

Abstract
Psoriasis is a chronic, inflammatory skin disease caused by a combination of environmental and genetic factors. The Tnip1 gene encodes ABIN-1 (A20 Binding and Inhibitor of NFκB-1) protein and is strongly associated with susceptibility to psoriasis in humans. ABIN-1, a widely expressed ubiquitin binding protein, restricts TNF and TLR induced signals. We report here that mice lacking ABIN-1 specifically in dendritic cells (DCs), ABIN-1Flox CD11c-Cre mice, exhibit perturbed immune homeostasis. ABIN-1 deficient DCs display exaggerated NFκB and MAP kinase signaling and produce more IL-23 than normal cells in response to TLR ligands. Challenge of ABIN-1Flox CD11c-Cre mice with topical TLR7 ligand leads to greater numbers of TH17 and TCRγδ T cells and exacerbated development of psoriaform lesions. These phenotypes are reversed by DC-specific deletion of the TLR adaptor MyD88. These studies link ABIN-1 with IL-23 and IL-17, and provide cellular and molecular mechanisms by which ABIN-1 regulates susceptibility to psoriasis.
Introduction
Psoriasis is a common immune mediated skin disorder whose complex pathophysiology involves environmental factors, including microbes, as well as host susceptibility factors, including genetically determined immunologic propensities (1, 2). Recent genome wide association studies (GWAS) have highlighted potential roles of specific proteins in disease pathogenesis, including HLA-C, IL-12B, TNIP1/ABIN-1, TNFAIP3/A20, IL-23A and IL-23R (3). Some of these polymorphisms are also linked to therapeutic responses of psoriasis patients to specific therapies (4). These findings emphasize the immunological nature of psoriasis and implicate specific immune functions, such as HLA-C mediated antigen presentation and IL-12/IL-23 dependent innate immune signals. As innate immune cells such as DCs are outstanding antigen presenting cells, and as IL-12 and IL-23 are secreted by innate immune cells to amplify T cell activation and differentiation events, these genetic clues suggest that aberrant DC and T cell functions are integral to psoriasis.
TNIP1, which encodes the ABIN-1 protein, is strongly linked to psoriasis in both European (combined genome-wide P value of 1 × 10−20) and Chinese (combined genome-wide P value of 3.8 × 10−21) populations (5–7). ABIN-1 restricts several NFκB signaling cascades and regulates cell survival (8–12). In vitro studies suggest that ABIN-1 can bind NEMO/IKKγ and inhibit TNF induced NFkB signaling (13). ABIN-1 can also bind ubiquitin chains, and ubiquitin binding by ABIN-1 is important for ABIN-1’s ability to restrict TNF and TLR signals (11, 12). Global loss or mutation of ABIN-1 leads to either embryonic lethality or spontaneous inflammation and autoimmunity (11, 12, 14). These studies indicated that ABIN-1 plays critical roles in regulating TNF and TLR signals. However, the mechanisms by which ABIN-1 regulates physiological immune homeostasis and psoriasis susceptibility are unknown.
DCs have long been recognized as important cells for triggering immune responses during overt immunizations or infections (15). Recent studies suggest that DCs also preserve immune homeostasis under basal conditions (16–21). DCs may regulate susceptibility to psoriasis by secreting type I interferons, TNF, or other pro-inflammatory cytokines, and stimulating skin T cells (22). Given the potential importance of DCs to immune homeostasis in the skin and the potential importance of ABIN-1 polymorphisms to psoriasis, we have investigated whether ABIN-1 expression in DCs may regulate psoriasis susceptibility.
Materials and Methods
Mice
The initial targeting of the Tnip1 (ABIN-1) gene in C57BL/6N PRX-B6T ES cells was previously described (11). ABIN-1 targeted ES cells were transfected with an EF1α-Cre expression construct (20), and colonies were screened for deletion of the neomycin gene and retention of ABIN-1 exons 12–15 flanked by LoxP sites (floxed allele). Genotypes were confirmed both by Southern blot analyses and by PCR (primers: TTGATTCCCCTTCGCCCATTCCAGC; CCTCAAACAGCAGAAGAGGAAAGC; and ATGGGTGGGTAGGCATAGGGATAG). MyD88FL mice were described previously (25). All mouse experiments were approved by the UCSF Institutional Care and Use Committee.
