

Abstract
CD4+ T cells developing towards a T helper 2 (Th2) fate express IL-4, IL-5, and IL-13 while inhibiting production of cytokines associated with other T helper types, such as the Th1 cytokine IFN- γ. IL-4-producing Th2 effector cells give rise to a long-lived memory population committed to re-activation of the Th2 cytokine gene expression program. However, re-activation of these effector-derived cells under Th1-skewing conditions leads to production of IFN-γ along with IL-4 in the same cell. We now show that this flexibility (“plasticity”) of cytokine expression is preceded by a loss of the repressive DNA methylation of the Ifng promoter acquired during Th2 polarization yet requires STAT4 along with T-bet. Surprisingly, loss of either STAT4 or T-bet increased Ifng promoter CpG methylation in both effector and memory Th2 cells. Taken together, our data suggest a model in which the expression of IFN-γ by Th2-derived memory cells involves attenuation of epigenetic repression in memory Th2 cells, combined with Th1-polarizing signals after their recall activation.
INTRODUCTION
CD4+ T cells play central roles in the ability of the immune system to adapt the nature of its responses to different types of pathogens. Upon exposure to their cognate antigen, signals from the T cell receptor and the local cytokine milieu drive naïve CD4+ T cells to develop into one of several effector programs. Each effector type, commonly referred to as a lineage or subset, produces a set of hallmark cytokines while inhibiting the expression of cytokine genes characteristic of other effector types (1). Development along an effector lineage is, in large part, directed by cytokines signaling through their membrane bound receptors at or around the time of antigen encounter (2, 3). The cell surface receptors for cytokines that can polarize populations of helper T cells activate immediate-early transcription factors in the family of STAT proteins (4, 5). These, in concert with other transcription factors and signaling pathways, induce subset-or lineage-specific transcription factors sometimes considered to be master regulators (6, 7). The combined signals from the TCR, cytokine receptors, and the activity of the master regulator transcription factors lead to the subset specific expression of cytokines that characterize the CD4 effector cells.
Two of the best studied effector programs, both in terms of physiological impact and the mechanisms leading to their development, are T helper one (Th1) and T helper two (Th2) (8, 9). Th1 effectors produce interferon gamma (IFN-γ), which activates macrophages to promote clearance of intracellular pathogens (10, 11) and is involved in the pathogenesis of autoimmune diseases such as type I diabetes (12). The importance of IFN-γ in human health is illustrated by the increased susceptibility to mycobacterial infections in patients lacking a functional IFN-γ receptor (13). Th1 polarization requires IL-12 receptor signaling via STAT4 and an induction of T-bet3 expression (4, 6, 7). These transcription factors direct histone post-translational modifications (14) along with chromatin remodeling of the Ifng gene (15) to allow for efficient gene transcription. In contrast, the Th2 effector program is characterized by repression of Ifng gene expression along with production of IL-4, IL-5, and IL-13 (8, 16). These Th2 cytokines direct responses against helminthic pathogens (17) and contribute to atopic diseases such as allergic asthma (18). Th2 polarization involves IL-4 receptor signaling though STAT6 (19, 20), followed by induction of the transcription factor GATA3 (21, 22), to enable transcription of the Th2 cytokine genes.
Beyond the activation of cytokine gene expression, each polarized T helper program also involves the silencing of genes from opposing transcriptional programs (23). For instance, GATA3 mediates the silencing of the genes encoding IFN-γ, IL-12 receptor beta, and STAT4 via repressive histone modifications during the course of Th2 polarization (24). Conversely, cells developing towards Th1 effector function inhibit the production of IL-4 through inhibition of GATA3 transcription (23, 25). The silencing of cytokines associated with other T helper subsets also involves repressive DNA methylation. Thus, the proximal promoter of the Ifng gene is CpG- methylated in Th2 clones (26) and primary effector cells (27–29) when compared to Th1 counterparts. Moreover, methylation of the evolutionarily conserved CpG at −53 in this proximal Ifng promoter sufficed to abrogate its activity (29). Together, these studies indicated that DNA methylation, especially at the −53 CpG of that Ifng promoter is a key aspect of repressing expression of this Th1 cytokine in Th2 effector cells.
Immunologic memory is a key feature of the adaptive immune system and provides protection against re-infection after a first exposure to a pathogen (30). After an antigen has been cleared, a fraction of responding cells survives as a long-lived population and appears to acquire a memory program differing from the effector state (31). Because cytokine production by memory cells upon antigen exposure can instruct a new generation of immune effectors, the profile of cytokines produced by recall responses of a memory CD4 population can dictate its protective value in repeat exposures to a given pathogen. For example, rapid production of IL-4 by memory-phenotype CD4+ T cells sufficed to guide CD4+ T cells to adopt a Th2 effector program following exposure to Leishmania major, leading to a failure to resolve the infection (32). In contrast, IFN-γ promotes resistance to such pathogens. Th2-derived memory cells arise from IL-4-producing Th2 effectors; after acquisition of a relatively quiescent state, Ag restimulation of these Th2-derived memory cells rapidly leads to IL-4 production (33, 34). After stimulation and growth in Th1-biased conditions in vitro or in vivo, these re-activated memory cells continue to produce IL-4, illustrating that Th2 memory cells retain a commitment to produce IL-4 (35).
While the production of IL-4 remains part of the programming for Th2-derived memory cells, restimulation under Th1 conditions also drove these cells to produce substantial amounts of IFN-γ (35–37). These findings revealed that the nature of gene silencing as part of the T helper program could be changed in memory cells, so that IFN-γ and the Th2 cytokine genes can be co-expressed within an individual CD4 lymphocyte. However, almost nothing is known about the molecular mechanisms for this plasticity of programming. Recent work indicated that, in addition to the Th1 master regulator T-bet, IL-12 was required for the induction of Ifng gene expression after re-activation of memory Th2 populations (38). The signal(s) downstream from IL-12 and essential for plasticity of Ifng regulation are not established. Moreover, a key unanswered question as to the mechanisms permitting IFN-γ production by Th2-derived memory cells is whether repressive epigenetic modifications of the Ifng promoter that occur during Th2 polarization are maintained in the memory phases. Here, we have tested if STAT4 impacts the ability of memory Th2 cells to express T-bet or IFN-γ in Th1 recall conditions. Further, we analyzed Ifng promoter DNA methylation in naïve, Th1, Th2, and Th2-derived memory CD4+ T cells, and explored the relationship between promoter methylation and the Th1-determining transcription factors STAT4 and T-bet.
MATERIALS AND METHODS
Mice
BALB/c Il4-IRES-Gfp (“4get”), DO11.10 mice were bred with BALB/c Tbx21 (T-bet) −/− (KO) or BALB/c Stat4 KO mice (Jackson labs). BALB/c-ByJ (Jackson labs) and athymic BALB/c nude (Foxn1/Foxn1) mice were used as recipients for transfer experiments. Recipients were 4–6 weeks old at the time of transfer. Mice were maintained in micro-isolator cages at a Vanderbilt University facility in accordance with Institutional Animal Care and Use Committee guidelines and an approved protocol.
Reagents
Fluorophore-conjugated and purified mAb were obtained from BD Pharmingen (San Jose, CA) and purified recombinant cytokines from Leinco (St. Louis, MO) unless otherwise indicated. Purified 11B11 anti-IL-4 and recombinant huIL-2 were obtained from the Biological Response Modifiers Program (NCI, Frederick MD). Anti-T-bet-eFluor 660 was obtained from eBioscience (San Diego, CA). Oligonucleotides were synthesized by Invitrogen (Grand Island, NY) unless otherwise stated.
