

Abstract
Breast cancer represents one of the most frequently diagnosed cancers and predominant causes of death in women worldwide. The value of preventive therapy to limit the devastating impact of breast cancer is well established. Various plant triterpenoids and their synthetic analogs have shown significant promise as potent chemopreventive agents in breast cancer. The current study was initiated to investigate mechanism-based chemopreventive potential of a novel synthetic oleanane triterpenoid (methyl-25-hydroxy-3-oxoolean-12-en-28-oate, AMR-Me) against 7,12-dimethylbenz(a)anthracene (DMBA)-initiated rat mammary carcinogenesis, an experimental rodent tumor model that closely resembles human mammary cancer. Rats were orally administered with AMR-Me (0.8, 1.2 and 1.6 mg/kg) three times per week for 18 weeks. Following two weeks of AMR-Me treatment, mammary carcinogenesis was initiated by oral administration of DMBA (50 mg/kg body weight). At the end of the study (16 weeks following DMBA exposure), AMR-Me exhibited a striking inhibition of DMBA-induced mammary tumor incidence, total tumor burden, average tumor weight and reversed histopathological alterations without toxicity. AMR-Me dose-dependently suppressed abnormal cell proliferation, induced apoptosis, up-regulated pro-apoptotic protein Bax and down-regulated anti-apoptotic protein Bcl-2 in mammary tumors. AMR-Me up-regulated the transcriptional levels of Bax, Bad, caspase-3, caspase-7 and poly(ADP-ribose) polymerase and down-regulated Bcl-2. These results clearly demonstrate for the first time that novel triterpenoid AMR-Me exerts chemopreventive efficacy in the classical DMBA model of breast cancer by suppressing abnormal cell proliferation and inducing apoptosis mediated through mitochondrial pro-apoptotic mechanisms. AMR-Me could be developed as a chemopreventive drug to reduce the risk of human breast cancer that remains a devastating disease.
Keywords: Chemoprevention, breast cancer, DMBA, oleanane triterpenoid, cell proliferation, apoptosis
Introduction
Breast cancer represents the most frequently diagnosed cancer and leading cause of cancer deaths in females worldwide. Approximately, 1.4 million new cases of breast cancer and 458,400 deaths in women due to this disease are estimated to have occurred in 2008.1 Highest rates of breast cancer occur in North America, western Europe and Australia, nevertheless, the incidence is increasing in all parts of the globe, especially in the developing countries. The preponderance of breast cancer is sporadic and the risk factors are predominantly linked to estrogen exposure and age. Steroid hormones, particularly estrogen, contribute to the development and progression of mammary gland neoplasia.2 Exposure to environmental xenobiotics during critical phases of early development has been found to play a vital role in breast cancer susceptibility during the latter periods of life.3 Genetic factors, including mutations in BRCA 1 and BRCA 2, contribute to approximately 10% of breast cancer incidence in the western world.4
Although there has been a substantial improvement in the outcome of breast cancer during the past several decades, primarily due to early diagnosis and increased use of hormonal and adjuvant chemotherapies, preventive approaches using pharmacological and other novel agents could be considered as the winning strategy in reducing the morbidity and mortality of breast cancer.5,6 A recently published consensus statement significantly underscores the value of preventive therapy to limit the devastating impact of breast cancer.7 Two selective estrogen receptor modulators, namely tamoxifen and raloxifene, are currently the only drugs approved by the United States Food and Drug Administration for the prevention of breast cancer in women with high risk for the development of the disease. Nevertheless, concern about serious side effects precludes the widespread and long-term use of these medical options.7,8 Hence, development of new breast cancer chemopreventive drugs with acceptable efficacy and toxicity suitable for use for protracted period of time is urgently needed.9
An impressive number of natural products, including phytoconstituents and dietary agents, have been shown to suppress the growth of breast tumor cells through modulation of cell proliferation, cellular differentiation, apoptosis, angiogenesis as well as several signal transduction pathways.10,11 The potential utility of several dietary supplements and natural products in breast cancer prevention has been tested by clinical intervention trials.12 Terpenoids (also known as terpenes or isoprenoids) constitute the largest group of natural compounds found in a variety of fruits, vegetables and medicinal plants. Interestingly, several terpenoids are structurally similar to human hormones. Natural terpenoids (e.g., monoterpenes, diterpenes, triterpenes and tetraterpenes) as well as their synthetic analogs have shown significant promise in the chemoprevention of breast cancer as demonstrated in preclinical studies.13,14
Amoorain (AMR), a triterpene acid (25-hydroxy-3-oxoolean-12-en-28-oic acid, Figure 1A-a) isolated from the stem bark of the Indian medicinal plant Amoora rohituka, exhibited cytostatic as well as cytotoxic effects in human neoplastic hematopoietic and epithelial cells of diverse origin.15 AMR has also been shown to possess growth inhibitory and apoptosis-inducing effects in several mammary carcinoma cells of human origin, namely MCF-7, MCF-7/TH and MDA-MB-468.16–18 In animal studies, AMR significantly inhibited the growth of xenografted SW 620 colon carcinoma in nude mice19 and reduced tumor size in N-methyl-N-nitrosourea-induced mammary adenocarcinoma in rats.20 Since the antitumor activity of plant-derived AMR is relatively weak, new analogs of this triterpenoid have been developed by chemical transformation in an attempt to yield more potent agents. Interestingly, one of the analogs, namely methyl-25-hydroxy-3-oxoolean-12-en-28-oate (amooranin-methyl ester or AMR-Me, Figure 1A-b), has been found to exhibit significant antiproliferative effect against MCF-7 breast cancer cells with superior potency to the parent compound.16 Ancillary studies have confirmed the potent cytotoxic effect of AMR-Me against MCF-7 cells through induction of apoptosis mediated by two different mitogen-activated protein kinase (MAPK) signaling pathways, namely p38 MAPK and c-jun N-terminal kinase (JNK).21 AMR-Me has also been shown to inhibit the growth of human acute lymphoblastic leukemia cells by pro-apoptotic mechanisms and improve the survival of mice bearing Dalton's lymphoma ascites tumor cells.22
Figure 1.

Amoorain compounds, experimental design and animal growth during the entire term of the study. (A) Structures of AMR (a) and AMR-Me (b); (B) Schematic representation of the experimental protocol of mammary carcinogenesis initiated with DMBA. (C) Effect of AMR-Me on body weight gain during DMBA-induced mammary tumorigenesis in rats. Each data point indicates mean±SEM. There were 6–11 animals in various groups. No significant difference in body weights was observed among various rat groups at any time point.
