

Abstract
Decorin, a small leucine-rich proteoglycan (SLRP), is involved in the pathophysiology of human congenital stromal corneal dystrophy (CSCD). This disease is characterized by corneal opacities and vision impairment. In reported cases, the human gene encoding decorin contains point mutations in exon 10, generating a truncated form of decorin lacking the C-terminal 33 amino acid residues. We have previously described a transgenic mouse model carrying a similar mutation in the decorin gene that leads to an ocular phenotype characterized by corneal opacities identical to CSCD in humans. We have also identified abnormal synthesis and secretion of various SLRPs in mutant mouse corneas. In the present study, we found that mutant C-terminal truncated decorin was retained in the cytoplasm of mouse keratocytes in vivo and of transfected human embryonic kidney cells. This resulted in endoplasmic reticulum stress and an unfolded protein response. Thus, we propose a novel cell-based mechanism underlying CSCD in which a truncated SLRP protein core is retained intracellularly, its accumulation triggering endoplasmic reticulum stress that results in abnormal SLRP synthesis and secretion, which ultimately affects stromal structure and corneal transparency.
Decorin is a multifunctional small leucine–rich proteoglycan (SLRP) that interacts with collagen fibrils and regulates fibrillogenesis in extracellular matrix assembly. It also interacts with a variety of growth factors and receptors and is involved in pathologic and physiologic processes such as fibrosis, tumor growth, and cell adhesion.1–5 Human congenital stromal corneal dystrophy (CSCD) is the only known human disease associated with a mutated decorin gene. Three different frameshift mutations have been reported, all leading to identical truncation of the C-terminal 33 amino acids of decorin.6–8 Decorin is an important regulator of matrix assembly in many connective tissues such as the cornea, sclera, and tendon.1 However, the only clinical manifestation of autosomal dominant human CSCD is a corneal stromal phenotype,9 which indicates that truncation interferes with corneal stromal assembly in a tissue-specific manner.
A transgenic mouse model (952delTDcn) faithfully recapitulates the phenotype of human CSCD.10 The mouse model exhibits opaque corneas with a similarly disrupted corneal stromal structure and abnormal keratocyte architecture, as observed in the disease in humans.6,10 In addition to the structural defects in the mouse model, altered expression of stromal extracellular matrix components, in particular, expression of SLRPs, is observed. The changes in these SLRPs are consistent with dysregulated fibrillogenesis and fibril packing, resulting in disrupted corneal stromal architecture and function. The mechanism by which C-terminal truncation of the decorin protein core alters SLRP expression is unknown.
Decorin, the most studied class I SLRP, has 12 leucine-rich repeats (LRRs) in the central domain, flanked by conserved cysteine-rich domains on either side.2,11–13 Soluble decorin as a monomer in solution binds and modulates various receptors and growth factors through different domains.14 The central LRR5-6 contain key binding sites for collagen I,15 the ectodomain of epidermal growth factor receptor,16 and low-density lipoprotein receptor–related protein 1.17 LRR12 at the C-terminus can bind to connective tissue growth factor.18 However, that the corneal stromal phenotypes in both the human disease and the 952delTDcn mouse model are distinct from that in the decorin-null mouse model suggests that the effects of the C-terminal truncation are not entirely explained by a loss-of-function mutation.19 It may also function in a dominant negative manner in which the truncated decorin transcribed from the mutant allele competes with the normal decorin transcribed from the normal allele. This could result in abnormal functioning of decorin extracellularly during collagen fibril assembly and/or intracellularly during maturation of the full proteoglycan.
Adjacent to the truncated 33 amino acids at the C-terminus is LRR11, the longest repeat that extends laterally from the main axis of the decorin protein core and is referred to as the ear repeat, a characteristic feature of SLRPs. The ear repeat is thought to participate in the protein folding of decorin and may also contribute to ligand recognition.20 Herein we demonstrate that absence of the C-terminal 33 amino acids from the decorin protein core leads to a misfolded and unstable/insoluble protein, indicating a pathogenic involvement of the ear repeat. Moreover, our findings implicate for the first time decorin-evoked endoplasmic reticulum (ER) stress that leads to the unfolded protein response. Thus, we propose a novel cell-based mechanism for the observed corneal opacity in which a truncated SLRP protein core is retained intracellularly, its accumulation inducing ER stress and resulting in abnormal SLRP synthesis and secretion, which ultimately affects stromal assembly and corneal transparency.
Materials and Methods
Animals
952delTDcn transgenic mice10 in Dcn+/− and Dcn−/− background21 and wild-type (WT) control mice were used in the present study. All animal studies were performed in compliance with animal protocols of the University of South Florida Institutional Animal Care and Use Committee.
Transmission Electron Microscopy
Corneal samples from WT and 952delTDcn transgenic mice were analyzed via transmission electron microscopy as previously described.22 In brief, three corneas per group were dissected and fixed in 4% paraformaldehyde, 2.5% glutaraldehyde, 0.1 mol/L sodium cacodylate (pH 7.4), and 8.0 mmol/L CaCl2 and were postfixed using 1% OsO4. The corneas were dehydrated in graded ethanol followed by propylene oxide. The tissue samples were infiltrated and embedded in a mixture of EMbed 812, DMP-30 (both from Leica Microsystems, Inc., Buffalo Grove, IL), nadicmethyl anhydride, and dodecenyl succinic anhydride. Thin sections, 80 nm, were cut using an ultramicrotome (Leica Microsystems, Inc., Buffalo Grove, IL) and poststained with 2% aqueous uranyl acetate and 1% phosphotungstic acid (pH 3.2). The sections were examined at 80 kV using a transmission electron microscope (JEOL 1400; JEOL USA, Inc., Peabody, MA) equipped with a digital camera (Ultrascan US1000 2K; Gatan, Inc., Warrendale, PA). For cell culture, HEK293 cells were grown on coverslips in 12-well plates and transfected with decorin or 952delT decorin. At 48 hours after transfection, cells were fixed in 4% paraformaldehyde, 2.5% glutaraldehyde, and 0.1 mol/L sodium cacodylate (pH 7.4), with 8.0 mmol/L CaCl2, and processed as described.23
Cell Culture and Reagents
A human embryonic kidney cell line (HEK293T) was purchased from ATCC (Manassas, VA). The cell line was maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 1× antibiotic-antimycotic solution, and 5 μg/mL gentamicin (Life Technologies Corp., Grand Island, NY) at 37°C in a humidified atmosphere containing 5% CO2/95% air. The cells were treated with different reagents, including 2.5 mmol/L MG132, 20 mmol/L NH4Cl, 10 μg/mL cycloheximide, and 2.5 μmol/L methyl-β-cyclodextrin (all from Sigma, St. Louis, MO), to disrupt homeostasis of cellular proteins in cell culture.
