

Abstract
Monocytes are critical effector cells of the innate immune system that protect the host by migrating to inflammatory sites, differentiating to macrophages and dendritic cells, eliciting immune responses, and killing pathogenic microbes. Monocyte chemoattractant protein 1(MCP-1), also known as CCL2, plays an important role in monocyte activation and migration. Chemotactic function of MCP-1 is mediated by binding to the CCR2 receptor, a member of the G protein-coupled receptor (GPCR) family. Desensitization of GPCR chemokine receptors is an important regulator of the intensity and duration of chemokine stimulation. G protein-coupled receptor kinases (GRKs) induce GPCR phosphorylation, and this leads to GPCR desensitization. Regulation of subcellular localization of GRKs is considered an important early regulatory mechanism of GRK function and subsequent GPCR desensitization. Chemokines and LPS are both present during Gram-negative bacterial infection, and LPS often synergistically exaggerates leukocyte migration in response to chemokines. In this study, we investigated the role of, and mechanism of, LPS-TLR4 signaling on the regulation of monocyte chemotaxis. We demonstrate that LPS augments MCP-1-induced monocyte migration. We also show that LPS, through p38 MAPK signaling, induces phosphorylation of GRK2 at serine 670, which, in turn, suppresses GRK2 translocation to the membrane, thereby preventing GRK2-initiated internalization and desensitization of CCR2 in response to MCP-1. This therefore results in enhanced monocyte migration. These findings reveal a novel function for TLR4 signaling in promoting innate immune cell migration.
Keywords: CCR2, GPCR, leukocyte migration, MCP-1
Introduction
Monocytes are critical effector cells of the innate immune system that protect the host by migrating to inflammatory sites, differentiating to macrophages and dendritic cells, eliciting immune responses, and killing pathogenic microbes (1, 2). Monocyte migration is tightly regulated by signaling mechanisms activated by chemokines, which act through chemokine receptors to direct monocyte migration along a concentration gradient (3).
Monocyte chemoattractant protein 1(MCP-1), also known as CCL2, is a 13 kD chemokine that plays an important role in monocyte activation and migration (4). MCP-1 is released from both hematopoietic and non-hematopoietic cells, including monocytes/macrophages, dendritic cells (DCs), astrocytes, endothelial cells and fibroblasts (5). MCP-1 derived from these MCP-1-produce cells contributes to the migration of monocytes into local tissue and organ during infection and inflammation (6). Physiological function of MCP-1 is mediated by binding to the CCR2 receptor, a member of the G protein-coupled receptor (GPCR) family (7).
Desensitization of GPCR family of chemokine receptors is an important determinant of the intensity and duration of agonist stimulation (8, 9). Receptor desensitization regulates not only the number of migrating cells, but also their motility and ability to stop upon contact with pathogens or target cells (8). G protein-coupled receptor kinases (GRKs) induce GPCR phosphorylation and thereby signal GPCR desensitization (10). GRKs constitute a family of seven mammalian serine and threonine protein kinases (11, 12). GRK2 is a member of the GRK family, which has been shown to modulate a variety of functions in leukocytes (13). Activity of GRKs is tightly regulated by three mechanisms: (i) subcellular localization, (ii) alterations in intrinsic kinase activity and (iii) alterations in GRK expression (14). Regulation of GRK subcellular localization therefore represents a logical means of regulating monocyte migration. However, the regulation of subcellular localization of GRKs in monocytes and their effects on monocytes behavior remain unknown.
LPS, a component of the outer membrane of Gram-negative bacteria, is a specific ligand for TLR4 and induces a range of inflammatory responses including production of pro-inflammatory mediators and induction of migration of innate immune cells to the site of infection (15). In this study, we investigated the role of, and the mechanism of, LPS-TLR4 signaling in regulating monocyte chemotaxis. We demonstrate that LPS augments MCP-1-induced monocyte migration. We also show that LPS, through p38 MAPK signaling, induces phosphorylation of GRK2 at Ser670, which in turn, suppresses GRK2 translocation to the membrane, thereby preventing GRK2-initiated internalization and desensitization of CCR2 in response to MCP-1. This therefore results in enhanced monocyte migration. These findings reveal a novel function for TLR4 signaling in promoting innate immune cell migration.
Materials and Methods
Animals
Male C57BL/6 wild-type (WT) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). TLR4 knockout (TLR4−/−) mice on a C57BL/6 background were bred in Dr. Billiar’s lab at the University of Pittsburgh. All experimental protocols involving animals were approved by Institutional Animal Care and Use Committee of VA Pittsburgh Healthcare System and University of Pittsburgh.
Cell culture
The preparation of murine bone marrow-derived monocytes (BMDM) was performed as previously described (16). Bone marrow cells were isolated by flushing femurs and tibias of 8- to 12-week-old C57BL/6 mice or TLR4−/− mice with Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS). Erythroid cells were lysed with red blood cells lysis buffer (eBioscience, San Diego, CA) and then were washed twice with DMEM, adjusted to a cell suspension of 1 × 106 cells/ml, and seeded in 6 cm ultra-low attachment surface plates (Corning Costar, Corning, NY). Cells were supplemented with 20 ng/ml rmM-CSF (colony-stimulating factor, Sigma-Aldrich, St. Louis, MO) and cultured in a humidified incubators at 37°C and 5% CO2 for 5 days. BMDMs were collected from the non-adherent cell population by centrifugation of cell culture supernatant at 1000rpm for 10 min. RAW 264.7 cells, a mouse macrophage cell line obtained from American Type Culture Collection (Rockville, MD), were also used in the experiments, and were cultured in DMEM containing 10% FBS.
Cell migration assay
Cell migration assay was performed in chemotaxis microchambers (Neuroprobe, Gaithersburg, Maryland) (15, 17). The lower well of the chemotaxis microchambers was filled with DMEM containing 0.2% BSA and different concentrations of recombinant mouse MCP-1 (BioLegend, San Diego, CA). The upper well was loaded with monocytes in suspension (50,000 cells) with or without LPS (100 ng/ml) (18). In some cases, monocytes were preincubated with a variety of MAPK inhibitors: p38 inhibitor SB203580 (10 μM, Cell Signaling Technology, Boston, MA); ERK inhibitor PD98059 (50 μM, Cell Signaling Technology, Boston, MA) (19); or JNK inhibitor SP600125 (10 μM, Sigma-Aldrich, St. Louis, MO) for 30 min (20). The chamber was incubated in humidified air with 5% CO2 at 37°C for the time as indicated. At the end of experiment, the membrane between the upper and lower wells was fixed and stained with Wright-Giemsa stain (Sigma-Aldrich, St. Louis, MO). Cell migration was determined by counting the number of cells that had migrated through the filter as determined by three random high-power microscope fields (x400). A chemotactic index (CI) was used to express chemotactic activity and was measured by calculating the number of monocytes that migrated across the membrane divided by the number of monocytes that migrated spontaneously towards blank medium in control group. All experiments were performed in triplicate.
Measurement of Cell Surface CCR2
Monocytes and RAW264.7 cells were pretreated with p38 inhibitor SB203580 (10 μM), ERK inhibitor PD98059 (50 μM) (19) or JNK inhibitor SP600125 (10 μM) for 30 min at 37°C and then were stimulated with MCP-1 (200 ng/ml) in the presence or absence of LPS (100 ng/ml) for the time as indicated. The treatment was stopped by addition of ten-times volume of ice-cold FACS buffer (0.1% sodium azide, 2% BSA, PBS). The cells were then incubated with CCR2 antibody for 1h on ice, followed by incubation with FITC-labeled secondary antibody (abcam, Cambridge, MA) and PE-labeled CD11b antibody (eBioscience, San Diego, CA) for 1 h on ice. The cells were washed and analyzed using flow cytometer (FACSCalibur; Becton Dickinson). The cutoff to define chemokine receptor-positive cells was set according to the staining with the isotype control antibody. Monocytes were identified by their light-scatter properties and expression of CCR2 and CD11b.
