

Abstract
Notch signaling pathway plays important roles in promoting the generation of marginal zone (MZ) B cells at the expense of follicular (FO) B cells during periphery B cell maturation, but the underlying molecular mechanisms are not well understood. We hypothesize that Notch favors the generation of MZ B cells by down-regulating E protein activity. Here, we demonstrated that expression of Id2 and ankyrin-repeat SOCS box-containing protein 2 (Asb2) was elevated in MZ B cells and by Notch signaling. Id2 inhibits the DNA binding activity of E proteins whereas Asb2 facilitates E protein ubiquitination. Next, we examined the phenotypes of splenic B cells in mice expressing constitutively active Notch1 and/or two gain-of-function mutants of E proteins that counteract Id2-mediated inhibition or Notch-induced degradation. We found that up-regulation of E proteins promoted the formation of FO B cells while it suppressed the maturation of MZ B cells. In contrast, excessive amounts of Notch1 stimulated the differentiation of MZ B cells and inhibited the production of FO B cells. More interestingly, the effects of Notch1 were reversed by gain of E protein function. Furthermore, high levels of Bcl-6 expression in FO B cells was shown to be diminished by Notch signaling and restored by E proteins. In addition, E proteins facilitated and Notch hindered the differentiation of transitional B cells. Taken together, it appears that Notch regulates peripheral B cell differentiation, at least in part, through opposing E protein function.
Introduction
Maturation of B cells predominantly occurs in the spleen (1). Immature B cells arrive in the spleen from the bone marrow as Transitional 1 (T1) cells, which then acquire the ability to recirculate and transform into Transitional 2 (T2) cells. Transitional 3 (T3) cells are believed to represent anergic B cells (2). T2 cells can differentiate into precursors of marginal zone (MZP) B cells, which then turn into mature marginal zone (mMZ) B cells. T2 B cells can also give rise to follicular (FO) B cells, which are subdivided into FO-I and FO-II cells, both of which are capable of recirculating between spleen and periphery (1, 3). MZ B cells, located at the marginal zone area between the red pulp and the white pulp in the spleen, are mainly responsible for clearance of blood borne pathogens (4). Follicular B cells, residing in splenic follicles and capable of recirculation, are primarily in charge of T cell-dependent immune responses. Nonetheless, important cell fate decisions are being made by transitional B cells with regard to the marginal zone versus follicular B lineage choices (1). It is well known that the signal strength from B cell receptors influence the choice, namely stronger signaling for FO and weaker ones for MZ B cells (1, 5). Furthermore, BAFF receptor-mediated signaling and NF-κB activation are also recognized to be important in distinguishing FO and MZ B cell fates (6–9).
A number of studies have indicated a key role of the Notch signaling pathway in the generation of marginal zone B cells. Ablation of the Notch2 gene resulted in a dramatic reduction in MZ B cell production, whereas Notch2 haploinsufficiency leads to impaired formation of MZP (10, 11). B cell specific deletion of the RBP-Jκ gene, which encodes the key mediator of Notch signaling resulted in the loss of MZ B cells with a concomitant increase in FO B cells (12). Likewise, expression of a dominant-negative mastermind-like-1 mutant inhibits Notch-mediated transcription and MZ B cell differentiation (13). Furthermore, elimination of the delta-like-1 ligand of Notch receptors abolished MZ B cell formation (14). Conversely, overexpression of a constitutively active form of Notch2, the intracellular domain (IC), resulted in a tremendous accumulation of MZ B cells at the expense of FO B cells (15). It has been established that these bona fide MZ B cells express surface markers and reside in anatomical locations that are consistent with the identities of MZ B cells. Taken together, these studies clearly pointed to the importance of Notch signaling in MZ B cell development. However, the downstream molecular mechanisms whereby Notch promotes the generation of MZ B cells remains largely unknown.
The basic helix-loop-helix (bHLH) family of transcription factors has also been implicated in the regulation of peripheral B cell maturation. This family includes products of E2A, HEB and E2-2 genes, which are collectively called E proteins and have similar biochemical properties (16–18). The function of E proteins can be diminished by a group of inhibitory molecules named Id1–4. E proteins are well known to play crucial roles in immunoglobulin gene expression and receptor editing (19, 20). A role for bHLH proteins in MZ B cell development has also been implicated by analyzing E2A+/− and Id3−/− mice (19). E2A+/− mice showed an increase in the proportion of marginal zone B cells with a concomitant decrease in the proportion of follicular B cells. In contrast, Id3-deficient splenocytes had a decline in the proportion of marginal zone B cells, which indicated that E protein activity contributes to cell fate determination of MZ and FO B cells (19). However, the effects of loss of E2A protein or Id3 genes appeared rather subtle, probably due to a functional redundancy among multiple E and Id proteins. Indeed, Id2 has also been shown to influence splenic B cell differentiation and Id2 deficiency results in a reduction in the number of MZ B cells (21).
We have previously shown that activation of Notch signaling promotes the degradation of E2A proteins trigged by their ubiquitination catalyzed by Cullin1-Skp1-F-box protein Skp2 (SCFSkp2) E3 ligases (22, 23). E2A ubiquitination depends on its phosphorylation by MAP kinases and mutation of the specific MAP kinase recognition sites renders the protein resistant to Notch-induced degradation in both cell cultures and in a knockin mouse model (22, 24). We have also found that Notch signaling stimulates the transcription of the gene encoding ankyrin-repeat SOCS-box containing protein 2 (Asb2) (25). Asb2 is capable of facilitating E2A ubiquitination by bridging the formation of non-canonical dimeric ubiquitin ligase complexes, which consist of both Cullin5-ElonginB/C and Cullin1-Skp1-Skp2 subunits (25). Therefore, it is possible that Notch signaling promotes the generation of MZ B cells by increasing the turnover of E proteins. To examine the interplay between Notch signaling pathways and E protein regulation by both Id-mediated inhibition and ubiquitin-mediated degradation, we first examined gene expression in both MZ and FO populations from wild type mice and mice expressing a constitutively active form of Notch1. We show that high levels of Id2 and Asb2 expression are associated with MZ B cells and Notch activation. Since Id2 and Asb2 can both down-regulate E protein function via different mechanisms, we utilized two mouse models to counteract Id-mediated inhibition of E proteins and Notch-induced degradation, respectively (24, 26). We demonstrate that restoration of E protein function in these two models diminishes the formation of MZ B cells on the wild type background and reverses the augmented proportion of MZ B cells caused by overexpression of constitutively active Notch1, N1-IC. Therefore, these data support our hypothesis that Notch signaling favors the MZ B-cell fate at least in part by opposing the function of E proteins, which shed light on the molecular regulation of MZ B cell differentiation.
Materials and Methods
Mice
The generation of ROSA26-N1-IC, ROSA26-ET2 and E2AM/M mice are as previously described (24, 26, 27). Mice used for phenotypic analyses were conducted at 8–12 weeks of age. All animals were on the C57BL/6J background and maintained in specific-pathogen-free conditions and handled according to protocols pre-approved by the Institutional Animal Care and Use Committee of Oklahoma Medical Research Foundation.