Cell preparation and analyses
Cell preparations and flow cytometric analyses were performed as previously described (20). Immunoblots were performed as described (11). Antibodies to surface markers (BD Biosciences), actin (Calbiochem), phospho-IκBα, IκBα, phospho-Erk, Erk, phospho-JNK, and JNK (Cell Signaling Technologies) were purchased.
Imiquimod (IMQ) treatment and scoring of skin inflammation
IMQ treatments were performed largely as previously described (26). Mice received a daily topical dose of 12.5 μg of IMQ cream (5%) (Perrigo) or lotion control for 12 consecutive days. Skin inflammation was scored using a previously described scoring system (26). Histological sections were prepared by the UCSF VAMC Pathology Core.
Results and Discussion
To investigate whether ABIN-1 expression in DCs regulates immune functions in vivo, we generated mice bearing LoxP sites flanking exons 12–15 of ABIN-1 (ABIN-1FL) mice and bred them with CD11c-Cre transgenic mice to create mice lacking ABIN-1 in DCs (Supplemental Figure 1A) (24). ABIN-1FL/FL CD11c-Cre mice appeared grossly normal for up to six months of age. Conventional DCs (cDCs, CD11chigh MHC-II+) and plasmacytoid DCs (pDCs, CD11cLo B220Hi) were present in slightly elevated numbers in spleens from ABIN-1FL/FL CD11c-Cre mice (Fig. 1A). These cells expressed relatively normal levels of activation markers, with the exception of minimally elevated levels of CD40 on cDCs and slightly lower CD80 levels on pDC (Supplemental Figure 1B–C). Thus, ABIN-1 expression in DCs is not required for DC development but modestly regulates DC activation under basal conditions. ABIN-1FL/FL CD11c-Cre mice developed splenomegaly and lymphadenopathy by 3–4 months of age, accumulating myeloid (CD11b+ Gr-1+) cells and memory phenotype T cells (Fig. 1B–D, and data not shown). Thus, ABIN-1 expression in DCs preserves myeloid and lymphoid immune homeostasis.
Figure 1. ABIN-1 expression in DCs is necessary to preserve immune homeostasis.
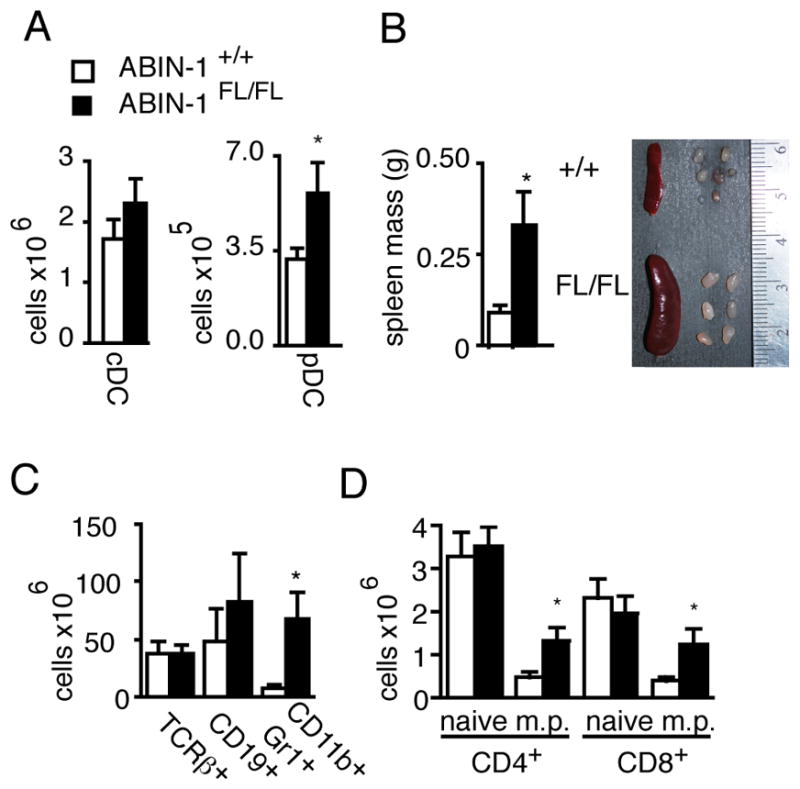
(A) Flow cytometric quantitation of MHC II+ CD11chi conventional DCs (cDCs) and MHC II+ CD11cdim B220+ plasmacytoid DCs (pDCs) in spleens from indicated mice. (B) Spleens from indicated mice. (C) Flow cytometric analyses of myeloid, T cell, and B cell splenic subpopulations. (D) Quantitation of naïve (CD44lo) and memory phenotype (CD44hi or m.p.) T cells. Error bars represent SEM. Open bars indicate ABIN-1+/+ CD11c-Cre+ mice; shaded bars indicate control ABIN-1FL/FL CD11c-Cre+ mice. Data are representative of 5–6 pairs of 8–16 week old mice in 3–7 independent experiments. * indicates p< 0.05 by Student’s t-test.