GRK2 RNA interference
Accell small interfering RNA (siRNA; Dharmacon, Lafayette, CO) was used to knockdown GRK2 (21). Briefly, RAW264.7 cells (3 × 105/well) were seeded in 6 cm plates and incubated for 16 h. The growth media was removed and cells were transfected with 25nM Adrbk1 Accell siRNA, according to the manufacturer’s instructions, and were incubated for additional 48 h. Cells were then collected for detection of GRK2 protein expression.
Membrane-bound GRK2 assay
BMDM were stimulated with MCP-1(200 ng/ml) in the presence or absence of LPS (100 ng/ml) for up to 1 h. Cells incubated with DMEM medium only were used as a negative control. In some cases, cells were pretreated with p38 inhibitor SB203580 (10 μM), ERK inhibitor PD98059 (50μM), or JNK inhibitor SP600125 (10 μM) for 30 min at 37°C before MCP-1 treatment. The reactions were quenched by addition of 10 ml of ice-cold PBS, and cell membrane proteins were extracted using ProteoExtract Native Membrane Protein Extraction Kit (Milipore, Billerica, MA) (22), and concentrated by utilizing ProteoExtract Protein Precipitation Kit (Milipore, Billerica, MA). The resultant proteins were resuspended in 2x Laemmli buffer and boiled for 10 min for SDS-PAGE. Rabbit polyclonal antibody to GRK2 (Santa Cruz Biotechnology, Inc. Santa Cruz, CA) was used for detection of membrane-bound GRK2. GRK2 and actin levels in the cell supernatant were also examined. The images presented in the results are representatives of at least three independent experiments. The densitometry analysis of blots was performed using the NIH-developed ImageJ software, and depicted as the mean ± SEM of the percentage changes in the ratio of membrane-bound GRK2/cytosal-GRK2 in the monocytes, which were normalized by the density of actin, from three experiments.
In vivo monocytes migration study
MCP-1-induced monocyte migration into the lungs was studied in vivo. MCP-1(50 ng per mouse) and/or LPS (25 ng per mouse) was intratracheally injected into WT or TLR4−/− mice, and at 6 h after MCP-1 and/or LPS injection, bronchoalveolar lavage fluid (BALF) was collected for monocyte counts.
Data Presentation and Statistical Analysis
The data are presented as mean ± SEM of the indicated number of experiments. Statistical significance among group means was assessed by ANOVA. Student Neuman-Keuls post-hoc test was performed. Differences were considered significant at p<0.01.
Results
LPS-TLR4 augments MCP-1-induced monocytes migration
Monocyte migration to the site of infection is an essential part of first line immune defenses. MCP-1 plays an important role in initiating monocyte activation and migration (23). LPS, however, promotes innate immune cell migration through an as yet unclear mechanism. In order to address the influence of LPS on chemokine-induced monocyte migration, we used a Boyden chamber assay (24), in which monocyte migration was driven by MCP-1. As shown in Fig. 1A, within a MCP-1 concentration range of 0 to 20 ng/ml, MCP-1 induced monocyte migration in a dose-dependent manner, whereas, a higher concentration of 200 ng/ml of MCP-1 decreased monocyte migration, possibly due to a rapid induction of chemokine receptor desensitization (15, 25). Importantly, while LPS alone failed to induce monocyte migration, LPS significantly augmented monocyte migration in response to MCP-1 in a concentration rage of 2ng/ml ~ 200 ng/ml (Fig. 1A). This effect of LPS was mediated through TLR4, since TLR4-deficiency completely prevented the LPS-enhanced monocyte migration in response to MCP-1 (Fig. 1B).
FIGURE 1.
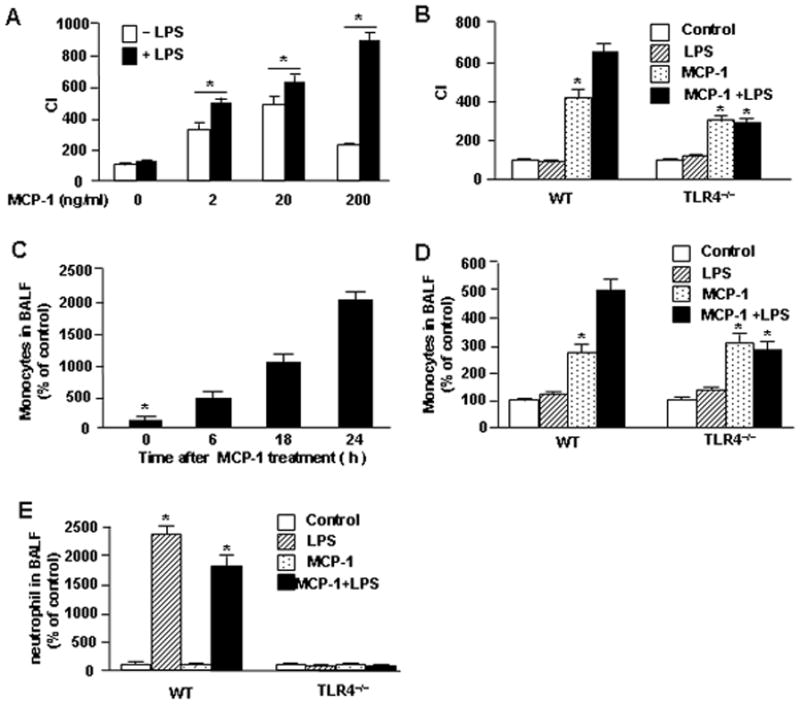
LPS-TLR4 augments MCP-1-induced monocyte migration. A. monocytes were loaded into the upper wells of the microchamber with or without 100 ng/ml LPS. The lower wells were filled with MCP-1 at concentration of 0, 2, 20, or 200 ng/ml. Cell migration was allowed for 2 h and cells that acrosd the filter were counted. B. monocytes isolated from C57BL/6 wild-type (WT) mice or TLR4 knockout (TLR4−/−) mice were loaded into the upper wells of the microchamber with or without 100 ng/ml LPS. The lower wells were filled with 200 ng/ml of MCP-1 or medium alone. Cells migration was induced for 2 h, and cells that crossed the filter were then counted. C. WT mice were given intratracheal (i.t.) instillation with MCP-1 (50 ng per mouse) for the time as indicated, and then the bronchoalveolar lavage fluid (BALF) was collected for cell counts. D and E.WT mice and TLR4−/− mice were given i.t. MCP-1 (50 ng per mouse) and/or LPS (25 ng per mouse) at the same time, and BALF was collected after 6 h for cell counts. The graph shows the mean and SEM of percentage of control from three independent experiments.
* p<0.01 compared with the groups labeled with no asterisk.
The augmenting effects of LPS on MCP-1-induced chemotaxis shown in vitro were also shown in vivo in mice. Intratracheal (i.t.) instillation of MCP-1 in WT mice, induced monocyte infiltration into the lungs in a time-dependent manner (Fig. 1C), and LPS given i.t. significantly enhanced this MCP-1-induced monocyte infiltration in the lungs at 6 h after MCP-1 stimulation (Fig. 1D). Also, consistent with the in vitro findings, genetic deletion of TLR4 prevented LPS-enhanced monocyte migration into the lungs (Fig. 1D). In addition, we observed using light microscopy that in WT mice LPS alone did not induce monocyte infiltration in the lungs, but instead induced neutrophil sequestration in the lungs (Fig. 1E). Taken together, these results indicate that LPS signals via TLR4 to augment, but not directly induce, monocyte migration in response to chemoattractants through a mechanism that is different from LPS-induced neutrophil migration.
