034 |
0.034 ± 0.041 |
13%↑ |
ns |
0.11 |
| STEP 33 kDa/β-actin |
0.55 ± 0.38 |
0.17 ± 0.13 |
69%↓ |
0.024b |
−1.63b |
| STEP 27 kDa/β-actin |
0.70 ± 0.67 |
0.53 ± 0.39 |
24%↓ |
ns |
−0.32 |
| β-actin |
10.2 ± 1.67 |
9.06 ± 3.1 |
11%↓ |
ns |
nd |
|
Children |
|
|
|
|
|
| RAC1/β-actin |
1.09 ± 0.545 |
1.1 ± 0.471 |
0.9%↑ |
ns |
0.01 |
| Homer 1/β-actin |
0.188 ± 0.13 |
0.218 ± 0.109 |
16%↑ |
ns |
0.26 |
| APP 120 kDa/β-actin |
0.071 ± 0.029 |
0.077 ± 0.033 |
8.4%↑ |
ns |
0.19 |
| APP 88 kDa/β-actin |
0.084 ± 0.055 |
0.100 ± 0.071 |
19%↑ |
ns |
0.25 |
| STEP 66 kDa/β-actin |
0.01 ± 0.008 |
0.059 ± 0.091 |
490%↑ |
ns |
0.64 |
| STEP 61 kDa/β-actin |
0.002 ± 0.002 |
0.023 ± 0.031 |
1050%↑ |
ns |
1.15 |
| STEP 46 kDa/β-actin |
0.0022 ± 0.0011 |
0.024 ± 0.024 |
991%↑ |
ns |
1.16 |
| STEP 33 kDa/β-actin |
0.057 ± 0.085 |
0.13 ± 0.17 |
128%↑ |
ns |
0.51 |
| STEP 27 kDa/β-actin |
0.70 ± 0.68 |
0.53 ± 0.38 |
24%↓ |
ns |
−0.72 |
| β-actin |
9.48 ± 1.11 |
9.52 ± 3.71 |
0.4%↑ |
ns |
nd |
|
Adults |
|
|
|
|
|
| RAC1/NSE |
0.23 ± 0.16 |
0.93 ± 0.49 |
304%↑ |
0.016b |
1.73b |
| Homer 1/NSE |
0.18 ± 0.24 |
0.099 ± 0.11 |
45%↓ |
ns |
−0.46 |
| APP 120 kDa/NSE |
0.062 ± 0.021 |
0.031 ± 0.008 |
50%↓ |
0.012b |
−2.40b |
| APP 88 kDa/NSE |
0.06 ± 0.05 |
0.02 ± 0.01 |
67%↓ |
ns |
−1.22 |
| STEP 66 kDa/NSE |
0.18 ± 0.14 |
0.032 ± 0.04 |
82%↓ |
0.018b |
−1.59b |
| STEP 61 kDa/NSE |
0.018 ± 0.035 |
0.004 ± 0.005 |
78%↓ |
ns |
−0.21 |
| STEP 46 kDa/NSE |
0.039 ± 0.041 |
0.037 ± 0.042 |
5.1%↓ |
ns |
−0.59 |
| STEP 33 kDa/NSE |
0.72 ± 0.49 |
0.21 ± 0.17 |
71%↓ |
0.020b |
−1.68b |
| STEP 27 kDa/NSE |
1.27 ± 0.82 |
0.44 ± 0.38 |
65%↓ |
0.038b |
−1.40b |
| NSE |
7.42 ± 1.26 |
7.69 ± 1.3 |
3.6%↑ |
ns |
nd |
|
Children |
|
|
|
|
|
| RAC1/NSE |
0.84 ± 0.51 |
0.69 ± 0.23 |
18%↓ |
ns |
−0.38 |
| Homer 1/NSE |
0.13 ± 0.09 |
0.16 ± 0.07 |
23%↑ |
ns |
0.33 |
| APP 120 kDa/NSE |
0.040 ± 0.025 |
0.056 ± 0.021 |
40%↑ |
ns |
0.70 |
| APP 88 kDa/NSE |
0.054 ± 0.025 |
0.072 ± 0.053 |
33%↑ |
ns |
0.42 |
| STEP 66 kDa/NSE |
0.014 ± 0.011 |
0.102 ± 0.165 |
628%↑ |
ns |
0.61 |
| STEP 61 kDa/NSE |
0.0024 ± 0.001 |
0.037 ± 0.054 |
1441%↑ |
ns |
0.96 |
| STEP 46 kDa/NSE |
0.003 ± 0.002 |
0.03 ± 0.047 |
900%↑ |
ns |
1.02 |
| STEP 33 kDa/NSE |
0.078 ± 0.12 |
0.23 ± 0.33 |
194%↑ |
ns |
0.55 |
| STEP 27 kDa/NSE |
0.36 ± 0.24 |
0.99 ± 0.93 |
175%↑ |
ns |
0.77 |
| NSE | 6.97 ± 0.84 | 6.23 ± 1.53 | 10.6%↓ | ns | nd |
Values for control and autistic groups are presented as mean ± SD aRAC1, ras-related C3 botulinum toxin substrate 1; APP, amyloid beta A4 precursor protein; STEP, striatal-enriched protein tyrosine phosphatase; NSE, neuronal specific enolase; nd, not determined; ns, not significant. bStatistically significant.
Figure 3.
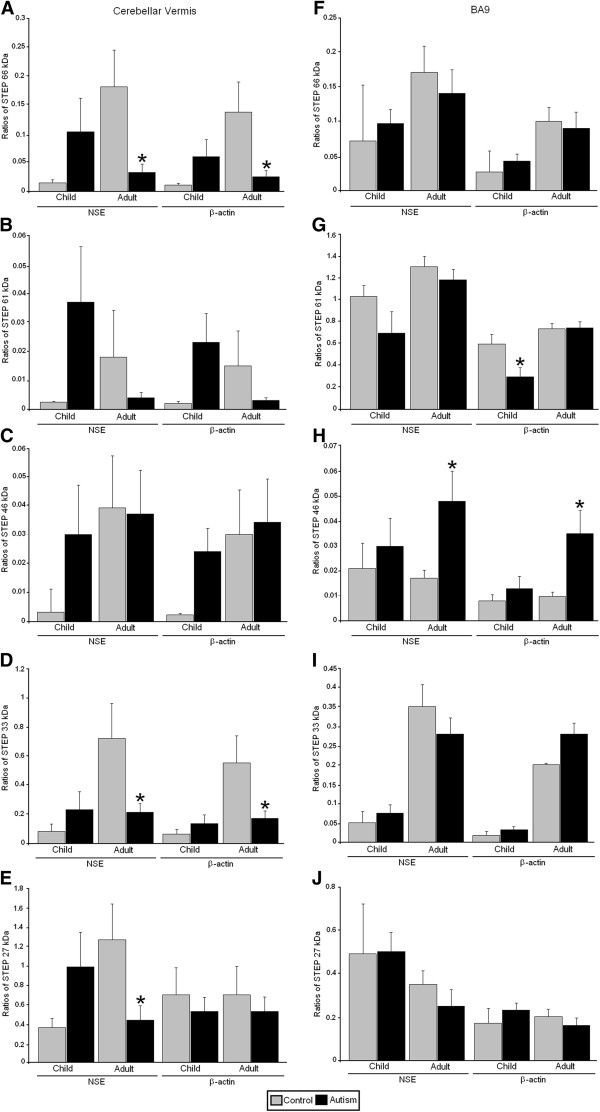
Expression of STEP in the cerebellar vermis and BA9 of subjects with autism and controls. Control group data are presented as gray histogram bars; autistic group data are presented as black histogram bars. (A) and (F) STEP 66 kDa. (B) and (G) STEP 61 kDa. (C) and (H) STEP 46 kDa. (D) and (I). STEP 33 kDa. (F) and (J) STEP 27 kDa. Error bars express standard error of the mean. *P <0.05. STEP, striatal-enriched protein tyrosine phosphatase; NSE, neuronal specific enolase; BA9, Brodmann Area 9.
In BA9 of adults with autism we observed significantly increased ratios for RAC1/β-actin (P <0.031, d = 1.32), RAC1/NSE (P <0.042, d = 1.24) and significantly reduced ratios for homer 1/β-actin (P <0.027, d = −1.37), homer 1/NSE (P <0.020, d = −1.43) (Figures 1 and 4, Table 3). STEP 46 kDa/β-actin ratio (P <0.012, d = 1.10) and STEP 46 kDa/NSE ratio (P <0.020, d = 1.02) were significantly increased in BA9 of adults with autism (Figures 1 and 3, Table 3). In BA9 of children with autism we observed significant increases in ratios for RAC1/β-actin (P <0.008, d = 2.74), RAC1/NSE (P <0.017, d = 2.09), APP 120 kDa/β-actin (P <0.032, d = 2.03), APP 120 kDa/NSE (P <0.017, d = 2.12), APP 88 kDa/β-actin (P <0.025, d = 2.54) and APP 88 kDa/NSE (P <0.012, d = 3.24) (Figures 1 and 4, Table 3). Finally, there was a significant reduction in STEP 61 kDa/β-actin ratio (P <0.036, d = −1.23) in BA9 of children with autism (Figures 1 and 3, Table 3).