Flow cytometry and cell sorting
For flow cytometric analyses, red blood cell–depleted cell suspensions from BM or spleen were prepared and stained at optimal dilutions of conjugated antibodies in Hanks balanced salt solution supplemented with 2% fetal calf serum (H2F). Antibodies used included PerCPCy5.5-phycoerythrin (PE)–, PE-Cy7–, allophycocyanin (APC)–, APC-Cy5.5–, APC-Cy7–conjugated monoclonal antibodies to the following cell surface antigens: B220 (RA3-6B2), CD23 (B3B4), CD21/35 (7E9), IgM (R6-60.2), Mac1 (M1/70), IgD (11-26C.2a), CD93 (AA4.1), and CD19 (1D3). Except antibodies against CD23 and CD19 which were from BD Pharmingen (San Diego, CA), all antibodies were purchased from Biolegend (San Diego, CA). Dead cells were excluded by propidium iodide staining (Molecular Probes, Eugene, OR). Relative fluorescence parameters were collected with a LSR II flow cytometer (BD Biosciences) and analyzed with Diva or FlowJo7.6.4 (TreeStar Inc) software. Cell sorting was performed on MoFLo (Beckman Coulter, Indianapolis IN).
Co-culture with OP9 stromal cells
Splenocytes were labeled with antibodies against Mac-1, Gr1 CD4, CD8, NK1.1 and Ter119. Labeled cells were then incubated with BioMag anti-Rat Ig conjugated magnetic particles (Qiagen; Valencia, CA). Unbound cells were recovered in alpha medium supplemented with 10% fetal calf serum for one before being plated onto OP9 stromal cells transduced with vector (OP9-V) or delta-like-1-expressing retrovirus (OP9-DL1) by a 30-min centrifugation and 2-hour incubation at 37°C (28). Non-adherent cells were then harvested for RNA isolation.
BrdU labeling
Cohorts of 10–12 weeks old mice were provided with 0.25 mg/ml of BrdU in drinking water for 7, 11 and 15 days. Splenocytes were analyzed by simultaneously staining for surface antigens with fluorochrome-label antibodies against B220, CD21, CD23, IgM and IgD and for BrdU incorporation using the APC-BrdU Flow kit (BD Biosciences) according to the manufacturer's instruction. BrdU positive gate was set and based on the criterion that the percentage of BrdU positive cells is less than 1% in each cognate cell population of wild type un-labeled mice.
Analyses of Gene expression
Total RNA was extracted from sorted cells using TRIzol reagent (Invitrogen). cDNA was synthesized using Moloney murine leukemia virus reverse-transcriptase (Invitrogen). PCR amplification was performed using SYBR GREEN PCR Master Mix (Qiagen, Valencia, CA) and ABI Prism 7500 real-time PCR machine (Applied Biosystems, Foster City, CA). Primers for Asb2 were purchased from Qiagen. Other Primers were synthesized by Integrated DNA Technologies (Coralville, IA) using the following sequences: Hes5, AGTCCCAAGGAGAAAAACCGA and GCTGTGTTTCAGGTAGCTGCTGAC; Hes1 CCAGCCAGTGTCAACACGA and AATGCCGGGAGCTATCTTTCT; Id3, CGACCGAGGAGCCTCTTAG and GCAGGATTTCCACCACCTGGCTA; Id2, ATGAAAGCCTTCAGTCCGGTG and AGCAGACTCATCGGGTCGT; Deltex1 CGCTCCATGCAAATGGTACA and AGGATGTGGTTCGGAGGTACAT; Bcl-6, CCGGCACGCTAGTGATGTT and TGTCTTATGGGCTCTAAACTGCT.
Statistical analyses
Unpaired t test was performed using either the Prism or excel software.
Results
MZ B cells expressed Id2 and Asb2 at much higher levels than FO B cells
To investigate the relationship between Notch activation and E protein activities, we examined levels of gene expression in sorted splenic B cells based on B220+CD21HiCD23int and B220+CD21intCD23Hi phenotypes, which are believed to enrich for MZ and FO B cells, respectively (3). To test the effects of Notch signaling, we utilized a mouse model where constitutively active Notch1 is expressed in B lineage cells. Specifically, the intracellular domain of Notch1 (N1-IC) was inserted downstream of the promoter of the ROSA26 gene along with a Floxed Stop sequence and EGFP which can be expressed from the internal ribosome entry site (27, 29, 30). When this strain of mice was crossed with CD19-Cre transgenic mice (31), expression of N1-IC as well as EGFP was efficiently induced (Figure 1A). We first examined the expression of well-known Notch target genes in wild type FO and MZ B cells. As expected, expression levels of Deltex1 and Hes5 was at least 10-fold higher in MZ than FO B cells. The level of Hes1 was also higher in MZ B cells albeit less dramatically (Figure 1B). These results confirm that Notch signaling is indeed activated in MZ B cells.
Figure 1. Gene expression in FO B cells and MZ B cells.
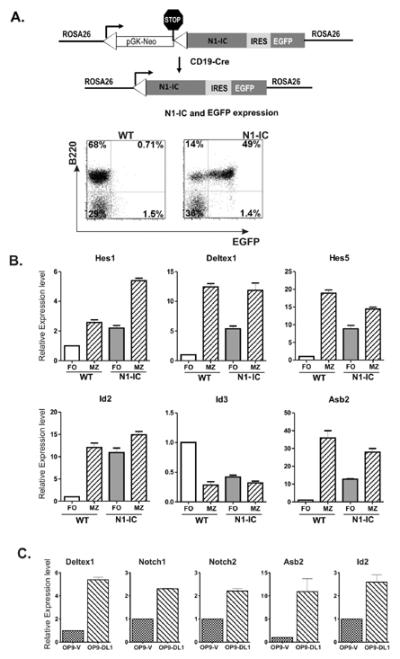
(A) Schematic diagrams of the ROSA26-N1-IC construct and the induction of gene expression with CD19-Cre, which can be indicated by EGFP expressed from the internal ribosome entry site (IRES). EGFP and B220 expression in N1-IC splenocytes is analyzed by FACS along with wild type control. (B) Total RNA was isolated from B220+CD21+CD23Hi (FO) and B220+CD21HiCD23Lo (MZ) cells of indicated strains of mice. For cells from N1-IC mice, GFP+FO or GFP+MZ B cells were used. Quantitative PCR was performed to determine the levels of indicated transcripts. Levels of expression are presented in relation to that detected in WT FO cells. Data shown are the averages of two independent experiments, each of which is done in triplicates. (C) Splenocytes depleted of Mac-1+, Gr1, CD4+, CD8+, Ter119+ or NK1.1+ cells were co-cultured with OP9-vector or OP9-DL1 stromal cells for 2 hours and harvested for total RNA isolation. Quantitative PCR was performed to determine the levels of indicated transcripts. Levels of expression in cells exposed to OP9-DL1 are presented in comparison to those in cells cultured with OP9-vector. Data shown are the averages of two independent experiments, each of which is done in duplicates or triplicates.
Since transcription of E protein genes is generally considered constitutive and ubiquitous, we assessed the expression of genes encoding products that are capable of regulating E protein function in post-transcriptional manners. These include Id proteins, which inhibits the DNA binding activity of E proteins, and Asb2 which facilitates ubiquitin-mediated degradation of E proteins (18, 25, 32). We found that MZ B cells exhibited a 12-fold higher level of Id2 than FO B cells, which is consistent with that described by Kin et al. (Figure 1B) (33). However, the level of Id3 in MZ B cells was lower than in FO cells. Significantly, the Asb2 gene was expressed at a 35-fold higher level in MZ B cells than in FO B cells (Figure 1B). Collectively, it appears that two separate mechanisms exist in MZ B cells to down-regulate E protein function, namely expression of Id2 and Asb2 proteins.