LPS-TLR4 augments MCP-1-induced monocyte migration by modulating cell surface expression of CCR2 receptors
Upon agonist activation of receptors, a rapid attenuation of receptor responsiveness, called desensitization, occurs through feedback mechanisms, which prevent acute and chronic over-stimulation of the receptor (26). Receptor internalization is a major mechanism of receptor desensitization (7, 27). It has been reported that MCP-1 induces CCR2 internalization, and therefore regulates the desensitization of its receptor in monocytes (28). In order to elucidate the effect of LPS on MCP-1-induced CCR2 internalization, we detected cell surface CCR2 expression on monocytes following MCP-1 and/or LPS treatment using flow cytometry. As shown in Fig. 2A, MCP-1 induced decrease in cell surface expression of CCR2 in a time-dependent manner. However, LPS prevented MCP-1-induced CCR2 internalization as shown in Fig. 2B. This effect of LPS was blocked by TLR4 deficiency (Fig. 2B). The data suggest that LPS-TLR4 augments MCP-1 induced monocyte migration by modulating cell surface expression of CCR2.
FIGURE 2.
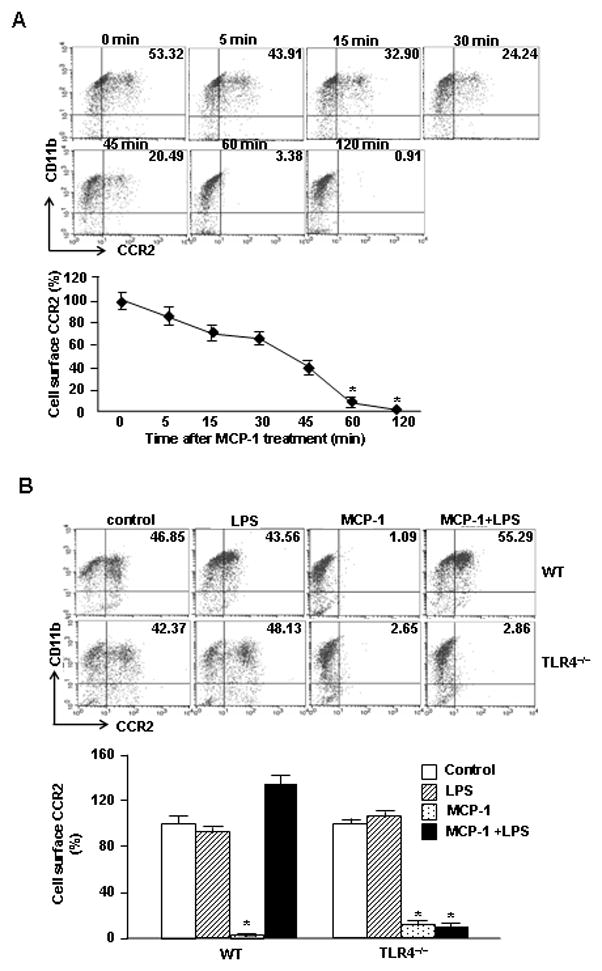
LPS-TLR4 augments MCP-1-induced monocyte migration by modulating cell surface expression of CCR2. A. Monocytes were incubated with 200 ng/ml of MCP-1 for the time as indicated, and the cell surface expression of CCR2 was then measured using flow cytometry analysis. B. Monocytes isolated from WT or TLR4−/− mice were stimulated with MCP-1 and/or LPS for 60 min, and the cell surface expression of CCR2 was then measured by flow cytometry analysis. Results are represented as percentage of control; mean ± SEM, n=3, * p<0.01 compared with the groups labeled with no asterisk.
GRK2 is a key molecule regulating MCP-1-induced CCR2 internalization
GRK2 is known to promote CCR2 desensitization through phosphorylation (29). To determine the role of GRK2 in mediating MCP-1-induced CCR2 internalization in monocytes/macrophages, we used an RNA interference approach to knockdown GRK2 in RAW264.7 cells, and analyzed the influence of GRK2 knockdown on cell surface expression of CCR2 and the ability of monocytes to migrate. Transfection of Accell small interfering RNA to GRK2 resulted in 85% decrease in GRK2 protein expression in RAW264.7 cells as shown in Fig. 3A. In these cells the MCP-1-induced decrease of cell surface expression of CCR2 was markedly suppressed (Fig. 3B), which was associated with increased cell migration in response to MCP-1 (Fig. 3C). Noteworthy, GRK2 knockdown also diminished the influence of LPS on cell surface CCR2 expression (Fig. 3B) as well as prevented the enhancing effect of LPS on the cell migration (Fig. 3C). Aggregately, the results demonstrate a critical role of GRK2 in mediating LPS/MCP-1 regulation of monocytes migration.
FIGURE 3.
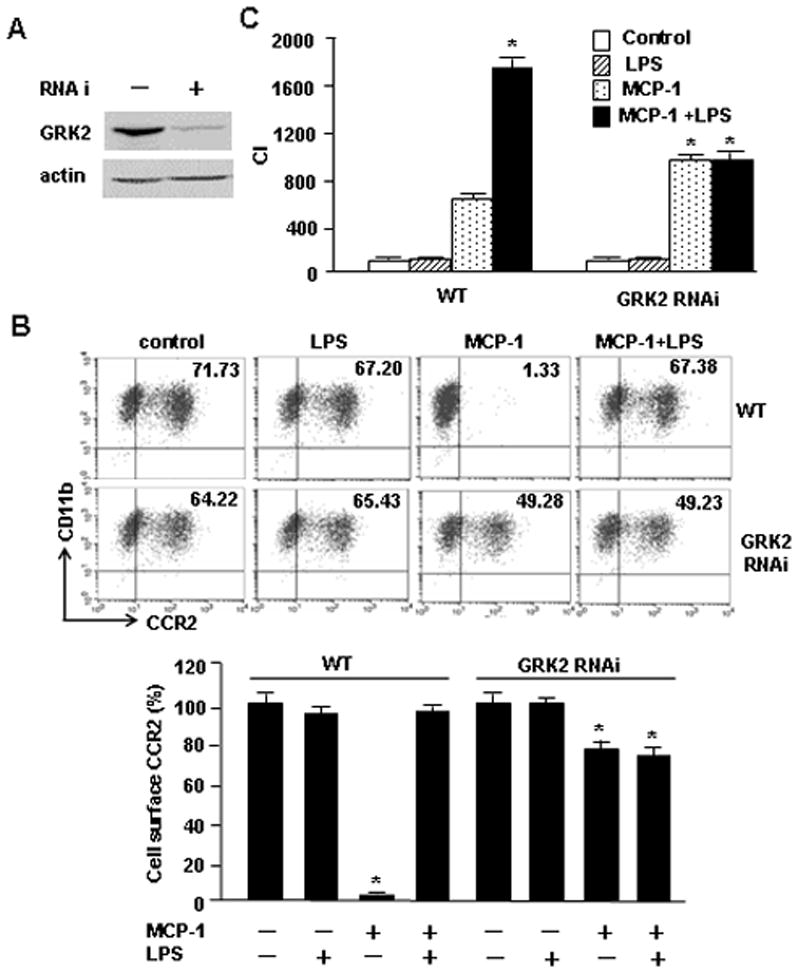
GRK2 mediates the crosstalk between LPS-TLR4 and MCP-1-CCR2 signaling. A. RAW264.7 cells were treated with Adrbk1 siRNA (siRNA to GRK2) for 48 h. The efficiency of GRK2 knockdown was measured by immunoblotting. The images are representatives of three independent experiments. B. RAW264.7 cells, or RAW264.7 cells with GRK2 knockdown (RNAi), were loaded into the upper wells of the microchamber in the presence or absence of 100 ng/ml of LPS. The lower wells were filled with 200 ng/ml of MCP-1 or medium only. Cells migration was induced for 6 h, and cells that crossed the filter were counted under microscope. C. RAW264.7 cells or RAW264.7 cells with GRK2 knockdown were stimulated with LPS and/or MCP-1 for 60 min, and the cell surface expression of CCR2 was then measured by flow cytometry analysis. Results are represented as percentage of control; mean ± SEM, n=3, *p<0.01 compared with the groups labeled with no asterisk.