Plasmid Construction and Transfection
A full-length mouse decorin cDNA24 was used as the template for generating the mutant mouse decorin cDNAs (952 delT, 926 delG, and 932 delG), using a QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene Corp., La Jolla, CA). WT decorin cDNA and mutant mouse decorin cDNAs were individually cloned into the appropriate restriction sites of pcDNA3 vectors (Invitrogen Corp., Carlsbad, CA). All plasmids were verified via gene sequencing. HEK293T cells were seeded on mouse collagen I–coated coverslips in 6- or 12-well plates. Transfections of HEK293T cells were performed according to the manufacturer's protocol using Lipofectamine 2000 dissolved in Opti-MEM I (both from Invitrogen), which was replaced with serum-free Dulbecco's modified Eagle's medium at 6 hours after transfection. At 60 hours after transfection, the medium and cell layers were harvested. To study the turnover of intracellular decorin, the cells were trypsinized, rinsed with PBS, and collected at different times after treatment with cycloheximide or cycloheximide plus MG132. A cotransfected plasmid containing green fluorescent protein was used as transfection control.
mRNA Analysis
Total RNA was isolated from transfected HEK293T cells using Ribopure TM kits (Ambion, Inc., Austin, TX). Independent cDNA preparations from different transfections were obtained via reverse transcription of a series dilution of RNA with random primers (High-Capacity cDNA Archive Kit; Applied Biosystems, Inc., Foster City, CA). Real-time RT-PCR was performed using the StepOnePlus system with the SYBR Green PCR Master Mix (both from Applied Biosystems). The primer sequences for decorin were as follows: forward primer, 5′-TGAGCTTCAACAGCATCACC-3′; reverse primer, 5′-AAGTCATTTTGCCCAACTGC-3′. Actin was used as an endogenous control to standardize the amount of sample RNA. The PCR parameters were 95°C for 20 seconds followed by 40 cycles at 95°C for 3 seconds and 60°C for 30 seconds.
Antibodies, Immunoblots, and Phos-Tag Gels
Rabbit anti-decorin LF113; provided by Dr. Larry Fisher (NIH, National Institute of Dental and Craniofacial Research, Bethesda, MD)25 was used at 1:500 dilution, rabbit anti-BiP, anti-PERK, and mouse anti-CHOP antibodies (Cell Signaling Technology, Inc., Danvers, MA) were used at 1:200 dilution, and rabbit anti–green fluorescent protein (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) was used at 1:500 dilution. Actin reactivity in each sample was detected using an anti-actin antibody (Chemicon International, Temecula, CA) at 1:500 dilution. Proteins from cell culture medium or cell layer lysates in radioimmunoprecipitation assay buffer were separated using 4% to 12% Bis-Tris gels (Invitrogen) and transferred to HyBond-C membrane (Amersham Biosiences Corp., Piscataway, NJ) for immunoblotting. Goat anti-rabbit or anti-mouse IgG-peroxidase (Amersham Biosciences) was used as secondary antibody at 1:3000 dilution with an ECL detection system (Pierce Chemical Co., Rockford, IL). To remove O-linked glycosaminoglycan (GAG) chains, serum-free medium from transfected cell cultures was digested with chondroitinase ABC (ChABC) (Seikagaku Corp., Tokyo, Japan) for 24 hours at 37°C. The samples were denatured and digested with peptide-N-glycosidase (PNGase F; ProZyme, Inc., San Leandro, CA) using the manufacturer's protocol to remove the N-glycosylated oligosaccharides from decorin. Phos-tag gels were performed as described for immunoblots, with the exceptions that 7.5% SDS-PAGE containing 50 μmol/L Phos-tag (Wako Chemical USA, Inc., Richmond, VA) and 50 μmol/L MnCl2 (Sigma) were used and the gels were incubated in 1 mmol/L EDTA for 10 minutes before transfer.
Immunofluorescence and Confocal Microscopy
Whole eyes, three to five per group, were embedded in optimal cutting temperature compound (O.C.T.; Tissue-Tek, Sakura Finetek USA Inc., Torrance, CA), frozen on dry ice, and stored at −80°C. Frozen sections, 4 μm, were cut using an HM505E cryostat (Themo Fisher Scientific, Inc., Hanover Park, IL). The slides were blocked with 5% bovine serum albumin in PBS, followed by addition of the primary antibodies. The secondary antibody was Alexa Fluor 568- or 488-conjugated goat anti-rabbit IgG (Molecular Probes, Inc., Eugene, OR) used at 1:100 dilution. Vectashield mounting solution with DAPI (Vector Laboratories, Inc., Burlingame, CA) was used as a nuclear marker. Images were captured using a Leica CTR 5500 microscope and Leica DFC 340 FX camera. Identical conditions and set integration times were used to facilitate comparisons between samples.
For immunocolocalization assays, transfected HEK293T cells seeded on collagen I–coated coverslips were fixed in 4% paraformaldehyde and permeabilized with 0.3% Triton X-100. Mouse monoclonal anti–protein disulfide isomerase and anti–Golgi 97 antibodies (Abcam, Cambridge, MA) were used at 1:200 dilution for immunocolocalization assays. Images were captured using a Leica TCS SP2 laser scanning confocal microscope with a 60 × 1.42 numerical aperture oil immersion lens. Samples were scanned sequentially using 405-, 488-, and 543-nm lasers, and emissions were collected with appropriate spectral slit settings.
