

Abstract
Xylonate is a valuable chemical for versatile applications. Although the chemical synthesis route and microbial conversion pathway were established decades ago, no commercial production of xylonate has been obtained so far. In this study, the industrially important microorganism Escherichia coli was engineered to produce xylonate from xylose. Through the coexpression of a xylose dehydrogenase (xdh) and a xylonolactonase (xylC) from Caulobacter crescentus, the recombinant strain could convert 1 g/L xylose to 0.84 g/L xylonate and 0.10 g/L xylonolactone after being induced for 12 h. Furthermore, the competitive pathway for xylose catabolism in E. coli was blocked by disrupting two genes (xylA and xylB) encoding xylose isomerase and xylulose kinase. Under fed-batch conditions, the finally engineered strain produced up to 27.3 g/L xylonate and 1.7 g/L xylonolactone from 30 g/L xylose, about 88% of the theoretical yield. These results suggest that the engineered E. coli strain has a promising perspective for large-scale production of xylonate.
Introduction
Xylonate is a five-carbon organic acid. In the past few years, xylonate has gained increasing interest due to its potential as an important platform chemical. Xylonate has extensively versatile applications similar to many other sugar acids such as gluconate, and can be used in food, chemical, and pharmaceutical industries [1]. In particular, xylonate could serve as a precursor for 1,2,4-butanetriol synthesis [2] and a concrete water reducer [3]. Xylonate might be produced from the non-food hemicellulose hydrolysate, which provides an inexpensive alternative to gluconate. In a report from the U.S. Department of Energy, xylonate is among the top 30 value-added chemicals manufactured from biomass [4].
Although chemical oxidation of xylose to produce xylonate could be obtained by using platinum or gold as the catalysts [5], the poor selectivity makes these synthetic routes not economically feasible for industrial purposes. Microbial conversion of xylose to xylonate, which was well characterized in previous studies, has become a research hotspot during recent years. Several bacterial strains, e.g., Enterobacter cloacae [6], Pseudomonas fragi [7], Gluconobacter oxydans [8], were able to produce xylonate in high yields. Compared with the chemical synthetic routes, microbial fermentation offers the potential for benign processing with high specificity and reduced manufacturing cost.
Along with the development of modern molecular biology techniques, the metabolic pathway from xylose to xylonate has been largely elucidated. Xylose is first converted to xylonolactone by xylose dehydrogenase or glucose oxidase, and xylonolactone is subsequently hydrolyzed spontaneously or enzymatically by xylonolactonase to yield xylonate [9]. Up to now, genes encoding xylose dehydrogenases have been identified and cloned from different bacterial and fungal strains [10]–[12]. Heterologous expression of these xylose dehydrogenase led to xylonate production in the corresponding hosts [13], [14]. However, few xylonolactonases have been characterized. Spontaneous hydrolysis of xylonolactone was relatively slow, and its accumulation in the culture broth greatly inhibited the growth of bacteria, thus hampering xylonate production [15].
Recently, the oxidative xylose utilization pathway was discovered in the fresh water bacterium Caulobacter crescentus. Enzymes including an NAD+ dependent xylose dehydrogenase (xdh) and a xylonolactonase (xylC) were identified in a xylose-inducible operon [16]. In this study, the two genes were expressed in a xylose catabolic deficient Escherichia coli mutant strain (knockout of xylA and xylB, encoding xylose isomerase and xylulose kinase) to construct a heterologous xylonate-producing system (Figure 1). Xylonate production by the engineered strain was further evaluated under fed-batch culture conditions.
Figure 1. The metabolic pathway from xylose to xylonate in engineered E. coli.