LPS-induced phosphorylation of GRK2 at serine 670 suppresses GRK2 translocation to cell membrane
It has been reported that GRK2 translocation to the cell membrane upon MCP-1 stimulation is a determinant for desensitization of the CCR2 receptor in monocytes (7, 16). We hypothesized that LPS may act to preventing GRK2 subcellular translocation and so suppress CCR2 internalization and subsequent desensitization in response to MCP-1, and therefore enhance monocyte migration. To test this hypothesis, we treated monocytes with MCP-1 and/or LPS for up to 60 min, and then extracted membrane proteins from the monocytes for detection of membrane-bound GRK2. As shown in Fig. 4A, MCP-1 increased the membrane-bound GRK2 in a time-dependent manner, which suggests induction of translocation of GRK2 to the cell membrane. LPS, however, prevented this increase in membrane-bound GRK2 in response to MCP-1 stimulation (Fig. 4A). Again, TLR4-deficiency prevented LPS-mediated effects on MCP-1-mediated translocation of GRK2 (Fig. 4B). These results suggest a critical inhibitory role for LPS in GRK2 translocation to cell membrane, which is an important step promoting CCR2 desensitization.
FIGURE 4.
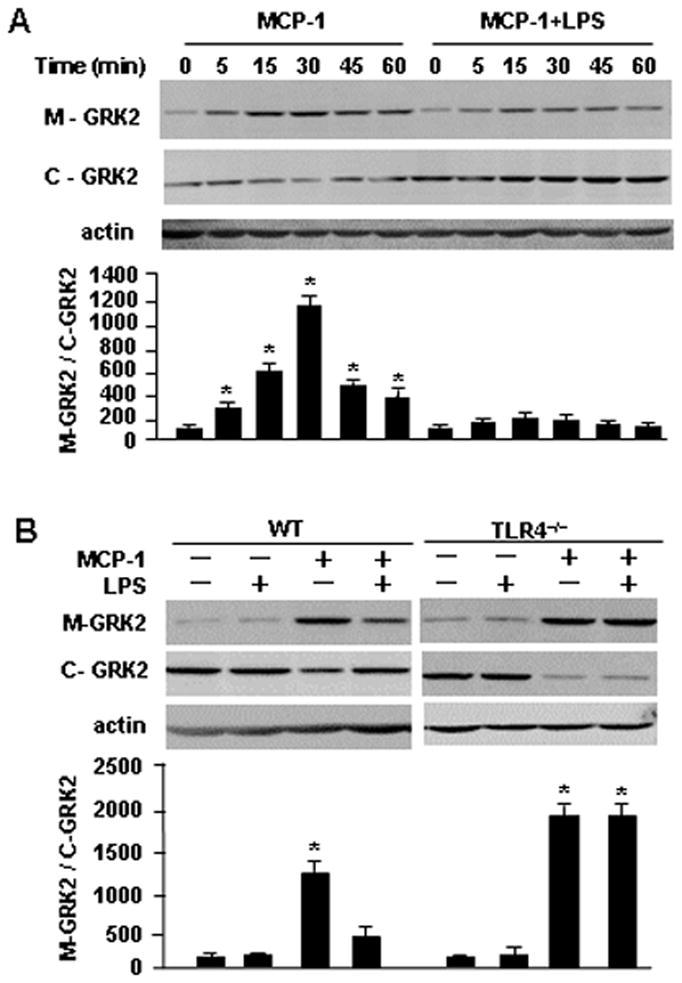
LPS inhibits the MCP-1-induced GRK2 translocation. A and B. Monocytes isolated from WT or TLR4−/− mice were incubated with MCP-1(200 ng/ml) and/or LPS (100 ng/ml) for the time as indicated, and cell membrane protein and plasma proteins were extracted and separated, and were subjected to Western blot analysis for detecting membrane-bound GRK2 (M-GRK2) and cytosolic GRK2 (C-GRK2). The images are representatives of three independent experiments. The densitometry analysis of blots was performed using the NIH-developed ImageJ software. The graph depicts the mean ± SEM of the percentage changes in the ratio of M-GRK2/C-GRK2 in the monocytes isolated from WT and TLR4−/− mice, which were normalized by the density of actin, from three experiments. * p<0.01 compared with the groups labeled with no asterisk.
Mitogen-activated protein kinases (MAPK) have been reported to regulate GRK2 activity. In vitro and in situ experiments have shown that ERK1 is able to phosphorylate recombinant GRK2 at serine 670 (Ser670), which lies within the Gβγ binding domain of GRK2 (30). Another study showed that MAPK phosphorylation at this site impairs the GRK2-Gβγ interaction, thereby inhibiting GRK2 translocation to the cell membrane, and subsequent induction of kinase activity and GPCR regulation (31). To elucidate whether LPS-TLR4, through modification of GRK2 phosphorylation, regulates GRK2 mobility, we detected specific phosphorylation of Ser670 in GRK2 in monocytes following stimulation of BMDM with MCP-1 and/or LPS. Western blotting for phospho-Ser670 on GRK2 showed that LPS, or LPS plus MCP-1, induced the phosphorylation of GRK2 at Ser670, whereas, MCP-1 alone failed to increase the phosphorylation level of Ser670 (Fig. 5A and 5B). Furthermore, LPS or LPS plus MCP-1 were unable to induce the phosphorylation of GRK2 on Ser670 in TLR4-deficient monocytes (Fig. 5B). These data demonstrate that LPS-TLR4 signaling modifies GRK2 phosphorylation at Ser670.
FIGURE 5.
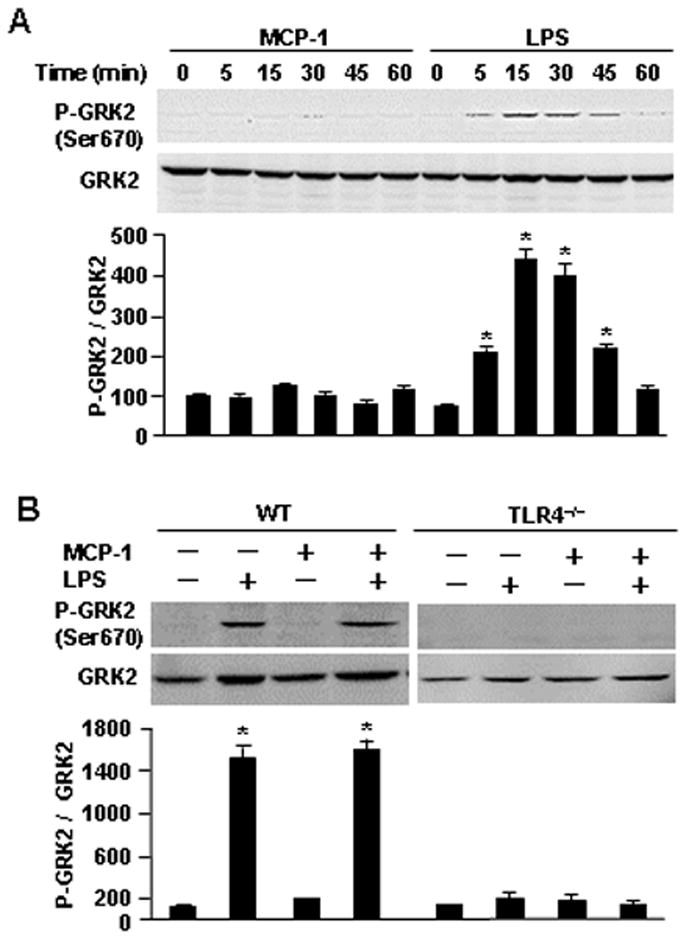
The translocation of GRK2 is regulated through phosphorylation of GRK2 at Ser670 induced by LPS. A. Monocytes isolated from WT mice were stimulated with MCP-1 (200 ng/ml) or LPS (100 ng/ml) for the time as indicated, and then the phosphorylation of GRK2 at Ser670 (P-GRK2 Ser670) was detected by immunoblotting analysis. The images are representatives of three independent experiments. The graph depicts the mean ± SEM of the percentage changes in the ratio of P-GRK2/total GRK2 in the monocytes from three experiments. * p<0.01 compared with the groups labeled with no asterisk. B. monocytes isolated from WT or TLR4−/− mice were stimulated with MCP-1 (200 ng/ml) and/or LPS (100 ng/ml) for 30 min, and then the phosphorylation of GRK2 at Ser670 (P-GRK2 Ser670) was detected by immunoblotting analysis. The graph depicts the mean ± SEM of the percentage changes in the ratio of P-GRK2/total GRK2 in the monocytes from three experiments. * p<0.01 compared with the groups labeled with no asterisk.