Despite interesting preliminary results on in vitro antitumor effects, mechanism-based chemopreventive potential of AMR-Me has not been tested against an experimentally validated preclinical animal model of human neoplastic disease primarily due to challenges involved in obtaining sufficient amount of this material required for chronic in vivo study. This obstacle has been removed by development of method for the synthesis of pure AMR-Me.23 The discovery of the method for large-scale synthesis of AMR-Me represents a key breakthrough as it eliminates the need of using plant material and hence the dependency on the nature. It also creates the opportunity for animal studies to explore the chemopreventive efficacy of AMR-Me. We report herein, for the first time, the chemopreventive effect of AMR-Me and related mechanisms using 7,12-dimethylbenz(a)anthracene (DMBA)-induced rat mammary carcinogenesis, a classical animal model that closely resembles human breast cancer.24,25
Materials and Methods
Materials
AMR-Me has been synthesized following a method described in elsewhere.23 The purity of this compound was >98% as determined by NMR. DMBA was purchased from Sigma-Aldrich (St. Louis, MO). Paraformaldehyde was obtained from Ted Pella (Redding, CA). Primary antibodies, such as proliferating cell nuclear antigen (PCNA, sc-56), Bax (sc-70407), Bcl-2 (sc-7382), Bcl-xL (SC-8392) and β-actin (sc-47778) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Quick RNA mini Prep kit was obtained from Zymo Research (Irvine, CA) and Verso cDNA synthesis kit was purchased from Thermo Fisher Scientific (Waltham, MA).
Animals and diet
Pathogen-free virgin female Sprague-Dawley rats (~36 days of age) were obtained from Harlan Laboratories (Indianapolis, IN). The animals were housed in a conventional animal facility accredited by the American Association for the Accreditation of Laboratory Animal Care. All animals were acclimatized to standard housing conditions (ambient temperature 22±2 °C, relative humidity 30–50%, and a 12-h dark-light cycle) in plastic cages (3–4 animals/cage) with solid bottom surface covered with special bedding (Cell-Sorb® Plus from Fangman, Cincinnati, OH) for one week before initiation of treatment. The animals had free access to a well-defined, Constant Nutrition® formula basal diet (LabDiet, St. Louis, MO) and drinking water. The entire animal study has been conducted using an animal protocol approved by the Northeast Ohio Medical University Institutional Animal Care and Use Committee (Rootstown, OH).
Experimental design for chemoprevention study
The experimental plan used in this study represents an adaptation of a protocol for DMBA-induced chemical rat mammary carcinogenesis as previously published by Bishayee and colleagues.26 Following an acclimatization period of one week, rats (~ 43 days of age) were randomly divided into six groups of 6–11 animals in each based on a power analysis. Various experimental groups are depicted in Figure 1B. Two groups (groups A and B) were maintained on the basal diet without any further treatment, whereas the remaining four groups (groups C, D, E and F) were fed with AMR-Me through oral gavage three times a week (Monday-Wednesday-Friday). AMR-Me was mixed in polyethylene glycol (Sigma-Aldrich, St. Louis, MO) and administered by gently securing the animal by holding the skin at the back of its head and delivering the test material slowly via an animal feeding needle (Popper and Sons, Inc., New Hyde Park, NY) attached to a 1-ml syringe (Becton Dickinson, Franklin Lakes, NJ). This technique ensured the accurate oral dosing of the test material. Three doses of AMR-Me were used: 0.8 mg/kg (for group C), 1.2 mg/kg (for group D) and 1.6 mg/kg (for groups E and F). These three doses (low, medium and high) represent 40, 60 and 80% of the calculated maximum tolerated dose (MTD) in rats, respectively. The rat MTD was calculated based on the MTD in mice (3 mg/kg/day)21 as well as the mouse-rat interspecies dose conversion scheme.27
Following two week of aforementioned treatment regimen, at ~57 days of age, mammary tumorigenesis was initiated in all animals belonging to groups B, C, D and E by a single administration of DMBA, an established mammary carcinogen, at 50 mg/kg body weight by oral gavage. The DMBA was dissolved in corn oil. This dose of DMBA was chosen so that substantial tumor incidence could be produced but not so high as to overwhelm the potential chemopreventive action of AMR-Me. Oral treatment of rats with AMR-Me in groups C, D, E and F were continued for 16 weeks following the DMBA administration (i.e., a total period of 18 weeks). Food and water intake as well as behavioral patterns were monitored daily and body weights of animals were recorded every alternate week. Animals were palpated along the milk line twice a week starting 4 weeks following DMBA treatment to detect the presence of mammary tumors. All animals were sacrificed at 16 weeks post-DMBA treatment (i.e., 18 weeks following the start of the experiment).
Tissue harvesting and assays
Following an overnight fast, animals from each group were anesthetized between 09:00 and 11:00 h by intramuscular injection of 40–87 mg/kg ketamine and 5–13 mg/kg xylazine. The skin was dissected out to expose mammary tumors. The tumors (approximated spheres) were separated from mammary gland parenchyma, carefully excised, rinsed with phosphate-buffered saline (pH 7.4) to flush out any blood, blotted dry on a paper towel, weighed, and photographed. Each tumor was measured in two perpendicular directions with a vernier caliper to the nearest mm to obtain an average diameter. The representative tumor tissue was harvested and either immediately flash frozen in liquid nitrogen for molecular work or fixed in 4% paraformaldehyde for histopathological and immunohistochemical studies. Later, serial tumor sections (~15-μm thick) were prepared using a microtome. These sections were used for histopathological assessment by hematoxylin and eosin (H&E) staining.
Immunohistochemical assessment
Serial sections of tumor tissue were used for immunohistochemical analysis. Cell proliferation was studied by immunohistochemical detection of PCNA as a proliferation marker following our established technique.28 The analysis of apoptotic cells in tumor sections was performed by using TdT-FragEL™ DNA fragmentation detection assay kit (EMD Biosciences, Inc., San Diego, CA) as we described earlier.28 The protein expression of Bax (apoptosis inducer) and Bcl-2 (apoptosis repressor) were determined by standard techniques.28 The immunohistochemical slides were visualized under a light microscope (BX43, Olympus, Center Valley, PA) and at least 1,000 tumor cells/animal were analyzed. Results were expressed as percentage of immunopositive cells.
Western blot analysis
Total proteins were extracted from tumor tissues with lysis buffer (containing 20 mM HEPES, 350 mM NaCl, 500 mM EDTA, 1mM MgCl2, 20% glycerol, and 1% NP-40), boiled and separated by 4–20% gradient sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The proteins were transferred to polyvinylidene fluoride membranes (Santa Cruz Biotechnology Inc., Santa Cruz, CA), and reacted with primary mouse monoclonal antibodies to Bax, Bcl-2, Bcl-xL and β-actin (dilution 1: 200) and subsequently with anti-mouse secondary antibodies (dilution 1:1,000) conjugated to horseradish peroxidase. The immunoreactions were detected by an enhanced chemiluminescence detection kit (Thermo Scientific, Rockford, IL) and relative intensities of bands on blots were quantitated by autoluminography using a Kodak digital imaging system. Normalization of Western blot was ensured by β-actin.