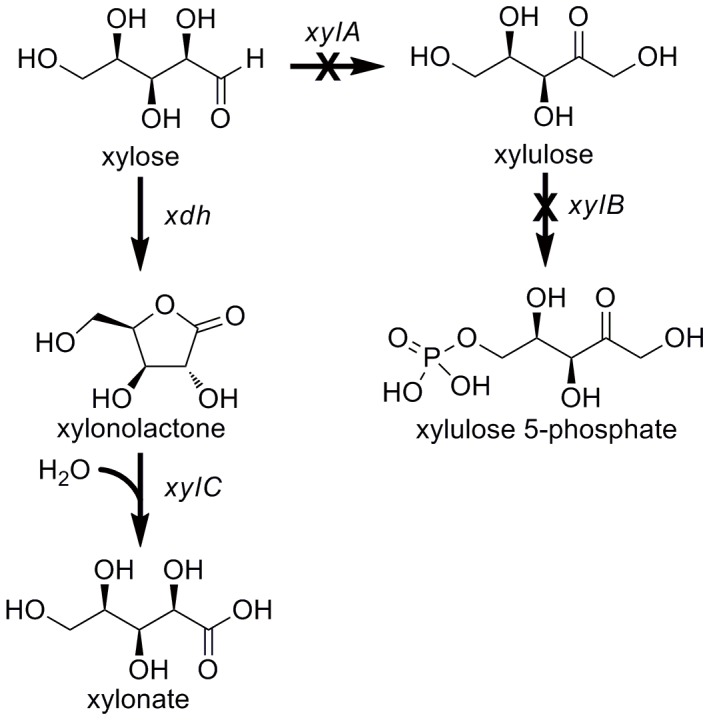
Enzymes encoded by the genes shown are: xdh, xylose dehydrogenase from C. crescentus; xylC, xylonolactonase from C. crescentus; xylA, native E. coli xylose isomerase; xylB, native E. coli xylulose kinase.
Materials and Methods
Bacterial Strains and Plasmids Construction
A list of bacterial strains and recombinant plasmids used in this study is presented in Table 1. The one-step gene inactivation strategy, previously described by Datsenko and Wanner [17], was applied to knock out the chromosomal genes in E. coli BL21 star(DE3). Oligonucleotide primers used for gene disruption are listed in Table S1. For the construction of strain BL21ΔxylAB, a linear DNA fragment containing the FRT-flanked kanamycin resistance cassette was amplified with primers xylAB_Del_F and xylAB_Del_R from plasmid pKD4. The obtained disrupting fragments were electrotransformed into E. coli competent cells that carried the Red recombinase expression vector pKD46 and integrated into its chromosome. Successfully disrupted colonies were then transformed with plasmid pCP20 and induced at 42°C to eliminate the kanamycin resistance. PCR verifications were performed using primer pairs designed according to the sequences up- and downstream of disrupted regions (xylAB_DelIden_F and xylAB_DelIden_R).
Table 1. Strains and plasmids used in this study.
| Strains or plasmids | Genotype/Description | Sources |
| Strains | ||
| E. coli BL21 star(DE3) | F– ompT hsdSB (rB – mB –) gal dcm rne131 (DE3) | Invitrogen |
| E. coli BL21 star(DE3) ΔxylAB | Knockout of xylA and xylB encoding xylose isomerase and xylulose kinase | This study |
| Plasmids | ||
| pACYCduet-1 | Cmr oriP15A lacIq T7p | Novagen |
| pA-xdh | pACYCduet-1 harboring C. crescentus xdh | This study |
| pA-xylC | pACYCduet-1 harboring C. crescentus xylC | This study |
| pA-xdhxylC | pACYCduet-1 harboring C. crescentus xdh and xylC | This study |
| pKD46 | Apr oriR101 repA(Ts) λ Red (γ, β and exo) | Coli Genetic Stock Center |
| pKD4 | FRT-Kanr-FRT oriR6K | Coli Genetic Stock Center |
| pCP20 | Apr Cmr repA(Ts) FLP | Coli Genetic Stock Center |
The xdh (GenBank Accession No.: NACL94329) and xylC (GenBank Accession No.: NACL94328) genes from C. crescentus were codon optimized, chemically synthesized and cloned into pGH vector by Generay Biotech Co., Ltd. (Shanghai, China). Then xdh and xylC were PCR amplified and cloned into the restriction sites NcoI/BamHI and NdeI/XhoI of vector pACYCduet-1, creating pA-xdh and pA-xylC, respectively. The primers for the amplification of xdh and xylC are also given in Table S1. The xdh gene was further cloned into pA-xylC between NcoI and BamHI sites leading to plasmid pA-xdhxylC, which was used for the coexpression of the two genes. Successful gene cloning was verified by colony PCR, restriction mapping and direct nucleotide sequencing.