Cell culture and purification of GFP+ IL-4 producing effectors and adoptive transfers
DO11.10 cells were activated with OVA323-339 peptide, and all cells were cultured, as described (35) with the following modifications. For Th1 culture conditions, cells were plated at 7 × 106 cells/ml and received 1 μg/ml OVA peptide, 5 ng/ml IL-12, and 3 μg/ml anti-IL-4 antibody 11B11. For Th2 cell cultures, cells were plated at 3.5 × 106 cells/ml and received 0.5 μg/ml OVA peptide, 7.5 ng/ml IL-4, 3 μg/ml anti-IFN-γ antibody, and 2 μg/ml anti-IL-12 antibody. Both Th1 and Th2 cell cultures were supplemented with IL-2 (50 units/ml 24 and 72 hours after Ag stimulation). GFP+ Th2 effector cells were purified for transfer as described (35). After four days of culture in Th2 conditions, 4get, DO11.10 cells (Tbx21 −/−, Stat4 −/−, or transcriptionally WT) were stained with APC-conjugated anti-CD4 and PE-conjugated KJ1-26 (anti-DO11.10 TCR) antibodies and flow sorted on a FACS Aria (BD, Franklin Lakes, NJ) to purify (> 98.5%) viable DO11.10 KJ1-26+ CD4+, GFP+ cells. Prior to transfer or DNA isolation for methyl-CpG analyses, these cells were cultured in Th2 conditions for 9–10 d after re-stimulation with APCs (4:1 with T cells) and 0.25 μg/ml OVA323-339 (13–14 d total).
Quantitation of cytokine production
T helper cell cultures plated with APCs at a 1:4 ratio, and single-cell suspensions of recipient spleens, were stimulated with 1 μg/ml OVA323-339. Cytokine concentrations in supernatant collected after 36 hr were quantified using a flow cytometric kit (Th1/Th2/Th17 cytokine bead array; BD Pharmingen). Intracellular cytokines were analyzed by stimulating cells 18 hr with OVA323-339 (1 μg/ml) in the presence of APCs, followed by Golgi-Stop (BD) (2–3 hr), and staining in the presence of anti-CD16/32 (Fc Block, BD Pharmingen) as previously described (35). Viable cells were selected based on forward and side scatter characteristics, and fluorescence signals representing intracellular cytokines were determined in cells positive for the DO11.10 TCR and CD4.
Bisulfite analysis of DNA methylation
DNA isolated from flow-purified naïve, effector (Th1, Th2, 13 d) and memory CD4 T cells, or from tissues, was digested with Bam H1 (New England Biolabs, Ipswich MA), bisulfite-modified using the Imprint bisulfite modifying kit (Sigma-Aldrich, St. Louis, MO), then used as template in quadruplicate PCR performed using primer pairs specific for each modified strand sequence in the Ifng promoter (Supplemental Table I) (29). The noncoding strand of the Ifng promoter was amplified with a single reaction, while the coding strand was amplified using a nested PCR. After pooling four tubes of separate amplification for each sample, specific PCR products were identified on agarose gels, extracted using the Qiaquick gel extraction kit (Qiagen, Valencia, CA), ligated with T-easy vector (Promega, Madison, WI), and then transformed into JM109 cells (Agilent, Santa Clara, CA). For each of three independent biological replicate cell samples and for each strand, eight to ten clones derived from each reaction pool were sequenced and scored for the frequency of unmodified C residues in the CpG dinucleotides; the modification frequency for C residues outside of CpG dyads was verified as > 99% for all sample sets.
EMSA
EMSAs were performed as reported (29, 39) except that extracts of Th1 cells developing from primary mouse CD4+ T cells were used. Methylated upper strand oligonucleotides were synthesized by Invitrogen (Grand Island, NY). The upper strand oligonucleotides were designated as unmethylated, meC(−53) hemimethylated, and tri-hemimethylated (meC at −53, −45, and −34) (Supplemental Table I). Each was annealed to an unmethylated lower strand oligonucleotide after radiolabeling with γ-[32P]-ATP (Perkin-Elmer, Waltham MA) and T4 polynucleotide kinase (New England Biolabs, Beverly MA). For competition assays, unlabeled competitor was added simultaneously with the labeled probe at molar ratios (competitor:probe) of 100, 10, and 1:1. Antibodies used for the super shift assays were CREB1 (sc-186), ATF2 (sc-187) and c-Jun (sc-45) (Santa Cruz Biotechnology, Santa Cruz, CA).
Transient transfection and reporter assays
Nucleofection was carried out via the Amaxa T cell kit (Lonza, Basel, Switzerland) using a minimal Ifng promoter reporter P1P2-Luc (40) along with pCMV-Sport6-CREB1 or pCMV-Sport6. All results were normalized to GFP expression from the pMAX-GFP plasmid (Lonza) measured via flow cytometry. Luciferase activity was measured using the Dual-glow luciferase assay system (Promega) according to manufacturer’s protocols.
Chromatin Immune Precipitation assays
Chromatin immunoprecipitation (ChIP) assays were performed essentially as described previously (15). After cross-linking with formaldehyde, primary Th1 and Th2 cells were sonicated in a Bioruptor (Diagenode, Denville, NJ) to produce an average sheered DNA length of 400 bp. Immune precipitation was carried out using anti-AcH3(K9) (Millipore, #DAM1813175), or CREB1 (Santa-Cruz, sc-186X), and the precipitates were analyzed by PCR using primers shown in Supplemental Table I.
Proliferation in vitro and in vivo
Proliferation studies using CFSE partitioning and BrdU incorporation were carried out as described (39). For CFSE partitioning in vivo, Th2 cells were grown for five days, labeled with CFDA-SE (Invitrogen) (2.5μM, 15 min) following manufacturer instructions, and then transferred into BALB/c recipients. Fluorescence was measured on donor-derived cells recovered 12 d after transfer with gating as described above. For CFSE partitioning assays in vitro, DO11.10 splenocytes were either labeled, Ag-stimulated, and cultured 2 d, or activated with Ag, cultured in Th2 conditions (11 d, with one interim Ag stimulation as for cells used in adoptive transfers), labeled with CFDA-SE as above, and then cultured 2 d in IL-2 supplemented medium before analysis by flow cytometry. For assays of BrdU incorporation into donor cells in vivo, recipient mice were injected twice (72, 24 h before harvest; 3 mg i.p. per injection) with BrdU (Sigma Aldrich) in sterile saline. Cells harvested 12 d post-transfer were then processed as described (39) to detect Alexa-647 anti-BrdU (Invitrogen) in donor- (KJ1-26+) and recipient-derived CD4+ T cells by flow cytometry. For in vitro assays, BrdU (1 μM) was added to Th2 cultures (days 2 and 13 after Ag activation) followed 4 h later by processing, direct immunofluorescent anti-BrdU staining, and flow cytometry of KJ1-26+ CD4+ cells.
Retroviral transduction
Retroviral transduction was carried out using as previously described (15). In brief, retrovirus-containing supernatants from ΦNX packaging cells transfected with MSCV-IRES-Thy1.1 (MiT), or MSCV-T-bet-IRES-Thy1.1 (MIT-T-bet) were used to transduce established GFP+ DO11.10 Th2 cells two days after restimulation. Cells were then cultured under Th1 or Th2 conditions, followed by measurements of Ag-stimulated cytokine production or intracellular cytokines as above.
RESULTS
Strand-biased acquisition of Ifng promoter methylation in Th2 effector cells
The finding that memory Th2 cells, unlike their effector-phase precursors, can respond to an inflammatory milieu by efficient induction of IFN-γ production (35, 36) raises questions about the fate of repressive DNA methylation observed in Th2 effectors once these cells transition to memory status. Accordingly, we used bisulfite modification of DNA followed by strand specific PCR to analyze CpG methylation in Ifng promoter DNA of naïve CD4+ lymphocytes, Th1, and Th2 T cells. Naïve CD4+ and Th1 effector cells showed little methylation of either strand of DNA upstream from the transcription start site (Fig. 1), whereas high methylation densities were found at two dinucleotides in exon 1, independent of T cell differentiation, as expected (41, 42). In effector Th2 cells (Fig. 1B, C), we found increased methylation of the coding strand of the Ifng promoter, with a majority of samples exhibiting modification of the key −53 CpG whose modification appeared to abrogate promoter activity (29). Surprisingly, however, the noncoding strand was reproducibly and significantly less methylated in Th2 effector cells relative to the coding strand (Figure 1D), particularly at the −53 CpG (Fig. 1E). In light of this unexpected result, we tested samples including DNA from a mouse brain and 3T3 cells, both of which would be expected to have symmetrical hypermethylation of the Ifng promoter, along with thymocytes and naïve CD4 lymphocytes. Brain and 3T3 cell DNA demonstrated a high density of symmetrical methylation across the surveyed region and in particular at the crucial −53 CpG, whereas thymocytes, like naïve CD4 T cells, exhibited little meC (Table I). The frequency of non-coding strand DNA methylation in Th2 cells was too low simply to represent a lack of modification on one chromosome, e.g., from mono-allelism (43). Separate analyses (Table I and later results) exclude a strand bias in the detection method as the basis for the observation. Accordingly, we infer from these data that Ifng promoters were hemimethylated in these Th2 effector cells. The asymmetry of CpG methylation might in principle be observed because a high proportion of the Th2 population was actively in cell cycle and rapidly undergoing DNA synthesis. However, virtually no divisions were observed in CFSE analyses of the effector Th2 cells in the 2 d prior to the time at which DNA samples were prepared (Fig. 1F). Moreover, only a small fraction of the population was in cell cycle as indicated by a low frequency of BrdU+ cells at this time 13 after the initial Ag activation (Fig. 1G). From these low frequencies of cycling cells, we conclude that at most a very small minority of asymmetric methylation observed in Th2 effectors could be due to DNA replication, whereas the remainder is an epigenetic feature of the Ifng locus at this stage in Th2 effectors.