DCs are activated by TLR ligands during overt immunizations and infections and may also respond to MyD88-dependent signals under basal conditions (20). We asked whether ABIN-1 preserves immune homeostasis by restricting MyD88-dependent signals in DCs by generating compound ABIN-1FL/FL MyD88FL/FL CD11c-Cre mice that lack both ABIN-1 and MyD88 specifically in DCs (25). Remarkably, in contrast to ABIN-1FL/FL CD11c-Cre mice, the spontaneous accumulation of myeloid cells and activated T lymphocytes observed in ABIN-1FL/FL CD11c-Cre mice was abrogated in ABIN-1FL/FL MyD88FL/FL CD11c-Cre mice (Fig. 2A–D, Supplemental Figure 2A–B). Thus, ABIN-1 restricts basal Myd88-dependent intracellular signals in DCs, thereby preserving immune homeostasis in unperturbed mice.
Figure 2. ABIN-1 restricts MyD88-dependent signals in DCs.
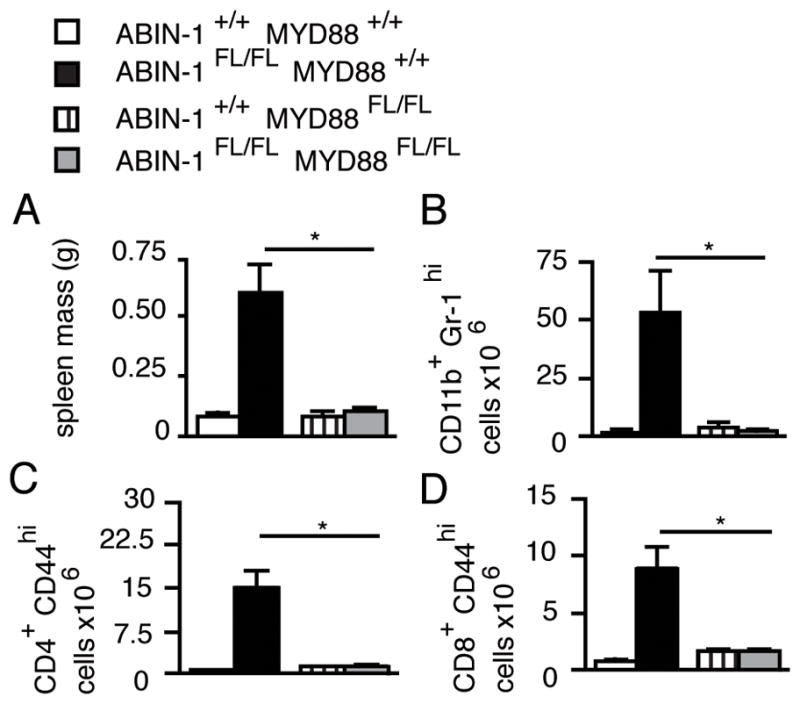
(A) Spleen mass of indicated genotypes. (B) Number of myeloid cells in spleens of indicated mice determined by flow cytometry. (C,D) Number of memory phenotype CD4+ (C) and CD8+ (D) T cells in pooled axillary, brachial, and inguinal lymph nodes. All mice are CD11c-Cre+. Error bars represent SEM. All data are representative of 3 independent experiments. * indicates p < 0.05 by Students t-test.