Reagents, Media and Culture Conditions
Calcium xylonate hydrate for reference standard was purchased from Toronto Research Chemicals Inc. (North York, Canada). Xylonolactone was prepared by autoclaving calcium xylonate in 1 M HCl at 121°C for 20 min [18]. Luria-Bertani (LB) medium (10 g/L tryptone, 5 g/L yeast extract, 10 g/L NaCl) or M9 mineral medium (6 g/L Na2HPO4, 3 g/L KH2PO4, 1 g/L NH4Cl, 0.5 g/L NaCl, 0.12 g/L MgSO4) supplemented with appropriate carbon sources were used for DNA manipulation, protein expression and liquid growth tests. All cultures were grown at 37°C in the presence of 34 µg/ml chloramphenicol and shaken at 180 rpm. 0.5 mM of isopropyl-β-D-thiogalactopyranoside (IPTG) was added at an OD600 of 0.6 to induce the expression of recombinant proteins. For xylonate production, xylose was added to the culture to a final concentration of 1 g/L at the same time. Sample was taken at different intervals and the supernatants were used for xylonate and xylonolactone determination.
Protein Expression and Enzyme Activity Determination
Recombinant E. coli strains harboring pA-xdh, pA-xylC or pA-xdhxylC were induced for 4 h to express the recombinant proteins. Cells were collected from 1 ml of bacteria cultures by centrifugation, dissolved in 100 µl sodium dodecyl sulfate (SDS) sample buffer, heated at 100°C for 10 min and then analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) [19]. The harvested bacterial cells were also suspended in phosphate buffer (pH 8.0) and subjected to ultrasonication. The mixture was centrifuged at 12,000 rpm and 4°C for 10 min, and the supernatant obtained was used for enzymatic activity determination. Generally, assays of xylose dehydrogenase were carried out in 1 ml of reaction system containing 50 mM phosphate buffer (pH 8.0), 10 mM xylose, 1 mM NAD+ and 10 µl crude protein extracts. The assay mixture containing 50 mM phosphate buffer (pH 8.0), 10 mM xylonolactone, and 10 µl crude protein extracts was used for xylonolactonase enzyme reaction. Activities of the two enzymes were measured by directly monitoring product formation by ion chromatography.
Fed-batch Fermentation
For large-scale production of xylonate, fed-batch cultures were carried out in a Biostat B plus MO5L fermentor (Sartorius, Germany) containing 3 L of growth medium (20 g/L of tryptone, 10 g/L yeast extract and 5 g/L NaCl) that was sterilized at 121°C for 20 min. Glycerol (10 g/L), K2HPO4·3H2O (5 g/L), MgSO4 (0.12 g/L) and trace elements (1 ml per liter, 3.7 g/L (NH4)6Mo7O24·4H2O, 2.9 g/L ZnSO4·7H2O, 24.7 g/L H3BO3, 2.5 g/L CuSO4·5H2O, 15.8 g/L MnCl2·4H2O) were autoclaved or filter-sterilized separately and added prior to initiation of the fermentation. 50 ml of inoculum was prepared by incubating the culture in shake flasks containing liquid LB medium overnight at 37°C. The fermentation was first operated in a batch mode and the control settings were: 37°C, stirring speed at 600 rpm, and airflow at 2 L/min. During the fermentation process, the pH was controlled at 7.0 via automated addition of 5 M NaOH. Antifoam 204 was added to prohibit foam development. The agitation was associated with the dissolved oxygen (DO) to maintain a DO concentration of above 20% saturation. After the initial glycerol was nearly exhausted, fed-batch mode was commenced by feeding a solution containing 50% of glycerol at appropriate rates and the residual glycerol was maintained at a very low level. When the cells were grown to an OD600 of about 20, IPTG was added to the culture at a concentration of 0.5 mM, xylose was added to a final concentration of 30 g/L, and the culture temperature was switching to 30°C. Samples of fermentation broth were removed at appropriate intervals to determine residual xylose, xylonate and xylonolactone production.