Figure 1. Asymmetric increase in CpG methylation of the Ifng promoter in Th2 cells.

Methylation of CpG residues was determined by sequencing individual subcloned products from strand-specific PCR of bisulfite-modified DNA from naïve CD4+ T cells (A), and Th1 and Th2 cells (B). Shown are diagrams illustrating representative individual sequences, from biological replicates for each type of sample, generated from naïve (A) or Th1 and Th2 cells (B). Filled circles represent methylated cytosine and open circles the unmethylated residue. Analyses included at least eight independent clones from each of three independent biological replicates per sample. (C) Data for the coding (left) and non-coding (right) strands are shown, with each bar representing the mean (±SEM) number of methylated CpGs per clone in all surveyed upstream CpGs (six total), the CpGs at −205, −190, −170, or the CpG triad at −53, −45, and −34. (D) Comparison of coding and non-coding strand methylation in Th2 effectors. (E) Methylation frequencies at the −53 CpG dinucleotide. (F) CFSE partitioning was measured forty-eight hours after labeling cells that were either acutely stimulated (two days total), or from eleven day Th2 cultures (thirteen days total). (G) Cells cultured under Th2 conditions for two days or thirteen days were pulsed with BrdU for four hours. The incorporation of BrdU into cellular DNA was measured by intracellular staining. Cells grown continuously in BrdU or unexposed to BrdU were used as staining controls. Results (F, G) are representative of two independent experiments. (* p<0.05, ** p<0.01, *** p<0.001; N.S., not statistically significant)
Table I.
CpG methylation in naïve CD4+ T cells, thymus, brain, and NIH 3T3 cells
| A. | ||||||||
|---|---|---|---|---|---|---|---|---|
| CpG positiona | ||||||||
| Sample | Strand | −205 | −190 | −170 | −53 | −45 | −34 | 16 |
| Naïve CD4 | Coding | 11.4 (3.6)b | 11.4 (3.6) | 14.4 (3.6) | 17.4 (3.9) | 14.4 (3.6) | 14.4 (3.6) | 59.8 (6.1) |
| Non-coding | 17 (3.6) | 10.8 (1.6) | 6.25 (2.3) | 17 (3.4) | 15.3 (2) | 15.3 (2) | 90.9 (3.6) | |
| Thymus | Coding | 0 | 0 | 0 | 0 | 0 | 0 | 72.2 (3.5) |
| Non-coding | 0 | 0 | 0 | 0 | 0 | 0 | 71.3 (3.5) | |
| Brain | Coding | 100 | 86.6 (2) | 86.6 (2) | 100 | 100 | 100 | 100 |
| Non-coding | 95 (2.7) | 76.7 (3) | 100 | 95 (2.7) | 100 | 91.7 (3.3) | 83.3 (4.9) | |
| NIH 3T3 | Coding | 78.9 (3.2) | 63.1 (3.8) | 52.6 (1.8) | 63.1 (0.9) | 63.1 (0.9) | 52.6 (3.5) | 68.4 (2.8) |
| Non-coding | 62.5 (4.2) | 36.3 (6.1) | 43.8 (3.0) | 56.3 (3.0) | 50 (0.0) | 50 (0.0) | 87.5 (4.2) | |
| B. | ||||
|---|---|---|---|---|
| Sample | Strand | Totalc | −205–170 | −53–34 |
| Naïve CD4 | Coding | 0.9 (0.25)d | 0.4 (0.18) | 0.52 (0.2) |
| Non-coding | 0.7 (0.34) | 0.25 (0.14) | 0.45 (0.22) | |
| Thymus | Coding | 0.11 (0.07) | 0 (0) | 0.11 (0.07) |
| Non-coding | 0 (0) | 0 (0) | 0 (0) | |
| Brain | Coding | 5.7 (0.12) | 2.7 (0.12) | 3 (0) |
| Non-coding | 5.6 (0.16) | 2.6 (0.15) | 2.9 (0.06) | |
| 3T3 | Coding | 3.8 (0.45) | 2.1 (0.27) | 1.8 (0.31) |
| Non-coding | 2.9 (0.53) | 1.4 (0.26) | 1.5 (0.35) | |
position is presented as the distance in bases from the transcription start site
data are presented as the mean (± SEM) percentage of samples methylated at a given position.
Cluster of CpG dinucleotides being assayed.
Mean (± SEM) number of methyl-CpGs per clone in a given cluster.
Asymmetrical methylation impacts transcription factor binding to the Ifng promoter
Based on the evidence that the Ifng promoter in many Th2 cells can be in a state of asymmetrical methylation, we investigated whether hemimethylation might impact transcription factor recruitment to the Ifng promoter. EMSA using nuclear extracts of primary Th1 cells were carried out using unmethylated or hemimethylated probes (Fig. 2A). Both hemimethylated probes impaired the formation of the slower migrating complex (indicated by filled arrow, Fig. 2B). Competition assays using unlabeled competitor DNA confirmed that the mobility shift bands represented sequence-specific binding; moreover, 10-fold more cold competitor was needed to attenuate the slower migrating complex to the WT as compared to hemimethylated probe (Fig. 2C). To characterize this complex, we performed Ab blocking/supershift assays with the unmethylated probe and antibodies against CREB/ATF family members. The upper band was impacted by anti-CREB1 (Fig. 2D) whereas antibodies against ATF2 and c-Jun had no discernible effect, leading us to conclude that the slower migrating complex is predominantly formed by CREB1. Consistent with the hemimethylation observed at the Ifng promoter having an impact on CREB1 recruitment in vivo, ChIPs performed using anti-CREB1 Ab showed greater promoter occupancy in Th1 cells than their Th2 counterparts (Fig. 2E). The decreased binding of CREB1 in effector-stage Th2 cells, in which the Ifng gene is not active, would be consistent with CREB1 function as a trans-activator. To test if CREB1 can increase activity of the Ifng promoter in primary Th1 cells, we performed nucleofections of developing Th1 cells using a minimal Ifng promoter reporter construct and either a CREB1 expression vector or an empty vector control (Fig. 2F). We found that CREB1 increased activity of the Ifng reporter construct. All together, these findings show that upper-strand hemimethylation of the CpG at −53 can impair binding of CREB1, a trans-activator of the Ifng promoter.
Figure 2. Asymmetric methylation suffices to inhibit transcription factor binding to the Ifng promoter.