To determine how ABIN-1 restricts MyD88 dependent signals in DCs, we tested the responses of ABIN-1−/− and ABIN-1+/+ bone marrow derived DCs (BMDCs) to the TLR4 ligand LPS. LPS stimulated ABIN-1−/− BMDCs secreted more TNF, IL-6, IL-12, and IL-23 than control BMDCs (Supplemental Figure 2D). This is consistent with a recent report showing ABIN-1 restricts TLR-induced IL-6 and TNF (12). Importantly, this new study implicates ABIN-1 in restricting IL-23 production by DCs. After LPS stimulation, ABIN-1−/− BMDCs also exhibited exaggerated NFκB, JNK and p38 (but not ERK) signaling when compared to control BMDCs (Supplemental Figure 2C). Hence, ABIN-1 regulates TLR responses in DCs by restricting TLR induced NFκB and MAP kinase signals.
Given the genetic linkage of ABIN-1 to psoriasis and the exaggerated production of IL-12 and IL-23 by ABIN−/− DCs, we asked whether ABIN-1 expression in DCs regulates susceptibility to experimental psoriasis. Topical treatment with the TLR7 ligand imiquimod can cause a psoriasis-like condition in humans and causes similar lesions in mice. This is now an established mouse model of psoriasis (26, 27). To interrogate the functions of radiation sensitive DCs, we generated radiation chimera bearing HSCs from ABIN-1FL/FL CD11c-Cre or ABIN-1+/+ CD11c-Cre mice. Treatment of mice with imiquimod caused markedly increased erythema, scaling, and skin thickening in ABIN-1FL/FL CD11c-Cre chimera, which combine to yield increased composite psoriasis scores in ABIN-1FL/FL CD11c-Cre mice compared to control chimera (Fig. 3A). Histologic examination of skin sections from these mice revealed epidermal hyperplasia, hypogranulosis, hyperkeratosis, and parakeratosis with neutrophils--all stereotypical histologic findings of human psoriasis—in ABIN-1FL/FL CD11c-Cre but not control mice (Fig. 3B–G). Treatment of mice with a topical emollient as control did not lead to significant clinical responses. Hence, ABIN-1 expression in DCs prevents susceptibility to experimental psoriasis.
Figure 3. ABIN-1 restricts imiquimod responses in DCs and prevents experimental psoriasis.

(A) Clinical scores of imiquimod induced skin inflammation in ABIN-1FL/FL CD11c-Cre+ and ABIN-1+/+ CD11c-Cre+ (control) mice at indicated days of imiquimod treatment. (B–G) Hematoxylin and eosin stained sections of back skin of mice of indicated genotypes from areas treated with imiquimod. Epithelial layer (“epi”) is indicated by brackets (B–G). Epidermal hyperplasia (thickening of epidermal layer) is evident in ABIN-1FL/FL (C,E,G), compared to ABIN-1+/+ (B,D,F). In (C), (E), and (G), open arrows show hyperkeratotic scaling. In (E), broad areas of hypogranulosis (abnormal loss of purple keratohyaline granules in the skin’s granular layer) are indicated by closed arrows. In (G), neutrophils (blue arrow) and parakeratosis (abnormal retention of nuclei in the outermost layer of skin, green arrow) are shown in Munro’s microabscess (dotted boxed area). For (D–G), scale bar = 0.1 mm. For (B–C), scale bar = 0.5 mm. (H) ELISA and multiplex Luminex analyses of cytokine production from BMDCs after treatment with the indicated doses of imiquimod. (I) Numbers of CD3+ and CD4+ T cells from skin draining lymph nodes from imiquimod treated mice of indicated genotypes. Numbers of TH17 (CD4+ IL-17+), TCRγ/δ+ (CD3+ GL-3+), and TH1 (CD4+ IFNγ+) T cells from skin draining lymph nodes from imiquimod treated mice. Distinct TH2 (CD4+ IL-4+) populations were not detected. All mice in (A–G) and (I) are CD11c-Cre+ radiation chimera. All data are representative of 3–7 independent experiments. Error bars represent SD. * indicates p < 0.05 by Student’s t-test.