Figure 4.

Expression of RAC1, APP, and homer 1 in BA9 of subjects with autism and controls. (A) Mean RAC1/β-actin, APP 120 kDa/β-actin, APP 88/β-actin, and homer 1/β-actin ratios for controls (gray histogram bars) and people with autism (black histogram bars) are shown for BA9. (B) Mean RAC1/NSE, APP 120 kDa/NSE, APP 88/NSE, and homer 1/NSE ratios for controls (gray histogram bars) and people with autism (black histogram bars) are shown for BA9. Error bars express standard error of the mean. *P <0.05. RAC1, Ras-related C3 botulinum toxin substrate 1; amyloid beta A4 precursor protein; NSE, neuronal specific enolase; BA9, Brodmann Area 9.
Table 3.
Western blotting results for RAC1, homer 1, APP, STEP, NSE, and β-actin and their ratios in BA9a
| Control | Autistic | Change | P-value | Cohen’s d | |
|---|---|---|---|---|---|
|
Adults |
|
|
|
|
|
| RAC1/β-actin |
0.834 ± 0.554 |
1.66 ± 0.65 |
99%↑ |
0.031b |
1.32b |
| Homer 1/β-actin |
0.512 ± 0.152 |
0.324± 0.132 |
37%↓ |
0.027b |
−1.37b |
| APP 120 kDa/β-actin |
0.41 ± 0.23 |
0.45 ± 0.24 |
9.8%↑ |
ns |
0.17 |
| APP 88 kDa/β-actin |
0.46 ± 0.19 |
0.53 ± 0.24 |
15%↑ |
ns |
0.29 |
| STEP 66 kDa/β-actin |
0.10 ± 0.07 |
0.09 ± 0.08 |
10%↓ |
ns |
−0.11 |
| STEP 61 kDa/β-actin |
0.73 ± 0.17 |
0.74 ± 0.18 |
1.3%↑ |
ns |
0.08 |
| STEP 46 kDa/β-actin |
0.0097 ± 0.006 |
0.035 ± 0.031 |
261%↑ |
0.012b |
1.10 |
| STEP 33 kDa/β-actin |
0.20 ± 0.13 |
0.18 ± 0.09 |
10%↓ |
ns |
−0.25 |
| STEP 27 kDa/β-actin |
0.20 ± 0.12 |
0.16 ± 0.12 |
20%↓ |
ns |
−0.29 |
| β-actin |
13.2 ± 2.41 |
13.1 ± 2.31 |
0.75%↓ |
ns |
nd |
|
Children |
|
|
|
|
|
| RAC1/β-actin |
1 ± 0.616 |
2.8 ± 0.7 |
180%↑ |
0.008b |
2.74b |
| Homer 1/β-actin |
0.46 ± 0.17 |
0.77 ± 0.31 |
67%↑ |
ns |
1.21 |
| APP 120 kDa/β-actin |
0.20 ± 0.07 |
0.63 ± 0.25 |
215%↑ |
0.032b |
2.03b |
| APP 88 kDa/β-actin |
0.22 ± 0.03 |
0.75 ± 0.26 |
241%↑ |
0.025b |
2.54b |
| STEP 66 kDa/β-actin |
0.027 ± 0.026 |
0.043 ± 0.031 |
59%↑ |
ns |
0.54 |
| STEP 61 kDa/β-actin |
0.59 ± 0.22 |
0.29 ± 0.24 |
51%↓ |
0.036b |
−1.23b |
| STEP 46 kDa/β-actin |
0.008 ± 0.006 |
0.013 ± 0.014 |
62%↑ |
ns |
0.45 |
| STEP 33 kDa/β-actin |
0.017 ± 0.026 |
0.033 ± 0.023 |
94%↑ |
ns |
0.64 |
| STEP 27 kDa/β-actin |
0.17 ± 0.17 |
0.23 ± 0.10 |
35%↑ |
ns |
0.45 |
| β-actin |
11.5 ± 1.31 |
11.7 ± 2.05 |
1.7%↑ |
ns |
nd |
|
Adults |
|
|
|
|
|
| RAC1/NSE |
0.50 ± 0.33 |
0.95 ± 0.37 |
90%↑ |
0.042b |
1.24 |
| Homer 1/NSE |
0.34 ± 0.12 |
0.19 ± 0.10 |
44%↓ |
0.020b |
−1.43 |
| APP 120 kDa/NSE |
0.25 ± 0.12 |
0.27 ± 0.16 |
8%↑ |
ns |
0.16 |
| APP 88 kDa/NSE |
0.28 ± 0.07 |
0.32 ± 0.14 |
14%↑ |
ns |
0.27 |
| STEP 66 kDa/NSE |
0.17 ± 0.13 |
0.14 ± 0.12 |
18%↓ |
ns |
−0.35 |
| STEP 61 kDa/NSE |
1.3 ± 0.34 |
1.18 ± 0.33 |
9.2%↓ |
ns |
−0.25 |
| STEP 46 kDa/NSE |
0.017 ± 0.011 |
0.048 ± 0.041 |
182%↑ |
0.020b |
1.02b |
| STEP 33 kDa/NSE |
0.35 ± 0.20 |
0.28 ± 0.14 |
20%↓ |
ns |
−0.46 |
| STEP 27 kDa/NSE |
0.35 ± 0.21 |
0.25 ± 0.17 |
29%↓ |
ns |
−0.53 |
| NSE |
9.14 ± 3.73 |
8.59 ± 1.41 |
6%↓ |
ns |
nd |
|
Children |
|
|
|
|
|
| RAC1/NSE |
0.61 ± 0.42 |
1.33 ± 0.21 |
118%↑ |
0.017b |
2.09b |
| Homer 1/NSE |
0.28 ± 0.11 |
0.40 ± 0.15 |
43%↑ |
ns |
0.94 |
| APP 120 kDa/NSE |
0.12 ± 0.04 |
0.33 ± 0.12 |
175%↑ |
0.017b |
2.12b |
| APP 88 kDa/NSE |
0.13 ± 0.004 |
0.39 ± 0.11 |
200%↑ |
0.012b |
3.24b |
| STEP 66 kDa/NSE |
0.072 ± 0.081 |
0.097 ± 0.072 |
34%↑ |
ns |
0.33 |
| STEP 61 kDa/NSE |
1.03 ± 0.25 |
0.69 ± 0.60 |
33%↓ |
ns |
−0.68 |
| STEP 46 kDa/NSE |
0.021 ± 0.021 |
0.030 ± 0.033 |
43%↑ |
ns |
0.37 |
| STEP 33 kDa/NSE |
0.050 ± 0.083 |
0.075 ± 0.054 |
50%↑ |
ns |
0.37 |
| STEP 27 kDa/NSE |
0.49 ± 0.57 |
0.50 ± 0.26 |
2%↑ |
ns |
0.031 |
| NSE | 6.58 ± 2.48 | 5.63 ± 1.3 | 14%↓ | ns | nd |
Values for control and autistic groups are presented as mean ± SD.aRAC1, ras-related C3 botulinum toxin substrate 1; APP, amyloid beta A4 precursor protein; STEP, striatal-enriched protein tyrosine phosphatase; NSE, neuronal specific enolase; BA9, Brodmann’s area 9; nd, not determined; ns, not significant. bStatistically significant.
In adults, we examined ethnicity, postmortem interval (PMI), intellectual disability, history of seizures, antidepressant, antipsychotic drug (APD), and anticonvulsant use in relation to the outcome measures. PMI, gender and intellectual disability were not significantly related to any of the values expressed as ratios of β-actin or NSE. Adults using anticonvulsants displayed significantly higher STEP 46 kDa/β-actin than all adults who did not use anticonvulsants (including autistic as well as nonautistic) (t(22) = 5.23, P <0.001). Adults with autism who were taking anticonvulsants displayed significantly increased expression of STEP 46 kDa/β-actin versus adults with autism who were not taking anticonvulsants (t(10) = 2.85, P <0.017). Moreover, adults with autism who were taking anticonvulsants displayed significantly increased expression of STEP 46 kDa/β-actin than adult controls (t(15) = 5.28, P <0.001). Finally, there was no significant difference in STEP 46 kDa/β-actin when adult controls were compared against adults with autism who were not taking anticonvulsants (t(17) = 1.44, P <0.17). Increased STEP 46 kDa expression was only seen in ratios compared to β-actin but not NSE, indicating that the difference was not specific to neuronal cells. By the same token, STEP 46 kDa/β-actin value in BA9 was also significantly different in those with seizure history versus those without (t(22) = 3.00, P <0.007). However, no significant difference existed between values in adults with autism with seizure disorder versus adults with autism who did not have seizure disorder, nullifying this association (mean 0.043 ± 0.034 versus 0.023 ± 0.027, P <0.29). Adults with autism who were taking anticonvulsants displayed significantly lower homer 1/NSE in BA9 than controls and adults with autism who were not taking anticonvulsants (t(15) = 2.69, P <0.017). While a comparison of homer 1/NSE values between adults with autism who took anticonvulsants (mean of 0.13 ± 0.086) versus those who did not (mean of 0.23 ± 0.085) did not show a significant difference (P <0.063), it is possible that the reduction of homer 1/NSE is at least partly due to anticonvulsant use.