(A) Schematic of the hemimethylated probes used. (B) Shown is an autoradiograph representative of EMSA results after using probes that are unmethylated (lane 1), methylated at the coding strand C(−53)pG (lane 2) or methylated at coding strand C(−53)pG, C(−45)pG, and C(−34)pG (lane 3). (C) EMSA using unmethylated probe in competition with oligonucleotides that were unmethylated (lanes 2–4), hemimethylated at C(−53) (lanes 5–7), or hemimethylated at all three upper strand residues (lanes 8, 9, 10), at 1, 10, and 100-fold excess of competitor over probe. Shown is one result representative of three replicate assays. (D) Antibody blocking/supershift assay against ATF2, c-Jun, CREB, or control (non-specific IgG). Shown is one result representative of three replicate assays. (E) ChIPs using antibodies specific for CREB1 or AcH3(K9). Eluted precipitates were analyzed for Ifng promoter sequences by PCR (left panel). Signals (mean ± SEM) from three independent biological replicates were quantified (right panel) as product relative to input as described in the Methods (** p ≤ 0.01; BLD, below limit of detection). (F) After nucleofection of primary Th1 cells with a minimal Ifng promoter-luciferase construct and pSport6 or pSport6-CREB1, luciferase activities were normalized to GFP expression. Shown are mean (± SEM) data from three independent experiments. (* p ≤ 0.05)
Loss of Ifng methylation in Th2-derived memory cells
Th2-derived memory cells can produce IFN-γ when exposed to Th1-skewing conditions during recall responses (35, 36). To investigate the relationship between this capacity and the repressive methylation observed in primary Th2 cells, we prepared DNA from purified effector cells and their memory Th2 descendants (Fig. 3A). As expected, cells in the donor-derived memory pool in each type of recipient underwent homeostatic divisions after transfer (Fig. 3B), and these memory cells produced IFN-γ after reactivation by Ag and growth in Th1 conditions (Fig. 3C). Weeks after transfers into normal or lymphopenic BALB/c mice, donor-derived cells were purified from the recipient lymphoid organs. Strand-specific PCR analyses of bisulfite-modified donor-derived cellular DNA showed that methylation of multiple sites decreased (Fig. 4B) and the −53 CpG of the Ifng promoter coding strand was almost completely unmethylated (Fig. 4A, C). These results were independent of whether the recipients had normal endogenous T cells or were lymphopenic (data not shown). These findings provide evidence of dynamic change in Ifng promoter methylation as the population of Th2 effectors yields a memory Th2 subset.
Figure 3. Elicitation of IFN-γ production from memory Th2 cells.
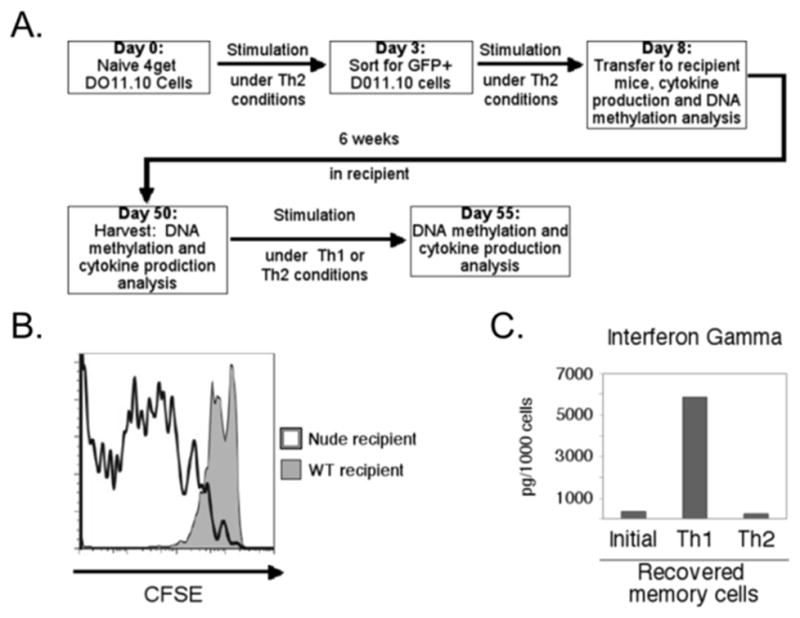
(A) Schematic overview of the experimental design and generation of Th2-derived memory cells. (B) CFSE partitioning assay for DO11.10 cells transferred into wildtype (shaded) or nude recipients (outline) and recovered after two weeks, gated on CD4+ DO11.10 TCR+ cells. (C) IFN-γ secretion data are shown for memory Th2 cells at the time of harvest and after recall activation and growth (6 d) under Th1 or Th2 polarizing conditions as indicated. Values in each individual sample were normalized to the frequencies of DO11.10 TCR+ CD4+ cell number to account for differences in cell prevalence. Shown are data from a single experiment representative of four independent replicates and consistent with (35, 36).
Figure 4. Altered DNA methylation of the Ifng promoter in effector-derived memory Th2 cells.

(A) Representative bisulfite sequencing results from freshly isolated memory cells are diagrammed as in Fig. 1A. (B) Mean methylated CpG dinucleotides per clone are shown as in Fig. 1C. The Th2 effector results are taken from Figure 1, and are shown for comparison. (C) Methylation of the Ifng promoter −53 CpG in effector versus memory Th2 cells is compared as per Figure 1E. (* p<0.05, ** p<0.01, *** p<0.001)
STAT4 is required for flexible IFN-γ production
The development of Th1 effector cells from naïve CD4+ T cell precursors is highly dependent on IL-12-induced STAT4 and, in most settings, on T-bet (4, 7, 44). IL-12 is required for the facultative induction of IFN-γ production by memory Th2 cells after recall stimulation in vitro and in vivo (35, 36, 38). However, the IL-12 receptor elicits multiple intracellular signals (45, 46), and which of these is essential for the plasticity of gene expression is not known. Accordingly, we compared the amounts of IFN-γ produced after recall stimulation and cultures of memory Th2 cells from Tbx21 −/− and Stat4 −/− T cells to that derived from parallel controls with normal transcriptional function (Fig. 5A and Supplemental Fig. S1A). When cytokine production was elicited one week after recall restimulation with peptide antigen and culture under Th1 and Th2 conditions, samples of each transcription factor-deficient memory Th2 population produced substantially less IFN-γ than the matched wild-type controls (Fig. 5A). IFN-γ production elicited after Th1-skewed recall was higher than background with each type of knockout cell type. To assess the extent to which double-producing (IL-4+ IFN-γ+) cells could be generated from memory Th2 cells, we used intracellular staining for these cytokines (Fig. 5B, C). Although subject to the likelihood that the limits of detection are more sensitive for secreted cytokine than intracellular staining, these analyses consistently revealed almost no IFN-γ+ donor-derived (KJ1-26+ CD4+) cells in the absence of either STAT4 or T-bet (Fig. 5C). In sharp contrast, ample IL-4+ IFN-γ+ CD4+ T cells were abundant (31% of donor T cells) when controls with normal transcription factor genes were used (Fig. 5C). These data indicate that STAT4 is required in support of the capacity for memory Th2 cells to turn on IFN-γ production to an extent similar to T-bet.
Figure 5. Impact of STAT4 and T-bet on potential for IFN-γ production by memory Th2 cells.

CD4+ Th2 cells of DO11.10 4get+ Tbx21−/− or DO11.10 4get+ Stat4−/− mice were generated in parallel with DO11.10 4get+ controls, flow-purified, and transferred into recipients as in Fig. 3A. Secreted IFN-γ and IL-4 were measured at the time of recovery and after recall activation and culture (6 d) under Th1 and Th2 conditions (panel A). Results are displayed as mean (±SEM) IFN-γ production by Tbx21 −/− or Stat4 −/− cells relative to wild-type controls after Ag re-stimulation with OVA323-339 peptide (* p<0.05, ** p<0.01, *** p<0.001). Intracellular cytokine staining was performed after Ag stimulation with OVA323-339 peptide at the time of cell recovery (B) and one week after recall stimulation and Th1 or Th2 culture as indicated (C). Cytokine staining profiles are shown for events within the donor-derived (KJ1-26+ CD4+) gate and represent consistent observations in three biological replicates.
T-bet induction in developing Th1 cells is driven by STAT1 and NF-κB (47, 48), but STAT4 regulates a later, IL-12-dependent phase of T-bet expression in the development of primary Th1 responses (49, 50). Therefore, we also tested if STAT4 is required for Ifng plasticity in memory Th2 cells because it is essential for T-bet induction. Consistent with the prior work (49, 50), intracellular stains detected T-bet immunofluorescence in STAT4-deficient Th1 effectors at levels equivalent to those observed for Th2 effectors (Fig. 6A, top panel). After recall activation and culture under Th1 conditions, however, STAT4-deficient memory Th2 cells displayed more T-bet protein expression, with at least half of the cells exhibiting induction to Th1 levels (Fig. 6A, middle panel). This finding indicates that the regulation of T-bet expression by STAT4 in this memory setting was not sufficient to explain the defect in Ifng induction. Together, the data show that the gene expression plasticity of memory Th2 cells, i.e., elicitation of IFN-γ, requires independent input from STAT4 as well as T-bet.
Figure 6. Impact of T-bet and STAT4 on DNA methylation at the Ifng promoter.