Results
Intracellular Localization and Increased Dilated Membranous Organelles in Keratocytes Indicate Retention of Mutant Decorin
Keratocytes are responsible for matrix synthesis and regulation of corneal stromal structure. Localization of mutant decorin was analyzed in the 952delTDcn mouse model of human CSCD with expression in a decorin-null background. The mutant decorin was intracellularly localized in keratocytes. Immunolocalization of mutant decorin revealed reactivity in the keratocyte cytoplasm, whereas reactivity in the extracellular matrix was comparable to that observed in the decorin-null genetic background (Figure 1, B and C). As previously shown,10 WT decorin was localized almost exclusively in the stromal extracellular matrix in the WT corneas (Figure 1A). These data indicate that mutant decorin might not be properly secreted and incorporated into the stromal matrix.
Figure 1.
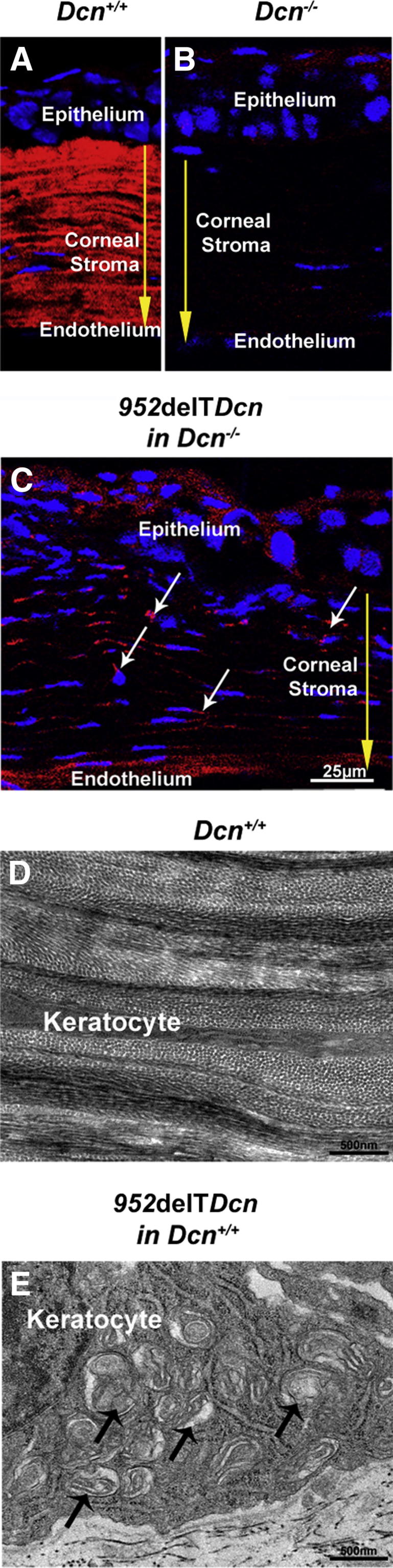
Increased dilated membranous organelles and higher cytoplasmic immunoreactivity for mutant decorin in 952delTDcn keratocytes. A: Decorin localized almost exclusively in the stromal extracellular matrix in WT corneas. B: Virtually no immunoreactivity of decorin was detected in the corneal stroma from decorin-deficient mice. C: Immunolocalization of the mutant decorin revealed higher reactivity of mutant decorin (white arrows) in the keratocyte cytoplasm than in the extracellular matrix in a mutant mouse with decorin-null background. Exposures in mice with decorin-null background were twofold those in WT mice. Yellow arrows indicate the corneal stroma. D: Ultrastructural analysis of the corneal stroma showed flat keratocytes along the lamellar layer in WT mice. E: In contrast, keratocytes were substantially enlarged, with increased dilated membranous organelles (arrows), in 952delTDcn mice.
Retention of mutant decorin was associated with enlarged keratocytes harboring markedly dilated membranous cytoplasmic organelles (Figure 1E). These organelles are consistent with Golgi and rough ER. In contrast, WT controls demonstrated a well-organized stromal structure with elongated flattened keratocytes between lamellae (Figure 1D). Keratocytes expressing mutant decorin were significantly larger than those in WT corneas, 1.34 ± 0.46 μm versus 0.61 ± 0.32 μm, respectively (P < 0.01). Widespread presence of dilated membranous organelles suggested retention of proteins in the synthetic/secretory machinery of the 952delTDcn mouse model.
Mutant Decorin Is Retained in Cytoplasm
To examine the secretion of mutant decorin, we used an in vitro system using transfected HEK293T cells. HEK293T cells were seeded on collagen I–coated coverslips before transfection and immunolocalization assays. The data demonstrated that most WT decorin was properly secreted and associated with collagen I (Figure 2A). In contrast, mutant decorin was localized almost exclusively in the cytoplasm of the transfected HEK293T cells, with immunoreactivity within the extracellular collagenous matrix below detection levels (Figure 2B). All immunofluorescence images were captured using the same intensity, gain, and exposure time. Secretion of mutant decorin was further determined by immunoblotting the media and cell layer fractions from cultures of HEK293T cells expressing mutant or WT decorin. Approximately 90% of WT decorin was secreted into the medium; however, we could not detect any released mutant decorin, even after longer exposure of the Western blots. All reactivity shown by the mutant decorin was associated with the cell layer (Figure 2C). Thus, the data clearly demonstrated that mutant decorin was not secreted but was retained in the cytoplasm.
Figure 2.

Mutant decorin is retained in the cytoplasm. A: HEK293T cells were seeded on collagen I–coated coverslips before transfection and immunolocalization assays. Most WT decorin was secreted and associated with collagen I. B: In contrast, compared with WT decorin, mutant decorin was not detectable outside the cell, and much higher immunoreactivity was observed in the cytoplasm (arrows). C: Distribution in medium and cell layer was different between WT and mutant decorin. Using cotransfected green fluorescent protein as a loading control, immunoblots showed that almost 90% of WT decorin was secreted into the medium. In contrast to the WT form, mutant decorin was undetectable in the medium, although levels of cell layer–associated decorin were similar between the two forms of decorin. These data clearly demonstrate that mutant decorin was retained in the cytoplasm.