For those using APDs, there were significantly reduced expression of APP 120 kDa/NSE in BA9 (t(15) = 2.20, P <0.044), APP 88 kDa/NSE in BA9 (t(14) = 2.63, P <0.02), and STEP 33 kDa/β-actin (t(22) = 2.18, P <0.040). However, none of these values showed any significant changes in adults with autism when compared with controls (P <0.8, P <0.55, and P <0.55, respectively). STEP 46 kDa/β-actin was significantly higher (t(22) = 2.79, P <0.011) in BA9 in subjects who took APD medication than in those who did not. However, comparison of values for STEP 46 kDa/β-actin in BA9 between subjects with autism taking APDs versus subjects with autism who did not take APDs was not statistically significant (mean 0.051 ± 0.031 versus 0.027 ± 0.03, respectively, P <0.22). For antidepressant use, the single individual taking antidepressants displayed significantly higher expression of APP 88 kDa/NSE in BA9 than the others (t(14) = 2.51, P <0.025) but this difference only reflects change in one subject and is not extendable to the whole group and thus, not amenable to statistical testing. There was a confounding effect of gender on homer 1/NSE in BA9 of adults (t(14) = 2.04, P <0.060). There was a significant reduction in homer 1/NSE in BA9 of adults with autism versus controls (P <0.020) (see Table 3). The mean for male subjects with autism was 0.262 ± 0.081 and the mean for female subjects with autism was 0.417 ± 0.15 (P <0.061), which was not significantly different. Due to having only two subjects for comparison, we were unable to compare female controls with female autistic subjects. Comparing male controls to male autistic subjects resulted in a significant reduction, similar to that reported in Table 3. It is therefore unlikely that gender has a meaningful effect on homer 1/NSE values. There was also a significantly higher expression of RAC1/NSE in Caucasian than African American subjects in BA9 (t(11) = 2.86, P <0.016). However, this comparison is not relevant as the African American sample size numbered only two subjects. Finally, there was an impact of anticonvulsant use on APP 88 kDa/NSE in BA9 of adults (t(14) = 2.12, P <0.052). However, there was no significant difference between adults with autism versus controls for APP 88 kDa/NSE in BA9 of adults, so this is not meaningful.
For children, ethnicity (Caucasian versus African American), history of seizures, antidepressant, APD, and anticonvulsant use were not significantly related to any of the β-actin or NSE measures. There was significant positive correlation between PMI and APP 88 kDa/NSE in BA9 of children (R = 0.734, P <0.060). However, PMI was not significantly different between control or autistic children in BA9, so this finding is not meaningful. For gender, one female subject displayed significantly higher APP 120 kDa/NSE in BA9 than male subjects (t(6) = 3.16, P <0.02). However, this significance could not be linked to a group of subjects, and thus, was not amenable to further statistical testing. Those with intellectual disability had significantly higher ratios on STEP 46 kDa/β-actin BA9 (t(13) = 2.81, P <0.015]. However, STEP 46 kDa/β-actin in BA9 was not significantly different between controls and those with autism.
To further investigate the effect of medications on our results we performed a second analysis comparing controls to people with autism who were not taking medication (Additional files 1: Table S1 and Additional file 2: Table S2). In the cerebellar vermis of adults, RAC1/NSE and RAC1/β-actin remained significant (Additional file 1: Table S1). In BA9 of adults, RAC1/NSE and RAC1/β-actin remained significant as did RAC1/β-actin in children along with APP 120 kDa/NSE, APP 120 kDa/β-actin, APP 88 kDa/NSE and APP 88 kDa/β-actin (Additional file 2: Table S2). Omitting people with autism who were on medication reduced the total number available for analysis so that seven comparisons could no longer be made, and in fifteen comparisons with a sample of three subjects for people with autism, power was reduced thus, affecting statistical significance.
Finally, we tested to see if there were statistical correlations between levels of FMRP or mGluR5 that we had previously reported [30,31] and levels of RAC1, STEP 66 kDa, STEP 61 kDa, STEP 46 kDa, STEP 33 kDa, STEP 27 kDa, APP120 kDa, APP 88 kDa, or homer 1. We did not find any significant statistical correlation for either FMRP or mGluR5 with any of the targets investigated in the current study (data not shown).
Discussion
The current studies demonstrated abnormal expression of several downstream biochemical targets of FMRP and mGluR5-mediated signaling in the brains of children and adults with autism. The most salient results included: 1) upregulation of RAC1 in BA9 and the vermis of adults with autism, and in BA9 only among children with autism; 2) upregulation of APP120 and 88 kDa species in BA9 of children with autism and downregulation of APP 120 kDa in the vermis of adults with autism; 3) upregulation of STEP 46 kDa in BA9 and downregulation of STEP 66 kDa, 33 kDa, and 27 kDa in the vermis of adults with autism; 4) downregulation of STEP 61 kDa in BA9 of children with autism; and 5) downregulation of homer 1 in BA9 of adults with autism. We observed more significant effects in BA9 than in the cerebellar vermis, consistent with previous findings in autism [36]. The results summarized above will be discussed further and the significant involvement of these proteins in autism will be analyzed in the following paragraphs.
RAC1 is a member of the Rho family of GTPases, a subfamily of the RAS superfamily of small guanine triphosphate (GTP)-binding proteins. Molecules in this superfamily regulate a number of cellular processes including kinase activation, cytoskeletal rearrangement, cell growth, differentiation, and survival [37]. Members of the Rho family have been implicated in cell proliferation, apoptosis, and regulation of gene expression [37]. RAC1 is involved in remodeling of the actin cytoskeleton [38] and plays an important role in the formation and maturation of dendritic spines as well as regulation of spine density and morphology [39-43]. Mutations in the Rho GTPase pathway and their regulators have been involved in intellectual disability-related disorders [43]. For example, RAC1 partners with oligophrenin-1 and synaptojanin which have been linked to X-linked intellectual disability and Down’s syndrome, respectively [43-45]. Overexpression of RAC1 in cultured hippocampal neurons younger than 11 days in vitro resulted in the formation of dendritic spines, clustering of 2-amino-3-(5-methyl-3-oxo-1,2- oxazol-4-yl)propanoic acid (AMPA) receptors and increased the amplitude of miniature excitatory postsynaptic currents, suggesting that RAC1 also enhances excitatory synaptic transmission [42].
RAC1 has been shown to interact with FMRP via cytoplasmic FMRP interacting proteins (CYFIP1/2) [46,47]. RAC1-induced actin remodeling is altered in mouse fibroblasts that lack FMRP, suggesting a role for FMRP in modulating actin dynamics [48]. More recently, RAC1 expression has been demonstrated to be increased in the hippocampus, cortex, brainstem, and cerebella of Fmr1 KO mice suggesting that FMRP acts as a negative regulator of RAC1 [49]. Our results of increased expression of RAC1 in the cerebellar vermis and BA9 of adults with autism, and in BA9 of children with autism, are novel and have never previously been reported in human postmortem brains of subjects with autism; they may be the result of reduced expression of FMRP in the same regions [30,31], and they mirror the same findings in Fmr1 KO mice. The overexpression of RAC1, particularly in BA9, may suggest increased excitatory signaling, which could impact synaptic transmission and ultimately cognitive function. This is especially true as studies using transgenic mice with a constitutively active form of RAC1 show overproduction of small abnormal supernumerary spines [43,50,51], indicating that overproduction of RAC1 in BA9 of both children and adults with autism is evidence of overabundance of active and abnormal neuronal spines in the brains of people with autism examined in this study. There is evidence in the literature that RAC1 may stimulate stellation of primary astrocytes [52] and increase migration of astrocytes [53]. Additionally, some residual RAC1 mRNA and protein activity has been detected in glial cells [49]. It is tempting to speculate that RAC1 overexpression in autism may also be responsible for increased glial fibrillary acidic protein (GFAP) immunoreactivity detected in the brains of subjects with autism [54]. Higher levels of RAC1 in Caucasian adults with autism compared to African American adults with autism is a novel finding that requires further investigation, and it may be due to genetic or epigenetic factors.
Studies have demonstrated significantly elevated levels of secreted APP and its cleavage products, including beta amyloid (Aβ), in individuals with autism [55-57]. Our current results, particularly the increased expression of APP 120 kDa and 88 kDa in BA9 of children with autism, verify these earlier findings. Children with severe autism and aggression have been shown to express at least twice as much APP when compared with control children and four times as much APP when compared to children with mild autism [55]. APP translation is normally repressed by FMRP, and mGluR5 activation releases this repression, which may explain why APP increases are seen only in children and not adults [58,59]. In support of this, levels of mGluR5 were also elevated in the same specimens as previously reported [30,31], further buttressing the notion that activation of mGluR5 in children may be the cause of our observed increases in APP 88 and 120 kDa. Antipsychotic and antidepressant drugs can affect levels of APP [60,61]. However, this scenario is unlikely, as evaluation of the impact of these medications on APP values proved not to be meaningful. APP is present presynaptically in the active zone where it has multiple potential interaction partners, including synaptic vesicle proteins, transporters and channels, and cell adhesion molecules [62]. APP is also present postsynaptically where it has been shown to coprecipitate with N-methyl-D-aspartate receptor (NMDAR) subunits [63], and may be involved in surface trafficking of NMDARs [64,65]. Thus, altered expression of APP may result in altered NMDA signaling, which is crucial for learning and memory. In Fmr1 KO mice, there are excess levels of secreted APP [59]. This excess of APP may result in decreased neural pruning and increased neural proliferation, helping to account for the presence of long, immature dendrites of neurons from patients with autism and FXS [59]. In Fmr1 KO mice that have been engineered to remove one App allele, there is a significant reduction in expression of Aβ and multiple phenotypes of FXS, including the presence of audiogenic seizures, the ratio of immature dendritic spines to mature spines, and long-term depression, were partially or fully corrected after removal of the App allele [66]. While the presence of APP in this report probably reflects neuronal activity, increases in brain APP have also been correlated with increased GFAP in both Alzheimer’s disease [67] and autism [54] and these could also reflect glial activity.