(A) Intracellular stain for T-bet in primary Th1 cells (top panel), and recall-activated Th2-derived memory cells cultured (5 d) in Th1 (middle) or Th2 conditions. Results gated on CD4+ DO11.10 TCR+ cells are shown. (B) WT, Tbx21−/−, and Stat4−/− Th2 cells were labeled with CFDA-SE and transferred into BALB/c nu/nu recipients for two weeks. Flow results show the indicated parameter in the gate for CD4+ DO11.10 TCR+ cells. (C) In vivo BrdU incorporation into WT, Tbx21−/−, and Stat4−/− Th2 cells five weeks after their transfer into BALB/c recipients. Shown are representative flow cytometry results of anti-BrdU signal in the CD4+ DO11.10 TCR+ lymphocyte gate. (D) Mean (±SEM) meCpG dinucleotides per clone are shown for Tbx21−/−, and Stat4−/− Th2 effector cells as in previous figures. WT Th2 effector results from Fig. 1 are shown for comparison. (E) Mean (±SEM) meCpG dinucleotides per sequenced clone for Tbx21−/−, and Stat4−/− Th2 memory cells are shown, with WT results from Fig. 4B reproduced for comparison. (F, G) Tbx21−/− DO11.10 4get+ CD4+ cells cultured in Th2 conditions for nine days were transduced with either MSCV-IRES-Thy1.1 (MIT) or MSCV-T-bet-IRES-Thy1.1 (MIT-T-bet), grown under Th1 conditions (1 wk), and analyzed as in Fig. 5 except that for intracellular cytokines were analyzed in the Thy1.1+, CD4+ and Thy1.1neg, CD4+ cells gates (F). (G) Secreted IFN-γ, measured as in Fig. 5A, was normalized to the number of Thy1.1+, CD4+, DO11.10 TCR+ cells in the culture. Analyses of total culture production of IFN-γ [MIT-T-bet, 164 (±35) ng/ml vs MIT control, 21 (±26) ng/ml; p=0.02] yielded the same conclusion. Results are representative of two (A–C), or three (D–G) independent biological replicates (* p<0.05, ** p<0.01, *** p<0.001).
T-bet and STAT4 alter Ifng promoter methylation pattern but not homeostatic divisions
Homeostatic divisions of cells in which asymmetric methyl-CpG marks were present could lead to descendants in which this repressive mark was absent from the Ifng promoter on both chromosomes. Accordingly, we tested if the rates of division were slower for Tbx21 −/− or Stat4 −/− Th2 cells in recipient mice. CFSE partitioning assays showed that there was no defect in rates of division for DO11.10 Th2 cells that were T-bet- or STAT4-deficient cells compared to controls that were wild-type with respect to the transcription factors (Fig. 6B). To compare proliferation of the transcriptionally deficient memory Th2 cells to WT controls long after the transfer, recipient mice received BrdU and its incorporation into DNA was measured. This analysis provided evidence that the transferred cells almost completely exited cell cycle and that low rates of S-phase entry were similar for all genotypes (Fig. 6C).
An alternative model is that the absence of T-bet or STAT4 led to a higher or more symmetric density of CpG methylation at the Ifng promoter during the development of Th2 effectors. Significant changes in upper strand me-CpG densities were not observed (Supplemental Fig. 1B), but the methylation frequency of the non-coding strand was strikingly higher for T-bet-null Th2 cells. In addition, STAT4-deficient cells had essentially symmetrical DNA methylation (Fig. 6D) and increased non-coding strand methylation, especially at the crucial −53 CpG (Supplemental Fig. 1C). Moreover, meCpG densities in the Ifng promoter DNA from transcription factor-deficient memory Th2 cells, recovered after several weeks in vivo, were higher than those from wild-type memory controls (Fig. 6E; supplemental Fig. S1D). Thus, T-bet and STAT4 each influenced Ifng promoter methylation in memory as well as effector Th2 lymphocytes.
Prior studies have supported several potential relationships between T-bet and the capacity to produce IFN-γ after Th2 differentiation. In one study, most human CD4 T cells could switch from polarized Th2 cytokine gene expression to turn on their IFNG gene; a subset of the helper cells unable to exhibit such flexibility was attributed to lack of T-bet expression (51). Parallel work indicated that differentiation progressively reduced the capacity of such Th2 cells to turn on IFN- γ expression in the presence of forced T-bet expression (52). In light of the failure of T-bet-deficient Th2 cells to exhibit flexibility (Fig. 5A) and their Ifng promoter methylation pattern, we explored the impact of forcing expression of this transcription factor after Th2 differentiation in its absence. Tbx21 −/− Th2 cells were transduced with a bicistronic retrovector (“MiT”) directing T-bet expression linked to Thy1.1, and compared to parallel transductions of the retrovector without T-bet cDNA. After culture in Th1 conditions and restimulation with APCs and Ag, intracellular staining for IL-4 and IFN-γ (Fig. 6F) revealed that high-level T-bet expression forced IFN-γ expression in Tbx21 −/− Th2 cells. Consistent with this finding, restimulation elicited substantial IFN- γ production by a T-bet-transduced Tbx21 −/− Th2 population as compared to controls. We conclude that T-bet at a sufficient level can overcome the block to IFN-γ production by established Tbx21 −/− Th2 cells. Based on the collective findings, we propose (see Discussion) that at least two barriers to Ifng gene expression are present in effector Th2 cells but reversible once cells have experienced a transition to memory state.
DISCUSSION
The capacity of memory cells derived from Th2 effectors to produce IFN-γ in recall responses represents a naturally occurring form of cellular reprogramming. Apart from a requirement for IL-12, type I interferons, and the transcription factor T-bet (35, 38), nothing is known about the molecular mechanisms by which this plasticity of gene expression is effected. We have found that the Ifng promoter exhibits asymmetric methylation in committed Th2 effectors. The coding strand DNA preferentially acquires significantly increased methylation relative to the low frequency of meCpG in naïve CD4+ T cells and on the non-coding strand. A hemimethylated state created by such asymmetry suffices to impair CREB1 binding to an Ifng promoter sequence that is highly conserved and strongly required for promoter activity. Consistent with these data, nucleofection assays provide evidence that CREB1 is a trans-activator of the Ifng promoter, and that this ubiquitously expressed transcription factor preferentially binds to the promoter in Th1 cells as compared to Th2 counterparts. Strikingly, CpG methylation of the Ifng promoter in memory Th2 cells was observed at a frequency little different from the naïve progenitor. Inasmuch as promoter methylation is a strongly repressive mark, these findings suggest that loss of meCpG marks contributes to the plasticity of Ifng gene expression upon recall activation. In investigating the transcription factor requirements for this facultative production of IFN-γ, we found that the IL-12-induced factor STAT4 is required along with T-bet. Surprisingly, increased densities of CpG methylation were observed in T-bet-deficient Th2 cells relative to WT controls, as well as in memory Th2 cells deficient in either of these essential transcription factors. We suggest that changes in the frequency of this repressive mark at promoters forms one – but not the only- part of the molecular basis for the reprogramming of gene expression in memory Th2 cells after recall activation.
Consistent with this component of our overall model, methylation of the coding stand of the Ifng promoter inhibited CREB1 binding, and CREB1 trans-activated Ifng promoter activity in primary Th1 cells. Previous work showed that Ifng promoter DNA methylation inhibited mobility shift complexes of the CREB/ATF family, and a more recent study used ChIP of the Th1 clone AE7 to implicate ATF2 as a major factor in this scenario (29, 53). One likely factor in a difference of results is the use of primary Th1 cells as opposed to a clone. The functional impact of CREB1 and other ATF transcription factors has been unclear, perhaps in part because of a paucity of analyses in primary cells. CREB1 appeared to inhibit (53, 54) or increase (55) Ifng transcription in T cells. However, CREB1 occupancy of the IFNG promoter enhanced transcription in human CD4+ T cells (56). Furthermore, exposure to M. tuberculosis induced CREB1 binding to the IFNG promoter in human CD4+ T cells, and RNAi-driven depletion of CREB1 in these cells decreased in IFN-γ production (57). Thus, the balance of evidence indicates that CREB1 promotes Ifng gene expression, so that asymmetric methylation at the highly conserved CREB/ATF binding site likely contributes to the inhibition of IFN-γ production in the developing Th2 cell. Loss of such hemimethylation, e.g., at the −53 site, would facilitate trans-activator interactions.