Imiquimod induced psoriasis involves IL-23 dependent production of IL-17 (26). We thus measured the levels of inflammatory cytokines produced by ABIN-1−/− BMDCs compared to control BMDCs in response to imiquimod. Imiquimod stimulated higher levels of IL-23, IL-6, IL-12p70 and TNF secretion from ABIN-1−/− BMDCs compared to wild type BMDCs, while IL-12p40 levels were similar (Fig. 3H). We next tested the induction of IL-17 expression in imiquimod treated mice. While the total numbers of T cells in skin draining lymph nodes were similar in ABIN-1FL/FL CD11c-Cre and ABIN-1+/+ CD11c-Cre chimera, increased numbers and percentages of CD4+ TH17 cells were observed in ABIN-1FL/FL CD11c-Cre mice (Fig. 3I). Many IL-17 producing T cells in imiquimod-treated mice are epidermal TCRγ/δ+ T cells (28). Consistent with this notion, increased percentages and numbers of TCRγ/δ+ T cells were noted in draining lymph nodes from ABIN-1FL/FL CD11c-Cre mice (Fig. 3I). In contrast, analyses of the skin draining lymph nodes from IMQ-treated ABIN-1FL/FL CD11c-Cre mice showed approximately normal TH1 (IFNγ+) cell numbers and no significant numbers of TH2 (IL-4+) cells (Fig. 3I and data not shown). Finally, consistent with IL-17’s role in supporting neutrophil recruitment, ABIN-1FL/FL CD11c-Cre mice contained increased epidermal neutrophilic microabscesses (Fig. 3G). Thus, ABIN-1 expression in DCs restricts IL-23 secretion, TH17 cell differentiation, neutrophilic inflammation, and psoriatic lesions after imiquimod treatment.
To determine whether ABIN-1 dependent regulation of MyD88 dependent signals in DCs was integral to disease pathogenesis, we tested the imiquimod responses of chimera generated from ABIN-1FL/FL MyD88FL/FL CD11c-Cre compound mutant and control hematopoietic stem cells. Double mutant ABIN-1FL/FL MyD88FL/FL CD11c-Cre mice exhibited much less psoriasis than ABIN-1FL/FL CD11c-Cre mice (Fig. 4A). Indeed, ABIN-1FL/FL MyD88FL/FL CD11c-Cre mice exhibited similar clinical responses to MyD88FL/FL CD11c-Cre and wild type chimera (Fig. 4A, Supplemental Figure 2E). Histological studies confirmed the reduced inflammation observed in ABIN-1FL/FL MyD88FL/FL CD11c-Cre mice compared with ABIN-1FL/FL CD11c-Cre mice (Fig. 4B). Thus, ABIN-1 dependent regulation of MyD88 dependent signals in DCs regulates susceptibility to experimental psoriasis.
Figure 4. ABIN-1 restricts MyD88 signals in DCs to prevent imiquimod induced psoriasis.

(A) Clinical skin inflammation scores in the indicated genotypes of mice at the indicated days of imiquimod treatment. (B) Hematoxylin and eosin stained sections of back skin of mice of indicated genotypes after treatment. Epithelial layer denoted by brackets and “epi.” Note that skin inflammation in ABIN-1FL/FL MYD88+/+ mice is abrogated in ABIN-1FL/FL MYD88FL/FL mice. All mice are CD11c-Cre+. Error bars represent SD; * indicates p <0.05 by Student’s t-test. Scale bar = 0.1 mm. Data are representative of 3 independent experiments.
Our findings indicate that ABIN-1 restricts MyD88-dependent signals in DCs. ABIN-1 expression in DCs restricts TLR induced NFκB and JNK signals, thereby limiting DC expression of IL-23 and other cytokines. IL-23 supports the accumulation of IL-17 and IL-22 producing T cells. IL-17 induces epidermal neutrophil infiltration, and IL-22 alters keratinocyte proliferation and differentiation. Thus, exaggerated IL-23 expression likely leads to characteristic dermal lesions of psoriasis (29). Our studies mechanistically link ABIN-1, a major psoriasis susceptibility gene, with IL-23A and IL-23R, two other major psoriasis susceptibility genes. This linkage suggests that ABIN-1 and IL-23-dependent inflammation may be part of a common dominant pathophysiological pathway leading to psoriasis. These cellular and molecular insights into how ABIN-1 prevents psoriasis provide mechanistic insights for the genetic suggestions about psoriasis pathophysiology. Moreover, mice bearing ABIN-1 mutations should be extremely valuable models for studying psoriasis pathophysiology and treatment.
Supplementary Material
Acknowledgments
We thank Sandra Huling and Ivy Hsieh of the UCSF VAMC Pathology Core for excellent assistance with histological studies.