Comparing gene expression between wild type and N1-IC expressing FO B cells revealed that enhanced Notch signaling not only stimulated the expression of Hes1, Deltex and Hes5 genes but also boosted the transcription of Id2 and Asb2 genes (Figure 1B). To further verify if Notch signaling stimulates the expression of Id2 and Asb2, we co-cultured splenocytes depleted with antibodies against Mac-1, gr1, CD4, CD8, Ter119 and NK1.1 on OP9-vector and OP9-DL1 stromal cells for 2 hours. We first tested the induction of known Notch target genes such as Deltex1 as well as Notch1 and Notch2, and found them to be up-regulated by 2–5 fold when exposed to the Notch ligand, DL1 (Fig. 1C). This suggested that Notch signaling pathways were activated under this condition. As a result, the stimulation of Asb2 and Id2 expression was on the average of 11 and 2.6 fold, respectively (Fig. 1C). Collectively, these results raised a possibility that Notch promotes MZ B cell differentiation, at least in part by down-regulating E protein function through Id2 and Asb2.
Animal models for gain of E protein function
To test the hypothesis that Notch-induced Id2 and Asb2 expression contributes to MZ B cell differentiation, we utilized two gain-of-E-protein-function animal models that we have previously generated. In one model, a chimeric protein called ET2 can be inducibly expressed from the ROSA26 promoter by Cre-mediated recombination (26, 34). ET2 is a fusion protein consisting of the N-terminus of E47 (one form of the E2A proteins) including the two known transcriptional activation domains and the C-terminus of Tal1 protein containing the bHLH domain for DNA binding and dimerization (Figure 2A) (35). Because the bHLH domain of Tal1 mediates homo-dimerization poorly, ET2 cannot activate transcription of E protein target genes unless it forms complexes with endogenous E proteins, which limits the impact of ET2 overexpression. Since the Tal1 bHLH domain has comparable affinities to E proteins as Id proteins (36), ET2 is able to compete with all Id proteins to bind to E proteins and neutralize their effects, thus overcoming the issue of functional redundancy among different Id proteins (26, 35). However, ET2 remains sensitive to Notch or Asb2-induced degradation since it contains not only the sequence signaling E2A degradation but also the one for Tal1 breakdown (22, 37). The second animal model possesses three point mutations in the E2A gene (E2AM/M) such that the encoded proteins lack three putative MAP kinase phosphorylation sites necessary for E2A protein ubiquitination and degradation (Figure 2B). E2A proteins from these knockin mice have previously been shown to be resistant to Notch or Asb2-induced degradation (24). However, the DNA binding activity of E2AM/M proteins can still be inhibited by Id proteins.
Figure 2. Animal models for gain of E protein or Notch function.
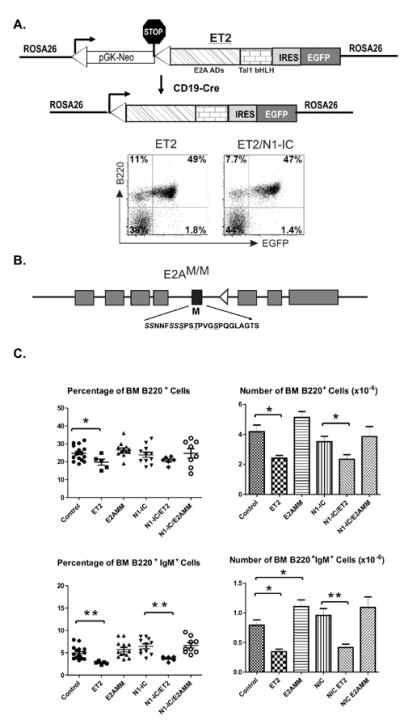
(A) Schematic diagram of the ROSA26-ET2constructs. Portions of the E2A and Tal1 proteins are labeled underneath the patterned boxes. EGFP and B220 expression in ET2 and ET2/N1-IC splenocytes are shown in FACS plots. (B) Schematic diagram of the E2AM/M allele. The black box indicates the exon containing MAP kinase site mutations, the sequence of which is shown below and the amino acids mutated to alanine are underlined. The triangle symbolizes a loxP site remained in the intron after the neo cassette was removed by cre-mediated recombination. (C) Analyses of the bone marrow of indicated strains. The average percentage and numbers of indicated cell populations are shown with SEM. (*p<0.05; **p<0.01).
While the E2AM/M allele is expressed similarly as the wild type E2A gene, expression of ET2, like that of N1-IC, was induced by crossing ROSA26-ET2 knockin mice with CD19-Cre mice (31). For the benefit of simplicity, ROSA26-ET2/CD19-Cre, ROSA26-N1-IC/CD19-Cre or ROSA26-ET2/ROSA26-N1-IC/CD19-Cre mice will be referred as ET2, N1-IC and N1-IC/ET2 mice hereafter, respectively. As the transcript encoding ET2 also includes EGFP downstream of a ribosomal entry site, induction of ET2 expression can be indicated by the fluorescence of EGFP. Efficient induction was detected in splenic B cells of ET2 or ET2/ N1-IC mice (Figure 2A). For analyses of ET2 and N1-IC expressing B cells, GFP+ gate was included.
Gross examination of B cell development in the bone marrow of ET2, E2AM/M and N1-IC-expressing mice revealed no major developmental defects except that the percentage and number of B220+ and B220+IgM+ cells in ET2 mice were significantly reduced (Figure 2C). In the spleen of ET2 and N1-IC mice, total counts of splenocytes and B cells were about 75% and 50% of those in wild type or E2AM/M mice (Table 1). This disparity in total cell counts combined with the necessity to identify GFP+ cells in some of the strains made the comparison of the numbers of a particular B cell population among the different strains of mice complicated and less informative. Therefore, we primarily focused on examining the percentages of B cell subsets and only compared the total cell numbers in appropriate groups of mice.
Table I.
Number of Splenocytes and B cell subsets in different strains of mice
| cell number (×106) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean (SEM) |
||||||||||
| Genotype | SP | Total B220+ | GFP+B220+ | T1 | T2 | T3 | Fo | MZ B | mMZ B | MZP B |
| Control | 132.1 (14.36) | 87.79 (9.67) | 3.12 (0.42) | 5.71 (0.56) | 4.38 (0.45) | 57.45 (7.32) | 11.38 (1.13) | 7.92 (1.06) | 2.67 (0.45) | |
| E2AMM | 156.9 (12.40) | 94.11 (8.72) | 4.17 (0.74) | 8.93 (1.06) | 5.59 (0.90) | 72.67 (4.87) | 7.52 (0.73) | 5.49 (0.60) | 3.97 (0.41) | |
| ET2 | 83.56 (10.20) | 42.45 (6.23) | 30.37 (5.64) | 2.16 (1.11) | 3.78 (0.55) | 2.9 (0.70) | 27.59 (4.85) | 4.96 (0.87) | 2.64 (0.93) | 2.02 (0.34) |
| N1-IC | 89.15 (4.18) | 45.62 (2.84) | 41.87 (4.80) | 4.64 (0.45) | 3.22 (0.20) | 0.95 (0.09) | 15.07 (0.94) | 16.93 (1.97) | 12.86 (1.53) | 1.54 (0.21) |
| N1-IC/E2AMM | 126.4 (7.47) | 66.88 (5.89) | 68.36 (10.57) | 4.53 (0.61) | 5.32 (0.69) | 1.36 (0.44) | 29.88 (3.32) | 22.31 (2.25) | 16.06 (2.95) | 3.98 (0.73) |
| N1-IC/ET2 | 111.2 (18.26) | 57.6 (9.17) | 42.15 (7.46) | 5.23 (1.00) | 5.08 (0.85) | 2.22 (0.62) | 20.88 (3.94) | 21.56 (4.73) | 13.53 (3.27) | 2.49 (0.51) |
Notch and E proteins have opposing effects on FO and MZ B cell differentiation
If Notch-dependent MZ B cell differentiation involves down-regulation of E protein activities, we would expect that neutralizing Id2-mediated E protein inhibition by ET2 or Asb2-induced E2A degradation by E2AM/M would each separately impact MZ versus FO lineage decisions. We examined the developmental profiles of splenic B cells in ET2 or E2AM/M mice using the parameters described by Allman and Pillai (3) and our general gating strategies are shown in Supplementary Fig. 1.