As in previous reports (26, 28, 29), our analyses indicate that thymocytes and naïve CD4 T cells have quite low levels of CpG methylation at the Ifng promoter. Thus, molecular processes that are part of Th2 differentiation include direction of de novo DNA methylation to this site. In general, DNA methyltransferases (DNMT) of the DNMT3 family (DNMT3a, b) appear to execute the process of adding new marks. T cell activation via TCR engagement increased DNMT3a expression, and a conditional loss-of-function study indicated that DNMT3a was important for repression of inappropriate cytokine genes in T helper differentiation (58). Intriguingly, although the memory cell generation and maintenance were not analyzed, in vitro analyses of DNMT3a-deficient cells detected plasticity of cytokine production somewhat akin to what memory Th2 cells are able to execute naturally (35, 36). Further analysis of this Dnmt3a0 model indicated that, similar to the low density of meCpG marking of the Ifng promoter in memory Th2 cells in our analyses, the de-repressed in vitro effectors lacking DNMT3a had levels of Ifng promoter methylation similar to those of naïve CD4 T cells (59). DNA replication naturally creates hemimethylation of CpG dyads, and establishment or enforcement of symmetry is predominantly executed by DNMT1 (60). Deletion of this DNMT early in T cell development led to a CD4 T cell population that produced over 10-fold more IFN-γ upon primary ex vivo activation (61), underlining the importance of DNA methylation in the restriction of cytokine expression in CD4+ T cells to a specific effector program. Surprisingly, we found that even among cells under Th2-differentiating conditions, absence of T-bet led to increased methylation of the coding strand and substantially greater symmetry (i.e., increased non-coding strand meCpG). These results suggest that, surprisingly, the low level of T-bet present early after activation of naïve CD4+ lymphocytes under Th2 conditions (6, 7, 44) directly or indirectly impedes access of DNMTs to the Ifng promoter. Beyond this unexpected function, we infer there is an additional aspect of T-bet in the molecular events underlying plasticity of Ifng gene expression. Although the promoter methylation and its symmetry increased in T-bet-deficient Th2 cells, a second impediment to IFN-γ production lies in a block to expression of T-bet when Th2 effectors are switched to Th1-promoting conditions without a period as memory cells. This would be consistent with the correlative data from switching experiments and measurements after single-cell cloning of human memory-phenotype CD4 T cells (51) as well as results of T-bet transduction [Fig. 6F, and (52)].
A complementary finding of our work identifies a functional importance of STAT4 equal to T-bet in allowing memory Th2 cells to produce IFN-γ along with Th2 cytokines after recall activation. The initial discovery of this flexibility indicated that IL-12 was a crucial factor for the process (35, 38). The IL-12 receptor signals through both STAT4-dependent (4) and -independent mechanisms (45, 62), each of which can promote Ifng gene expression. STAT4 is vital for Th1 differentiation, but IL-12 also activates a PI 3-kinase - mTOR – FoxO signaling pathway that culminates in de-repression of the Tbx21 gene encoding T-bet (46). Studies using conditional alleles will be needed to test if there is an additional contribution from the mTOR-Akt-FoxO pathway, but our data establish that STAT4 is required for the flexible production of IFN-γ by Th2-derived memory cells. Interestingly, this mechanism appears distinct from the primary effector phase, as we confirmed a vital role for STAT4 in maintaining T-bet protein levels in late (day 5) effector cells under Th1 conditions (49) but found little such effect in the memory Th2 cells. Thus, the requirement for STAT4 in memory Th2 cells’ plasticity is independent of an effect on T-bet expression.
A key finding of the work presented here was the unexpected dynamism in methylation density of promoter DNA as effector Th2 cells became a population with the low frequency of replicating (BrdU+) cells characteristic of the memory subset. Previous work on Ifng promoter regulation in memory or memory-phenotype T cells as compared to their naïve or effector counterparts has focused most on the CD8 lineage. Pioneering work indicated that a low level of CpG methylation present in naïve cells was lost upon cell activation (63–65). Surprisingly, resting memory-phenotype (total CD44hi) CD8+ T cells had substantial CpG methylation at their Ifng promoters, which was quickly lost upon recall stimulation (65). One potential model, which cannot be rigorously tested for a population of effector cells yielding memory, would involve active demethylation of the meCpG. Although remaining a controversial area, especially from the standpoint of molecular mechanisms, rapid loss of Il2 promoter DNA methylation scored by endonuclease sensitivity was found to occur without CD4 cell division (66), and other work also supports the existence of active DNA demethylation (67). Nonetheless, our findings suggest a straightforward passive mechanism by which the observed dynamism can be effected. Although Th2 clones were reported to exhibit almost uniform methylation of CpG at the Ifng promoter (26), under the conditions used for the present study we observed meCpG frequencies well below 100% at every site on the coding strand and an asymmetry in which a high fraction of coding strand meCpG dinucleotides exist in a base quartet in which the coding strand is paired with an unmethylated CpG. This indicates that the functional capacity of DNMT1 to establish symmetry after initial deoxycytosine methylation was insufficient.
Memory cell homeostasis is maintained by periodic divisions after DNA replication. As a consequence, the frequency of daughter strands lacking meCpG at each site would, in the setting of hemimethylation, increase as divisions progressed. While likely not the entire mechanistic explanation, the greater symmetry of Ifng promoter DNA methylation observed in the absence of T-bet or STAT4 in the primary Th2 effectors is associated with higher meCpG densities in the memory population. T-bet deficient, STAT4 deficient, and wild-type CD4 T cells had comparable cycling and division rates. Accordingly, we infer that the requirement for these transcription factors in memory Th2 cell plasticity stems not from an impact on rates of memory cell division, but rather in part from their impact on promoter methylation and from requirements for them in trans-activation.
Supplementary Material
Acknowledgments
We thank E. Volanakis, S. Joyce, J.W. Thomas, R. O’Brien, and E. Ruley for discussions and comments, and L. Williams for consultation and statistical expertise.
Footnotes
Supported by bridge funding from Vanderbilt University and by National Institute of Health grants R01 AI077528 to M.B. and T32 DK07563 to C.L.W.
DISCLOSURES:
The authors have no competing interest.
Abbreviations used: T-bet, T-box expressed in T cells; AcH3(K9), acetyl-lysine 9 of histone H3; me-, methyl-; CpG, deoxycytosine-deoxyguanine dinucleotide; IRES, internal ribosomal entry sequence; CFDA-SE, 5 (and 6)-carboxyfluorescein diacetate-succinimidyl ester; ChIP, chromatin immunoprecipitation; DNMT, DNA methyltransferase; FoxO, Forkhead box O-class.