STEP is a phosphatase that is implicated in multiple cellular processes. Kim et al. [68] have found increased tyrosine phosphatase activity in response to mGluR5 stimulation in Fmr1 KO mice, leading to reduced phosphorylated extracellular signal-regulated kinases 1 and 2 (ERK1/2), molecules implicated in induction and maintenance of synaptic plasticity [69]. This tyrosine phosphatase is thought to be STEP, as STEP translation is initiated in response to mGluR stimulation [70]. Alternative splicing of the protein tyrosine phosphatase, nonreceptor type 5 (PTPN5) gene, which encodes STEP, produces four isoforms, however, only two - STEP 61 kDa and STEP 46 kDa - have phosphatase activity [71,72]. STEP 61 kDa is primarily membrane-bound and expressed in the postsynaptic density extrasynaptic sites, and the endoplasmic reticulum [73]. STEP 46 kDa is primarily cytosolic [73]. Other isoforms of STEP include STEP 66 kDa, STEP 27 kDa, and STEP 33 kDa. STEP 33 kDa is a cleavage product of STEP 61 kDa [74]. Recent investigations have implicated STEP in the etiology of neuropsychiatric disorders [74]. Carty et al. [75] reported significant increases in STEP 61 kDa in the anterior cingulate and dorsolateral prefrontal cortices of subjects with schizophrenia. STEP has roles in mediating NMDA and AMPA receptor endocytosis [70,76], suggesting that it has a role in mGluR5-mediated long-term depression (LTD), a phenomenon that is enhanced in Fmr1 KO mice [77].
Basal levels of STEP are elevated in Fmr1 KO mice [73]. Recent studies have shown that genetic reduction of STEP improves cognition, synaptic plasticity, and NMDA receptor subunit expression in mouse models of Alzheimer’s disease [78] and reduced seizures, anti-social, and anxiety-related behaviors in Fmr1 KO mice [73]. We observed reduced expression patterns for STEP 66 kDa, 61 kDa, 33 kDa, and 27 kDa with only STEP 46 kDa showing a significant increase in BA9 of adults with autism. As there was an increase in one of the active forms of STEP and a decrease in the other active form, further experiments are needed to determine what role STEP might play in the pathology of autism. However, nonsignificant trends for increase in several isoforms of STEP were observed in the vermis of both children and adults with autism, with large effect sizes. Thus, it is possible that these increases are due to compensatory processes in response to reductions observed in FMRP. Evaluation of the impact of various confounders such as APDs, anticonvulsant drugs, and seizure disorder on STEP values did not prove to be meaningful, with the exception of an anticonvulsant effect on STEP46 kDa/β-actin in BA9. Data from Hasegawa et al. [79] show there is also minimal presence of STEP 46 kDa in reactive astrocytes of gerbil hippocampi and STEP 46 kDa/NSE data reflect STEP 46 kDa species in neuronal cells, which do not show any association with anticonvulsant use. However, evidence for seizure increasing STEP expression remains controversial [73,80], Thus, one cannot rule out the possibility of multiple factors modulating the levels of STEP molecules [75].
Homer 1 is a postsynaptic density (PSD) scaffolding protein that interacts with mGluR5 to anchor it in the postsynaptic membrane by linking mGluRs with inositol triphosphate, Shank, and PSD-95/Guanylate kinase associated protein (GKAP) [81,82]. Giuffrida et al. [83] found that in the postsynaptic membrane of neurons from Fmr1 KO mice there was a reduction of mGluR5 in the insoluble fraction of the membrane as well as a reduction in phosphorylated homer 1. The interaction between homer 1 and mGluR5 regulates the cell surface expression, lateral mobility, and signaling of mGluRs [84-87]. Moreover, disruption of homer 1-mGluR5 interactions blocks mGluR5-induced long-term depression and protein synthesis in wild-type animals but not in Fmr1 KO mice [87]. The authors suggest that in Fmr1 KO mice, interactions between homer 1 and mGluR5 are sufficiently reduced for homer 1 to no longer have a role in the regulation of synaptic plasticity [87]. The observed reduction in homer 1 expression in BA9 of adults with autism may have functional consequences resulting in altered glutamatergic expression and cognitive deficits associated with both FXS and autism. A recent report [88] showed that homer 1 is a risk gene in autism, thus validating our novel finding. Recent evidence indicates that treatment with lithium or valproic acid causes reductions in expression of homer 1b/1c [89]. Thus, we cannot rule out a potential confounding effect of anticonvulsant drug use on homer 1/NSE in BA9 of adults, consistent with this previous finding.
Reduced FMRP expression has also been shown to cause reduction in expression of gamma aminobutyric acid (GABA) receptor subunits (α1, α3, α4, β1, β2, β3, γ1, γ2, δ) in Fmr1 KO mice [90-93]. Moreover, in the Drosophila melanogaster fragile X model (dFMR1−/−), there are significant reductions in three receptors that have a high homology with ionotropic GABA receptors: resistant to dieldrin (Rdl), GABA and glycine-like receptor of Drosophila (Grd), and ligand-gated chloride channel homolog 3 (Lcch3) [91]. Our laboratory has found significantly reduced protein expression of various GABA(A) and GABA(B) receptor subunits in three brain regions (namely, the cerebellum, parietal cortex and BA9) of subjects with autism [32-34]. In BA9 we observed significantly reduced expression of GABAA receptor subunit α1 (GABRα1), GABAA receptor subunit α4 (GABRα4), GABAA receptor subunit α5 (GABRα5), GABAA receptor subunit β1 (GABRβ1), and GABAB receptor subunit 1 (GABBR1) [32-34]. In the parietal cortex we observed significantly reduced expression of GABRα1, GABAA receptor subunit α2 (GABRα2), GABAA receptor subunit α3 (GABRα3), GABAA receptor subunit β3 (GABRβ3) and GABBR1 [32-34]. In the cerebellum we observed significant reduction in expression of GABRα1, GABRβ3, GABBR1 and GABAB receptor subunit 2 (GABBR2) [32-34]. More recently, using a different set of tissue samples, we observed reduced expression of GABRβ3 in the cerebellar vermis of adults with autism [31]. Our consistent findings of reduced GABA receptor protein expression in multiple brain regions of people with autism can be explained by the reduced expression of FMRP.
A further connection to this pathway includes the reduction in Reelin protein in sera and blood from individuals with autism [94,95]. Recently, mRNA for Reelin, a glycoprotein with multiple roles during development (namely, neuronal cell migration and brain lamination) and in adults (namely, modulating synaptic function), has been identified as a target of FMRP [20]. Reelin has long been considered a potential biomarker for autism due to its roles during development. Our laboratory has demonstrated reduced Reelin protein expression in sera, BA9, and the cerebella of adults with autism [94-96], supporting the idea that Reelin deficits contribute to the pathology of autism. This reduction in Reelin expression may be due to dysregulation of FMRP expression in people with autism.
Building upon the mGluR5 theory of FXS [26] and our own findings using postmortem brain tissue from people with autism, we hypothesize that activation of mGluR5 in the brains of children with autism [30,31,97] leads to subsequent translocation of protein for FMRP from neuronal cell bodies to dendrites, and eventual decrease in FMRP at synapses [25]. Alternatively, an unknown mechanism of reduced expression of FMRP leads to increased mGluR5 signaling. These events may also lead to impaired GABAergic and Reelin signaling, ultimately leading to the morphological, cognitive, and behavioral deficits associated with autism (Figure 5). Deficits in the inhibitory GABAergic system could cause a perturbation in the excitatory/inhibitory balance in brain circuitry, potentially leading to the presence of seizures as well as impaired cognitive function. Deficits in Reelin function would also lead to abnormal brain development and abnormal synaptic transmission. Further evidence for impaired synaptic plasticity comes from altered expression of other targets of FMRP and mGluR5 signaling (namely APP, RAC1, and STEP), resulting in altered dendritic spine morphology and function as well as altered AMPA and NMDA receptor subunit composition (Figure 5). Taken together, abnormal FMRP and mGluR5 signaling in people with autism who do not have a diagnosis of FXS could explain the multiple pathologies of autism. We performed statistical correlations between levels of FMRP and mGluR5 previously reported [30,31] against RAC1, APP, STEP, and homer 1, which did not result in any significant values (data not shown). However, as relationships between these proteins are neither simple, biochemically well understood, nor stoichiometrically in a 1:1 ratio, this absence of correlation does not minimize the significance of findings in this report. Moreover, as recently shown by Darnell and Klann [98], FMRP target levels may appear unchanged or even decreased due to factors such as age, brain site, increased protein turnover and protein activation state, potentially offsetting target protein increases.