Opposite roles of p38 MAPK and ERK in regulating monocyte migration induced by LPS and MCP-1
MAPK, including ERK, JNK and p38, are important regulatory kinases involved in cell migration (20, 32). MCP-1-induced monocyte migration has been reported to be dependent on p38MAPK (19, 33). However, the role of MAPK in LPS-enhanced monocyte migration has not yet been elucidated. We therefore investigated the role of ERK, JNK and p38 in LPS-enhanced monocyte migration in response to MCP-1 using pharmacological inhibitor approaches. We found, using the Boyden chamber migration assay, that ERK inhibitor PD98059 significantly increased monocyte migration induced by LPS/MCP-1 (Fig. 6A). In contrast, p38 inhibitor SB203580 abrogated the LPS/MCP-1-induced monocyte migration (Fig. 6A), and JNK inhibitor SP600125 had no effect on the LPS/MCP-1-induced monocyte migration.
FIGURE 6.
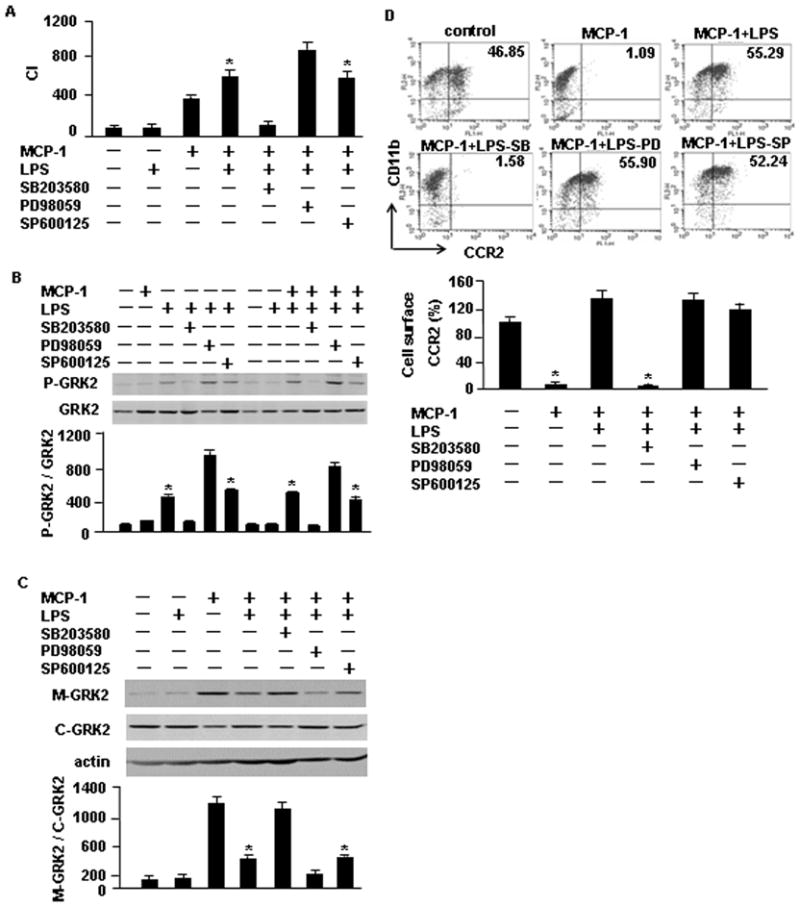
ERK and p38 play opposite roles in regulating monocyte migration induced by LPS and MCP-1. A. WT monocytes were pre-treated with p38 inhibitor SB203580 (SB; 10 μM), ERK inhibitor PD98059 (PD; 50) or JNK inhibitor SP600125 (SP; 10 μM) for 30 min, and were then loaded into the upper wells of the microchamber with or without 100 ng/ml LPS. The lower wells were filled with MCP-1 at a concentration of 200 ng/ml. Cell migration was allowed for 2 h and the cells that crossed the filter were counted. The graph shows the mean and SEM of percentage of control from three independent experiments. * p<0.01 compared with the groups labeled with no asterisk. B, C, and D. WT monocytes were pre-treated with p38 inhibitor SB203580 (SB; 10 μM), ERK inhibitor PD98059 (PD; 50) or JNK inhibitor SP600125 (SP; 10 μM) for 30 min, and then stimulated with LPS (100 ng/ml) and/or MCP-1 (200 ng/ml) for additional 30 min. B. The phosphorylation of GRK2 at Ser670 was detected by immunoblotting analysis. The images are representatives of three independent experiments. The graph depicts the mean and SEM of the percentage changes in the ratio of P-GRK2/total GRK2 in the monocytes from three experiments. * p<0.01 compared with the groups labeled with no asterisk. C. Cell membrane protein and plasma proteins were also extracted and separated, and subjected to immunoblotting analysis for detecting membrane-bound GRK2 (M-GRK2) and cytosolic GRK2 (C-GRK2). The images are representatives of three independent experiments. The graph depicts the mean ± SEM of the percentage changes in the ratio of M-GRK2/C-GRK2 in the monocytes, which were normalized by the density of actin, from three experiments. * p<0.01 compared with the groups labeled with no asterisk. D. The cell surface expression of CCR2 was measured by using flow cytometry analysis. Results are presented as percentage of control; mean ± SEM, n=3, * p<0.01 compared with the groups labeled with no asterisk.
To further address the mechanism by which ERK and p38 regulate monocyte migration, we determined the effect of ERK and p38 on the phosphorylation level of GRK2 at Ser670. Monocytes were pretreated with MAPK inhibitor for 30 min, followed by MCP-1 and/or LPS stimulation for additional 30 min. As shown in Fig. 6B, Western blotting demonstrated that p38 inhibitor SB203580 prevented LPS-induced phosphorylation of GRK2 at Ser670. However, ERK inhibitor PD98059 appeared to increase phosphorylation of GRK2 at Ser670. These data suggest that p38 enhances LPS-induced Ser670 phosphorylation, whereas ERK signaling negatively regulates Ser670 phosphorylation. These alterations in the phosphorylation of GRK2 at Ser670 are associated with consistent changes in levels of cell membrane-bound GRK2 (Fig. 6C) as well as cell surface expression of CCR2 (Fig. 6D). In monocytes pretreated with p38 inhibitor SB203580, which decreased the phosphorylation of GRK2 at Ser670, the membrane-bound level of GRK2 was increased (Fig. 6C) and cell surface expression of CCR2 was decreased (Fig. 6D). In contrast, in the cells pretreated with ERK inhibitor PD98059, which increased the Ser670 phosphorylation, the membrane-bound level of GRK2 was reduced (Fig. 6C) and cell surface expression of CCR2 was elevated (Fig. 6D).