Reverse transcription-polymerase chain reaction
Total RNA from 20 mg of tumor sample was extracted using Quick RNA mini Prep kit following the vendor's protocol. The expression levels of apoptotic and anti-apoptotic genes were monitored by reverse transcription-polymerase chain reaction (RT-PCR) using the cDNA verso kit with a temperature scale of 42 °C for 30 min for reverse transcription, and 32 cycles of 94 °C for 30 s, 56 °C for 30 s, and 72 °C for 30 s. The RT-PCR was carried out using the primers: BAD-F – 5'-GAGCTGACGTACAGCGTTGA-3', BAD-R – 5'-GGGTAGGGTGTGTGGAAAAC-3'; BAX-F – 5'-AGGGGCCTTTTTGTTACAGG-3', BAX-R – 5'-ACGTCAGCAATCATCCTCTG-3'; CASP3-F – 5'-AGGGTGCTACGATCCACCAGCA-3', CASP3-R – 5'-CCATGGCTCTGCTCCGGCTC-3'; CASP7-F – 5'-GCCATGCCCAGGACAAGCCA-3', CASP7-R – 5'-GCACGCCGGAGGACATGGTT-3'; PARP-F – 5'-CGACACGTTAGCGGAGCGGAC-3', PARP-R – 5'-GCGCCCGCTCTTAGCGTACT - 3'; and GAPDH-F – 5'-AGACAGCCGCATCTTCTTGT-3'; GAPDH-R – 5'-TACTCAGCACCAGCATCACC-3'. These primer sequences were designed utilizing the Primer3 online program and synthesized by Eurofins MWG Operon (Huntsville, AL). The PCR products were analyzed on 1% agarose gel and visualized by ethidium bromide staining.
Statistical analysis
Results are presented as mean±SEM unless indicated otherwise. Significant differences among various groups were detected by one-way ANOVA. Post hoc analysis was performed by the Student-Neuman-Keuls test. The criterion for statistical significance was set at P < 0.05. The commercial software SigmaStat 3.1 (Systat software, Inc., San Jose, CA) was used for all statistical analysis.
Results
General observations
No differences in food and water intakes were observed in the various experimental groups during the entire term of the study. Similarly, no behavioral changes were apparent among different animal groups. The growth of animals was not affected during the entire study by any of the compounds as no significant difference was detected in the body weight between normal and any treated group at any time-point (Figure 1C).
Effect of AMR-Me on DMBA-induced mammary tumorigenesis
While there were no visible mammary tumors in normal (group A) as well as AMR-Me control (group F), macroscopic tumors of various sizes arose from the mammary glands of DMBA-treated groups. Table 1 summarizes tumor incidence, total tumor burden and average tumor weight of DMBA-initiated groups with or without AMR-Me treatment. Oral AMR-Me at a dose of 0.8 mg/kg (group C) exhibited 20% reduction in observable tumor incidence compared to the DMBA control (group B), but the result did not reach the level of statistical significance. A significantly (P<0.05) reduced tumor incidence (54%) was observed in the group that received AMR-Me at a dose of 1.2 mg/kg (group D) or 1.6 mg/kg (group E) as compared to DMBA control (group B). Oral administration of AMR-Me reduced the total cumulative tumor burden (31–99%) in various DMBA-induced groups in a dose-responsive fashion. The average tumor weight was found to be 45–98% smaller in all AMR-Me treatment groups, but a statistically significant (P<0.05) result was observed in groups D and E compared to group B.
Table 1.
Effect of AMR-Me on DMBA-induced mammary tumorigenesis in Sprague-Dawley rats.
| Groups | No. of rats with tumors/total rats | Tumor Incidence (%) | Total tumor burden (g) | Inhibition (%) | Average tumor weight (g) | Inhibition (%) |
|---|---|---|---|---|---|---|
| B. DMBA | 9/11 | 82 | 83.3 | - | 20.8±4.6 | - |
| C. AMR-Me (0.8 mg/kg) + DMBA | 5/8 | 62 | 57.5 | 31 | 11.5±4.9 | 45 |
| D. AMR-Me (1.2 mg/kg) + DMBA | 2/7 | 28a | 9.6 | 88 | 3.2±1.5b | 85 |
| E. AMR-Me (1.6 mg/kg) + DMBA | 2/7 | 28a | 0.8 | 99 | 0.38±0.03b | 98 |
Rats from normal (group A) and AMR-Me (1.6 mg/kg) control group (group E) did not show any visible mammary tumor.
P<0.05 compared with DMBA (group B) by Fisher's exact probability test.
P<0.05 compared with DMBA (group B) by Student's t-test.
In rats treated with DMBA only, most of the tumors were large (Figure 2A). Treatment of rats with AMR-Me at low dose (0.8 mg/kg) reduced the size of tumors in DMBA-exposed animals (Figure 2B). A further decrease in the size of tumor was observed in animals given AMR-Me at medium dose (1.2 mg/kg) (Figure 2C). Interestingly, the tumors from the high dose (1.6 mg/kg) AMR-Me plus DMBA group showed striking reduction in size compared to those from any other DMBA-administered animals (Figure 2D).
Figure 2.

Chemoprevention of DMBA experimental rat mammary carcinogenesis by AMR-Me. Effects of AMR-Me on mammary tumorigenesis (A-D), histopathological indices (E-H), immunohistochemical profiles of Bax (I-L), and Bcl-2 (M-P). The rats were treated with oral AMR-Me (0.8, 1.2 or 1.6 mg/kg; three times a week) 2 weeks prior to and 16 weeks following DMBA administration. All animals were sacrificed 16 weeks following DMBA exposure. The mammary tumors were subjected to morphological observation as well as histopathological (H&E) and immunohistochemical analysis using anti-Bax or anti-Bcl-2 antibody. Arrows indicate cytoplasmic expression of Bax or Bcl-2. Various treatment groups are: (A, E, I and M) DMBA control; (B, F, J and N) AMR-Me (0.8 mg/kg body weight) plus DMBA; (C, G, K and O) AMR-Me (1.2 mg/kg body weight) plus DMBA; and (D, H, L and P) AMR-Me (1.6 mg/kg body weight) plus DMBA. Magnification: × 100 for H&E and × 200 for Bax and Bcl-2.