Figure 5.

Reduction of FMRP expression has multiple consequences. In the absence or with reductions of FMRP, mGluR5-mediated signaling and protein synthesis is accelerated, resulting in increased expression of RAC1, APP, and STEP. Increased RAC1 and APP results in altered dendritic morphology, including presence of increased dendritic spines with a long, immature appearance. Increased STEP activity dephosphorylates multiple targets, including AMPA receptors and NMDA receptors, resulting in receptor internalization and altered synaptic transmission. Reduced homer 1 expression, reduces homer 1-mGluR5 interactions, facilitating long term depression. Reduced FMRP also results in reduced expression of GABAA and GABAB receptor subunits, resulting in impaired GABAergic signaling. Reduced FMRP expression may also impact Reelin expression causing further effects on synaptic transmission. The overall effect of these changes is impaired synaptic transmission and ultimately the cognitive and behavioral deficits associated with autism. FMRP, fragile X mental retardation protein; mGluR5, metabotropic glutamate receptor 5; RAC1, Ras-related C3 botulinum toxin substrate 1; amyloid beta A4 precursor protein; STEP, striatal-enriched protein tyrosine phosphatase; AMPA, 2-amino-3-(5-methyl-3-oxo-1,2- oxazol-4-yl)propanoic acid; NDMA, N-methyl-D-aspartate; GABA, gamma aminobutyric acid.
Our results also may open new ways of treating people with autism. Drugs that target metabotropic glutamate receptors have been shown to reverse morphological and behavioral deficits in Fmr1 KO mice. AFQ056, 2-methyl-6-(phenylethynyl)-pyridine (MPEP), and 2-chloro-4-((2,5-dimethyl-1-(4-(trifluoromethoxy)phenyl)-1H-imidazol-4-yl)ethynyl)pyridine (CTEP), antagonists of mGluR5, have been shown to reduce the number of dendritic protrusions of cultured hippocampal neurons and primary visual cortex neurons from Fmr1 KO mice [99-101]. In Fmr1 KO mice, treatment with MPEP or AFQ056 rescues deficits in prepulse inhibition (PPI) [99,100], a sensorimotor gating behavior that is disrupted in subjects with autism, schizophrenia, and other psychiatric disorders. CTEP, a newer anti-mGluR5 inhibitor that is more potent and long lasting than MPEP, has been shown to correct for audiogenic seizures, hippocampal long-term depression, and cognitive deficits in Fmr1 KO mice [101]. The application of Rac1 inhibitor NSC23766 to hippocampal slices from Fmr1 KO mice reduced long-term depression, suggesting that modulation of RAC1 could ameliorate synaptic transmission deficits associated with FXS [49]. Minocycline, a tetracycline derivative, inhibits Aβ-induced neuronal cell death, Aβ fibril formation, and microglial activation [102,103]. Minocycline can also reverse FXS phenotypes, including dendritic spine immaturity [104]. The latter therapies represent more targeted approaches, and as there are currently no approved glutamatergic inhibitors for use in humans, treatments that focus on mGluR5, RAC1 or APP may provide safe, effective means of ameliorating the behavioral deficits of autism.
Conclusions
Taken together, the results of current postmortem studies provide novel evidence that targets of FMRP and mGluR5 signaling (RAC1, homer 1, APP, and STEP) display altered expression in the cerebellar vermis and BA9 of people with autism. There was a significant confounding effect of anticonvulant drugs on STEP 46 kDa/β-actin and a potential effect on homer 1/NSE in BA9 of adults with autism. These results are consistent with results from FXS animal models, despite the fact that none of the individuals included in the current study were diagnosed with FXS. The altered expression of proteins that may contribute to altered dendritic protein translation (homer 1), altered dendritic morphology (APP and RAC1), and receptor subunit expression (STEP), potentially contribute to the cognitive and behavioral impairments associated with autism and FXS. Our data tie in with our previous experiments, in which we have found reductions in protein expression for Reelin and GABA receptor subunit expression in multiple brain regions of people with autism. We hypothesize that reduction in FMRP and increase in mGluR5 may contribute to the dysregulation of these proteins in subjects with autism, resulting in multiple brain structural and behavioral deficits.
Abbreviations
Aβ: Beta amyloid; AMPA: 2-Amino-3-(5-methyl-3-oxo-1,2- oxazol-4-yl)propanoic acid; APD: Antipsychotic drug; APP: Amyloid beta A4 precursor protein; BA9: Brodmann area 9; CDC: Centers for disease control and prevention; CTEP: 2-Chloro-4-((2,5-dimethyl-1-(4-(trifluoromethoxy)phenyl)-1H-imidazol-4-yl)ethynyl)pyridine; CYFIP: Cytoplasmic FMRP interacting proteins; ERK: Extracellular signal-regulated kinases; FMR1: Fragile X mental retardation 1; FRMP: Fragile X mental retardation protein; FXS: Fragile X syndrome; GABA: Gamma aminobutyric acid; GABRα: GABAA receptor subunit alpha; GABRβ: GABAA receptor subunit beta; GFAP: Glial fibrillary acidic protein; GKAP: Guanylate kinase associated protein; GRD: Glycine-like receptor of Drosophila; GTP: Guanine triphosphate; KO: Knockout; LCCH3: Ligand-gated chloride channel homolog 3; LTD: Long-term depression; mGluR5: Metabotropic glutamate receptor 5; MPEP: 2-Methyl-6-(phenylethynyl)-pyridine; NMDA: N-methyl-D-aspartate; NSE: Neuronal specific enolase; PBS: Phosphate-buffered saline; PMI: Postmortem interval; PSD: Postsynaptic density; RAC1: Ras-related C3 botulinum toxin substrate 1; RDL: Resistant to dieldrin; RT: Room temperature; STEP: Striatal-enriched protein tyrosine phosphatase; TCI: Transitory cognitive impairment.
Competing interests
The authors declare that they have no competing interests. SH Fatemi has several patents on the use of Reelin as a diagnostic marker for neuropsychiatric disorders but has not derived any financial gains from these patents.
Authors’ contributions
SHF conceived the study, participated in its design, supervised the conduct of all experiments, and contributed to the drafting of the manuscript and its revisions. SBL, REK, and MKH performed western blotting experiments. TDF performed western blotting experiments and contributed to the drafting of the manuscript. PDT contributed to statistical analyses of data. All authors read and approved the final manuscript.
Supplementary Material
Western blotting results for RAC1, homer 1, APP, STEP, NSE, and β-actin and their ratios in the cerebellar vermis: controls versus people with autism not on medicationsa (anticonvulsant, antidepressant, and antipsychotic drugs). RAC1, Ras-related C3 botulinum toxin substrate 1; APP, amyloid beta A4 precursor protein; STEP, striatal-enriched protein tyrosine phosphatase; NSE, neuronal specific enolase.
Western blotting results for RAC1, homer 1, APP, STEP, NSE, and β-actin and their ratios in BA9: controls versus people with autism not on medicationsa (anticonvulsant, antidepressant, and antidepressant drugs). RAC1, Ras-related C3 botulinum toxin substrate 1; APP, amyloid beta A4 precursor protein; STEP, striatal-enriched protein tyrosine phosphatase; NSE, neuronal specific enolase; BA9, Brodmann Area 9.
Contributor Information
S Hossein Fatemi, Email: fatem002@umn.edu.
Timothy D Folsom, Email: folso013@umn.edu.
Rachel E Kneeland, Email: knee0030@umn.edu.
Mahtab K Yousefi, Email: mkarkhan@umn.edu.
Stephanie B Liesch, Email: lies0065@umn.edu.
Paul D Thuras, Email: pthuras@yahoo.com.
Acknowledgments
Human tissue was obtained from the NICHD Brain and Tissue Bank for Developmental Disorders, University of Maryland, Baltimore, MD (the role of the NICHD Brain and Tissue Bank is to distribute tissue, and therefore, cannot endorse the studies performed or the interpretation of results); the Harvard Brain Tissue Resource Center, which is supported in part by Public Health Service grant number R24 MH068855; the Brain Endowment Bank, which is funded in part by the National Parkinson Foundation, Inc., Miami, Florida; and the Autism Tissue Program, and is gratefully acknowledged. Grant support by the National Institute of Child Health and Human Development (#5R01HD052074-01A2 and 3R01HD052074-03S1) and the Minnesota Medical Foundation Alfred and Ingrid Lenz Harrison Autism Initiative Fund to SHF is gratefully acknowledged. The funding body had no role in the study design, collection, analysis and interpretation of data, in the writing of the manuscript or in the decision to submit the manuscript for publication. SH Fatemi is also supported by the Bernstein Endowed Chair in adult psychiatry.