Considered as a whole, phosphorylation of Ser670 of GRK2 is a critical regulatory target for LPS-TLR4 regulation of GRK2 subcellular translocation, in which, p38 mediates LPS-TLR4 signaling and this is negatively regulated by ERK.
Discussion
In the current study we observed that crosstalk between TLR4 and CCR2 in monocytes results in significant augmentation of MCP-1-driven monocyte migration. LPS activation of TLR4 induces phosphorylation of Ser670 of GRK2 through a mechanism mediated by p38 MAPK, and thereby suppresses GRK2 translocation to the cell membrane and the subsequent initiation of CCR2 internalization and desensitization. This then leads to enhanced monocyte migration (Fig. 7). Thus, the present study demonstrates a novel function for TLR4 signaling in driving innate immune responses that control monocyte migratory responses towards sites of infection by regulating the availability of cell surface chemokine receptors.
FIGURE 7.
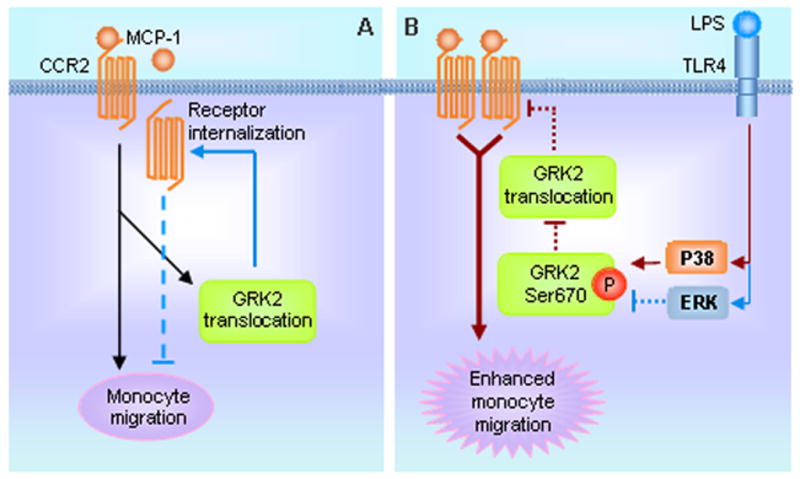
Model of TLR4 and chemokine receptor crosstalk. A. MCP-1 binding to CCR2 induces monocytes migration, as well as GRK2 translocation to membrane which results in CCR2 internalization and desensitization (blue arrow and dotted line), thereby negatively regulating monocyte migration. B. LPS acting through the TLR4 signaling and p38 MAPK pathway induces the phosphorylation of GRK2 at Ser670 inhibits the translocation of GRK2 to membrane, thereby suppressing the internalization and desensitization of CCR2, which leads to augmented monocyte migration (red arrows and dotted lines). TLR4 signaling-activated ERK plays a negative regulatory role in the phosphorylation of GRK2 at Ser670; this role can be observed while ERK is inhibited.
MCP-1 is a major chemokine that directs monocyte migration to the site of infection and activates monocytes functions, such as endocytosis (5, 23). Monocyte migration is the net result of multiple mediators, including chemokines and LPS that are both present during Gram-negative bacterial infection. Although LPS alone fails to induce monocyte migration, as seen in the present study, previous reports showed that LPS is able to augment monocyte migration in response to MCP-1 through the induction of IL-8/CXCL8 expression (18) or through the activation of MARCKS-related protein (MRP) (25), both of which synergistically amplify MCP-1-induced monocytes migration. The current study, however, provides a new mechanism by which LPS directly enhances CCR2 availability through the suppression of GRK2-induced CCR2 desensitization by internalization. The role of TLR4 signaling in mediating the LPS-enhanced monocyte migration was shown using TLR4−/− monocytes and in vivo studies using TLR4−/− mice. Genetic deletion of TLR4 completely blocked the effect of LPS on augmenting monocyte migration both in vitro and in vivo. Noteworthy, the current finding may represent a rapid regulation mechanism that may play a particularly important role in the early stages of inflammation. Alternative regulatory mechanisms may take place at later time points, for instance LPS regulation of GRK mRNA (9) or CCR2 mRNA expression (34, 35) to further influence leukocyte migration behavior.
For a large number of related GPCRs, rapid desensitization is initiated through agonist-promoted receptor phosphorylation by GRKs (36–38). To date, there are seven members of the GRK family (39, 40). GRK2 is the most widely studied member of the GRK family, is known to modulate multiple cellular responses in various physiological contexts (41, 42). Based on the previous report showing an important role for GRK2 in regulating MCP-1-induced CCR2B receptor desensitization (3), we focused our study on the role of GRK2 in the cross-talk between TLR4 and CCR2. Subcellular localization of GRK represents a significant early regulatory mechanism of GPCR desensitization (31). Ser670 lies within the Gβγ binding domain of GRK2 (31, 40), and phosphorylation at this site impairs the GRK2/Gβγ interaction thereby inhibiting the translocation of the kinase and its subsequent catalytic kinase activity at receptor membrane substrates (31, 43). This signaling pathway therefore represents a negative feedback loop that inhibits the active pool of GRK2 engaged in GPCR regulation (31), although the signaling that modulates phosphorylation of Ser670 on GRK2 was not fully elucidated in previous studies. ERK1 is known to be activated by MCP-1 (44), and previous studies implicated ERK1 in induction of phosphorylation of GRK2 on Ser670 (30, 45). In the present study, we did not see MCP-1-induced phosphorylation of GRK2 on Ser670, but we observed that LPS alone induced the phosphorylation of GRK2 at this site. We further found that p38 plays a role in mediating this LPS-TLR4-induced phosphorylation of GRK2 on Ser670, with ERK, however, playing a negative regulatory role in this process.
The phosphorylation of GRK2 on Ser670 induced by LPS results in a profound impact on CCR2 internalization and availability as well as subsequent effects on monocyte migration. As evidenced by the results from the current study, LPS-induced phosphorylation of GRK2 on Ser670 inhibited GRK2 translocation to the cell membrane and subsequent CCR2 internalization, which was associated with enhanced monocyte migration. These effects of LPS could be reversed by prevention of the phosphorylation of GRK2 on Ser670 by inhibiting p38. Furthermore, the negative regulatory effects of ERK were suppressed by ERK inhibitor PD98059, with enhanced LPS-induced phosphorylation of GRK2 on Ser670 and an augmented cell surface expression of CCR2 and increased cell migration as shown in Figure 6.
It was observed in the study that knockdown of GRK2 did not significantly enhance MCP-1-induced monocyte migration. This observation may be interpreted by the following facts: First, it is important to note that using an siRNA approach could not completely knockdown GRK2 expression in the RAW264.7 cells (Fig. 3A), and thus, MCP-1 may not be able to induce enhanced cell migration in GRK2 knockdown cells to the same degree as that induced by LPS+MCP-1 in WT cells; Second, the cell migration data from the microchamber experiments were a net end result at 6 h after cell stimulation, whereas, the cell surface expression of CCR2 detected by flow cytometry reflected the changes at 1 h after cell stimulation. The expression level of CCR2 at this time point may not entirely match the alteration in cell migration; Third, analysis of cell surface expression of CCR2 shows that in WT cells LPS+MCP-1 decreases cell surface CCR2 by 6% as compared to the control group, while, MCP-1 in GRK2 knockdown cells declined cell surface CCR2 by 23%. This significant difference may also contribute to the difference in cell migration between these groups; Finally, although the data demonstrate a predominant role of GRK2 in regulating monocyte migration, effects from other GRKs may exist as well, which leads to a more complicated outcome.