Effect of AMR-Me on mammary tumor histopathology
The administration of DMBA produced ductal hyperplasia characterized by marked proliferation in the lumen of the mammary duct as evidenced from H&E staining of tumor tissue. The analysis of morphological characteristics of tumor section reveals alteration and enlargement of alveolus with cells arranged in cribriform pattern. The cellular architecture shows uniformly neoplastic ductal epithelial cells with nuclear pleomorphism, marked by nuclear enlargement, hyperchromatinization and clumping of chromatids (Figure 2E). Although the low dose (0.8 mg/kg) of AMR-Me did not modify tumor histopathological features (Figure 2F), a medium (1.2 mg/kg) (Figure 2G) or high (1.6 mg/kg) dose of AMR-Me (Figure 2H) exhibited a moderate and substantial improvement of cellular architecture in tumor tissue, respectively. The latter group showed almost normal ductal and alveolar structure of breast tissue with uniform epithelial cells and no signs of hyperplasia or abnormal proliferation (Figure 2H).
Effects of AMR-Me on tumor cell proliferation and apoptosis
To determine whether AMR-Me affects cell proliferation in mammary tumors induced by DMBA, the expression of PCNA was analyzed by immunohistochemical technique in tumor sections originating from several experimental groups. Tumor samples from DMBA control animals had an abundance of PCNA-positive cells (data not shown), indicating active cell proliferation. Although a marginal reduction in the proliferation of tumor cells was observed in low dose AMR-Me group, a substantial inhibition of cell proliferation in medium and high dose of AMR-Me-treated group indicate antiproliferative potential of this compound. As presented in Figure 3A, the mean PCNA labeling index (LI) was found to be smaller in all AMR-Me-treated animals. Interestingly, a statistically significant (P<0.001) decrease in PCNA LI was observed in medium or high dose AMR-Me group exposed to DMBA compared to DMBA control.
Figure 3.
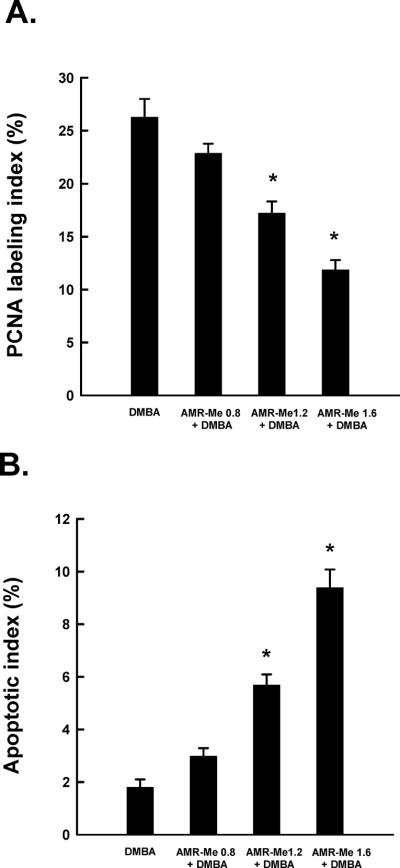
Quantitative analysis of tumor cell proliferation and apoptosis during DMBA mammary carcinogenesis in rats in the presence or absence of AMR-Me. Effects of AMR-Me on intra-tumor PCNA labeling index (LI) as determined by immunohistochemistry (A) and apoptotic index (AI) as measured by DNA fragmentation (B). The LI or AI was expressed as the number of immunopositive cells × 100/total number of cells analyzed. Results are expressed as mean±SEM (n = 4). *P<0.001 compared with DMBA control.
We have used TdT-FragEL™ DNA fragmentation detection assay to investigate the extent of apoptosis in tumor samples. The chromagen-generated brown staining was used to identify apoptotic cells. While the presence of any positive staining was extremely rare in samples from DMBA control or low dose AMR-Me plus DMBA group, we observed a large number of brown staining overlapping the condensed chromatin of apoptotic bodies in medium or high dose AMR-Me plus DMBA group (data not shown). Figure 3B illustrates the apoptotic index (AI) of each experimental group. While we did not notice any difference in this index between two DMBA control and AMR-Me (low dose) plus DMBA group, there was a significant increase (P<0.001) in AI in tumor samples obtained from two experimental groups that received AMR-Me at medium or high dose compared to DMBA control.
Effects of AMR-Me on apoptosis-related gene expressions
To investigate a possible mechanism of apoptosis induction by AMR-Me, the expression of the apoptosis-related proteins, namely Bax and Bcl-2, in mammary tumor sections was studied by immunohistochemical staining. The frequency of Bax-immunopositive cells was extremely low in tumors from DMBA-treated animals (Figure 2I) or low dose of AMR-Me plus DMBA group (Figure 2J). On the other hand, a dose-dependent increase in the expression of Bax was noticeable in the cytoplasm of tumor sections obtained from medium (Figure 2K) or high dose of AMR-Me (Figure 2L). These two doses of AMR-Me significantly (P<0.001) increased Bax-positive cells in DMBA-initiated rats compared to rats exposed to DMBA alone (Figure 4A). Tumor sections from DMBA control animals exhibited substantial expression of cytoplasmic Bcl-2 (Figure 2M) which was not altered by the low dose of AMR-Me (Figure 2N). Other two doses of AMR-Me displayed considerable attenuation of Bcl-2 immunopositivity (Figures 2O and 2P) with almost absence of Bcl-2 positive cells observed in the tumor tissue from animals treated with the highest dose (Figure 2P). The quantitative analysis of Bcl-2-positive cells revealed a significant (P<0.05 or 0.001) reduction in immunopositive cells in tumor samples from rats received medium or high dose of AMR-Me, respectively (Figure 4B). Moreover, these two doses of AMR-Me elevated the Bax/Bcl-2 ratio in a dose-responsive manner (Figure 4C). Nevertheless, a striking result (P<0.001) was obtained with the Bax/Bcl-2 ratio in the group that received the highest dose of AMR-Me compared to DMBA control.
Figure 4.
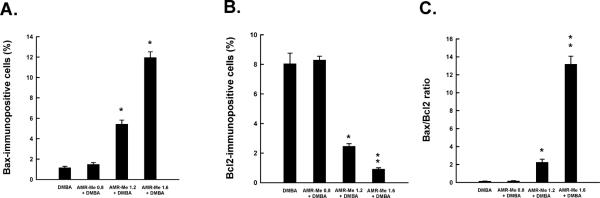
Quantitative analysis of Bax-immunopositive cells (A), Bcl-2-immunopositive cells (B) and Bax/Bcl-2 ratio (C) in mammary tumors induced by DMBA in rats. Each bar represents the mean±SEM (n = 4). (A) *P<0.001 and (B and C) *P<0.05 and **P<0.001 as compared to DMBA control.
Tumor samples harvested from various experimental groups were used to perform Western blotting to confirm our immunohistochemical data on apoptosis-related proteins. As shown in Figure 5A, very limited expression of Bax was noticed in tumors separated from DMBA control animals. Pre- and post-treatment of AMR-Me at all doses elevated the Bax expression. In contrast, a considerable expression of Bcl-2 was observed in tumor tissue originated from DMBA control animals. Although treatment with AMR-Me at low or medium dose did not modify Bcl-2 expression, a complete abrogation in the expression of this protein was achieved with the high dose. Similar results were obtained with another protein, namely Bcl-xL. Figure 5B represents the gene expression data determining the mRNA levels of aforementioned as well as additional apoptosis-regulated genes. A clear up-regulation of BAX, BAD, CASP 3, CASP 7 and PARP and down-regulation of BCL2 represent the salient feature of the gene expression study. All these results explain the pro-apoptotic mechanisms involved in AMR-Me-mediated prevention of mammary tumorigenesis.