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4. Washington, DC: APA Press; 1994. [Google Scholar]
- Casanova MF, Buxhoeveden DP, Switala AE, Roy E. Minicolumnar pathology in autism. Neurology. 2002;58:428–432. doi: 10.1212/WNL.58.3.428. [DOI] [PubMed] [Google Scholar]
- Palmen SJ, van Engeland H, Hof PR, Schmitz C. Neuropathological findings in autism. Brain. 2004;127:2572–2583. doi: 10.1093/brain/awh287. [DOI] [PubMed] [Google Scholar]
- McCaffrey P, Deutsch CK. Macrocephaly and the control of brain growth in autistic disorders. Prog Neurobiol. 2005;77:38–56. doi: 10.1016/j.pneurobio.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Autism and Developmental Disabilities Monitoring Network Surveillance Year 2008 Principal Investigators; Centers for Disease Control and Prevention. Prevalence of autism spectrum disorders-Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008. MMWR Surveill Summ. 2012;61:1–19. [PubMed] [Google Scholar]
- Canitano R. Epilepsy in autism spectrum disorders. Eur Child Adolesc Psychiatry. 2006;16:61–66. doi: 10.1007/s00787-006-0563-2. [DOI] [PubMed] [Google Scholar]
- Aarts JH, Binnie CD, Smit AM, Wilkins AJ. Selective cognitive impairment during focal and generalized epileptiform EEG activity. Brain. 1984;107:293–308. doi: 10.1093/brain/107.1.293. [DOI] [PubMed] [Google Scholar]
- Binnie CD. Cognitive impairment during epileptiform discharges: is it ever justifiable to treat the EEG? Lancet Neurol. 2003;2:725–730. doi: 10.1016/S1474-4422(03)00584-2. [DOI] [PubMed] [Google Scholar]
- Geschwind DH. Autism: many genes, common pathways? Cell. 2008;135:391–395. doi: 10.1016/j.cell.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai Y, Shaw CA, Dawson BC, Dugas DV, Al-Mohtaseb Z, Hill DE, Zoghbi HY. Protein interactome reveals converging molecular pathways among autism disorders. Sci Transl Med. 2011;3:86ra49. doi: 10.1126/scitranslmed.3002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numis AL, Major P, Montenegro MA, Muzykewicz DA, Pulsifer MB, Thiele EA. Identification of risk factors for autism spectrum disorders in tuberous sclerosis complex. Neurology. 2011;76:981–987. doi: 10.1212/WNL.0b013e3182104347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman R, Au J, Hagerman P. FMR1 premutation and full mutation molecular mechanisms related to autism. J Neurodev Disord. 2011;3:211–224. doi: 10.1007/s11689-011-9084-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loat CS, Curran S, Lewis CM, Duvall J, Geschwind D, Bolton P, Craig IW. Methyl-CpG-binding protein 2 polymorphisms and vulnerability to autism. Genes Brain Behav. 2008;7:754–760. doi: 10.1111/j.1601-183X.2008.00414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonati MT, Russo S, Finelli P, Valsecchi MR, Cavalleri F, Roberts W, Elia M, Larizza L. Evaluation of autism traits in Angelman syndrome: a resource to unfold autism genes. Neurogenetics. 2007;8:169–178. doi: 10.1007/s10048-007-0086-0. [DOI] [PubMed] [Google Scholar]
- Hatton DD, Sideris J, Skinner M, Mankowski J, Bailey DB Jr, Roberts J, Mirrett P. Autistic behavior in children with Fragile X syndrome: prevalence, stability, and the impact of FMRP. Am J Med Genet A. 2006;140A:1804–1813. doi: 10.1002/ajmg.a.31286. [DOI] [PubMed] [Google Scholar]
- Kauffman WE, Cortell R, Kau A, Bukelis I, Tierney E, Gray R, Cox C, Capone G, Stanard P. Autism spectrum disorder in fragile X syndrome: Communication, social interaction, and specific behaviors. Am J Med Genet A. 2004;129A:225–234. doi: 10.1002/ajmg.a.30229. [DOI] [PubMed] [Google Scholar]
- Hagerman RJ, Staley LW, O’Conner R, Lugenbeel K, Nelson D, McLean SD, Taylor A. Learning-disabled males with a fragile X CGG expansion in the upper premutation size range. Pediatrics. 1996;97:122–126. [PubMed] [Google Scholar]
- Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devys D, Lutz Y, Rouyer N, Bellocq JP, Mandel JL. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet. 1993;4:335–340. doi: 10.1038/ng0893-335. [DOI] [PubMed] [Google Scholar]
- Pacey LK, Doering LC. Developmental expression of FMRP in the astrocyte lineage: implications for fragile X syndrome. Glia. 2007;55:1601–1609. doi: 10.1002/glia.20573. [DOI] [PubMed] [Google Scholar]
- Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K, Comery TA, Patel B, Eberwine J, Greenough WT. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci U S A. 1997;94:5395–5400. doi: 10.1073/pnas.94.10.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker CE, de Diego OY, Bontekoe C, Raghoe P, Luteijn T, Hoogeveen AT, Oostra BA, Willemsen R. Immunocytochemical and biochemical characterization of FMRP, FXR1P, and FXR2P in the mouse. Exp Cell Res. 2000;258:162–170. doi: 10.1006/excr.2000.4932. [DOI] [PubMed] [Google Scholar]
- Krueger DD, Bear MF. In: Autism Spectrum Disorders. Amaral DG, Dawson G, Geschwind DH, editor. New York: Oxford University Press, USA; 2011. The mGluR theory of fragile X syndrome; pp. 1239–1258. [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of Fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Grossman AW, Elisseou NM, McKinney BC, Greenough WT. Hippocampal pyramidal cells in adult Fmr1 knockout mice exhibit an immature-appearing profile of dendritic spines. Brain Res. 2006;1084:158–164. doi: 10.1016/j.brainres.2006.02.044. [DOI] [PubMed] [Google Scholar]
- Hutsler JJ, Zhang H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010;1309:83–94. doi: 10.1016/j.brainres.2009.09.120. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, Kooy F, Willems PJ, Cras P, Kozlowski PB, Swain RA, Weiler IJ, Greenough WT. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet. 2001;98:161–171. doi: 10.1002/1096-8628(20010115)98:2<161::AID-AJMG1025>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Folsom TD. Dysregulation of fragile X mental retardation protein and metabotropic glutamate receptor 5 in superior frontal cortex of subjects with autism: a postmortem brain study. Mol Autism. 2011;2:6. doi: 10.1186/2040-2392-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Folsom TD, Kneeland RE, Liesch SB. Metabotropic glutamate receptor 5 upregulation in children with autism is associated with underexpression of both Fragile X mental retardation protein and GABAA receptor beta 3 in adults with autism. Anat Rec. 2011;294:1635–1645. doi: 10.1002/ar.21299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Reutiman TJ, Folsom TD, Thuras PD. GABA(A) Receptor downregulation in brains of subjects with autism. J Autism Dev Disord. 2009;39:223–230. doi: 10.1007/s10803-008-0646-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Folsom TD, Reutiman TJ, Thuras PD. Expression of GABA(B) receptors is altered in brains of subjects with autism. Cerebellum. 2009;8:64–69. doi: 10.1007/s12311-008-0075-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Reutiman TJ, Folsom TD, Rooney RJ, Patel DH, Thuras PD. mRNA and Protein levels for GABA(A) alpha 4, alpha 5, beta 1, and GABA(B)R1 receptors are altered in brains from subjects with autism. J Autism Dev Disord. 2010;40:743–750. doi: 10.1007/s10803-009-0924-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J. A power primer. Psychol Bull. 1992;112:155–159. doi: 10.1037//0033-2909.112.1.155. [DOI] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustelo XR, Sauzeau V, Berenjeno IM. GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays. 2007;29:356–370. doi: 10.1002/bies.20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaibuchi K, Kuroda S, Amano M. Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu Rev Biochem. 1999;68:459–486. doi: 10.1146/annurev.biochem.68.1.459. [DOI] [PubMed] [Google Scholar]
- Corbetta S, Gualdoni S, Ciceri G, Monari M, Zuccaro E, Tybulewicz VL, de Curtis I. Essential role of Rac1 and Rac3 GTPases in neuronal development. FASEB J. 2009;23:1347–1357. doi: 10.1096/fj.08-121574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negishi M, Katoh H. Rho family GTPases and dendrite plasticity. Neuroscientist. 2005;11:187–191. doi: 10.1177/1073858404268768. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Yuste R. Regulation of dendritic spine motility and stability by Rac1 and Rho kinase: evidence for two forms of spine motility. Mol Cell Neurosci. 2004;26:429–440. doi: 10.1016/j.mcn.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Wiens KM, Lin H, Liao D. Rac1 induces the clustering of AMPA receptors during spinogenesis. J Neurosci. 2005;25:10627–10636. doi: 10.1523/JNEUROSCI.1947-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R. Dendritic Spines. Cambridge, MA: The MIT Press; 2010. [Google Scholar]