Abbreviations
- ABIN-1
A20 Binding and Inhibitor of NFkB-1
- DC
dendritic cell
- cDC
conventional DC
- pDC
plasmacytoid DC
Footnotes
This work was supported by grants from the NIH (A.M.) and by the Histopathology and Immunology cores of the UCSF Liver Center. G.E.H. was supported by T32 DK007007. J.A.C. was partially supported by an NSF predoctoral fellowship.
References
- 1.Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361:496–509. doi: 10.1056/NEJMra0804595. [DOI] [PubMed] [Google Scholar]
- 2.Capon F, Burden AD, Trembath RC, Barker JN. Psoriasis and other complex trait dermatoses: from Loci to functional pathways. J Invest Dermatol. 2012;132:915–922. doi: 10.1038/jid.2011.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson-Huang LM, Lowes MA, Krueger JG. Putting together the psoriasis puzzle: an update on developing targeted therapies. Dis Model Mech. 2012;5:423–433. doi: 10.1242/dmm.009092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tejasvi T, Stuart PE, Chandran V, Voorhees JJ, Gladman DD, Rahman P, Elder JT, Nair RP. TNFAIP3 gene polymorphisms are associated with response to TNF blockade in psoriasis. J Invest Dermatol. 2012;132:593–600. doi: 10.1038/jid.2011.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, Gudjonsson JE, Li Y, Tejasvi T, Feng BJ, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strange A, Capon F, Spencer CC, Knight J, Weale ME, Allen MH, Barton A, Band G, Bellenguez C, Bergboer JG, et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun LD, Cheng H, Wang ZX, Zhang AP, Wang PG, Xu JH, Zhu QX, Zhou HS, Ellinghaus E, Zhang FR, et al. Association analyses identify six new psoriasis susceptibility loci in the Chinese population. Nat Genet. 2010;42:1005–1009. doi: 10.1038/ng.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heyninck K, De Valck D, Vanden Berghe W, Van Criekinge W, Contreras R, Fiers W, Haegeman G, Beyaert R. The zinc finger protein A20 inhibits TNF-induced NF-kappaB-dependent gene expression by interfering with an RIP- or TRAF2-mediated transactivation signal and directly binds to a novel NF-kappaB-inhibiting protein ABIN. J Cell Biol. 1999;145:1471–1482. doi: 10.1083/jcb.145.7.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wullaert A, Wielockx B, Van Huffel S, Bogaert V, De Geest B, Papeleu P, Schotte P, El Bakkouri K, Heyninck K, Libert C, Beyaert R. Adenoviral gene transfer of ABIN-1 protects mice from TNF/galactosamine-induced acute liver failure and lethality. Hepatology. 2005;42:381–389. doi: 10.1002/hep.20785. [DOI] [PubMed] [Google Scholar]
- 10.Wagner S, Carpentier I, Rogov V, Kreike M, Ikeda F, Lohr F, Wu CJ, Ashwell JD, Dotsch V, Dikic I, Beyaert R. Ubiquitin binding mediates the NF-kappaB inhibitory potential of ABIN proteins. Oncogene. 2008;27:3739–3745. doi: 10.1038/sj.onc.1211042. [DOI] [PubMed] [Google Scholar]
- 11.Oshima S, Turer EE, Callahan JA, Chai S, Advincula R, Barrera J, Shifrin N, Lee B, Benedict Yen TS, Woo T, Malynn BA, Ma A. ABIN-1 is a ubiquitin sensor that restricts cell death and sustains embryonic development. Nature. 2009;457:906–909. doi: 10.1038/nature07575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nanda SK, Venigalla RK, Ordureau A, Patterson-Kane JC, Powell DW, Toth R, Arthur JS, Cohen P. Polyubiquitin binding to ABIN1 is required to prevent autoimmunity. J Exp Med. 2011;208:1215–1228. doi: 10.1084/jem.20102177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mauro C, Pacifico F, Lavorgna A, Mellone S, Iannetti A, Acquaviva R, Formisano S, Vito P, Leonardi A. ABIN-1 binds to NEMO/IKKgamma and cooperates with A20 in inhibiting NF-kappaB. J Biol Chem. 2006;281:18482–18488. doi: 10.1074/jbc.M601502200. [DOI] [PubMed] [Google Scholar]