Figure 5.

Effects of AMR-Me on the expression of anti- and pro-apoptotic genes in the tumors isolated from rats exposed to DMBA. (A). Representative Western blot analysis of Bax, Bcl-2 and Bcl-xL. Total cellular protein was separated and blotted with specific antibodies. (B) Representative RT-PCR analysis of apoptosis-related genes in various groups of rats. Total RNA was isolated from tumor samples, subjected to reverse transcription, and resulting cDNA was subjected to RT-PCR analysis using specific primer sequence. The GAPDH was used as the housekeeping gene.
Discussion
The DMBA rat mammary tumor is one of the most popular preclinical animal models of breast cancer and has been extensively used in chemopreventive drug development. DMBA is a polycyclic aromatic hydrocarbon (PAH) known to cause mammary tumors in rats. PAHs are common organic environmental pollutants derived from incomplete combustion of fossil fuels and also present in tobacco smoke and various foods. These chemicals represent a predominant class of carcinogens responsible for the development of breast tumor in humans.29 Interestingly, the DMBA-induced rat mammary tumor model was utilized in the development of breast cancer chemopreventive drug tamoxifen30 and has been extensively used by various laboratories worldwide to develop potential breast cancer preventive agents.26,31,32 In this model, mammary tumors can be reproducibly induced with high frequency. This estrogen-dependent breast cancer model is relevant to human cancer especially with respect to origin; both cancers arise from ductal epithelial cells.33 Moreover, histogenesis, morphology and progression of hyperplastic premalignant and malignant lesions are similar to those of human breast cancer.24 In addition, rat mammary tumors induced by DMBA express many biochemical and molecular markers that are also expressed in human mammary tumors.25
Our present study provides substantial evidence for the first time that a novel triterpenoid AMR-Me exerts a remarkable chemopreventive activity against DMBA-induced mammary tumorigenesis in female Sprague-Dawley rats. Interestingly, breast tumor inhibitory effect of AMR-Me has been found to be achieved in a dose-responsive fashion during the 18-week study. While the low dose (0.8 mg/kg) reduces mammary carcinogenesis in statistically insignificant manner, the chemopreventive effect of medium (1.2 mg/kg) or high dose (1.6 mg/kg) has been found to statistically significant and comparable. An inhibitory effect of AMR-Me has been clearly visualized by the reduced incidence of mammary tumors induced by DMBA. The observation of a significant lower percentage of rats with tumors following AMR-Me treatment could be explained in the virtue of the fact that the carcinogenic effect even though initiated was suppressed to a substantial extent. The potential chemopreventive response of AMR-Me has also been reflected in the reduced total tumor burden as well as average tumor weight in DMBA-exposed animals. The observed tumor growth inhibitory effect of AMR-Me may represent a selective toxic manifestation to proliferating cells in the light of the fact that these cells are rapidly proliferating compared to a relatively non-proliferative environment and thereby eventually suppress the development of breast cancer. Our histopathological results reveal hyperplasia in rats exposed to DMBA and its reversal by AMR-Me treatment. Since a close connection between hyperplasia and subsequent steps leading to malignancy has long been established34, the protective effect of AMR-Me against mammary carcinogenesis may be construed as a substantial suppression of the occurrence of hyperplasia thereby reducing the development of mammary tumors. Our in vivo results reported here are in concurrence with previous in vitro data showing selective and dose-dependent inhibitory effects of AMR-Me against human breast cancer cells.21
A prominent finding of this study is the uniform and consistent growth of all experimental animal groups as evident from similar body weight gains during the entire course of the investigation. This is an important aspect of AMR-Me function that suggests unaltered nutritional status of the treated animals. It is well known that dietary restriction as well as nutritional deprivation resulting in body weight loss is implicated in the inhibition of tumor growth.35,36 The ability of dietary and caloric restriction in reducing tumor incidence and arresting tumor growth has been established in various animal models of breast cancer.37,38 Since the animals in this study grew equally irrespective of specific treatment, it may be concluded that the observed mammary tumor inhibitory effect of AMR-Me is not linked to an impairment of nutritional status of DMBA-initiated animals. Our results are in accord with a previous study that reported complete lack of toxicity of orally administered AMR-Me at 3 mg/kg/day for 28 days in female BABL/c mice.21
To investigate the mechanism by which AMR-Me inhibited mammary tumorigenesis, we examined the extent of cell proliferation in tumors isolated from DMBA-exposed rats with or without AMR-Me treatment. Cell proliferation has been used clinically for assessment of tumor prognosis due to its effect on malignancy and for analysis of response of cancer cells to clinical interventions.39 Since cell proliferation plays a vital role in the progression of rat mammary gland tumorigenesis, exploration of agents that can affect tumor cell proliferation has immense value in chemoprevention of breast cancer. PCNA, a 36-kDa nuclear protein with functions as a cofactor of DNA polymerase δ, serves as an important proliferative marker in mammary carcinogenesis.40,41 We have detected the expression of PCNA by immunohistochemical technique. A substantially elevated expression of PCNA in mammary tumors of DMBA control animals is reminiscence of massive proliferation of tumor cells. The reduced expression of PCNA in conjunction with lower PCNA LI in tumor tissues of rats treated with AMR-Me strongly suggests antiproliferative mechanisms involved in the observed chemopreventive efficacy of this novel triterpenoid.
The programmed cell death, also known as apoptosis, is a fundamental form of physiological cell destruction driven by a distinct cellular mechanism characterized by cellular morphological alterations, chromatin condensation, DNA cleavage, and generation of apoptotic bodies. The process of carcinogenesis selects against apoptosis to initiate, promote, and perpetuate the malignant phenotype and drug-induced apoptosis in tumors has been widely accepted as novel therapeutic and preventive strategy against cancer. In the present study, intratumor apoptosis was evaluated by DNA fragmentation assay using immunohistochemical technique to gain insight of the mechanism of suppression of tumor cell proliferation by AMR-Me. Our results demonstrate a gradual increase in DNA fragmentation with increasing doses of AMR-Me indicating an increment in cell death. Incidentally, this is the first study demonstrating apoptosis-inducing activity of AMR-Me in an experimental in vivo breast tumor model and complimentary to previously reported proapoptotic property of this synthetic oleanane triterpenoid against breast carcinoma cells.21
The cellular machinery associated with apoptosis is highly conserved and mutations in genes that regulate apoptosis pathways, including the Bcl-2 family members, are common in human cancers, and underscore the importance of apoptosis resistance in carcinogenesis.42 Several members of the Bcl-2 family which are overexpressed in a number of human malignancies (e.g., Bcl-2 and Bcl-xL) are known to block cell death, whereas other members (including Bax and Bad) are promoters of apoptosis with diminished levels found in several types of cancers.43 In this context, it is noteworthy that apoptosis-inducing ability with an increase in the expression ratio between pro-apoptotic and anti-apoptotic and genes of Bcl-2 family has become a primary factor in considering the efficacy of a chemopreventive agent.28 Based on our immunohistochemical analysis, continuous treatment with AMR-Me increased Bax expression and decreased Bcl-2 expression in mammary tumors with a resultant elevation of Bax/Bcl-2 ratio, providing ample evidence of the involvement of the members of Bcl-2 family in the induction of apoptosis during rat mammary carcinogenesis. Our Western blot and RT-PCR data lend strong support to immunohistochemical observations and confirm Bcl-2 family proteins as the target of AMR-Me during mammary gland carcinogenesis.