Levels of CD21 and CD23 surface markers can largely be used to separate B220+ splenocytes into two populations enriched of FO and MZ cells. In comparison to the control group consisting of wild type mice or mice which carry either the CD19-Cre, N1-IC or ET2 allele, ET2-expressing mice had a small but statistically significant increase in the proportion of FO B cells and a corresponding decrease in MZ B cells (Figure 3A). In E2AM/M mice, a more profound difference was observed; the percentage of MZ B cells was reduced by 2 fold (Figure 3B). This is consistent with the decreases in the number of MZ B cells comparing the control to E2AM/M mice, which have similar counts of total splenic B cells (Table 1).
Figure 3. Opposing effects of N1-IC and E proteins on FO and MZ B cell differentiation.

CD21 and CD23 profiles of B220+ splenocytes from (A) ET2 and/or N1-IC-expressing mice and (B) E2AM/M and/or N1-IC mice. Control mice (Con) include littermates which carry wild type, ROSA26-ET2 only, N1-IC only or CD19-Cre only genotypes. Gates show the populations enriched of MZ or FO cells and numbers indicate the percentage of cells within the gate. Average percentages of FO or MZ B cells are shown below the FACS plots. (*p<0.05; **p<0.01).
Hampel et al. have recently shown that overexpression of Notch2-IC (N2-IC) from the CAGGS (CMV early enhancer/chicken β-actin/rabbit globin) promoter forcefully drives MZ B cell differentiation. They have also demonstrated that these phenotypic MZ B cells have expected anatomical location and function. Therefore, we made use of our N1-IC mice to enhance Notch signaling in B lineage cells and assess the effects of ET2 and E2AM/M on Notch-driven MZ B cell development. Similar to the phenotypes of N2-IC expressing mice, albeit less dramatic, expression of N1-IC also results in a significant increase (4 fold) in the proportion of MZ B cells, which was accompanied by a decrease in the percentage of FO B cells (Figure 3A and B). Although the total number of splenocytes was reduced by 36% in N1-IC mice compared to control mice, the number of MZ cells was increased by 50% while that of FO cells dropped about 4 fold (Table 1). These results suggest that increased Notch function indeed enhanced MZ B cell production and suppressed FO B cell differentiation. They also showed that Notch1 can to some extent, substitute Notch2 function when it is ectopically expressed. Interestingly, introducing ET2 or E2AM/M into N1-IC expressing mice each led to a partial rescue of the phenotype caused by N1-IC (Figure 3A and B). Again, the effect of E2AM/M appeared stronger than that of ET2.
The follicular B cell population consists of FO-I and FO-II subsets, which are characterized as B220+CD93−CD21+IgDHiIgMlo and B220+CD93−CD21+IgDHiIgMHi, respectively. While FO-I includes the majority of FO cells, FO-II B cells are believed to serve as a reservoir for MZ B cells when they are depleted upon blood-borne pathogen infection (38). Despite the increases of MZ B cells by enhanced Notch activity in N1-IC mice, we did not observe any increase in the number of FO-II B cells although the percentage of FO-II B cells was slightly increased. In contrast, a marked reduction in the fraction of FO-I B cells was detected (supplementary Figures 1 and 2). Elevation of E protein activities also did not drastically affect the proportion of FO-II B cells. The fluctuation in the numbers of FO-I and FO-II B cells are consistent with that of total number of FO B cells (Table 1).
Down-regulation of E proteins is necessary for MZ B cell maturation
Another important checkpoint in MZ B cell development is the transition from MZP to mMZ B cells, which are characterized as B220+CD93−CD21HiIgMHi plus CD23+IgDHi and CD23−IgDLo respectively (3, 39). Expression of ET2 and E2AM/M both impaired the maturation from MZP to mMZ stages, resulting in an increased proportion of MZP cells and decreased percentages of mMZ B cells (Figure 4A and B). Unlike the situation with MZ versus FO differentiation shown in Figure 3, ET2 exerted a greater block of MZP maturation than E2AM/M. Conversely, production of N1-IC facilitated mMZ production and co-expression of ET2 or E2AM/M reversed the trend (Figure 4A and B). These results suggest that Notch and E proteins play opposing roles during the maturation of MZ B lineage cells.
Figure 4. Down-regulation of E proteins is necessary for MZ B cell maturation.
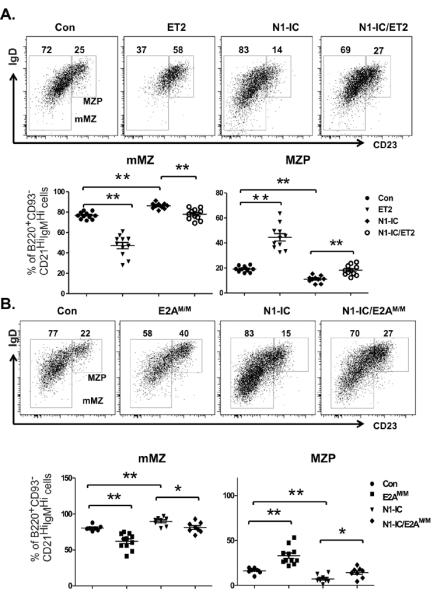
IgD and CD23 profile of B220+CD93−CD21highIgMhigh splenocytes from (A) ET2 and/or N1-IC-expressing mice and (B) E2AM/M and/or N1-IC mice. Gates for marginal zone precursors (MZP) and mature MZ cells (mMZ) are as labeled and numbers indicate the percentage of cells within the adjacent gate. Average percentages of MZP and mMZ B cells are shown below the FACS plots. (*p<0.05; **p<0.01).
Effects of Notch on FO and MZ B cell production
To closely monitor the production of FO and MZ B cells, we performed an in vivo BrdU labeling assay. Cohorts of Control, N1-IC, ET2 and E2AM/M mice were maintained with BrdU-containing drinking water for 7, 11 or 15 days. BrdU incorporation in FO (B220+CD21intCD23HiIgMint) and MZ (B220+CD21HiCD23intIgMHi) B cells were then analyzed. In terms of the percentage of BrdU uptake in FO or MZ B cells at each time point, there was no significant difference among the different strains except that FO cells in N1-IC and ET2 mice showed significant increases on Day 11 or Day 15 (Fig. 5A). This is likely due to a reduction in total counts of splenocytes in these mice, which might elevate homeostatic proliferation of FO B cells. It is important to note that BrdU incorporation in MZ B cells was not significantly increased in N1-IC mice, which suggested that the increase in the number of MZ B cells in N1-IC mice was not due to an expansion of this population of cells.
Figure 5. In vivo BrdU-labeling assays.