Retention of Mutant Decorin in ER Is Associated with Dilated ER
As observed in our mouse model, retention of mutant decorin was associated with enlarged keratocytes harboring markedly dilated membranous cytoplasmic organelles. To determine whether this morphologic change is a cellular dysfunction or a secondary response to the disrupted extracellular matrix, the ultrastructural morphologic change also was studied using transfected HEK293T cells in vitro. In agreement with our transgenic mouse model, compared with WT transfected cells (Figure 3A), the 952delTDcn-transfected 293 cells exhibited widespread dilated ER (Figure 3B). Enlarged or dilated lysosomes and/or phagosomes were not observed.
Figure 3.
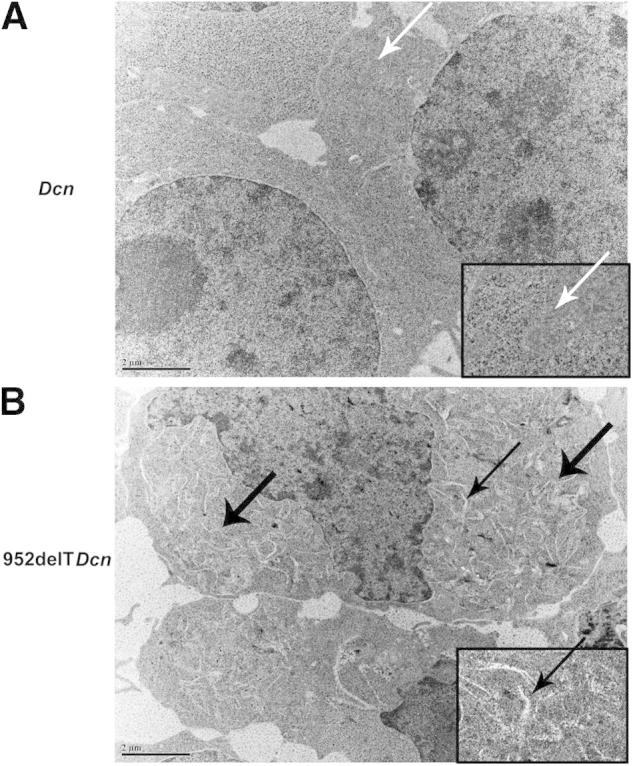
Retention of mutant decorin in the ER is associated with dilated ER. HEK293T cells were transfected with either WT or mutant decorin cDNA. In agreement with our transgenic mouse model, compared with WT transfected cells (A, arrows), the 952delTDcn transfected 293 cells have widely spread dilated membrane structures (B, thick arrows). Insets: Magnification of areas indicated by arrow in A and thin arrow in B. Scale bars: 2 μm (A and B).
Expression of Mutant Decorin Results in Lack of GAG Chain Addition in the Golgi Apparatus
Decorin has a single O-linked GAG chain at the N-terminus, whereas the central domain has three N-glycosylated oligosaccharides. One N-glycosylation site is near the truncated C-terminus. The GAG chain and the N-linked oligosaccharides are important for protein folding and efficient secretion. To investigate whether the truncated C-terminus affected decorin glycosylation, three mutant decorin constructs were created and transfected into HEK293T cells. This yielded cells expressing the mutations comparable to those reported in families with human CSCD. A tag was not used, to eliminate possible influences on trafficking and the function of the expressed proteins. To this end, we transfected HEK293T cells with empty vector alone or with vectors carrying either WT or mutant decorin cDNAs. Decorin mRNA was undetectable in the cells transfected with vector only. The mRNA levels of both WT and mutant decorin were similar, indicating comparable transfection efficiencies of the two vectors (Figure 4A). Significant decorin expression was observed in HEK293T cells transfected with either WT or 952delT decorin (Figure 4B). Next, we sequentially digested the samples with ChABC, which removes dermatan/chondroitin sulfate, and PNGase F, which removes N-linked oligosaccharides. As a consequence, expression of the decorin protein core (Figure 4B) increased. In contrast, there was no change in the 952delT decorin protein core after ChABC treatment, indicating absence of glycanation of the mutant form. N-deglycosylation by PNGase F led to a shift of all of the O-deglycosylated bands on the immunoblots. The distance between WT and truncated decorin bands after O-deglycosylation remained constant on the immunoblots after sequential deglycosylation with ChABC and PNGase F (Figure 4B). Proteoglycans are modified with N-linked oligosaccharides in the ER and with O-linked glycosaminoglycan chains in the Golgi apparatus. Our data indicated that the truncated C-terminus of decorin affected O-glycosylation but not N-glycosylation, further suggesting a specific deficiency of GAG chain attachment and polymerization, presumed due to lack of transport into the Golgi apparatus.
Figure 4.

Mutant decorin lacks O-linked GAG chains. A: HEK293T cells were transfected with vector only, vector carrying WT decorin cDNA, or vector carrying mutant decorin cDNA. Decorin mRNA was undetectable in cells transfected with vector only. mRNA levels of decorin were comparable between cells transfected with either WT or mutant decorin, indicating similar transfection efficiency between the two groups. B: At 60 hours after transfection, proteins were collected from the cells. There was virtually no endogenous decorin expression in HEK293 cells transfected with vector only. Substantial decorin expression was observed in cells transfected with either WT or 952delT decorin. The samples were digested sequentially with ChABC and PNGase F. WT decorin bands became sharper and thicker after removing the O-linked GAG chain with ChABC; however, there was no change in the 952delT decorin band, indicating lack of a GAG chain in the 952delT mutant decorin. Furthermore, N-deglycosylation by PNGase F induced a shift in all of the O-deglycosylated bands on the immunoblots. The distance between the WT and truncated decorin bands after O-deglycosylation remained constant on the immunoblots after sequential deglycosylation with ChABC and PNGase F.