It is well known that both pro- and anti-apoptotic members of the Bcl-2 family regulate mitochondrial membrane permealization and thereby control the release of apoptotic factors from the mitochondrial intermembrane space. The release of cyt. c causes downstream initiation of caspase cascade by activating downstream effectors, such as caspase-3 and caspase-7, resulting in cleavage cascade of a variety of cellular proteins to facilitate apoptotic events.44 The long-term treatment of DMBA-exposed rats with AMR-Me has been successful in up-regulating the transcriptional levels of caspase-3, caspase-7 and PARP in mammary tumors which may account for elevated apoptotic activities and eventual inhibition of tumor growth. It is noteworthy to mention that the growth inhibitory and apoptosis-inducing activities of AMR-Me against breast cancer cells has been paralleled with increase in Bax and decrease in Bcl-2, cyt. c release, and subsequent induction of pro-caspase-9 and -7 and PARP cleavage.21
The carcinogenic effect of DMBA, an indirect acting carcinogen, is mediated through its hepatic and extra-hepatic (mammary glands) metabolism catalyzed by phase I enzymes, namely cytochrome P-450 (CYP) and epoxide hydrolase, followed by subsequent epoxidation by CYP isozyme CYP1A1 and CYP1B1 to yield the ultimate carcinogen DMBA-3,4-dihydro-diol-1,2-epoxide capable of forming DNA adducts. The detoxification of DMBA epoxide and other phenolic metabolites is mediated by phase II enzymes, including glutathione S-transferase. Hence, the observed mammary tumor inhibitory action of AMR-Me treatment started 2 weeks prior to DMBA initiation could be effected by alteration of DMBA metabolism through modulation of phase I and phase II enzymes. Nevertheless, future studies are necessary to confirm this and other possible chemopreventive mechanisms of action of AMR-Me.
Several triterpenoid analogues of oleanolic acid, including 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid (CDDO) and methyl ester of CDDO (CDDO-Me), have been synthesized, which are shown to suppress the growth of several human breast carcinoma cells in vitro45–48 and inhibited or suppressed tumor formation in various breast cancer models in vivo46–50 Two recently completed phase I clinical trials evaluated antitumor activities of both CDDO (NCT00322140) and CDDO-Me (NCT00508807) in adult patients with solid tumors and lymphomas (http://clinicaltrials.gov/). Like CDDO-Me, the structure of AMR-Me resemble steroids and other isoprenoid molecules (Figure 1A–b). In view of this background, results derived from the present study showing inhibition of rat mammary tumors through pharmacological modulation of several cellular and molecular regulatory pathways by chemical modification of a naturally occurring triterpenoid scaffold may offer great potential for the prevention of human breast cancer.
In conclusion, the results of our present investigation clearly demonstrate for the first time that the novel triterpenoid AMR-Me exhibits a striking chemopreventive efficacy in DMBA classical animal model of breast cancer in a dose-responsive fashion. The chemopreventive effect of AMR-Me has been reflected in the ability of this compound to diminish the development of DMBA-induced mammary tumors and significantly reduce tumor burden. Furthermore, our study provides substantial evidence that breast tumor-inhibitory effect of AMR-Me could be achieved, at least in part, though interference with key hallmark capabilities of tumor cells, such as abnormal cell proliferation and evasion of apoptosis. Finally, AMR-Me-mediated pro-apoptotic signal during experimentally-induced mammary carcinogenesis could be propagated through an up-regulation of pro-apoptotic proteins and down-regulation of anti-apoptotic proteins of the mitochondrial apoptotic pathway. These interesting results coupled with a safety profile underscore the development of AMR-Me as chemopreventive drug for breast cancer which is a devastating disease.
Novelty and impact.
We investigated mechanism-based chemopreventive potential of a novel synthetic oleanane triterpenoid (methyl amooranin, AMR-Me) against clinically relevant 7,12-dimethylbenz(a)anthracene (DMBA)-initiated rat mammary tumorigenesis. Our results demonstrate for the first time that AMR-Me exerts a striking chemopreventive efficacy in classical DMBA breast cancer model by suppressing abnormal cell proliferation and inducing apoptosis mediated through mitochondrial pro-apoptotic mechanisms. This study may benefit the development of AMR-Me as a chemopreventive drug to reduce the risk of human breast cancer.
Acknowledgments
This work was supported by the award R03CA136014 from the National Cancer Institute (NCI)/National Institutes of Health (NIH). The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of NCI or NIH. A portion of this study was conducted at the Northeast Ohio Medical University (Rootstown, OH). The authors thank Qiwen Shi for assistance with Western blot analysis, and Sharon Reuben for technical support with RT-PCR studies.
Grant sponsor: National Institutes of Health; Grant number: R03CA136014.