- Billuart P, Bienvenu T, Ronce N, Des Portes V, Vinet MC, Zemni R, Roest Crollius H, Carrié A, Fauchereau F, Cherry M, Briault S, Hamel B, Fryns JP, Beldjord C, Kahn A, Moraine C, Chelly J. Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retardation. Nature. 1998;392:923–926. doi: 10.1038/31940. [DOI] [PubMed] [Google Scholar]
- Arai Y, Ijuin T, Takenawa T, Becker LE, Takashima S. Excessive expression of synaptojanin in brains with Down syndrome. Brain Dev. 2002;24:67–72. doi: 10.1016/S0387-7604(01)00405-3. [DOI] [PubMed] [Google Scholar]
- Schenck A, Bardoni B, Moro A, Bagni C, Mandel JL. A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP-related proteins FXR1P and FXR2P. Proc Natl Acad Sci U S A. 2001;98:8844–8849. doi: 10.1073/pnas.151231598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenck A, Bardoni B, Langmann C, Harden N, Mandel JL, Giangrande A. CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein. Neuron. 2003;38:887–898. doi: 10.1016/S0896-6273(03)00354-4. [DOI] [PubMed] [Google Scholar]
- Castets M, Schaeffer C, Bechara E, Schenck A, Khandjian EW, Luche S, Moine H, Rabilloud T, Mandel JL, Bardoni B. FMRP interferes with the Rac1 pathway and controls actin cytoskeleton dynamics in murine fibroblasts. Hum MolGenet. 2005;14:835–844. doi: 10.1093/hmg/ddi077. [DOI] [PubMed] [Google Scholar]
- Bongmba OY, Martinez LA, Elhardt ME, Butler K, Tejada-Simon MV. Modulation of dendritic spines and synaptic function by Rac1: A possible link to fragile X syndrome pathology. Brain Res. 2011;1399:79–95. doi: 10.1016/j.brainres.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Hensch TK, Ackerman L, Barbel S, Jan LY, Jan YN. Differential effects of the Rac GTPase on Purkinje cell axons and dendritic trunks and spines. Nature. 1996;379:837–840. doi: 10.1038/379837a0. [DOI] [PubMed] [Google Scholar]
- Nakayama AY, Harms MB, Luo L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 2000;20:5329–5338. doi: 10.1523/JNEUROSCI.20-14-05329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racchetti G, D’Alessandro R, Meldolesi J. Astrocyte stellation, a process dependent on Rac1 is sustained by the regulated exocytosis of enlargeosomes. Glia. 2012;60:465–475. doi: 10.1002/glia.22280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourguignon LY, Gilad E, Peyrollier K, Brightman A, Swanson RA. Hyaluronan-CD44 interaction stimulates Rac1 signaling and PKN gamma kinase activation leading to cytoskeleton function and cell migration in astrocytes. J Neurochem. 2007;101:1002–1017. doi: 10.1111/j.1471-4159.2007.04485.x. [DOI] [PubMed] [Google Scholar]
- Laurence JA, Fatemi SH. Glial fibrillary acidic protein is elevated in superior frontal, parietal and cerebellar cortices of autistic subjects. Cerebellum. 2005;4:201–210. doi: 10.1080/14734220500208846. [DOI] [PubMed] [Google Scholar]
- Sokol DK, Chen D, Farlow MR, Dunn DW, Maloney B, Zimmer JA, Lahiri DK. High levels of Alzheimer beta-amyloid precursor protein (APP) in children with severely autistic behavior and aggression. J Child Neurol. 2006;21:444–449. doi: 10.1177/08830738060210062201. [DOI] [PubMed] [Google Scholar]
- Bailey AR, Giunta BN, Obregon D, Nikolic WV, Tian J, Sanberg CD, Sutton DT, Tan J. Peripheral biomarkers in autism: Secreted amyloid precursor protein-alpha as a probable key player in early diagnosis. Int J Clin Exp Med. 2008;1:338–344. [PMC free article] [PubMed] [Google Scholar]
- Ray B, Long JM, Sokol DK, Lahiri DK. Increased secreted amyloid precursor protein-α (sAPPα) in severe autism: proposal of a specific, anabolic pathway and putative biomarker. PLoS One. 2011;6:e20405. doi: 10.1371/journal.pone.0020405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol DK, Maloney B, Long JM, Ray B, Lahiri DK. Autism, Alzheimer disease, and fragile X: APP, FMRP, and mGluR5 are molecular links. Neurology. 2011;76:344–1352. doi: 10.1212/WNL.0b013e3182166dc7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmark CJ, Malter JS. FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 2007;5:e52. doi: 10.1371/journal.pbio.0050052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Xu H, Dyck LE, Li XM. Olanzapine and quetiapine protect PC12 cells from beta-amyloid peptide(25–35)-induced oxidative stress and the ensuing apoptosis. J Neurosci Res. 2005;81:572–580. doi: 10.1002/jnr.20570. [DOI] [PubMed] [Google Scholar]
- Chavant F, Deguil J, Pain S, Ingrand I, Milin S, Fauconneau B, Pérault-Pochat MC, Lafay- Chebassier C. Imipramine, in part through tumor necrosis factor alpha inhibition, prevents cognitive decline and beta-amyloid accumulation in a mouse model of Alzheimer’s disease. J Pharmacol Exp Ther. 2010;332:505–514. doi: 10.1124/jpet.109.162164. [DOI] [PubMed] [Google Scholar]
- Volknandt W, Karas M. Proteomic analysis of the presynaptic active zone. Exp Brain Res. 2012;217:449–461. doi: 10.1007/s00221-012-3031-x. [DOI] [PubMed] [Google Scholar]
- Innocent N, Cousins SL, Stephenson FA. NMDA receptor/amyloid precursor protein interactions: a comparison between wild-type and amyloid precursor protein mutations associated with familial Alzheimer’s disease. Neurosci Lett. 2012;515:131–136. doi: 10.1016/j.neulet.2012.03.029. [DOI] [PubMed] [Google Scholar]
- Cousins SL, Hoey SE, Stephenson FA, Perkinton MS. Amyloid precursor protein 695 associates with assembled NR2A- and NR2B-containing NMDA receptors to result in the enhancement of their cell surface delivery. J Neurochem. 2009;111:1501–1513. doi: 10.1111/j.1471-4159.2009.06424.x. [DOI] [PubMed] [Google Scholar]
- Hoe HS, Fu Z, Makarova A, Lee JY, Lu C, Feng L, Pajoohesh-Ganji A, Matsuoka Y, Hyman BT, Ehlers MD, Vicini S, Pak DT, Rebeck GW. The effects of amyloid precursor protein on postsynaptic composition and activity. J Biol Chem. 2009;284:8495–8506. doi: 10.1074/jbc.M900141200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmark CJ, Westmark PR, O’Riordan KJ, Ray BC, Hervey CM, Salamat MS, Abozeid SH, Stein KM, Stodola LA, Tranfaglia M, Burger C, Berry-Kravis EM, Malter JS. Reversal of fragile X phenotypes by manipulation of AβPP/Aβ levels in Fmr1KO mice. PLoS One. 2011;6:e26549. doi: 10.1371/journal.pone.0026549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Ingelsson M, Fukumoto H, Ramasamy K, Kowa H, Frosch MP, Irizarry MC, Hyman BT. Expression of APP pathway mRNAs and proteins in Alzheimer’s disease. Brain Res. 2007;1161:116–123. doi: 10.1016/j.brainres.2007.05.050. [DOI] [PubMed] [Google Scholar]
- Kim SH, Markham JA, Weiler IJ, Greenough WT. Aberrant early-phase ERK inactivation impedes neuronal function in fragile X syndrome. Proc Natl Acad Sci U S A. 2008;105:4429–4434. doi: 10.1073/pnas.0800257105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canal F, Palygin O, Pankratov Y, Corrêa SA, Müller J. Compartmentalization of the MAPK scaffold protein KSR1 modulates synaptic plasticity in hippocampal neurons. FASEB. 2011;25:2362–2372. doi: 10.1096/fj.10-173153. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Venkitaramani DV, Gladding CM, Zhang Y, Kurup P, Molnar E, Collingridge GL, Lombroso PJ. The tyrosine phosphatase STEP mediates AMPA receptor endocytosis after metabotropic glutamate receptor stimulation. J Neurosci. 2008;28:10561–10566. doi: 10.1523/JNEUROSCI.2666-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma E, Zhao F, Bult A, Lambroso PJ. Identification of two alternatively spliced transcripts of STEP: a subfamily of brain-enriched protein tyrosine phosphatases. Brain Res Mol Brain Res. 1995;32:87–93. doi: 10.1016/0169-328X(95)00066-2. [DOI] [PubMed] [Google Scholar]
- Bult A, Zhao F, Dirkx R Jr, Raghunathan A, Solimena M, Lombroso PJ. STEP: a family of brain-enriched PTPs. Alternative splicing produces transmembrane, cytosolic and truncated isoforms. Eur J Cell Biol. 1997;72:337–344. [PubMed] [Google Scholar]