In summary, we have identified a novel interaction between TLR4 and chemokine receptors that can control monocyte migration, an essential component of the innate immune response. The results support a new model, as shown in Figure 7, in which LPS-TLR4 signaling augments monocyte migration by modulating the cell surface expression of chemokine receptors in a GRK2-dependent manner. Intervention in the specific signaling pathway that involving in the regulation of GRK2 subcellular translocation may represent a therapeutic strategy for controlling inflammation during bacterial infection.
Acknowledgments
This work was supported by the National Institutes of Health Grant R01-HL-079669 (J.F. and M.A.W.), National Institutes of Health Center Grant P50-GM-53789 (T.R.B. and J.F.), National Institutes of Health Grant R01-GM-50441 (T.R.B.), a VA Merit Award (J.F.), National Key Basic Research (973) Program of China No. 2011CB510200 (Y.J.), National Natural Science Foundation of China No.81030055 (Y.J.), NSFC-Guangdong Joint Fund No. U0632004 (Y.J.), Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT) No. IRT0731 (Y.J.).
Abbreviations used in this paper
- BMDM
Bone marrow-derived monocyte
- CCL2
CC chemokine ligand 2
- CCR2
CC chemokine receptor 2
- DMEM
Dulbecco’s modified Eagle’s medium
- ERK
extracellular signal-regulated kinase
- FACS
fluorescence-activated cell sorter
- FBS
fetal bovine serum
- GPCR
G protein-coupled receptor
- GRK2
G protein-coupled receptor kinase 2
- JNK
c-Jun N-terminal kinase
- LPS
lipopolysaccharide
- MCP-1
Monocyte chemoattractant protein 1
- MAPK
mitogen-activated protein kinase
- M-CSF
macrophage CSF
- PBS
phosphate-buffered saline
- PE
phycoerythrin
- siRNA
small interfering RNA
- TLR4
Toll-like receptor 4
Footnotes
The contents do not represent the views of the Department of Veterans Affairs or the United States Government.
References
- 1.Kamei M, Carman CV. New observations on the trafficking and diapedesis of monocytes. Current Opinion in Hematology. 2010;17:43–52. doi: 10.1097/MOH.0b013e3283333949. [DOI] [PubMed] [Google Scholar]
- 2.Steevels TA, Meyaard L. Immune inhibitory receptors: essential regulators of phagocyte function. European Journal of Immunology. 2011;41:575–587. doi: 10.1002/eji.201041179. [DOI] [PubMed] [Google Scholar]
- 3.Weber C, Schober A, Zernecke A. Chemokines: key regulators of mononuclear cell recruitment in atherosclerotic vascular disease. Arteriosclerosis, Tthrombosis, and Vascular Biology. 2004;24:1997–2008. doi: 10.1161/01.ATV.0000142812.03840.6f. [DOI] [PubMed] [Google Scholar]
- 4.Cochran BH, Reffel AC, Stiles CD. Molecular cloning of gene sequences regulated by platelet-derived growth factor. Cell. 1983;33:939–947. doi: 10.1016/0092-8674(83)90037-5. [DOI] [PubMed] [Google Scholar]
- 5.Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. Journal of Interferon & Cytokine Research: the Official Journal of the International Society for Interferon and Cytokine Research. 2009;29:313–326. doi: 10.1089/jir.2008.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Zoelen MA, Verstege MI, Draing C, de Beer R, van’t Veer C, Florquin S, Bresser P, van der Zee JS, Ate Velde A, von Aulock S, van der Poll T. Endogenous MCP-1 promotes lung inflammation induced by LPS and LTA. Molecular Immunology. 2011;48:1468–1476. doi: 10.1016/j.molimm.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 7.Aragay AM, Mellado M, Frade JM, Martin AM, Jimenez-Sainz MC, Martinez AC, Mayor F., Jr Monocyte chemoattractant protein-1-induced CCR2B receptor desensitization mediated by the G protein-coupled receptor kinase 2. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:2985–2990. doi: 10.1073/pnas.95.6.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lefkowitz RJ. G protein-coupled receptors. III. New roles for receptor kinases and beta-arrestins in receptor signaling and desensitization. The Journal of Biological Chemistry. 1998;273:18677–18680. doi: 10.1074/jbc.273.30.18677. [DOI] [PubMed] [Google Scholar]
- 9.Sun L, Ye RD. Role of G protein-coupled receptors in inflammation. Acta Pharmacologica Sinica. 2012;33:342–350. doi: 10.1038/aps.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gainetdinov RR, Bohn LM, Walker JK, Laporte SA, Macrae AD, Caron MG, Lefkowitz RJ, Premont RT. Muscarinic supersensitivity and impaired receptor desensitization in G protein-coupled receptor kinase 5-deficient mice. Neuron. 1999;24:1029–1036. doi: 10.1016/s0896-6273(00)81048-x. [DOI] [PubMed] [Google Scholar]
- 11.Benovic JL, DeBlasi A, Stone WC, Caron MG, Lefkowitz RJ. Beta-adrenergic receptor kinase: primary structure delineates a multigene family. Science. 1989;246:235–240. doi: 10.1126/science.2552582. [DOI] [PubMed] [Google Scholar]
- 12.Penn RB, Pronin AN, Benovic JL. Regulation of G protein-coupled receptor kinases. Trends in Cardiovascular Medicine. 2000;10:81–89. doi: 10.1016/s1050-1738(00)00053-0. [DOI] [PubMed] [Google Scholar]
- 13.Penela P, Ribas C, Aymerich I, Mayor F., Jr New roles of G protein-coupled receptor kinase 2 (GRK2) in cell migration. Cell Adhesion & Migration. 2009;3:19–23. doi: 10.4161/cam.3.1.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Penn RB, Benovic JL. Structure of the human gene encoding the beta-adrenergic receptor kinase. The Journal of Biological Chemistry. 1994;269:14924–14930. [PubMed] [Google Scholar]
- 15.Fan J, Malik AB. Toll-like receptor-4 (TLR4) signaling augments chemokine-induced neutrophil migration by modulating cell surface expression of chemokine receptors. Nature Medicine. 2003;9:315–321. doi: 10.1038/nm832. [DOI] [PubMed] [Google Scholar]
- 16.Francke A, Herold J, Weinert S, Strasser RH, Braun-Dullaeus RC. Generation of mature murine monocytes from heterogeneous bone marrow and description of their properties. The journal of histochemistry and cytochemistry: official journal of the Histochemistry Society. 2011;59:813–825. doi: 10.1369/0022155411416007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hintzen C, Quaiser S, Pap T, Heinrich PC, Hermanns HM. Induction of CCL13 expression in synovial fibroblasts highlights a significant role of oncostatin M in rheumatoid arthritis. Arthritis and Rheumatism. 2009;60:1932–1943. doi: 10.1002/art.24602. [DOI] [PubMed] [Google Scholar]
- 18.Chun KR, Bae EM, Kim JK, Suk K, Lee WH. Suppression of the lipopolysaccharide-induced expression of MARCKS-related protein (MRP) affects transmigration in activated RAW264.7 cells. Cellular Immunology. 2009;256:92–98. doi: 10.1016/j.cellimm.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 19.Ashida N, Arai H, Yamasaki M, Kita T. Differential signaling for MCP-1-dependent integrin activation and chemotaxis. Annals of the New York Academy of Sciences. 2001;947:387–389. doi: 10.1111/j.1749-6632.2001.tb03969.x. [DOI] [PubMed] [Google Scholar]