References
- 1.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787– 793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 2.Carding SR, West J, Woods A, Bottomly K. Differential activation of cytokine genes in normal CD4-bearing T cells is stimulus dependent. European Journal of Immunology. 1989;19:231–238. doi: 10.1002/eji.1830190203. [DOI] [PubMed] [Google Scholar]
- 3.Zhu J, Paul WE. Peripheral CD4 T cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev. 2010;238:247–262. doi: 10.1111/j.1600-065X.2010.00951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacobson NG, Szabo SJ, Weber-Nordt RM, Zhong Z, Schreiber RD, JED, Murphy KM. Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat)3 and Stat4. J Exp Med. 1995;181:1755–1762. doi: 10.1084/jem.181.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaplan MH, Grusby MJ. Regulation of T helper cell differentiation by STAT molecules. J Leukoc Biol. 1998;64:2–5. doi: 10.1002/jlb.64.1.2. [DOI] [PubMed] [Google Scholar]
- 6.Lighvani AA, Frucht DM, Jankovic D, Yamane H, Aliberti J, Hissong BD, Nguyen BV, Gadina M, Sher A, Paul WE, O’Shea JJ. T-bet is rapidly induced by interferon-γ in lymphoid and myeloid cells. Proc Natl Acad Sci U S A. 2001;98:15137–15142. doi: 10.1073/pnas.261570598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 8.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition According to Profiles of Lymphokine Activities and Secreted Proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 9.Scott P, Natovitz P, Coffman RL, Pearce E, Sher A. Immunoregulation of cutaneous leishmaniasis. T cell lines that transfer protective immunity or exacerbation belong to different T helper subsets and respond to distinct parasite antigens. J Exp Med. 1988;168:1675–1684. doi: 10.1084/jem.168.5.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carl JP, Chung CL, Mayor SEH, Subramanyam JM, Goldman SJ, Sieburth DS, Wolf SF, Schaub RG. Resolution of cutaneous leishmaniasis: interleukin 12 initiates a protective T helper type 1 immune response. J Exp Med. 1993;177:1797–1802. doi: 10.1084/jem.177.6.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 12.Katz JD, Benoist C, Mathis D. T helper cell subsets in insulin-dependent diabetes. Science. 1995;268:1185–1188. doi: 10.1126/science.7761837. [DOI] [PubMed] [Google Scholar]
- 13.Dorman SE, Picard C, Lammas D, Heyne K, van Dissel JT, Baretto R, Rosenzweig SD, Newport M, Levin M, Roesler J, Kumararatne D, Casanova JL, Holland SM. Clinical features of dominant and recessive interferon gamma receptor 1 deficiencies. Lancet. 2004;364:2113–2121. doi: 10.1016/S0140-6736(04)17552-1. [DOI] [PubMed] [Google Scholar]
- 14.Oestreich KJ, Weinmann AS. Transcriptional mechanisms that regulate T helper 1 cell differentiation. Current Opinion in Immunology. 2012;24:191–195. doi: 10.1016/j.coi.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang F, Boothby M. T helper type 1–specific Brg1 recruitment and remodeling of nucleosomes positioned at the IFN-γ promoter are Stat4 dependent. J Exp Med. 2006;203:1493–1505. doi: 10.1084/jem.20060066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Avni O, Lee D, Macian F, Szabo SJ, Glimcher LH, Rao A. TH cell differentiation is accompanied by dynamic changes in histone acetylation of cytokine genes. Nature Immunology. 2002;3:643–651. doi: 10.1038/ni808. [DOI] [PubMed] [Google Scholar]
- 17.Svetic A, Madden KB, Zhou XD, Lu P, Katona IM, Finkelman FD, Urban JF, Gause WC. A primary intestinal helminthic infection rapidly induces a gut-associated elevation of Th2-associated cytokines and IL-3. J Immunol. 1993;150:3434–3441. [PubMed] [Google Scholar]
- 18.Kuperman D, Schofield B, Wills-Karp M, Grusby MJ. Signal Transducer and Activator of Transcription Factor 6 (Stat6)-deficient mice are protected from antigen-induced airway hyperresponsiveness and mucus production. J Exp Med. 1998;187:939–948. doi: 10.1084/jem.187.6.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hou J, Schindler U, Henzel WJ, Ho TC, Brasseur M, McKnight SL. An interleukin-4-induced transcription factor: IL-4 Stat. Science. 1994;265:1701–1706. doi: 10.1126/science.8085155. [DOI] [PubMed] [Google Scholar]
- 20.Kubo M, Ransom J, Webb D, Hashimoto Y, Tada T, Nakayama T. T-cell subset-specific expression of the IL-4 gene is regulated by a silencer element and STAT6. EMBO J. 1997;16:4007–4020. doi: 10.1093/emboj/16.13.4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 22.Zhu J, Min B, Hu-Li J, Watson CJ, Grinberg A, Wang Q, Killeen N, Urban JF, Guo L, Paul WE. Conditional deletion of Gata3 shows its essential function in TH1-TH2 responses. Nature Immunology. 2004;5:1157–1165. doi: 10.1038/ni1128. [DOI] [PubMed] [Google Scholar]
- 23.Grogan JL, Mohrs M, Harmon B, Lacy DA, Sedat JW, Locksley RM. early transcription and silencing of cytokine genes underlie polarization of T helper cell subsets. Immunity. 2001;14:205–215. doi: 10.1016/s1074-7613(01)00103-0. [DOI] [PubMed] [Google Scholar]
- 24.Ouyang W, Ranganath SH, Weindel K, Bhattacharya D, Murphy TL, Sha WC, Murphy KM. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity. 1998;9:745–755. doi: 10.1016/s1074-7613(00)80671-8. [DOI] [PubMed] [Google Scholar]
- 25.Usui T, Preiss JC, Kanno Y, Yao ZJ, Bream JH, O’Shea JJ, Strober W. T-bet regulates Th1 responses through essential effects on GATA-3 function rather than on IFNG gene acetylation and transcription. J Exp Med. 2006;203:755–766. doi: 10.1084/jem.20052165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Young HA, Ghosh P, Ye J, Lederer J, Lichtman A, Gerard JR, Penix L, Wilson CB, Melvin AJ, McGurn ME. differentiation of the T helper phenotypes by analysis of the methylation state of the IFN-gamma gene. J Immunol. 1994;153:3603–3610. [PubMed] [Google Scholar]
- 27.Yano S, Ghosh P, Kusaba H, Buchholz M, Longo DL. Effect of promoter methylation on the regulation of IFN-γ gene during in vitro differentiation of human peripheral blood T cells into a Th2 population. J Immunol. 2003;171:2510–2516. doi: 10.4049/jimmunol.171.5.2510. [DOI] [PubMed] [Google Scholar]
- 28.Winders BR, Schwartz RH, Bruniquel D. A distinct region of the murine IFN-γ promoter is hypomethylated from early T cell development through mature naive and Th1 cell differentiation, but is hypermethylated in Th2 cells. J Immunol. 2004;173:7377–7384. doi: 10.4049/jimmunol.173.12.7377. [DOI] [PubMed] [Google Scholar]
- 29.Jones B, Chen J. Inhibition of IFN-gamma transcription by site-specific methylation during T helper cell development. EMBO J. 2006;25:2443–2452. doi: 10.1038/sj.emboj.7601148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cerottini JC, MacDonald HR. The cellular basis of T-cell memory. Annu Rev Immunol. 1989;7:77–89. doi: 10.1146/annurev.iy.07.040189.000453. [DOI] [PubMed] [Google Scholar]
- 31.Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 2002;111:837–851. doi: 10.1016/s0092-8674(02)01139-x. [DOI] [PubMed] [Google Scholar]
- 32.Himmelrich H, Parra-Lopez C, Tacchini-Cottier F, Louis JA, Launois P. The IL-4 rapidly produced in BALB/c mice after infection with Leishmania major down-regulates IL-12 receptor β2-chain expression on CD4+ T cells resulting in a state of unresponsiveness to IL-12. J Immunol. 1998;161:6156–6163. [PubMed] [Google Scholar]
- 33.Swain SL. Generation and in vivo persistence of polarized Th1 and Th2 memory cells. Immunity. 1994;1:543–552. doi: 10.1016/1074-7613(94)90044-2. [DOI] [PubMed] [Google Scholar]
- 34.London CA, Lodge MP, Abbas AK. Functional responses and costimulator dependence of memory CD4+ T Cells. J Immunol. 2000;164:265–272. doi: 10.4049/jimmunol.164.1.265. [DOI] [PubMed] [Google Scholar]
- 35.Adeeku E, Gudapati P, Mendez-Fernandez Y, Van Kaer L, Boothby M. Flexibility accompanies commitment of memory CD4 lymphocytes derived from IL-4 locus-activated precursors. Proc Natl Acad Sci U S A. 2008;105:9307–9312. doi: 10.1073/pnas.0704807105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Löhning M, Hegazy AN, Pinschewer DD, Busse D, Lang KS, Höfer T, Radbruch A, Zinkernagel RM, Hengartner H. Long-lived virus-reactive memory T cells generated from purified cytokine-secreting T helper type 1 and type 2 effectors. J Exp Med. 2008;205:53–61. doi: 10.1084/jem.20071855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krawczyk CM, Shen H, Pearce EJ. Functional plasticity in memory T helper cell responses. J Immunol. 2007;178:4080–4088. doi: 10.4049/jimmunol.178.7.4080. [DOI] [PubMed] [Google Scholar]