- Goebel-Goody SM, Wilson-Wallis ED, Royston S, Tagliatela SM, Naegele JR, Lombroso PJ. Genetic manipulation of STEP reverses behavioral abnormalities in a fragile X syndrome mouse model. Genes Brain Behav. 2012;11:586–600. doi: 10.1111/j.1601-183X.2012.00781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick CJ, Lombroso P. The Role of Striatal-Enriched Protein Tyrosine Phosphatase (STEP) in Cognition. Front Neuroanat. 2011;5:47. doi: 10.3389/fnana.2011.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carty NC, Xu J, Kurup P, Brouillette J, Goebel-Goody SM, Austin DR, Yuan P, Chen G, Correa PR, Haroutunian V, Pittenger C, Lombroso PJ. The tyrosine phosphatase STEP: implications in schizophrenia and the molecular mechanism underlying antipsychotic medications. Transl Psychiatry. 2012;2:e137. doi: 10.1038/tp.2012.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurup P, Zhang Y, Xu J, Venkitaramani DV, Haroutunian V, Greengard P, Nairn AC, Lombroso PJ. Abeta-mediated NMDA receptor endocytosis in Alzheimer’s disease involves ubiquitination of the tyrosine phosphatase STEP61. J Neurosci. 2010;30:5948–5957. doi: 10.1523/JNEUROSCI.0157-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Acad Natl Sci U S A. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kurup P, Xu J, Carty N, Fernandez SM, Nygaard HB, Pittenger C, Greengard P, Strittmatter SM, Nairn AC, Lombroso PJ. Genetic reduction of striatal-enriched tyrosine phosphatase (STEP) reverses cognitive and cellular deficits in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A. 2010;107:19014–19019. doi: 10.1073/pnas.1013543107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa S, Morioka M, Goto S, Korematsu K, Okamura A, Yano S, Kai Y, Hamada JI, Ushio Y. Expression of neuron specific phosphatase, striatal enriched phosphatase (STEP) in reactive astrocytes after transient forebrain ischemia. Glia. 2000;29:316–329. doi: 10.1002/(SICI)1098-1136(20000215)29:4<316::AID-GLIA3>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Briggs SW, Walker J, Asik K, Lombroso P, Naegele J, Aaron G. STEP regulation of seizure thresholds in the hippocampus. Epilepsia. 2011;52:497–506. doi: 10.1111/j.1528-1167.2010.02912.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Yuan JP, Lanahan AA, Leoffert K, Li M, Linden DJ, Worley PF. Homer binds a novel prolin-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron. 1998;21:717–726. doi: 10.1016/S0896-6273(00)80589-9. [DOI] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, Worley PF. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density protein. Neuron. 1999;23:583–592. doi: 10.1016/S0896-6273(00)80810-7. [DOI] [PubMed] [Google Scholar]
- Giuffrida R, Musumeci S, D’Antoni S, Bonaccorso CM, Giuffrida-Stella AM, Oostra BA, Catania MV. A reduced number of metabotropic glutamate subtype 5 receptors are associated with constitutive homer proteins in a mouse model of fragile X syndrome. J Neurosci. 2005;25:8908–8916. doi: 10.1523/JNEUROSCI.0932-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ango F, Robbe D, Muller T, Tu JC, Xiao B, Worley PF, Pin JP, Bockaert J, Fagni L. Homer-dependent cell surface expression of metabotropic glutamate receptor type 5 in neurons. Mol Cell Neurosci. 2002;20:323–329. doi: 10.1006/mcne.2002.1100. [DOI] [PubMed] [Google Scholar]
- Kammermeier PJ. Endogenous homer proteins regulate metabotropic glutamate receptor signaling in neurons. J Neurosci. 2008;28:8560–8567. doi: 10.1523/JNEUROSCI.1830-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergé A, Fourgeaud L, Hémar A, Choquet D. Receptor activation and Homer differentially control the lateral mobility of metabotropic glutamate receptor 5 in the neuronal membrane. J Neurosci. 2002;22:3910–3920. doi: 10.1523/JNEUROSCI.22-10-03910.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi JA, Huber KM. Homer interactions are necessary for metabotropic glutamate receptor-induced long-term depression and translational activation. J Neurosci. 2008;28:543–547. doi: 10.1523/JNEUROSCI.5019-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ 3rd, Geigenmüller U, Hovhannisyan H, Trautman E, Pinard R, Rathmell B, Carpenter R, Margulies D. High-throughput sequencing of mGluR signaling pathway genes reveals enrichment of rare variants in autism. PLoS One. 2012;7:e35003. doi: 10.1371/journal.pone.0035003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bartolomeis A, Tomasetti C, Cicale M, Yuan PX, Manji HK. Chronic treatment with lithium or valproate modulates the expression of Homer1b/c and its related genes Shank and Inositol 1,4,5-trisphosphate receptor. Eur Neuropsychopharmacol. 2012;22:527–535. doi: 10.1016/j.euroneuro.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Idrissi A, Ding XH, Scalia J, Trenkner E, Brown WT, Dobkin C. Decreased GABAA receptor expression in the seizure-prone fragile X mouse. Neurosci Lett. 2005;377:141–146. doi: 10.1016/j.neulet.2004.11.087. [DOI] [PubMed] [Google Scholar]
- D’Hulst C, De Geest N, Reeve SP, Van Dam D, De Deyn PP, Hassan BA, Kooy RF. Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 2006;1121:238–245. doi: 10.1016/j.brainres.2006.08.115. [DOI] [PubMed] [Google Scholar]
- Gantois I, Vandescompele J, Speleman F, Reyniers E, D’Hooge R, Severijnen LA, Willemsen R, Tassone F, Kooy RF. Expression profiling suggests underexpression of the GABAA receptor subunit delta in the fragile X knockout mouse model. Neurobiol Dis. 2006;21:346–357. doi: 10.1016/j.nbd.2005.07.017. [DOI] [PubMed] [Google Scholar]
- Hong A, Zhang A, Ke Y, El Idrissi A, Shen CH. Downregulation of GABA(A) β subunits is transcriptionally controlled by Fmr1p. J Mol Neurosci. 2012;46:272–275. doi: 10.1007/s12031-011-9531-5. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Stary JM, Halt AR, Realmuto GR. Dysregulation of Reelin and Bcl-2 proteins in autistic cerebellum. J Autism Dev Disord. 2001;31:529–535. doi: 10.1023/A:1013234708757. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Stary JM, Egan EA. Reduced blood levels of reelin as a vulnerability factor in pathophysiology of autistic disorder. Cell Mol Neurobiol. 2002;22:139–152. doi: 10.1023/A:1019857620251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Snow AV, Stary JM, Araghi-Niknam M, Reutiman TJ, Lee S, Brooks AI, Pearce DA. Reelin signaling is impaired in autism. Biol Psychiatry. 2005;57:777–787. doi: 10.1016/j.biopsych.2004.12.018. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Folsom TD. The role of fragile X mental retardation protein in major mental disorders. Neuropharmacology. 2012;60:1221–1226. doi: 10.1016/j.neuropharm.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Klann E. The translation of translational control by FMRP: therapeutic targets for FXS. Nat Neurosci. 2013. [DOI] [PMC free article] [PubMed]
- de Vrij FM, Levenga J, van der Linde HC, Koekkoek SK, De Zeeuw CI, Nelson DL, Oostra BA, Willemsen R. Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol Dis. 2008;31:127–132. doi: 10.1016/j.nbd.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenga J, Hayashi S, de Vrij FM, Koekkoek SK, van der Linde HC, Nieuwenhuizen I, Song C, Buijsen RA, Pop AS, Gomezmancilla B, Nelson DL, Willemsen R, Gasparini F, Oostra BA. AFQ506, a new mGluR5 antagonist for treatment of fragile X syndrome. Trends Mol Med. 2011;16:516–527. doi: 10.1016/j.nbd.2011.01.022. [DOI] [PubMed] [Google Scholar]
- Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, Jaeschke G, Bear MF, Lindemann L. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron. 2012;74:49–56. doi: 10.1016/j.neuron.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Familian A, Boshuizen RS, Eikelenboom P, Veerhuis R. Inhibitory effect of minocycline on amyloid beta fibril formation and human microglial activation. Glia. 2006;53:233–240. doi: 10.1002/glia.20268. [DOI] [PubMed] [Google Scholar]
- Ryu JK, Franciosi S, Sattayaprasert P, Kim SU, McLarnon JG. Minocycline inhibits neuronal death and glial activation induced by beta-amyloid peptide in rat hippocampus. Glia. 2004;48:85–90. doi: 10.1002/glia.20051. [DOI] [PubMed] [Google Scholar]
- Bilousova TV, Dansie L, Ngo M, Aye J, Charles JR, Ethell DW, Ethell IM. Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet. 2009;46:94–102. doi: 10.1136/jmg.2008.061796. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Western blotting results for RAC1, homer 1, APP, STEP, NSE, and β-actin and their ratios in the cerebellar vermis: controls versus people with autism not on medicationsa (anticonvulsant, antidepressant, and antipsychotic drugs). RAC1, Ras-related C3 botulinum toxin substrate 1; APP, amyloid beta A4 precursor protein; STEP, striatal-enriched protein tyrosine phosphatase; NSE, neuronal specific enolase.
Western blotting results for RAC1, homer 1, APP, STEP, NSE, and β-actin and their ratios in BA9: controls versus people with autism not on medicationsa (anticonvulsant, antidepressant, and antidepressant drugs). RAC1, Ras-related C3 botulinum toxin substrate 1; APP, amyloid beta A4 precursor protein; STEP, striatal-enriched protein tyrosine phosphatase; NSE, neuronal specific enolase; BA9, Brodmann Area 9.