- 14.Zhou J, Wu R, High AA, Slaughter CA, Finkelstein D, Rehg JE, Redecke V, Hacker H. A20-binding inhibitor of NF-kappaB (ABIN1) controls Toll-like receptor-mediated CCAAT/enhancer-binding protein beta activation and protects from inflammatory disease. Proc Natl Acad Sci U S A. 2011;108:E998–1006. doi: 10.1073/pnas.1106232108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol. 2012;30:1–22. doi: 10.1146/annurev-immunol-100311-102839. [DOI] [PubMed] [Google Scholar]
- 16.Chen M, Wang YH, Wang Y, Huang L, Sandoval H, Liu YJ, Wang J. Dendritic cell apoptosis in the maintenance of immune tolerance. Science. 2006;311:1160–1164. doi: 10.1126/science.1122545. [DOI] [PubMed] [Google Scholar]
- 17.Stranges PB, Watson J, Cooper CJ, Choisy-Rossi CM, Stonebraker AC, Beighton RA, Hartig H, Sundberg JP, Servick S, Kaufmann G, Fink PJ, Chervonsky AV. Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunity. 2007;26:629–641. doi: 10.1016/j.immuni.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Birnberg T, Bar-On L, Sapoznikov A, Caton ML, Cervantes-Barragan L, Makia D, Krauthgamer R, Brenner O, Ludewig B, Brockschnieder D, Riethmacher D, Reizis B, Jung S. Lack of conventional dendritic cells is compatible with normal development and T cell homeostasis, but causes myeloid proliferative syndrome. Immunity. 2008;29:986–997. doi: 10.1016/j.immuni.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 19.Ohnmacht C, Pullner A, King SB, Drexler I, Meier S, Brocker T, Voehringer D. Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med. 2009;206:549–559. doi: 10.1084/jem.20082394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hammer GE, Turer EE, Taylor KE, Fang CJ, Advincula R, Oshima S, Barrera J, Huang EJ, Hou B, Malynn BA, Reizis B, DeFranco A, Criswell LA, Nakamura MC, Ma A. Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat Immunol. 2011;12:1184–1193. doi: 10.1038/ni.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kool M, van Loo G, Waelput W, De Prijck S, Muskens F, Sze M, van Praet J, Branco-Madeira F, Janssens S, Reizis B, Elewaut D, Beyaert R, Hammad H, Lambrecht BN. The ubiquitin-editing protein A20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity. 2011;35:82–96. doi: 10.1016/j.immuni.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 22.Nestle FO, Di Meglio P, Qin JZ, Nickoloff BJ. Skin immune sentinels in health and disease. Nat Rev Immunol. 2009;9:679–691. doi: 10.1038/nri2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cantisani C, Lazic T, Richetta AG, Clerico R, Mattozzi C, Calvieri S. Imiquimod 5% cream use in dermatology, side effects and recent patents. Recent Pat Inflamm Allergy Drug Discov. 2012;6:65–69. doi: 10.2174/187221312798889301. [DOI] [PubMed] [Google Scholar]
- 24.Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8− dendritic cells in the spleen. J Exp Med. 2007;204:1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hou B, Reizis B, DeFranco AL. Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity. 2008;29:272–282. doi: 10.1016/j.immuni.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Fits L, Mourits S, Voerman JS, Kant M, Boon L, Laman JD, Cornelissen F, Mus AM, Florencia E, Prens EP, Lubberts E. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol. 2009;182:5836–5845. doi: 10.4049/jimmunol.0802999. [DOI] [PubMed] [Google Scholar]
- 27.Fanti PA, Dika E, Vaccari S, Miscial C, Varotti C. Generalized psoriasis induced by topical treatment of actinic keratosis with imiquimod. Int J Dermatol. 2006;45:1464–1465. doi: 10.1111/j.1365-4632.2006.02980.x. [DOI] [PubMed] [Google Scholar]
- 28.Cai Y, Shen X, Ding C, Qi C, Li K, Li X, Jala VR, Zhang HG, Wang T, Zheng J, Yan J. Pivotal role of dermal IL-17-producing gamma delta T cells in skin inflammation. Immunity. 2011;35:596–610. doi: 10.1016/j.immuni.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pantelyushin S, Haak S, Ingold B, Kulig P, Heppner FL, Navarini AA, Becher B. Ror gammat+ innate lymphocytes and gammadelta T cells initiate psoriasiform plaque formation in mice. J Clin Invest. 2012;122:2252–2256. doi: 10.1172/JCI61862. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.