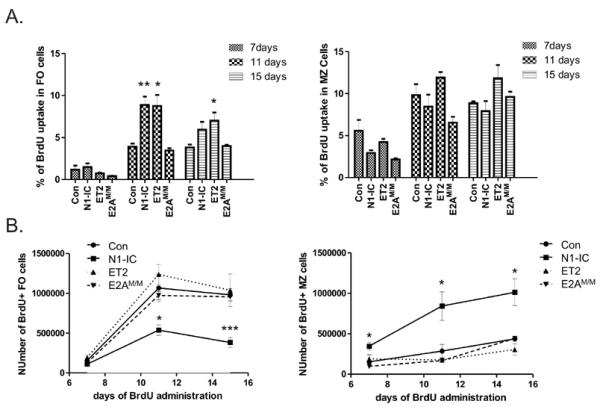
Cohorts of indicated strains of mice (n=3 to 6) at the age of 10–12 weeks were provided with BrdU containing drinking water for indicated lengths of time. Splenocytes were analyzed by intracellular staining for BrdU along with antibodies against surface markers. FO and MZ B cells are defined as B220+CD21intCD23HiIgMint and B220+CD21HiCD23lowIgMHi, respectively. (A) Average percentages with SEM of BrdU+ cells in the FO or MZ population of indicated strains of mice at indicated time points are shown in each bar graph. (B) Time course of BrdU labeling of FO and MZ B cells of the indicated strains of mice (*p<0.05; **p< 0.01, p<0.001).
By plotting the number of BrdU labeled cells over time, we found that the rate of accumulation of BrdU+ FO cells in N1-IC mice was significantly slower compared to the control mice (Fig. 5B), even though enhanced homeostatic proliferation had lead to a higher percentage of BrdU labeled FO B cells in N1-IC mice (Fig. 5A). In contrast, the rate of BrdU+ MZ cells in N1-IC mice was obviously higher than the control. These results suggest that N1-IC expression inhibits FO B cell production while augments the generation of MZ B cells. However, it should be cautioned that the differences in the survival of BrdU labeled cells at each time point may also contribute to the numbers of BrdU+ cells detected. We also noticed that BrdU incorporation in FO B cells appeared to have reached the plateau by Day 15. The reduction in the numbers of BrdU+ FO B cells in N1-IC mice comparing Day 11 and Day 15 was statistically insignificant (Fig. 5B).
In ET2 and E2AM/M mice, the rate of BrdU incorporation was comparable to the control mice (Fig. 5B). However, we did detect a small reduction in BrdU+ MZ B cells in both ET2 and E2AM/M mice on Day 11, even though the differences failed to reach statistical significance based on the limited numbers of mice analyzed. This trend would be consistent with the decrease in steady-state numbers of MZ B cells seen in these mice (Fig. 3).
High levels of Bcl-6 expression in FO B cells was diminished by Notch and restored by E proteins
To evaluate E protein activities in FO and MZ B cells, we selected several potential target genes including Bcl-6, FOXO1, KLF3 and Bcl-2, which were found to be bound by E proteins during the transition from Pre-pro B to Pro B cells using genome-wide ChIP-seq assays (40). We recognize that the regulation of these genes may be completely different in the context of FO and MZ B cells. Nevertheless, we found that while expression of the FOXO1, KLF3 and Bcl-2 genes was similar in FO and MZ B cells (data not shown), Bcl-6 levels were 2-fold higher in FO B cells than that in MZ B cells (Figure 6), which is consistent with that previously described (33). Expression of ET2 or E2AM/M indeed elevated Bcl6 expression in FO and MZ B cells (Figure 6). Interestingly, expression of N1-IC led to reduction of Bcl-6 expression by 3 and 4.5 fold in FO and MZ B cells, respectively. However, expression of E2AM/M and ET2 together with N1-IC significantly restored Bcl-6 levels in FO B cells compared to N1-IC alone (Figure 6). These results suggest that Bcl-6 expression is associated with E protein function and Notch activation diminishes E protein activity in FO B cells.
Figure 6. Bcl-6 expression in FO and MZ B cells of indicated stain of mice.
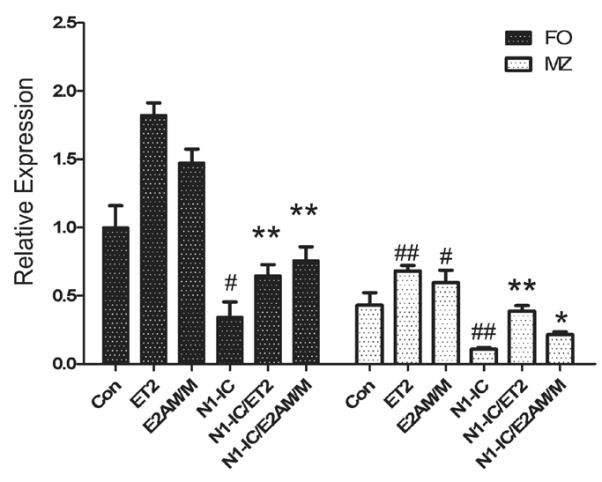
Total RNA was isolated from FO and MZ cells of indicated strains of mice. For N1-IC and ET2-expressing mice, GFP+ cells were sorted. Quantitative PCR was performed to determine the levels of indicated transcripts. Levels of expression are presented in relation to that detected in WT FO cells. # indicates the comparison between control and ET2, E2AM/M or N1-IC. *designates the comparison between N1-IC and N1-IC/ET2 or N1-IC/E2AM/M. (# or *p<0.05; ## or **p<0.01).
Effects of Notch and E proteins on transitional B cell differentiation
Immature B cells migrate from the bone marrow to the spleen as T1 cells, which then differentiate into T2 B cells. Whether E proteins play a role in this transition is not known. We found that the percentage of T1 (B220+CD93+IgM+CD23−) B cells was reduced in ET2 or E2AM/M mice, which is accompanied by a significant increase in the proportion of T2 (B220+CD93+IgM+CD23+) B cells (Figure 7A and B). These results suggest that E protein activity promotes B cell maturation from the T1 to the T2 stages. However, E protein activities did not alter the proportion of T3 B cells.
Figure 7. Effects of Notch and E proteins on the transitional B cell differentiation.
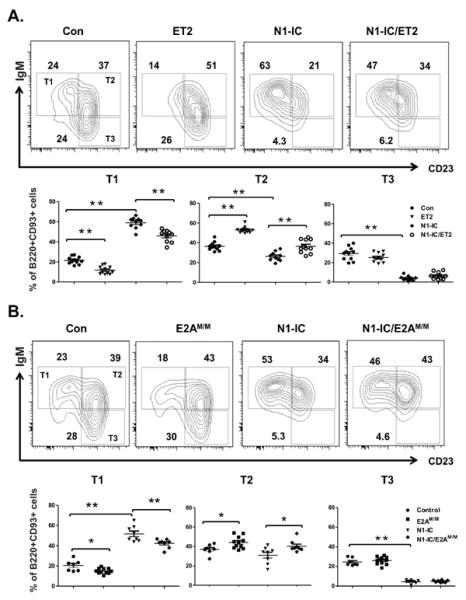
IgM and CD23 profile of B220+CD93+splenocytes from (A) ET2 and/or N1-IC-expressing mice and (B) E2AM/M and/or N1-IC mice. The gates for T1, T2 and T3 B cells are labeled within the gates for control mice. Numbers indicate the percentage of cells within their adjacent gate. Average percentages of T1, T2 and T3 B cells are shown below the FACS plots. (* p<0.05; ** p<0.01).