- 20.Liu X, Ma B, Malik AB, Tang H, Yang T, Sun B, Wang G, Minshall RD, Li Y, Zhao Y, Ye RD, Xu J. Bidirectional regulation of neutrophil migration by mitogen-activated protein kinases. Nature Immunology. 2012;13:457–464. doi: 10.1038/ni.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, Zhu N, Wang Q, Wang J, Ma Y, Qiao C, Li Y, Li X, Su B, Shen B. MEKK3 overexpression contributes to the hyperresponsiveness of IL-12-overproducing cells and CD4+ T conventional cells in nonobese diabetic mice. Journal of Immunology. 2010;185:3554–3563. doi: 10.4049/jimmunol.1000431. [DOI] [PubMed] [Google Scholar]
- 22.Bunger S, Roblick UJ, Habermann JK. Comparison of five commercial extraction kits for subsequent membrane protein profiling. Cytotechnology. 2009;61:153–159. doi: 10.1007/s10616-009-9249-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yadav A, Saini V, Arora S. MCP-1: chemoattractant with a role beyond immunity: a review. Clinica Chimica Acta; International Journal of Clinical Chemistry. 2010;411:1570–1579. doi: 10.1016/j.cca.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 24.Ebihara N, Yamagami S, Yokoo S, Amano S, Murakami A. Involvement of C-C chemokine ligand 2-CCR2 interaction in monocyte-lineage cell recruitment of normal human corneal stroma. Journal of Immunology. 2007;178:3288–3292. doi: 10.4049/jimmunol.178.5.3288. [DOI] [PubMed] [Google Scholar]
- 25.Gouwy M, Struyf S, Verbeke H, Put W, Proost P, Opdenakker G, Van Damme J. CC chemokine ligand-2 synergizes with the nonchemokine G protein-coupled receptor ligand fMLP in monocyte chemotaxis, and it cooperates with the TLR ligand LPS via induction of CXCL8. Journal of Leukocyte Biology. 2009;86:671–680. doi: 10.1189/jlb.1008638. [DOI] [PubMed] [Google Scholar]
- 26.Moser E, Kargl J, Whistler JL, Waldhoer M, Tschische P. G protein-coupled receptor-associated sorting protein 1 regulates the postendocytic sorting of seven-transmembrane-spanning G protein-coupled receptors. Pharmacology. 2010;86:22–29. doi: 10.1159/000314161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marchese A, Paing MM, Temple BR, Trejo J. G protein-coupled receptor sorting to endosomes and lysosomes. Annual review of Pharmacology and Toxicology. 2008;48:601–629. doi: 10.1146/annurev.pharmtox.48.113006.094646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mack M, Cihak J, Simonis C, Luckow B, Proudfoot AE, Plachy J, Bruhl H, Frink M, Anders HJ, Vielhauer V, Pfirstinger J, Stangassinger M, Schlondorff D. Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. Journal of Immunology. 2001;166:4697–4704. doi: 10.4049/jimmunol.166.7.4697. [DOI] [PubMed] [Google Scholar]
- 29.Otten JJ, de Jager SC, Kavelaars A, Seijkens T, Bot I, Wijnands E, Beckers L, Westra MM, Bot M, Busch M, Bermudez B, van Berkel TJ, Heijnen CJ, Biessen EA. Hematopoietic G-protein-coupled receptor kinase 2 deficiency decreases atherosclerotic lesion formation in LDL receptor-knockout mice. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2013;27:265–276. doi: 10.1096/fj.12-205351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elorza A, Sarnago S, Mayor F., Jr Agonist-dependent modulation of G protein-coupled receptor kinase 2 by mitogen-activated protein kinases. Molecular Pharmacology. 2000;57:778–783. doi: 10.1124/mol.57.4.778. [DOI] [PubMed] [Google Scholar]
- 31.Penela P, Ribas C, Mayor F., Jr Mechanisms of regulation of the expression and function of G protein-coupled receptor kinases. Cellular Signalling. 2003;15:973–981. doi: 10.1016/s0898-6568(03)00099-8. [DOI] [PubMed] [Google Scholar]
- 32.Yeh TM, Liu SH, Lin KC, Kuo C, Kuo SY, Huang TY, Yen YR, Wen RK, Chen LC, Fu TF. Dengue Virus Enhances Thrombomodulin and ICAM-1 Expression through the Macrophage Migration Inhibitory Factor Induction of the MAPK and PI3K Signaling Pathways. PloS One. 2013;8:e55018. doi: 10.1371/journal.pone.0055018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arefieva TI, Kukhtina NB, Antonova OA, Krasnikova TL. MCP-1-stimulated chemotaxis of monocytic and endothelial cells is dependent on activation of different signaling cascades. Cytokine. 2005;31:439–446. doi: 10.1016/j.cyto.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 34.Sica A, Saccani A, Borsatti A, Power CA, Wells TN, Luini W, Polentarutti N, Sozzani S, Mantovani A. Bacterial lipopolysaccharide rapidly inhibits expression of C-C chemokine receptors in human monocytes. The Journal of Experimental Medicine. 1997;185:969–974. doi: 10.1084/jem.185.5.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou Y, Yang Y, Warr G, Bravo R. LPS down-regulates the expression of chemokine receptor CCR2 in mice and abolishes macrophage infiltration in acute inflammation. Journal of Leukocyte Biology. 1999;65:265–269. doi: 10.1002/jlb.65.2.265. [DOI] [PubMed] [Google Scholar]
- 36.Lefkowitz RJ. G protein-coupled receptor kinases. Cell. 1993;74:409–412. doi: 10.1016/0092-8674(93)80042-d. [DOI] [PubMed] [Google Scholar]
- 37.Lohse MJ, Krasel C, Winstel R, Mayor F., Jr G-protein-coupled receptor kinases. Kidney International. 1996;49:1047–1052. doi: 10.1038/ki.1996.153. [DOI] [PubMed] [Google Scholar]
- 38.Premont RT, Inglese J, Lefkowitz RJ. Protein kinases that phosphorylate activated G protein-coupled receptors. FASEB Journal: official publication of the Federation of American Societies for Experimental Biology. 1995;9:175–182. doi: 10.1096/fasebj.9.2.7781920. [DOI] [PubMed] [Google Scholar]
- 39.Penela P, Murga C, Ribas C, Lafarga V, Mayor F., Jr The complex G protein-coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. British journal of Pharmacology. 2010;160:821–832. doi: 10.1111/j.1476-5381.2010.00727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ribas C, Penela P, Murga C, Salcedo A, Garcia-Hoz C, Jurado-Pueyo M, Aymerich I, Mayor F., Jr The G protein-coupled receptor kinase (GRK) interactome: role of GRKs in GPCR regulation and signaling. Biochimica et Biophysica Acta. 2007;1768:913–922. doi: 10.1016/j.bbamem.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 41.Evron T, Daigle TL, Caron MG. GRK2: multiple roles beyond G protein-coupled receptor desensitization. Trends in Pharmacological Sciences. 2012;33:154–164. doi: 10.1016/j.tips.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Penela P, Murga C, Ribas C, Salcedo A, Jurado-Pueyo M, Rivas V, Aymerich I, Mayor F., Jr G protein-coupled receptor kinase 2 (GRK2) in migration and inflammation. Archives of Physiology and Biochemistry. 2008;114:195–200. doi: 10.1080/13813450802181039. [DOI] [PubMed] [Google Scholar]
- 43.Gurevich EV, Tesmer JJ, Mushegian A, Gurevich VV. G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacology & Therapeutics. 2012;133:40–69. doi: 10.1016/j.pharmthera.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jimenez-Sainz MC, Fast B, Mayor F, Jr, Aragay AM. Signaling pathways for monocyte chemoattractant protein 1-mediated extracellular signal-regulated kinase activation. Molecular Pharmacology. 2003;64:773–782. doi: 10.1124/mol.64.3.773. [DOI] [PubMed] [Google Scholar]
- 45.Pitcher JA, Tesmer JJ, Freeman JL, Capel WD, Stone WC, Lefkowitz RJ. Feedback inhibition of G protein-coupled receptor kinase 2 (GRK2) activity by extracellular signal-regulated kinases. The Journal of Biological Chemistry. 1999;274:34531–34534. doi: 10.1074/jbc.274.49.34531. [DOI] [PubMed] [Google Scholar]