- 38.Hegazy AN, Peine M, Helmstetter C, Panse I, Fröhlich A, Bergthaler A, Flatz L, Pinschewer DD, Radbruch A, Löhning M. Interferons direct Th2 cell reprogramming to generate a stable GATA-3+T-bet+ cell subset with combined Th2 and Th1 cell functions. Immunity. 2010;32:116–128. doi: 10.1016/j.immuni.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 39.Lee K, Gudapati P, Dragovic S, Spencer C, Joyce S, Killeen N, Magnuson MA, Boothby M. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity. 2010;32:743–753. doi: 10.1016/j.immuni.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soutto M, Zhang F, Enerson B, Tong Y, Boothby M, Aune TM. A minimal IFN-γ promoter confers Th1 selective expression. J Immunol. 2002;169:4205–4212. doi: 10.4049/jimmunol.169.8.4205. [DOI] [PubMed] [Google Scholar]
- 41.Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, Turecki G, Delaney A, Varhol R, Thiessen N, Shchors K, Heine VM, Rowitch DH, Xing X, Fiore C, Schillebeeckx M, Jones SJM, Haussler D, Marra MA, Hirst M, Wang T, Costello JF. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deaton AM, Webb S, Kerr ARW, Illingworth RS, Guy J, Andrews R, Bird A. Cell type–specific DNA methylation at intragenic CpG islands in the immune system. Genome Res. 2011;21:1074–1086. doi: 10.1101/gr.118703.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bix M, Locksley RM. Independent and epigenetic regulation of the Interleukin-4 alleles in CD4+ T cells. Science. 1998;281:1352–1354. doi: 10.1126/science.281.5381.1352. [DOI] [PubMed] [Google Scholar]
- 44.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in Th1 lineage commitment and IFN-γ production in CD4 and CD8 T cells. Science. 2002;295:338–342. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 45.Yoo JK, Cho JH, Lee SW, Sung YC. IL-12 provides proliferation and survival signals to murine CD4+ T Cells through phosphatidylinositol 3-Kinase/Akt signaling pathway. J Immunol. 2002;169:3637–3643. doi: 10.4049/jimmunol.169.7.3637. [DOI] [PubMed] [Google Scholar]
- 46.Rao RR, Li Q, Bupp MRG, Shrikant PA. Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8+ T cell differentiation. Immunity. 2012;36:374–387. doi: 10.1016/j.immuni.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murphy KM, Ouyang W, Farrar JD, Yang J, Ranganath S, Asnagli H, Afkarian M, Murphy TL. Signaling and transcription in T helper development. Annual Review of Immunology. 2000;18:451–494. doi: 10.1146/annurev.immunol.18.1.451. [DOI] [PubMed] [Google Scholar]
- 48.Corn RA, Hunter C, Liou HC, Siebenlist U, Boothby MR. Opposing roles for RelB and Bcl-3 in regulation of T-box expressed in T cells, GATA-3, and Th effector differentiation. J Immunol. 2005;175:2102–2110. doi: 10.4049/jimmunol.175.4.2102. [DOI] [PubMed] [Google Scholar]
- 49.White SJ, Underhill GH, Kaplan MH, Kansas GS. Cutting Edge: Differential requirements for Stat4 in expression of glycosyltransferases responsible for selectin ligand formation in Th1 cells. J Immunol. 2001;167:628–631. doi: 10.4049/jimmunol.167.2.628. [DOI] [PubMed] [Google Scholar]
- 50.Schulz EG, Mariani L, Radbruch A, Höfer T. Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-γ and interleukin-12. Immunity. 2009;30:673–683. doi: 10.1016/j.immuni.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 51.Messi M, Giacchetto I, Nagata K, Lanzavecchia A, Natoli G, Sallusto F. Memory and flexibility of cytokine gene expression as separable properties of human TH1 and TH2 lymphocytes. Nature Immunology. 2002;4:78–86. doi: 10.1038/ni872. [DOI] [PubMed] [Google Scholar]
- 52.Sundrud MS, Grill SM, Ni D, Nagata K, Alkan SS, Subramaniam A, Unutmaz D. Genetic Reprogramming of Primary Human T Cells Reveals Functional Plasticity in Th Cell Differentiation. J Immunol. 2003;171:3542–3549. doi: 10.4049/jimmunol.171.7.3542. [DOI] [PubMed] [Google Scholar]
- 53.Penix LA, Sweetser MT, Weaver WM, Hoeffler JP, Kerppola TK, Wilson CB. The proximal regulatory element of the interferon-γ promoter mediates selective expression in T cells. J Biol Chem. 1996;271:31964–31972. doi: 10.1074/jbc.271.50.31964. [DOI] [PubMed] [Google Scholar]
- 54.Zhang F, Wang DZ, Boothby M, Penix L, Flavell RA, Aune TM. Regulation of the activity of IFN-γ promoter elements during Th cell differentiation. J Immunol. 1998;161:6105–6112. [PubMed] [Google Scholar]
- 55.Cippitelli M, Sica A, Viggiano V, Ye J, Ghosh P, Birrer MJ, Young HA. Negative transcriptional regulation of the interferon-gamma promoter by glucocorticoids and dominant negative mutants of c-Jun. J Biol Chem. 1995;270:12548–12556. doi: 10.1074/jbc.270.21.12548. [DOI] [PubMed] [Google Scholar]
- 56.Samten B, Howard ST, Weis SE, Wu S, Shams H, Townsend JC, Safi H, Barnes PF. Cyclic AMP response element-binding protein positively regulates production of IFN-γ by T cells in response to a microbial pathogen. J Immunol. 2005;174:6357–6363. doi: 10.4049/jimmunol.174.10.6357. [DOI] [PubMed] [Google Scholar]
- 57.Samten B, Townsend JC, Weis SE, Bhoumik A, Klucar P, Shams H, Barnes PF. CREB, ATF, and AP-1 transcription factors regulate IFN-γ secretion by human T cells in response to mycobacterial antigen. J Immunol. 2008;181:2056–2064. doi: 10.4049/jimmunol.181.3.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gamper CJ, Agoston AT, Nelson WG, Powell JD. Identification of DNA methyltransferase 3a as a T cell receptor-induced regulator of Th1 and Th2 differentiation. J Immunol. 2009;183:2267. doi: 10.4049/jimmunol.0802960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thomas RM, Gamper CJ, Ladle BH, Powell JD, Wells AD. De novo DNA methylation is required to restrict T helper lineage plasticity. The Journal of Biological Chemistry. 2012 doi: 10.1074/jbc.M111.312785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robertson KD, Uzvolgyi E, Liang G, Talmadge C, Sumegi J, Gonzales FA, Jones PA. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucl Acids Res. 1999;27:2291–2298. doi: 10.1093/nar/27.11.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Pérez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, Cherry SR, Tsai JH, Tucker SM, Weaver WM, Kelso A, Jaenisch R, Wilson CB. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15:763–774. doi: 10.1016/s1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- 62.Garcia CA, Wang H, Benakanakere MR, Barrett E, Kinane DF, Martin M. c-Jun controls the ability of IL-12 to induce IL-10 production from human memory CD4+ T cells. J Immunol. 2009;183:4475–4482. doi: 10.4049/jimmunol.0901283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fitzpatrick DR, Shirley KM, McDonald LE, Bielefeldt-Ohmann H, Kay GF, Kelso A. Distinct methylation of the Interferon γ (IFN-γ) and interleukin 3 (IL-3) genes in newly activated primary CD8+ T lymphocytes: Regional IFN-γ promoter demethylation and mRNA expression are heritable in CD44highCD8+ T cells. J Exp Med. 1998;188:103–117. doi: 10.1084/jem.188.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fitzpatrick DR, Shirley KM, Kelso A. Cutting Edge: Stable epigenetic inheritance of regional IFN-γ promoter demethylation in CD44highCD8+ T lymphocytes. J Immunol. 1999;162:5053–5057. [PubMed] [Google Scholar]
- 65.Kersh EN, Fitzpatrick DR, Murali-Krishna K, Shires J, Speck SH, Boss JM, Ahmed R. Rapid demethylation of the IFN-γ gene occurs in memory but not naive CD8 T cells. J Immunol. 2006;176:4083–4093. doi: 10.4049/jimmunol.176.7.4083. [DOI] [PubMed] [Google Scholar]
- 66.Bruniquel D, Schwartz RH. Selective, stable demethylation of the interleukin-2 gene enhances transcription by an active process. Nature Immunology. 2003;4:235–240. doi: 10.1038/ni887. [DOI] [PubMed] [Google Scholar]
- 67.Bhutani N, Burns DM, Blau HM. DNA demethylation dynamics. Cell. 2011;146:866–872. doi: 10.1016/j.cell.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.