In contrast, expression of N1-IC caused a marked accumulation of T1 B cells and a slight reduction in T2 B cells (Figure 7). However, the most dramatic effect of N1-IC expression on transitional B cells was the diminution of T3 anergic B cells. Compared to controls, N1-IC mice had a 6 fold reduction in the percentage of T3 B cells and a corresponding decrease in the number of such cells (Figure 7 and Table 1). Together, these results suggest that E proteins and Notch impact different steps of transitional B cell maturation.
Discussion
The differentiation of follicular and marginal zone B cells in the spleen is influenced by multiple signaling pathways including those triggered by BCR and BAFF receptors. Notch signaling has also been thought to regulate the FO versus MZ lineage decision, which may be achieved by processes involving crosstalks between Notch and BCR or BAFF signaling pathways. Likewise, the accumulation of FO and MZ B cells in the spleen is dictated by the net results of cell differentiation, survival and homeostasis. In this study, we focused on the role of Notch signaling and its downstream effectors in the development of FO and MZ B cells.
The essential role of Notch signaling pathway in marginal zone B cell development is well established. However, what Notch-mediated MZ B cell differentiation entails remains largely unknown. One widely recognized idea is that Notch influences the FO vs. MZ B cell fate, which is consistent with its known function in controlling lineage choices in numerous situations during the development of organisms ranging from Drosophila to human (41, 42). It is possible that Notch signaling suppresses the FO B cell differentiation, thus allowing the same precursors such as T2 cells to commit to the MZ lineages. The other possibility is that Notch drives the expansion of MZ B cells. Our data shows that a dramatic reduction in the percentage of FO B cells was accompanied by a corresponding increase in the proportion of MZ B cells. Comparison of the absolute numbers of FO and MZ B cells in N1-IC mice to those of control mice revealed that N1-IC mice gained about 5×106 MZ B cells but lost 4×107 FO B cells (Table 1). This deficit could account for the reduction in total B cell counts in the spleen of N1-IC mice, which suggests that N1-IC exerts a powerful suppression of FO B cell differentiation or impairment of the survival of these cells. In contrast, a relatively modest increase in the total number of MZ B cells was found in N1-IC mice and this could not compensate for the loss of all FO B cells. To more closely monitor FO and MZ B cell production, we performed an in vivo BrdU labeling assay and obtained a time course of FO and MZ B cell production over a two-week period. We found a decreased rate of FO B cell production and an increased rate of MZ B cell output in N1-IC expressing mice. Since the percentage of BrdU uptake by MZ B cells were comparable between control and N1-IC mice, it is unlikely that Notch simply drives the expansion of MZ B cells. Because a higher percentage of BrdU-labeled FO B cells was found in N1-IC mice, the possibility of defective FO B cell proliferation was also ruled out. Our data would thus be consistent with the notion that Notch promotes MZ B cell differentiation at the expense of FO B cell fate. However, it remains possible that Notch also causes excessive death of FO B cells or increases the exit of FO B cells from the spleen.
There is also a possibility that some of the effects of Notch on FO and MZ B cells are separable. Several findings are in support of this idea. First, Both N1-IC and N2-IC mice have increased numbers of T1 B cells, which has been suggested to directly give rise to MZ B cells (15, 43). Second, both Notch receptors have the ability to drive MZ B cell maturation from MZ progenitors. Finally, comparison of the phenotypes of N1-IC and N2-IC mice revealed that while the suppression of FO B cell production is similar in these two strains of mice, the number of MZ B cells generated in N2-IC mice was much larger than that seen N1-IC mice. It appears that the N2-IC mice exhibited more dramatic bias towards MZ B cell differentiation compared to the N1-IC mice. This may be attributed to a possibly higher level of N2-IC expression than N1-IC, because the former is driven by a CMV-based promoter whereas the latter is transcribed off the promoter of ROSA26 gene even though both were induced by CD19-Cre-mediated recombination. More likely, intrinsic difference exists between N1-IC and N2-IC proteins in their ability to direct MZ B cell differentiation. Regardless of the reasons for the differences between N1-IC and N2-IC mice, the fact that both N1-IC and N2-IC have similar capacities in suppressing FO B cell differentiation but have different effects on MZ B cell production would suggest that inhibition of FO differentiation and promotion of MZ B cell formation are two separate events mediated by Notch signaling. This is reminiscent of the situation in Notch-regulated B versus T lineage decision, where Notch inhibits B cell development and promotes T cell differentiation (24, 44, 45). Whether Notch exerts its effects on the same or different precursors of B and T cells is also an issue of intense debate.
With regard to the molecular mechanisms whereby Notch controls FO and MZ B cell differentiation, we specifically investigated whether down-regulation of E protein function by Notch plays a significant role in this process. Notch signaling stimulates the transcription of the Id2 and Asb2 genes, which encode products capable of down-regulating E protein function by either inhibiting its DNA binding activity or by promoting its degradation, respectively. We utilized two animal models, which express ET2 or E2AM/M. While ET2 counteracts the effects of Id2, E2AM/M proteins are resistant to Asb2-induced degradation. Indeed, we show that although elevated Notch activity promotes MZ B cell maturation while diminishing FO B cell development, expression of ET2 or E2AM/M led to reduced accumulation of MZ B cells to a varying degree, suggesting that ET2 and E2AM/M can oppose the effects of Notch signaling and the mechanism whereby Notch supports MZ B cell differentiation at least in part includes down-regulation of E protein function. However, because ET2 is vulnerable to Asb2-mediated breakdown and E2AM/M can be inactivated by Id2, a combination of the properties of ET2 and E2AM/M proteins might have been able to exert more dramatic opposing effects. This would require the establishment of a new mouse model to express a modified version of ET2 in which the degradation signals of both E2A and Tal1 are deleted (22, 37). It should also be noted that down-regulation of E proteins is unlikely the only mechanism by which Notch regulates MZ B cell differentiation.
The transcriptional programs involved in Notch-controlled FO and MZ B cell differentiation are also not well understood. Kearney and colleagues performed a comprehensive analysis of gene expression profiles in FO and MZ B cells but the functional significance of the differentially expressed genes needs to be verified (33). Our study provided evidence to suggest that attenuation of E protein function is downstream of Notch signaling. Indeed, we showed that restoring E protein function has opposing effects to Notch signaling on FO B cell differentiation and MZ B cell maturation, as well as the progression of newly arrived immature B cells through the transition stages. E proteins are essential transcription factors for B cell development in the bone marrow and regulate the transcription of many B cell specific genes important for early stages of B cell differentiation. They were originally identified based on their ability to bind to the intronic enhancer of the immunoglobulin heavy chain locus and are thus responsible for immunoglobulin gene expression, which dictates the density of B cell receptors (BCR) on the surface of the cells (46–48). Since strong BCR signaling is thought to favor FO B cell differentiation, it is conceivable that E proteins promote FO B cell formation by enhancing BCR signaling (1). Indeed, levels of IgD transcripts have been shown to be significantly higher in FO than MZ B cells (33). However, other crucial target genes of E proteins in FO and MZ cell development are not known.
We examined the expression patterns of several genes which have the potential to play roles in splenic B cell development and are known to be controlled by E proteins in pro-B cells (40). Interestingly, the Bcl-6 gene is highly expressed in FO B cells in comparison to MZ B cells. N1-IC inhibits Bcl-6 expression in FO B cells whereas ET2 or E2AM/M restores Bcl-6 levels. Thus, these expression patterns would suggest a potential link between Bcl-6 and Notch-mediated down-regulation of E protein activities. Bcl-6 is considered a master regulator of germinal center reaction and key oncogene in B cell lymphomagenesis (49, 50). However, its roles in FO and MZ B cell differentiation have not been established. Given the mutually antagonistic relationship between Bcl-6 and NF-κB (51), a transcription factor required for MZ B cell differentiation (7, 8), the role of Bcl-6 in FO versus MZ B cell fate decisions warrants further investigation.