Determination of Xylose, Xylonolactone and Xylonate by Ion Chromatography
The determination of xylose, xylonolactone and xylonate in the culture broth was performed on an ICS-3000 (Dionex, Sunnyvale, CA, USA) ion chromatography (IC) system. The IC was equipped with an IonPac AS11 anion chromatography column (4.0 mm×250 mm) and an AG-11 guard column (4.0 mm×50 mm). Suppression was achieved with anion suppressor (ASRS 300 4 mm). Peaks were detected using electrochemical detector. A mixture of 250 mM NaOH (2%) and H2O (98%) was used for elution at a flow rate of 1 mL/min. For sample analysis, another elution step with 80% of 250 mM NaOH was employed to remove the residual components. Data collection and handling were carried out by Dionex Chromeleon software.
Results and Discussion
Inactivation of Native E. Coli Xylose Catabolic Pathways
E. coli BL21 star(DE3) was chosen as the host for xylonate production in this work. Unlike E. coli K12, strain BL21 and its derivatives lack the requisite xylonate dehydratase activity (potentially encoded by yjhG and yagF). Therefore, they cannot consume xylonate as the carbon source and would be useful for the accumulation of xylonate in the culture broth. In addition, strain BL21 star(DE3) carries a mutated rne gene which encodes a truncated RNase E enzyme and lacks the ability to degrade mRNA, resulting in an increase in mRNA stability. This might be helpful to the Red recombination enzyme activities and thus this strain has been successfully used for gene knockout [20]. E. coli could efficiently utilize xylose as a carbon source for growth. The first two genes responsible for xylose catabolism have been recognized as xylose isomerase (encoded by xylA) and xylulose kinase (encoded by xylB) [21]. In order to block the native xylose catabolic pathway, we disrupted xylA and xylB in strain BL21 star(DE3) chromosome using the Red recombination method. Successful gene disruptions were confirmed by PCR amplification (Figure S1). Liquid growth tests on M9 mineral medium supplemented with xylose as the sole carbon source further proved that the xylose metabolism-blocked strain BL21ΔxylAB grew much more poorly than its parent strain.
Heterologous Expression of Xylose Dehydrogenase and Xylonolactonase
With the aim to express the xylose dehydrogenase and xylonolactonase enzymes, we cloned the coding region of xdh and xylC from C. crescentus into pACYCduet-1 expression vector under T7 promoter, respectively. The recombinant plasmids pA-xdh, pA-xylC and pA-xdhxylC were confirmed by restriction enzyme digestion and DNA sequencing. To verify the expression levels of the recombinant proteins, strain BL21ΔxylAB was transformed by the expression vectors including different genes and grown in liquid LB medium followed by induction using 0.5 mM IPTG. Figure 2 showed the gel electrophoresis patterns of samples from different recombinant strains analyzed with coomassie brilliant blue staining. We noted distinct bands of the expected size from protein extracts of the recombinant strains compared with the control strain harboring pACYCduet-1. SDS-PAGE analysis of the recombinant strain carrying pA-xdh and pA-xylC revealed the recombinant proteins of different sizes (corresponding to the bands of molecular weight 26.6 kDa and 31.6 kDa) while the lysate of strain BL21ΔxylAB/pA-xdhxylC gave both of the two bands.
Figure 2. Expression of the recombinant xylose dehydrogenase and xylonolactonase from C. crescentus.