Mutant Decorin Is Retained in ER with Abnormal Transport/Secretion
Like all secreted glycoproteins and proteoglycans, WT decorin protein core is synthesized in the ER; packaged in the Golgi, where most of the posttranslational modifications occur; transported in secretory vesicles; and finally secreted into the extracellular matrix. It is also noteworthy that secreted decorin can be endocytosed and transferred to the lysosome for degradation.26,27 In HEK293T cells transfected with WT decorin, immunocolocalization assays showed low decorin immunoreactivity in the ER and occasional scattered punctate reactivity that did not colocalize with the ER marker, consistent with cytoplasmic transport vesicles. The distribution of WT decorin in the cytoplasm was consistent with a constitutive secretory pathway (Figure 5, A–C). Conversely, mutant decorin showed a distinct localization pattern in transfected HEK293T cells. Compared with WT decorin, mutant decorin exhibited substantially greater colocalization with the ER, indicating retention and abnormal intracellular trafficking of mutant decorin in the cytoplasm (Figure 5, D–F).
Figure 5.
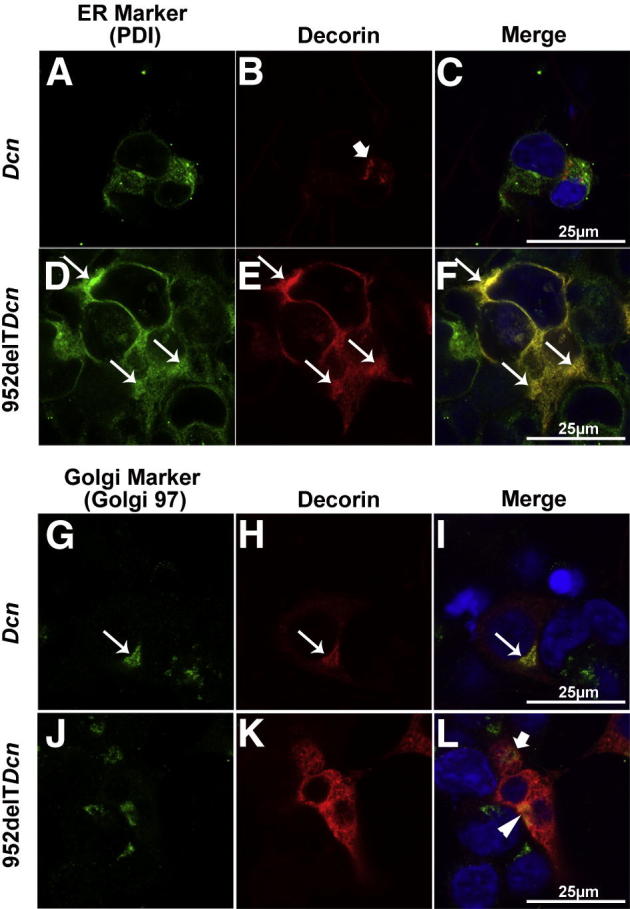
Mutant decorin is retained in the ER with abnormal transport/secretion. A–C: HEK293T cells were seeded on collagen I–coated coverslips and transfected with either WT or 952delT decorin cDNA. Immunocolocalization demonstrated low immunoreactivity of decorin in the ER and higher scattered punctate immunoreactivity that did not colocalize with the ER marker (arrow in B) in cells transfected with WT decorin. D–F: Substantially higher decorin immunoreactivity was colocalized with the ER (arrows) in cells transfected with 952delT decorin. G–I: Immunocolocalization indicated that punctate WT decorin colocalized with the Golgi marker (arrows). J–L: In HEK293T cells transfected with mutant decorin, although mutant decorin was highly expressed in the cytoplasm compared with the controls, it did not colocalize with the Golgi marker. There was limited reactivity against mutant decorin that colocalized with the Golgi marker (yellow pixels indicated by the arrowhead in L). A part of the Golgi is not colocalized with decorin (green pixels indicated by the arrow in L). Scale bars: 25 μm (all panels).
The Golgi is critical for determining the destination of proteins, in particular, its secretion. Our immunocolocalization studies have indicated that the Golgi in WT decorin-transfected cells was always associated with punctate decorin (Figure 5, G–I). In HEK293T cells transfected with mutant decorin, although much higher immunoreactivity of mutant decorin was diffusely localized in the cytosol, only a small fraction was associated with the Golgi (Figure 5, J–L). These data suggest that mutant decorin had an intrinsic deficiency in entering the Golgi apparatus, corroborating the biochemical data showing deficiency in O-glycosylation.
Mutant Decorin Is Degraded through the Ubiquitin-Proteasome Pathway
The findings presented indicated differential localization/transport of WT and mutant decorin within the ER/Golgi boundaries. Thus, we investigated the intracellular turnover of WT and mutant decorin by monitoring the behavior of intracellular decorin over time after blocking newly synthesized decorin with cycloheximide. We found that WT decorin levels decreased over time, with an estimated t1/2 of 4.5 hours (Figure 6B), consistent with a combination of intracellular lysosomal degradation and extracellular secretion. Of note, although the mutant decorin was retained in the ER, its signal also decreased over time, with a t1/2 of 7 hours, which suggests that alternative degradative pathways were involved (Figure 6B).
Figure 6.