In this study, we have shown that Asb2 expression in MZ B cells is 35-fold higher than in FO B cells and N1-IC can significantly elevate Asb2 levels in FO B cells. Asb2 is a newly identified Notch target and facilitates the ubiquitination of a variety of substrates besides E2A proteins (25). Therefore, Asb2 may emerge to be an important regulator by controlling the turnover of crucial proteins for MZ B cell differentiation and function. Investigations using gain and loss-of-function mutants of the Asb2 gene will further shed light on the molecular mechanisms controlling MZ B cell formation. Knowledge of these control mechanisms is necessary for our understanding of B cell-mediated immunity, autoimmunity and leukemia.
Supplementary Material
Acknowledgements
We are grateful to the animal and flow cytometry facilities at the Oklahoma Medical Research Foundation for technical support.
This work was supported by the grants to XHS from the National Institute of Health (AI56129). XHS holds the Lew and Myra Chair in Biomedical Research.
Reference
- 1.Pillai S, Cariappa A. The follicular versus marginal zone B lymphocyte cell fate decision. Nat.Rev.Immunol. 2009;9:767–777. doi: 10.1038/nri2656. [DOI] [PubMed] [Google Scholar]
- 2.Merrell KT, Benschop RJ, Gauld SB, Aviszus K, Decote-Ricardo D, Wysocki LJ, Cambier JC. Identification of anergic B cells within a wild-type repertoire. Immunity. 2006;25:953–962. doi: 10.1016/j.immuni.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 3.Allman D, Pillai S. Peripheral B cell subsets. Curr.Opin.Immunol. 2008;20:149–157. doi: 10.1016/j.coi.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin F, Kearney JF. Marginal-zone B cells. Nat.Rev.Immunol. 2002;2:323–335. doi: 10.1038/nri799. [DOI] [PubMed] [Google Scholar]
- 5.Pillai S, Cariappa A, Moran ST. Positive selection and lineage commitment during peripheral B-lymphocyte development. Immunol.Rev. 2004;197:206–218. doi: 10.1111/j.0105-2896.2003.097.x. [DOI] [PubMed] [Google Scholar]
- 6.Batten M, Groom J, Cachero TG, Qian F, Schneider P, Tschopp J, Browning JL, Mackay F. BAFF mediates survival of peripheral immature B lymphocytes. J.Exp.Med. 2000;192:1453–1466. doi: 10.1084/jem.192.10.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cariappa A, Liou HC, Horwitz BH, Pillai S. Nuclear factor kappa B is required for the development of marginal zone B lymphocytes. J.Exp.Med. 2000;192:1175–1182. doi: 10.1084/jem.192.8.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moran ST, Cariappa A, Liu H, Muir B, Sgroi D, Boboila C, Pillai S. Synergism between NF-kappa B1/p50 and Notch2 during the development of marginal zone B lymphocytes. J.Immunol. 2007;179:195–200. doi: 10.4049/jimmunol.179.1.195. [DOI] [PubMed] [Google Scholar]
- 9.Schiemann B, Gommerman JL, Vora K, Cachero TG, Shulga-Morskaya S, Dobles M, Frew E, Scott ML. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science. 2001;293:2111–2114. doi: 10.1126/science.1061964. [DOI] [PubMed] [Google Scholar]
- 10.Saito T, Chiba S, Ichikawa M, Kunisato A, Asai T, Shimizu K, Yamaguchi T, Yamamoto G, Seo S, Kumano K, Nakagami-Yamaguchi E, Hamada Y, Aizawa S, Hirai H. Notch2 is preferentially expressed in mature B cells and indispensable for marginal zone B lineage development. Immunity. 2003;18:675–685. doi: 10.1016/s1074-7613(03)00111-0. [DOI] [PubMed] [Google Scholar]
- 11.Witt CM, Won WJ, Hurez V, Klug CA. Notch2 haploinsufficiency results in diminished B1 B cells and a severe reduction in marginal zone B cells. J.Immunol. 2003;171:2783–2788. doi: 10.4049/jimmunol.171.6.2783. [DOI] [PubMed] [Google Scholar]
- 12.Tanigaki K, Han H, Yamamoto N, Tashiro K, Ikegawa M, Kuroda K, Suzuki A, Nakano T, Honjo T. Notch-RBP-J signaling is involved in cell fate determination of marginal zone B cells. Nat.Immunol. 2002;3:443–450. doi: 10.1038/ni793. [DOI] [PubMed] [Google Scholar]
- 13.Maillard I, Weng AP, Carpenter AC, Rodriguez CG, Sai H, Xu L, Allman D, Aster JC, Pear WS. Mastermind critically regulates Notch-mediated lymphoid cell fate decisions. Blood. 2004;104:1696–1702. doi: 10.1182/blood-2004-02-0514. [DOI] [PubMed] [Google Scholar]
- 14.Hozumi K, Negishi N, Suzuki D, Abe N, Sotomaru Y, Tamaoki N, Mailhos C, Ish-Horowicz D, Habu S, Owen MJ. Delta-like 1 is necessary for the generation of marginal zone B cells but not T cells in vivo. Nat.Immunol. 2004;5:638–644. doi: 10.1038/ni1075. [DOI] [PubMed] [Google Scholar]
- 15.Hampel F, Ehrenberg S, Hojer C, Draeseke A, Marschall-Schroter G, Kuhn R, Mack B, Gires O, Vahl CJ, Schmidt-Supprian M, Strobl LJ, Zimber-Strobl U. CD19-independent instruction of murine marginal zone B-cell development by constitutive Notch2 signaling. Blood. 2011;118:6321–6331. doi: 10.1182/blood-2010-12-325944. [DOI] [PubMed] [Google Scholar]
- 16.Murre C. Helix-loop-helix proteins and lymphocyte development. Nat.Immunol. 2005;6:1079–1086. doi: 10.1038/ni1260. [DOI] [PubMed] [Google Scholar]
- 17.Kee BL. E and ID proteins branch out. Nat.Rev.Immunol. 2009;9:175–184. doi: 10.1038/nri2507. [DOI] [PubMed] [Google Scholar]
- 18.Sun XH. Multitasking of helix-loop-helix proteins in lymphopoiesis. Adv.Immunol. 2004;84:43–77. doi: 10.1016/S0065-2776(04)84002-1. [DOI] [PubMed] [Google Scholar]
- 19.Quong MW, Martensson A, Langerak AW, Rivera RR, Nemazee D, Murre C. Receptor editing and marginal zone B cell development are regulated by the helix-loop-helix protein, E2A. J.Exp.Med. 2004;199:1101–1112. doi: 10.1084/jem.20031180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beck K, Peak MM, Ota T, Nemazee D, Murre C. Distinct roles for E12 and E47 in B cell specification and the sequential rearrangement of immunoglobulin light chain loci. J.Exp.Med. 2009;206:2271–2284. doi: 10.1084/jem.20090756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Becker-Herman S, Lantner F, Shachar I. Id2 negatively regulates B cell differentiation in the spleen. J.Immunol. 2002;168:5507–5513. doi: 10.4049/jimmunol.168.11.5507. [DOI] [PubMed] [Google Scholar]