Lane M, prestained protein ladder; lane 1, BL21ΔxylAB harboring pACYCduet-1; lane 2, BL21ΔxylAB harboring pA-xdh; lane 3, BL21ΔxylAB harboring pA-xylC; lane 4, BL21ΔxylAB harboring pA-xdhxylC.
Establishment of a Xylose, Xylonate and Xylonolactone Analysis Method for the Determination of Xylose Dehydrogenase and Xylonolactonase Activities
In several previous studies, a general high performance liquid chromatography (HPLC) method equipped with an ion exchange column (Aminex HPX-87H, Bio-Rad) was used to determine the extracellular metabolites including xylose, xylonate and xylonolactone. However, these three chemicals displayed similar retention times under the corresponding separation conditions, leading to the inaccuracy of measurement. Here, we developed an ion chromatography (IC) method to separate the three metabolites. Under such a condition, they were generally well separated, except that xylose was eluted only 0.14 min after xylonolactone (Figure 3A–C). Then the established IC analysis method was applied to determine the metabolites concentrations in the fermentation culture and the enzymatic products of xylose dehydrogenase and xylonolactonase. As the original xylose dehydrogenase activity has been measured by determining the absorption of NADH at 340 nm [16], we briefly detected the enzymatic reaction products of the enzyme by the established IC method. As shown in Figure 3D, the reaction mixture of xylose dehydrogenase gave an apparent peak of xylonolactone when xylose was supplemented as the substrate. And the reaction mixture of xylonolactonase showed the peaks of xylonolactone and xylonate, respectively (Figure 3E). These results demonstrated that both of the enzymes were expressed in their active forms in the recombinant E. coli strains.
Figure 3. Detection of xylose, xylonate and xylonolactone by ion chromatography.

A, 1 ppm xylose, corresponding to the retention time of 2.75 min; B, 200 ppm xylonolactone, corresponding to the retention time of 2.61 min; C, 100 ppm xylonate, corresponding to the retention time of 3.43 min; D, detection of the enzymatic product of xylose dehydrogenase; E, detection of the enzymatic product of xylonolactonase; F, ion chromatogram of the extracellular metabolites of strain BL21ΔxylAB/pA-xdhxylC after being induced for 12 h. Both the enzymatic reaction mixtures and culture broth supernatant were appropriately diluted for ion chromatography analysis.
Comparison of Xylonate Productivities of Different Engineered Strains
Because the native xylonate-producing strains are not suitable for an industrial process, several heterologous systems have been developed for xylonate production, including Saccharomyces cerevisiae [22] and Kluyveromyces lactis [14]. Compared with these yeast strains, E. coli genome encodes several xylose transporters [23] and xylonate transport across E. coli cell membrane is also efficient, which might facilitate the consumption of xylose and the accumulation of xylonate. However, E. coli lacks the requisite xylose dehydrogenase activity, and thus wild-type E. coli strain could not convert xylose to xylonate. To construct a xylonate-producing strain, the xylose dehydrogenase from C. crescentus was heterologously expressed in E. coli BL21 star(DE3). As expected, accumulations of xylonolactone and xylonate were found in the cultures of the recombinant strain. In order to reduce the inhibiting effects of xylonolactone and facilitate the IC analysis of metabolites, the substrate xylose was supplemented at a relatively low concentration (1 g/L). As shown in Figure 4, 0.57 g/L xylonolactone and 0.12 g/L xylonate were produced after being induced for 12 h and the initial xylose was nearly completely exhausted. This productivity was much lower than the theoretical value because E. coli could catabolize xylose through the native phosphate pentose pathway [24]. To reduce the conversion of xylose to biomass, we further disrupted native E. coli xylA and xylB genes, which encode the first two enzymes responsible for xylose utilization [25]. When the native xylose catabolic pathway was blocked in the strain BL21ΔxylAB, the final titers of xylonolactone and xylonate were enhanced to 0.72 g/L and 0.16 g/L, respectively. However, most of the fermentation products were xylonolactone in the strain expressing the xylose dehydrogenase solely. In order to increase the hydrolysis rate of xylonolactone, we further constructed the engineered strain BL21ΔxylAB/pA-xdhxylC. The ion chromatograph of extracellular metabolites of this strain after 12 h-induction was shown in Figure 3F. We could find most of the xylonolactone was converted to xylonate by this strain. The final titers of xylonolactone and xylonate reached 0.10 g/L and 0.84 g/L and the molar yield of xylonolactone and xylonate on xylose also reached 86.0%.