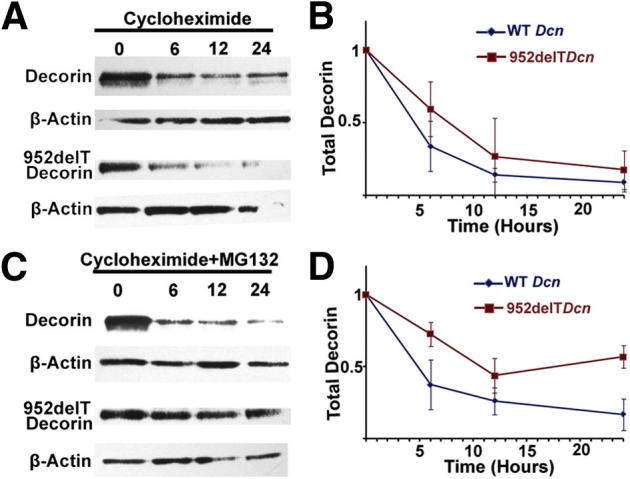
Mutant decorin is degraded through the ubiquitin-proteasome pathway. HEK293T cells were transfected with either WT or mutant decorin cDNA. At 36 hours after transfection, the cells were treated with cycloheximide without (A and B) or with (C and D) MG132. Cells were trypsinized, washed with PBS, and collected at 0, 6, 12, and 24 hours after treatment. A and B: Immunoblots showed that intracellular decorin levels decreased over time after treatment with cycloheximide. C and D: Combined treatment with cycloheximide and MG132 substantially increased intracellular decorin levels in cells transfected with 952delT decorin when compared with those treated with WT decorin.
The degradation pathway of mutant decorin was further investigated through disrupting protein homeostasis using various agents. MG132, a specific proteasome inhibitor, substantially decreased mutant decorin degradation but did not affect the levels of WT decorin (Figure 6D). Moreover, neither methyl-β-cyclodextrin, an endocytosis inhibitor, nor NH4Cl, a lysosomal inhibitor, interfered with mutant decorin degradation (Supplemental Figure S1). Collectively, our results demonstrate for the first time that a truncated C-terminal form of decorin is primarily degraded through the ubiquitin-proteasome pathway.
Retention of Mutant Decorin in the ER Leads to ER Stress
The ubiquitin-proteasome degradation pathway is often a cellular response to various forms of stress. Because mutant decorin is retained in the ER, we hypothesized that the unfolded protein response or ER stress could be directly implicated in the pathogenesis of CSCD. ER stress is necessary in clearing improperly folded proteins through enhancing the folding process or guiding unfolded proteins to ubiquitin-proteasome degradation. The unfolded protein response activates transcription factors through ATF6, PERK, and IRE1 sensors, increasing synthesis of chaperones and altering protein expression.28 The ER stress markers Chop, BiP, and phospho-PERK in HEK293T cells were substantially overexpressed when mutant rather than WT decorin was expressed de novo (Figure 7). Next, transfected HEK293T cell layers were collected, lysed, and loaded onto Phos-tag gels. Phospho-PERK levels were substantially elevated in mutant decorin–transfected cells compared with controls, indicating increased phosphorylation (Figure 7). Thus, our data indicated that intracellularly retained decorin protein core lacking the C-terminal ear repeat caused ER stress.
Figure 7.
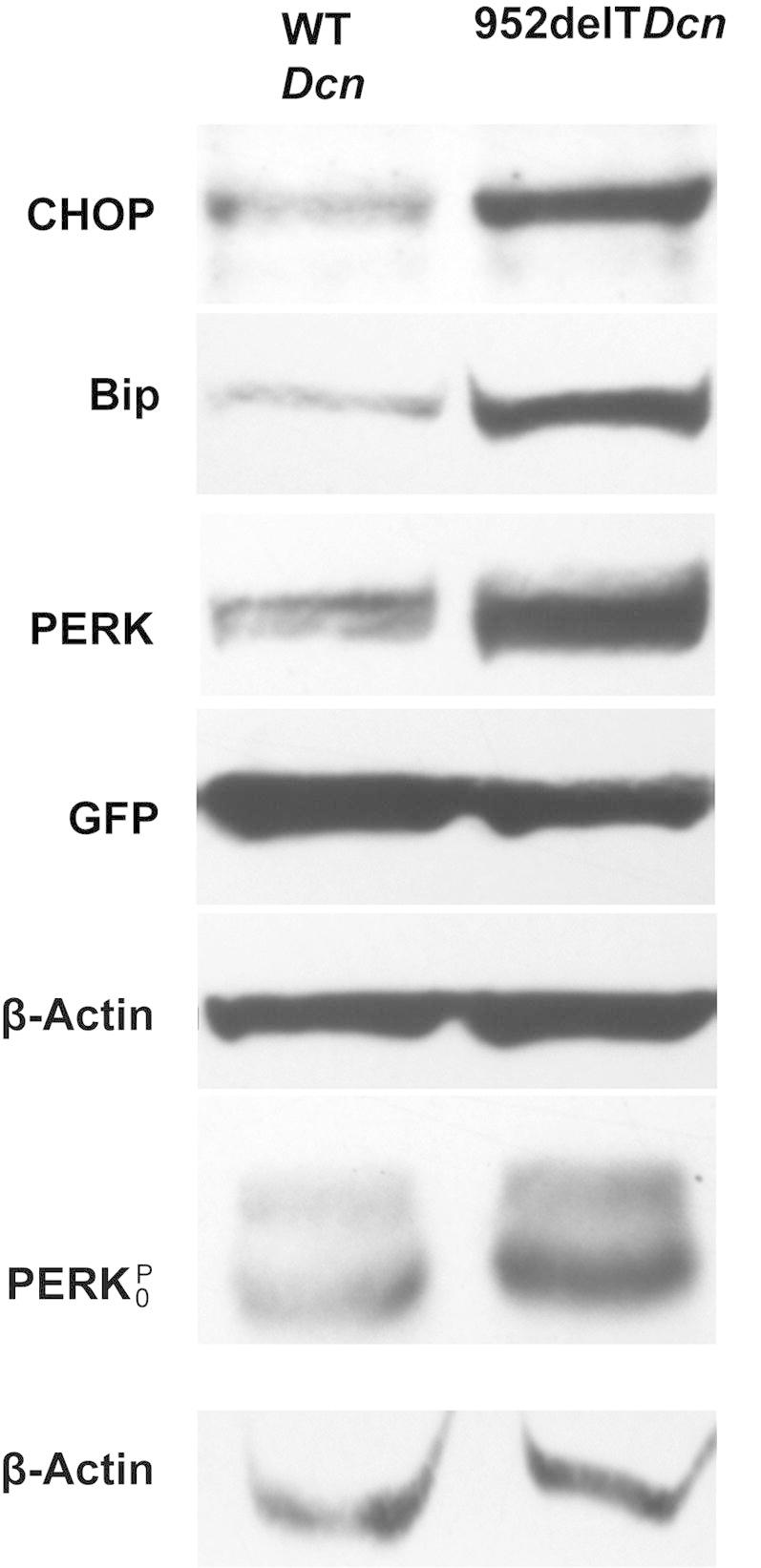
Retention of mutant decorin in the ER leads to ER stress. Immunoblots demonstrate substantially increased expression of CHOP, BiP, and PERK in HEK293T cells transfected with mutant decorin compared with those with WT decorin. Anti-actin bands were used as loading controls, and anti–green fluorescent protein bands as transfection controls. Transfected HEK293 cells were collected, lyzed, and loaded onto Phos-tag gels. Anti–phospho-PERK immunoblots show substantially increased reactivity in cells transfected with 952delT decorin compared with those transfected with WT decorin.