- 22.Nie L, Xu M, Vladimirova A, Sun XH. Notch-induced E2A ubiquitination and degradation are controlled by MAP kinase activities. EMBO J. 2003;22:5780–5792. doi: 10.1093/emboj/cdg567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang Z, Nie L, Xu M, Sun XH. Notch-induced E2A degradation requires CHIP and Hsc70 as novel facilitators of ubiquitination. Mol.Cell Biol. 2004;24:8951–8962. doi: 10.1128/MCB.24.20.8951-8962.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nie L, Perry SS, Zhao Y, Huang J, Kincade PW, Farrar MA, Sun XH. Regulation of lymphocyte development by cell-type-specific interpretation of notch signals. Mol.Cell Biol. 2008;28:2078–2090. doi: 10.1128/MCB.00844-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nie L, Zhao Y, Wu W, Yang YZ, Wang HC, Sun XH. Notch-induced Asb2 expression promotes protein ubiquitination by forming non-canonical E3 ligase complexes. Cell Res. 2011;21:754–769. doi: 10.1038/cr.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cochrane SW, Zhao Y, Welner RS, Sun XH. Balance between Id and E proteins regulates myeloid-versus-lymphoid lineage decisions. Blood. 2009;113:1016–1026. doi: 10.1182/blood-2008-06-164996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang HC, Peng V, Zhao Y, Sun XH. Enhanced notch activation is advantageous but not essential for T cell lymphomagenesis in id1 transgenic mice. PLoS.One. 2012;7:e32944. doi: 10.1371/journal.pone.0032944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmitt TM, Zuniga-Pflucker JC. Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity. 2002;17:749–756. doi: 10.1016/s1074-7613(02)00474-0. [DOI] [PubMed] [Google Scholar]
- 29.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat.Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 30.Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC.Dev.Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rickert RC, Roes J, Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 1997;25:1317–1318. doi: 10.1093/nar/25.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruzinova MB, Benezra R. Id proteins in development, cell cycle and cancer. Trends Cell Biol. 2003;13:410–418. doi: 10.1016/s0962-8924(03)00147-8. [DOI] [PubMed] [Google Scholar]
- 33.Kin NW, Crawford DM, Liu J, Behrens TW, Kearney JF. DNA microarray gene expression profile of marginal zone versus follicular B cells and idiotype positive marginal zone B cells before and after immunization with Streptococcus pneumoniae. J.Immunol. 2008;180:6663–6674. doi: 10.4049/jimmunol.180.10.6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zambrowicz BP, Imamoto A, Fiering S, Herzenberg LA, Kerr WG, Soriano P. Disruption of overlapping transcripts in the ROSA beta geo 26 gene trap strain leads to widespread expression of beta-galactosidase in mouse embryos and hematopoietic cells. Proc.Natl.Acad.Sci.U.S.A. 1997;94:3789–3794. doi: 10.1073/pnas.94.8.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park ST, Sun XH. The Tal1 oncoprotein inhibits E47-mediated transcription. Mechanism of inhibition. J.Biol Chem. 1998;273:7030–7037. doi: 10.1074/jbc.273.12.7030. [DOI] [PubMed] [Google Scholar]
- 36.Sun X-H, Copeland NG, Jenkins NA, Baltimore D. Id proteins, Id1 and Id2, selectively inhibit DNA binding by one class of helix-loop-helix proteins. Mol.Cell.Biol. 1991;11:5603–5611. doi: 10.1128/mcb.11.11.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nie L, Wu H, Sun XH. Ubiquitination and degradation of Tal1/SCL are induced by notch signaling and depend on Skp2 and CHIP. J.Biol.Chem. 2008;283:684–692. doi: 10.1074/jbc.M704981200. [DOI] [PubMed] [Google Scholar]
- 38.Cariappa A, Boboila C, Moran ST, Liu H, Shi HN, Pillai S. The recirculating B cell pool contains two functionally distinct, long-lived, posttransitional, follicular B cell populations. J.Immunol. 2007;179:2270–2281. doi: 10.4049/jimmunol.179.4.2270. [DOI] [PubMed] [Google Scholar]
- 39.Srivastava B, Quinn WJ, III, Hazard K, Erikson J, Allman D. Characterization of marginal zone B cell precursors. J.Exp.Med. 2005;202:1225–1234. doi: 10.1084/jem.20051038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin YC, Jhunjhunwala S, Benner C, Heinz S, Welinder E, Mansson R, Sigvardsson M, Hagman J, Espinoza CA, Dutkowski J, Ideker T, Glass CK, Murre C. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat.Immunol. 2010;11:635–643. doi: 10.1038/ni.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 42.Radtke F, Wilson A, MacDonald HR. Notch signaling in hematopoiesis and lymphopoiesis: lessons from Drosophila. Bioessays. 2005;27:1117–1128. doi: 10.1002/bies.20315. [DOI] [PubMed] [Google Scholar]
- 43.Tan JB, Xu K, Cretegny K, Visan I, Yuan JS, Egan SE, Guidos CJ. Lunatic and manic fringe cooperatively enhance marginal zone B cell precursor competition for delta-like 1 in splenic endothelial niches. Immunity. 2009;30:254–263. doi: 10.1016/j.immuni.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 44.Radtke F, Fasnacht N, MacDonald HR. Notch signaling in the immune system. Immunity. 2010;32:14–27. doi: 10.1016/j.immuni.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 45.Allman D, Punt JA, Izon DJ, Aster JC, Pear WS. An invitation to T and more: notch signaling in lymphopoiesis. Cell. 2002;109(Suppl):S1–11. doi: 10.1016/s0092-8674(02)00689-x. [DOI] [PubMed] [Google Scholar]
- 46.Schlissel M, Voronova A, Baltimore D. Helix-loop-helix transcription factor E47 activates germ-line immunoglobulin heavy-chain gene transcription and rearrangement in a pre-T-cell line. Genes Dev. 1991;5:1367–1376. doi: 10.1101/gad.5.8.1367. [DOI] [PubMed] [Google Scholar]
- 47.Murre C, McCaw PS, Baltimore D. A new DNA binding and dimerization motif in immunoglobin enhancer binding, daughterless, MyoD and myc proteins. Cell. 1989;56:777–783. doi: 10.1016/0092-8674(89)90682-x. [DOI] [PubMed] [Google Scholar]
- 48.Kadesch T. Helix-loop-helix proteins in the regulation of immunoglobulin gene transcription. Immunol.Today. 1992;13:31–36. doi: 10.1016/0167-5699(92)90201-h. [DOI] [PubMed] [Google Scholar]
- 49.Pasqualucci L, Bereschenko O, Niu H, Klein U, Basso K, Guglielmino R, Cattoretti G, Dalla-Favera R. Molecular pathogenesis of non-Hodgkin's lymphoma: the role of Bcl-6. Leuk.Lymphoma. 2003;44(Suppl 3):S5–12. doi: 10.1080/10428190310001621588. [DOI] [PubMed] [Google Scholar]
- 50.Basso K, Dalla-Favera R. BCL6: master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv.Immunol. 2010;105:193–210. doi: 10.1016/S0065-2776(10)05007-8. [DOI] [PubMed] [Google Scholar]
- 51.Barish GD, Yu RT, Karunasiri M, Ocampo CB, Dixon J, Benner C, Dent AL, Tangirala RK, Evans RM. Bcl-6 and NF-kappaB cistromes mediate opposing regulation of the innate immune response. Genes Dev. 2010;24:2760–2765. doi: 10.1101/gad.1998010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.