Figure 4. Comparison of xylonate and xylonolactone production of several different strains.

Data were obtained after each strain was induced for 12 h in liquid LB medium supplemented with 1 g/L xylose. BL21/pA-xdh, strain BL21 star(DE3) expressing C. crescentus xylose dehydrogenase; BL21ΔxylAB/pA-xdh, knockout of native xylA and xylB while expressing C. crescentus xylose dehydrogenase; BL21ΔxylAB/pA-xdhxylC, knockout of native xylA and xylB while coexpressing C. crescentus xylose dehydrogenase and xylonolactonase.
Xylonate Production Under Fed-batch Cultivation
Among the industrially important microorganisms, E. coli has been used for various biotechnological processes and production of many valuable chemicals [26]. To investigate the feasibility for larger-scale production of xylonate, the finally engineered E. coli strain BL21ΔxylAB/pA-xdhxylC was cultured in a 5 L-scale laboratory fermenter. Cell density, xylose utilization, and products accumulation were monitored over the course of the experiment. Figure 5 shows the time profiles for cell density, residual xylose, xylonolactone and xylonate concentrations during the fermentation processes. For approximately 16 h post-induction, the bacteria grew very fast. Xylonate accumulated rapidly in the culture media while xylonolactone remained at a relatively low level. The highest xylonate production was obtained after 16 h induction, that is, 27.3 g/L. At this time, the final titer of xylonolactone reached 1.7 g/L and the initial xylose was completely exhausted. The volumetric productivity of xylonate and xylonolactone was 1.8 g/(L·h). The molar yield of xylonate and xylonolactone on xylose was about 88.0%, which was comparable to the shake-flask scale. Both of the titers of xylonate and xylonolactone remained stable during the following culture processes. In contrast, the recombinant strain only expressing xylose dehydrogenase (BL21ΔxylAB/pA-xdh) grew much slower after being induced with IPTG and cell growth ceased at an OD600 of around 40 even though glycerol was still being consumed (data not shown).
Figure 5. Time profiles for cell density (OD600), residual xylose, xylonate and xylonolactone concentrations in the culture broth during fed-batch culture of the finally engineered strain BL21ΔxylAB/pA-xdhxylC.

In two previous studies using yeast as the host organism, S. cerevisiae could only accumulate up to 3.8 g/L xylonate [13] while K. lactis produced 19 g/L xylonate from 40 g/L xylose [14]. Considering that yeast does not have an efficient pentose transport system, xylose uptake and metabolism might be limited in these strains. As for the native xylonate producers, the highest production was reported to be 92 g/L achieved by P. fragi [7], which was much higher than the E. coli strains and the yield of xylonate on xylose reached 92%, which was also comparable to the current study. This might be due to that the wild-type strains could endure high concentrations of xylonate. Therefore, the current strains have the potential to be further engineered to enhance their resistance to xylonate, thus leading to even higher production. On the other hand, E. coli grows much faster in cheap culture medium than the native producers [27]. The whole fermentation process only requires less than 24 h for the engineered strain in this work, while the native producing strains always take several days to reach the maximum titer. This would contribute to reducing the production cost during large-scale fermentation.
Conclusions
In this study, a robust xylonate-producing E. coli strain was successfully constructed. Three distinct genetic alterations targeted at the xylose or xylonate metabolic pathways were introduced into the host strain BL21 star(DE3), including knockout of the endogenous xylA and xylB genes, which encodes the xylose isomerase and xylulose kinase, to block the native xylose catabolism; heterologous expression of a xylose dehydrogenase to render E. coli capable of converting xylose to xylonolactone; and further introducing a xylonolactonase to hydrolyze the intermediate xylonolactone to xylonate. Under fed-batch conditions, up to 27.3 g/L xylonate and 1.7 g/L xylonolactone were produced out of 30 g/L xylose by the finally engineered strain BL21ΔxylAB/pA-xdhxylC. The volumetric productivity was as high as 1.8 g/(L·h). This engineered E. coli has a promising application for the industrial-scale production of xylonate.