Footnotes
Disclosure of Potential Conflicts of Interest No potential conflicts of interest were disclosed.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Henderson BE, Feigelson HS. Hormonal carcinogenesis. Carcinogenesis. 2000;21:427–33. doi: 10.1093/carcin/21.3.427. [DOI] [PubMed] [Google Scholar]
- 3.Lamartiniere CA. Timing of exposure and mammary cancer risk. J Mammary Gland Biol Neoplasia. 2002;7:67–76. doi: 10.1023/a:1015722507237. [DOI] [PubMed] [Google Scholar]
- 4.Lillie SE, Brewer NT, O'Neill SC, Morrill EF, Dees EC, Carey LA, Rimer BK. Retention and use of breast cancer recurrence risk information from genomic tests: the role of health literacy. Cancer Epidemiol Biomarkers Prev. 2007;16:249–55. doi: 10.1158/1055-9965.EPI-06-0525. [DOI] [PubMed] [Google Scholar]
- 5.Kelloff GJ, Lippman SM, Dannenberg AJ, Sigman CC, Pearce HL, Reid BJ, Szabo E, Jordan VC, Spitz MR, Mills GB, Papadimitrakopoulou VA, Lotan R, et al. Progress in chemoprevention drug development: the promise of molecular biomarkers for prevention of intraepithelial neoplasia and cancer – a plan to move forward. Clin Cancer Res. 2006;12:3661–97. doi: 10.1158/1078-0432.CCR-06-1104. [DOI] [PubMed] [Google Scholar]
- 6.Jordan VC. Chemoprevention of breast cancer with selective oestrogen-receptor modulators. Nat Rev Cancer. 2007;7:46–53. doi: 10.1038/nrc2048. [DOI] [PubMed] [Google Scholar]
- 7.Cuzick J, DeCensi A, Arun B, Brown PH, Castiglione M, Dunn B, Forbes JF, Glaus A, Howell A, von Minckwitz G, Vogel V, Zwierzina H. Preventive therapy for breast cancer: a consensus statement. Lancet Oncol. 2011;12:496–503. doi: 10.1016/S1470-2045(11)70030-4. [DOI] [PubMed] [Google Scholar]
- 8.Bozovic-Spasojevic I, Azambuja E, McCaskill-Stevens W, Dinh P, Cardoso F. Chemoprevention of breast cancer. Cancer Treat Rev. 2012;38:329–39. doi: 10.1016/j.ctrv.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 9.Uray IP, Brown PH. Chemoprevention of hormone receptor-negative breast cancer: new approaches needed. Recent Results Cancer Res. 2011;188:147–62. doi: 10.1007/978-3-642-10858-7_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reuben SC, Gopalan A, Petit DM, Bishayee A. Modulation of angiogenesis by dietary phytoconstituents in the prevention and intervention of breast cancer. Mol Nutr Food Res. 2012;56:14–29. doi: 10.1002/mnfr.201100619. [DOI] [PubMed] [Google Scholar]
- 11.Vadodkar AS, Suman S, Lakshmanaswamy R, Damodaran C. Chemoprevention of breast cancer by dietary compounds. Anticancer Agents Med Chem. 2012 doi: 10.2174/187152012803833008. in press. PMID:22583403. [DOI] [PubMed] [Google Scholar]
- 12.Kado K, Forsyth A, Patel PR, Schwartz JA. Dietary supplements and natural products in breast cancer trials. Front Biosci. 2012;4:546–67. doi: 10.2741/399. [DOI] [PubMed] [Google Scholar]
- 13.Rabi T, Bishayee A. Terpenoids and breast cancer chemoprevention. Breast Cancer Res Treat. 2009;115:223–39. doi: 10.1007/s10549-008-0118-y. [DOI] [PubMed] [Google Scholar]
- 14.Bishayee A, Ahmed S, Brankov N, Perloff M. Triterpenoids as potential agents for the chemoprevention and therapy of breast cancer. Front Biosci. 2011;16:980–96. doi: 10.2741/3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramachandran C, Rabi T, Fonseca HB, Melnick SJ, Escalon EA. Novel plant triterpenoid drug amooranin overcomes multidrug resistance in human leukemia and colon carcinoma cell lines. Int J Cancer. 2003;105:784–9. doi: 10.1002/ijc.11180. [DOI] [PubMed] [Google Scholar]
- 16.Rabi T, Karunagaram D, Nair MK, Bhattathiri VN. Cytotoxic activity of amooranin and its derivatives. Phytother Res. 2002;16:S84–S6. doi: 10.1002/ptr.803. [DOI] [PubMed] [Google Scholar]
- 17.Rabi T, Ramachandran C, Fonseca HB, Nair RP, Alamo A, Melnick SJ, Escalon E. Novel drug amooranin induces apoptosis through caspase activity in human breast carcinoma cell lines. Breast Cancer Res Treat. 2003;80:321–30. doi: 10.1023/A:1024911925623. [DOI] [PubMed] [Google Scholar]
- 18.Rabi T, Wang L, Banerjee S. Novel triterpenoid 25-hydroxy-3-oxoolean-12-en-28-oic acid induces growth arrest and apoptosis in breast cancer cells. Breast Cancer Res Treat. 2007;101:27–36. doi: 10.1007/s10549-006-9275-z. [DOI] [PubMed] [Google Scholar]
- 19.Ramachandran C, Nair PK, Alamo A, Cochrane CB, Escalon E, Melnick SJ. Anticancer effects of amooranin in human colon carcinoma cell line in vitro and in nude mice xenografts. Int J Cancer. 2006;119:2443–54. doi: 10.1002/ijc.22174. [DOI] [PubMed] [Google Scholar]
- 20.Rabi T. Antitumor activity of amooranin from Amoora rohituka stem bark. Curr Sci. 1996;70:80–1. [Google Scholar]
- 21.Rabi T, Banerjee S. Novel synthetic triterpenoid methyl 25-hydroxy-3-oxoolean-12-en-28-oate induces apoptosis through JNK and p38 MAPK pathways in human breast adenocarcinoma MCF-7 cells. Mol Carcinog. 2008;47:415–23. doi: 10.1002/mc.20399. [DOI] [PubMed] [Google Scholar]
- 22.Rabi T, Banerjee S. Novel semisynthetic triterpenoid AMR-Me inhibits telomerase activity in human leukemic CEM cells and exhibits in vivo antitumor activity against Dalton's lymphoma ascites tumor. Cancer Lett. 2009;278:156–63. doi: 10.1016/j.canlet.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 23.Bishayee A, Martirosian A, Yeranosyan A. Amooranin compounds and analogs thereof, and related methods of use. US Provisional Patent Application No. 61/580,449, 2011
- 24.Witty JP, Cempka T, Coffe RJ, Jr, Matrisian LM. Decreases tumor formation in 7,12-dimethylbenz(a)anthracene-treated stromelysin-1 transgenic mice is associated with alterations in mammary epithelial cells apoptosis. Cancer Res. 1995;55:1401–6. [PubMed] [Google Scholar]
- 25.Costa I, Solanas M, Escrich E. Histopathological characterization of mammary neoplastic lesions induced with 7,12-dimethylbenz(a)anthracene in the rat: a comparative analysis with human breast tumors. Arch Pathol Lab Med. 2002;126:915–27. doi: 10.5858/2002-126-0915-HCOMNL. [DOI] [PubMed] [Google Scholar]