Discussion
Decorin is a ubiquitous SLRP with extensive functions. Its role in a specific biological context is complicated but also finely tuned by its trafficking, secretion, and retention. These processes determine its duration/concentration/sequence of interactions with various molecules and, in turn, its tissue-specific functions. CSCD is the only disease in humans that is known to be associated with a mutated decorin gene and clinically exhibits only a corneal stromal phenotype.9 The present study demonstrates that truncation of the decorin C-terminus results in a misfolded protein and ER stress, providing a possible cell-based mechanism for human CSCD. Moreover, it provides the first evidence for the importance of intracellular trafficking in regulating decorin function and the influence of ER stress on matrix assembly in the corneal stroma.
The ER is an important part of the protein trafficking pathway and a major surveillance system that assists in protein modification, folding, and assembly. The ER response to the burden of misfolded proteins involves generation of stress signals activated by three ER stress sensors: ATF6, PERK, and IRE1. The induced ER stress signals regulate numerous genes to maintain protein homeostasis.28 However, if the ER stress signals are too strong to be mitigated, apoptosis may be induced. ER stress is involved in many physiologic and pathologic conditions.29 The present study indicates that retention of mutant decorin in the ER increases ER stress substantially, which could be a possible mechanism underlying human CSCD. In normal conditions, ER stress is involved in normal clearing of improperly folded proteins. In the heterozygous condition, keratocytes synthesize both WT and mutant decorin, which substantially increases ER stress. Responding to the increased ER stress signals, keratocytes may reprogram themselves and become dedifferentiated; therefore, the cells may change their expression of many extracellular matrix components such as SLRPs. Keratocytes could survive ER stress in development but lose their tissue-specific functions. In the homozygous condition, increased mutant decorin in the ER may result in strong ER stress signals that induce apoptosis, causing cell death. In development, this may result in an embryonic lethal phenotype. Compared with other connective tissues, keratocytes synthesize large amounts of decorin and also secrete other SLRPs into the corneal stroma.30 Therefore, the corneal stroma, compared with other connective tissues, is vulnerable to decorin mutation. The present study provides a logical explanation for the cornea-specific phenotype resulting from decorin mutation in human CSCD.
The corneal stroma is uniquely organized, with regularly packed, homogeneous, small-diameter fibrils arranged as orthogonal lamellae required for transparency.31 Cooperative interactions involving decorin and other SLRPs are important in tightly regulating the fibrillogenesis required for assembly of the corneal stroma and, therefore, function.19,32–34 However, the sites where these regulatory interactions occur, ie, intracellular, extracellular or both, and the roles of cooperative interactions involving various SLRPs, have not been defined. In SLRP-deficient mouse models, the frequently observed phenotype is heterogeneous larger collagen fibrils with cauliflower profiles in the corneal stroma.1 The phenotype suggests extracellular regulation of the lateral growth of collagen fibrils through interactions with dynamic SLRPs influenced by local concentration and diffusing. Our 952delTDcn mouse model demonstrated a disrupted collagen fibril assembly that was distinct from those in SLRP-deficient mouse models.10
Procollagens are folded and assembled in the ER. The process is regulated by the ER enzyme prolyl 4-hydroxylase and the molecular chaperone HSP47.35,36 Differential proteomic analysis confirmed the association of highly expressed decorin with endoplasmin, an ER chaperone, in breast cancer.37 Whether retention of mutant decorin in the ER disturbs procollagen folding and assembly requires further investigation. Mutant decorin could disrupt the interactions between these chaperones and procollagen directly or indirectly through ER stress signals. Alternatively, retained mutant decorin in the ER may interact with or increase the retention/degradation of other SLRPs. We have previously demonstrated that expression of mutant truncated decorin led to decreased synthesis of KS-lumican and KS-keratocan in keratocytes, which could partially explain the altered collagen matrix observed in patients with CSCD.10,22,38,39
The N-terminus of decorin harbors the signal peptide that is important for decorin secretion. However, the possibility that the truncated C-terminus could affect the N-terminus is remote. The frameshift mutations neither introduce nor expose the ER retention sequence KDEL (Lys-Asp-Glu-Leu) in the C-terminus after truncation. Of note, N-terminal truncations of decorin lacking the 14-amino acid propeptide generate decorin isoforms with shorter GAG chains,40 indicating that the protein core with N-terminal truncation can undergo glycanation in the Golgi apparatus. N-glycosylation is important for secretion41; however, the present study did not show its absence in truncated decorin. The reported three frameshift mutations all affect the ear repeat, which is proposed to maintain the conformation of the protein core and influence ligand binding. The C-terminal cysteine-rich domain is critical for stability of the ear repeat, and a milder form of CSCD involves a nucleotide substitution (c.1036T>G) in this region, resulting in a substitution of 346 cysteine with glycine.42 The observed milder phenotype would be consistent with less severe disruption of the regional structure influencing protein core structure less, resulting in less ER stress. The mechanism by which mutant decorin is retained in the ER with altered ability to enter the Golgi requires further investigation to develop strategies to relieve ER stress in human CSCD.