Supporting Information
Identification of the xylA and xylB knockout E. coli strains. PCR verifications were performed with primers xylAB_DelIden_F and xylAB_DelIden_R (Table S1) corresponding to sequences up- and downstream of disrupted regions. Lane M, DNA molecular weight markers; lane 1, the original strain BL21 star(DE3); lane 2, the strain after introducing the kanamycin resistant disrupting cassette; lane 3, the final strain BL21/ΔxylAB by eliminating the kanamycin resistance by plasmid pCP20.
(TIF)
Primers used in this study for gene disruption, verification and plasmids construction.
(DOC)
Acknowledgments
The authors would like to thank Dr. Haiyan Yang and Dr. Yun Fa for ion chromatography analysis.
Funding Statement
This work was supported by the National Defense Innovation Foundation of Chinese Academy of Sciences (No. CXJJ-11-M56), National Natural Science Foundation of China (No. 21202179), Sci-Tech Development Project of Qingdao (No. 12-4-1-45-nsh), ‘Twelfth Five-Year Plan’ in National Science and Technology for the Rural Development (No. 2012BAD32B06-2) and National High-Tech R&D Program of China (No. SS2013AA050703-2). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ramachandran S, Fontanille P, Pandey A, Larroche C (2006) Gluconic acid: Properties, applications and microbial production. Food Technol Biotechnol 44: 185–195. [Google Scholar]
- 2. Niu W, Molefe MN, Frost JW (2003) Microbial synthesis of the energetic material precursor 1,2,4-butanetriol. J Am Chem Soc 125: 12998–12999. [DOI] [PubMed] [Google Scholar]
- 3. Chun BW, Dair B, Macuch PJ, Wiebe D, Porteneuve C, et al. (2006) The development of cement and concrete additive. Appl Biochem Biotechnol 131: 645–658. [DOI] [PubMed] [Google Scholar]
- 4.Werpy T, Petersen G (2004) Top value added chemicals from biomass. Volume 1: Results of screening for potential candidates from sugars and synthesis gas. U.S. Department of Energy, Oak Ridge, TN, USA. 76 p.
- 5. Governo AT, Proença L, Parpot P, Lopes MIS, Fonseca ITE (2004) Electro-oxidation of D-xylose on platinum and gold electrodes in alkaline medium. Electrochim Acta 49: 1535–1545. [Google Scholar]
- 6. Ishizaki H, Ihara T, Yoshitak J, Shimamur M, Imai T (1973) D-Xylonic acid production by Enterobacter cloacae . J Agric Chem Soc Jpn 47: 755–761. [Google Scholar]
- 7. Buchert J, Viikari L, Linko M, Markkanen P (1986) Production of xylonic acid by Pseudomonas fragi . Biotechnol Lett 8: 541–546. [Google Scholar]
- 8. Buchert J, Viikari L (1988) Oxidative D-xylose metabolism of Gluconobacter oxydans . Appl Microbiol Biotechnol 29: 375–379. [Google Scholar]
- 9. Toivari M, Nygård Y, Penttilä M, Ruohonen L, Wiebe M (2012) Microbial D-xylonate production. Appl Microbiol Biotechnol 96: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Berghäll S, Hilditch S, Penttilä M, Richard P (2007) Identification in the mould Hypocrea jecorina of a gene encoding an NADP+: D-xylose dehydrogenase. FEMS Microbiol Lett 277: 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Johnsen U, Schönheit P (2004) Novel xylose dehydrogenase in the halophilic archaeon Haloarcula marismortui . J Bacteriol 186: 6198–6207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnsen U, Dambeck M, Zaiss H, Fuhrer T, Soppa J, et al. (2009) D-Xylose degradation pathway in the halophilic archaeon Haloferax volcanii . J Biol Chem 284: 27290–27303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Toivari M, Ruohonen L, Richard P, Penttilä M, Wiebe M (2010) Saccharomyces cerevisiae engineered to produce D-xylonate. Appl Microbiol Biotechnol 88: 751–760. [DOI] [PubMed] [Google Scholar]