- 26.Bishayee A, Oinam S, Basu M, Chatterjee M. Vanadium chemoprevention of 7,12-dimethylbenz(a)anthracene-induced rat mammary carcinogenesis: probable involvement of representative hepatic phase I and II xenobiotic metabolizing enzymes. Breast Cancer Res Treat. 2000;63:133–45. doi: 10.1023/a:1006476003685. [DOI] [PubMed] [Google Scholar]
- 27.Morris TH. Dose estimation among species. In: Hawk CT, Leary SL, editors. Formulary for Laboratory Animals. Iowa State University Press; Ames: 1999. pp. 3–14. [Google Scholar]
- 28.Bishayee A, Dhir N. Resveratrol-mediated chemoprevention of diethylnitrosamine-initiated hepatocarcinogenesis: inhibition of cell proliferation and induction of apoptosis. Chem Biol Interact. 2009;179:131–44. doi: 10.1016/j.cbi.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 29.Tsuchiya Y, Nakajima M, Yokoi T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005;227:115–24. doi: 10.1016/j.canlet.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 30.Jordan VC, Morrow M. Tamoxifen, raloxifene, and the prevention of breast cancer. Endocr Rev. 1999;20:253–78. doi: 10.1210/edrv.20.3.0368. [DOI] [PubMed] [Google Scholar]
- 31.Brown NM, Belles CA, Lindley SL, Zimmer-Nechemias LD, Zhao X, Witte DP, Kim MO, Setchell KD. The chemopreventive action of equol enantiomers in a chemically induced animal model of breast cancer. Carcinogenesis. 2010;31:886–93. doi: 10.1093/carcin/bgq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamdy SM, Latif AK, Drees EA, Soliman SM. Prevention of rat breast cancer by genistein and selenium. Toxicol Ind Health. 2012;28:746–57. doi: 10.1177/0748233711422732. [DOI] [PubMed] [Google Scholar]
- 33.Russo J, Gusterson BA, Rogers AE, Russo IH, Wellings SR, van Zwieten MJ. Comparative study of human and rat mammary tumorigenesis. Lab Invest. 1990;62:244–78. [PubMed] [Google Scholar]
- 34.Farber E. Clonal adaptation carcinogenesis. Biochem Pharmacol. 1990;39:1837–46. doi: 10.1016/0006-2952(90)90599-g. [DOI] [PubMed] [Google Scholar]
- 35.Waitzberg DL, Gonçalves EL, Faintuch J, Bevilacqua LR, Rocha CL, Cologni AM. Effect of diets with different protein levels on the growth of Walker 256 carcinosarcoma in rats. Braz J Med Biol Res. 1989;22:447–55. [PubMed] [Google Scholar]
- 36.Pollack M. Do cancer cells care if their host is hungry? Cell Metab. 2009;9:401–3. doi: 10.1016/j.cmet.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 37.Macrae FA. Fat and calories in colon and breast cancer: from animal studies to controlled clinical trials. Prev Med. 1993;22:750–66. doi: 10.1006/pmed.1993.1069. [DOI] [PubMed] [Google Scholar]
- 38.Rogozina OP, Bonorden MJ, Grande JP, Cleary MP. Serum insulin-like growth factor-I and mammary tumor development in ad libitum-fed, chronic calorie-restricted, and intermittent calorie-restricted MMTV-TGF-alpha mice. Cancer Prev Res. 2009;2:712–9. doi: 10.1158/1940-6207.CAPR-09-0028. [DOI] [PubMed] [Google Scholar]
- 39.Christov K, Grubbs C, Shikaitis A, Juliana MM, Lubet RA. Short-term modulation of cell proliferation and apoptosis and preventive/therapeutic efficacy of various agents in a mammary cancer model. Clin Cancer Res. 2007;13:5488–96. doi: 10.1158/1078-0432.CCR-07-0404. [DOI] [PubMed] [Google Scholar]
- 40.Leonardi E, Girlando S, Serio G, Mauri FA, Perrone G, Scampini S, Dalla Palma P, Barbareschi M. PCNA and Ki67 expression in breast carcinoma: correlations with clinical and biological variables. J Clin Pathol. 1992;45:416–9. doi: 10.1136/jcp.45.5.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Al-Dhaheri WS, Hassouna I, Al-Salam S, Karam SM. Characterization of breast cancer progression in the rat. Ann N Y Acad Sci. 2008;1138:121–31. doi: 10.1196/annals.1414.018. [DOI] [PubMed] [Google Scholar]
- 42.Hockenbery D, Nunez G, Milliman C. Bcl2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348:334–6. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- 43.Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci. 2009;122:437–41. doi: 10.1242/jcs.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen M, Wang J. Initiator caspases in apoptosis signaling pathways. Apoptosis. 2002;7:313–9. doi: 10.1023/a:1016167228059. [DOI] [PubMed] [Google Scholar]
- 45.Place AE, Suh N, Williams CR, Risingsong R, Honda T, Honda Y, Gribble GW, Leesnitzer LM, Stimmel JB, Willson TM, Rosen E, Sporn MB. The novel synthetic triterpenoid, CDDO-imidazolide, inhibits inflammatory response and tumor growth in vivo. Clin Cancer Res. 2003;9:2798–806. [PubMed] [Google Scholar]
- 46.Lapillonne H, Konopleva M, Tsao T, Gold D, McQueen T, Sutherland RL, Madden T, Andreeff M. Activation of peroxisome proliferator-activated receptor γ by a novel synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid induces growth arrest and apoptosis in breast cancer cells. Cancer Res. 2003;63:5926–39. [PubMed] [Google Scholar]
- 47.Konopleva M, Zhang W, Shi YX, McQueen T, Tsao T, Abdelrahim M, Munsell MF, Johansen M, Yu D, Madden T, Safe SH, Hung MC, et al. Synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid induces growth arrest in HER2-overexpressing breast cancer cells. Mol Cancer Ther. 2006;5:317–28. doi: 10.1158/1535-7163.MCT-05-0350. [DOI] [PubMed] [Google Scholar]
- 48.Ling X, Konopleva M, Zeng Z, Ruvolo V, Stephens LC, Schober W, McQueen T, Dietrich M, Madden TL, Andreeff M. The novel triterpenoid C-28 methyl ester of 2-cyano-3, 12-dioxoolen-1, 9-dien-28-oic acid inhibits metastatic murine breast tumor growth through inactivation of STAT3 signaling. Cancer Res. 2007;67:4210–8. doi: 10.1158/0008-5472.CAN-06-3629. [DOI] [PubMed] [Google Scholar]
- 49.Liby K, Risingsong R, Royce DB, Williams CR, Yore MM, Honda T, Gribble GW, Lamph WW, Vannini N, Sogno I, Albini A, Sporn MB. Prevention and treatment of experimental estrogen receptor-negative mammary carcinogenesis by the synthetic triterpenoid CDDO-methyl ester and the rexinoid LG100268. Clin Cancer Res. 2008;14:4556–63. doi: 10.1158/1078-0432.CCR-08-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim EH, Deng C, Sporn MB, Royce DB, Risingsong R, Williams CR, Liby KT. CDDO-methyl ester delays breast cancer development in BRCA1-mutated mice. Cancer Prev Res. 2012;5:89–97. doi: 10.1158/1940-6207.CAPR-11-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]