- 14. Nygård Y, Toivari MH, Penttilä M, Ruohonen L, Wiebe MG (2011) Bioconversion of D-xylose to D-xylonate with Kluyveromyces lactis . Metab Eng 13: 383–391. [DOI] [PubMed] [Google Scholar]
- 15. Buchert J, Viikari L (1988) The role of xylonolactone in xylonic acid production by Pseudomonas fragi . Appl Microbiol Biotechnol 27: 333–336. [Google Scholar]
- 16. Stephens C, Christen B, Fuchs T, Sundaram V, Watanabe K, et al. (2007) Genetic analysis of a novel pathway for D-xylose metabolism in Caulobacter crescentus . J Bacteriol 189: 2181–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97: 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lien OG (1959) Determination of gluconolactone, galactonolactone, and their free acids by the hydroxamate method. Anal Chem 31: 1363–1366. [Google Scholar]
- 19. Cao YJ, Jiang XL, Zhang RB, Xian M (2011) Improved phloroglucinol production by metabolically engineered Escherichia coli . Appl Microbiol Biotechnol 91: 1545–1552. [DOI] [PubMed] [Google Scholar]
- 20. Chemler JA, Fowler ZL, McHugh KP, Koffas MAG (2010) Improving NADPH availability for natural product biosynthesis in Escherichia coli by metabolic engineering. Metab Eng 12: 96–104. [DOI] [PubMed] [Google Scholar]
- 21. David JD, Wiesmeye H (1970) Control of xylose metabolism in Escherichia coli . Biochim Biophys Acta 201: 497–499. [DOI] [PubMed] [Google Scholar]
- 22. Toivari M, Nygård Y, Kumpula EP, Vehkomäki ML, Benčina M, et al. (2012) Metabolic engineering of Saccharomyces cerevisiae for bioconversion of D-xylose to D-xylonate. Metab Eng 14: 427–436. [DOI] [PubMed] [Google Scholar]
- 23. Shimada T, Fujita N, Yamamoto K, Ishihama A (2011) Novel roles of cAMP receptor protein (CRP) in regulation of transport and metabolism of carbon sources. PLoS ONE 6: e20081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Desai TA, Rao CV (2010) Regulation of arabinose and xylose metabolism in Escherichia coli . Appl Environ Microbiol 76: 1524–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rosenfeld S, Stevis P, Ho NY (1984) Cloning and characterization of the xyl genes from Escherichia coli . Molec Gen Genet 194: 410–415. [DOI] [PubMed] [Google Scholar]
- 26. Vickers C, Klein-Marcuschamer D, Krömer J (2012) Examining the feasibility of bulk commodity production in Escherichia coli . Biotechnol Lett 34: 585–596. [DOI] [PubMed] [Google Scholar]
- 27. Yang F, Cao Y (2012) Biosynthesis of phloroglucinol compounds in microorganisms–review. Appl Microbiol Biotechnol 93: 487–495. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Identification of the xylA and xylB knockout E. coli strains. PCR verifications were performed with primers xylAB_DelIden_F and xylAB_DelIden_R (Table S1) corresponding to sequences up- and downstream of disrupted regions. Lane M, DNA molecular weight markers; lane 1, the original strain BL21 star(DE3); lane 2, the strain after introducing the kanamycin resistant disrupting cassette; lane 3, the final strain BL21/ΔxylAB by eliminating the kanamycin resistance by plasmid pCP20.
(TIF)
Primers used in this study for gene disruption, verification and plasmids construction.
(DOC)