In conclusion, C-terminal truncation of decorin induced misfolded, unstable, or insoluble decorin that was retained in the ER and could not progress to the next physiologic compartment such as the cis-Golgi, thereby leading to a block in its secretion. The intracellularly retained and misfolded decorin induced ER stress. These in vitro findings correlate well with our in vivo observations of enlarged keratocytes, increased dilated membranous organelles, and intracellular immunoreactivity of mutant decorin in the corneas of transgenic mice expressing the same mutant decorin. Therefore, the ER stress induced by truncated decorin is a novel cell-based mechanism for human CSCD. Future work to understand how the distribution of decorin is regulated in a tissue-specific context, such as corneal stromal matrix assembly, is of great pathophysiologic importance.
Acknowledgments
We thank Sheila Adams for technical assistance with transmission electron microscopy and Qingmei Yao for maintenance of the mouse lines.
Footnotes
Supported by grant NIHEY05129 (D.E.B.).
Supplemental Data
Mutant decorin is not degraded through the endosome-lysosome pathway. HEK293T cells were transfected with either WT or mutant decorin cDNA. At 36 hours after transfection, the cells were treated with 20 mmol/L NH4Cl to inhibit lysosomal digestion or with 2.5 μmol/L methyl-β-cyclodextrin to inhibit caveolin-dependent endocytosis. Cells were trypsinized, washed with PBS, and collected at 24 hours after treatment. Immunoblots show that intracellular decorin levels did not change after either treatment.
References
- 1.Birk D.E., Bruckner P. Collagens, suprastructures, and collagen fibril assembly. In: Mecham R.P., editor. The Extracellular Matrix: an Overview. Springer; New York: 2011. pp. 77–115. [Google Scholar]
- 2.Iozzo R.V. The biology of the small leucine-rich proteoglycans: functional network of interactive proteins. J Biol Chem. 1999;274:18843–18846. doi: 10.1074/jbc.274.27.18843. [DOI] [PubMed] [Google Scholar]
- 3.Iozzo R.V., Goldoni S., Berendsen A., Young M. Small leucine-rich proteoglycans. In: Mecham R.P., editor. The Extracellular Matrix: an Overview. Springer; New York: 2011. pp. 197–232. [Google Scholar]
- 4.Iozzo R.V., Karamanos N. Proteoglycans in health and disease: emerging concepts and future directions. FEBS J. 2010;277:3863. doi: 10.1111/j.1742-4658.2010.07796.x. [DOI] [PubMed] [Google Scholar]
- 5.Iozzo R.V., Schaefer L. Proteoglycans in health and disease: novel regulatory signaling mechanisms evoked by the small leucine-rich proteoglycans. FEBS J. 2010;277:3864–3875. doi: 10.1111/j.1742-4658.2010.07797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bredrup C., Stang E., Bruland O., Palka B.P., Young R.D., Haavik J., Knappskog P.M., Rødahl E. Decorin accumulation contributes to the stromal opacities found in congenital stromal corneal dystrophy. Invest Ophthalmol Vis Sci. 2010;51:5578–5582. doi: 10.1167/iovs.09-4933. [DOI] [PubMed] [Google Scholar]
- 7.Kim J.H., Ko J.M., Lee I., Kim J.Y., Kim M.J., Tchah H. A novel mutation of the decorin gene identified in a Korean family with congenital hereditary stromal dystrophy. Cornea. 2011;30:1473–1477. doi: 10.1097/ICO.0b013e3182137788. [DOI] [PubMed] [Google Scholar]
- 8.Rødahl E., Van Ginderdeuren R., Knappskog P.M., Bredrup C., Boman H. A second decorin frame shift mutation in a family with congenital stromal corneal dystrophy. Am J Ophthalmol. 2006;142:520–521. doi: 10.1016/j.ajo.2006.03.064. [DOI] [PubMed] [Google Scholar]
- 9.Bredrup C., Knappskog P.M., Majewski J., Rødahl E., Boman H. Congenital stromal dystrophy of the cornea caused by a mutation in the decorin gene. Invest Ophthalmol Vis Sci. 2005;46:420–426. doi: 10.1167/iovs.04-0804. [DOI] [PubMed] [Google Scholar]
- 10.Chen S., Sun M., Meng X., Iozzo R.V., Kao W.W., Birk D.E. Pathophysiological mechanisms of autosomal dominant congenital stromal corneal dystrophy: C-terminal-truncated decorin results in abnormal matrix assembly and altered expression of small leucine-rich proteoglycans. Am J Pathol. 2011;179:2409–2419. doi: 10.1016/j.ajpath.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schaefer L., Iozzo R.V. Biological functions of the small leucine-rich proteoglycans: from genetics to signal transduction. J Biol Chem. 2008;283:21305–21309. doi: 10.1074/jbc.R800020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iozzo R.V. The family of the small leucine-rich proteoglycans: key regulators of matrix assembly and cellular growth. Crit Rev Biochem Mol Biol. 1997;32:141–174. doi: 10.3109/10409239709108551. [DOI] [PubMed] [Google Scholar]
- 13.Chen S., Birk D.E. Focus on molecules: decorin. Exp Eye Res. 2010;92:444–445. doi: 10.1016/j.exer.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldoni S., Owens R.T., McQuillan D.J., Shriver Z., Sasisekharan R., Birk D.E., Campbell S., Iozzo R.V. Biologically active decorin is a monomer in solution. J Biol Chem. 2004;279:6606–6612. doi: 10.1074/jbc.M310342200. [DOI] [PubMed] [Google Scholar]
- 15.Kalamajski S., Aspberg A., Oldberg A. The decorin sequence SYIRIADTNIT binds collagen type I. J Biol Chem. 2007;282:16062–16067. doi: 10.1074/jbc.M700073200. [DOI] [PubMed] [Google Scholar]
- 16.Santra M., Reed C.C., Iozzo R.V. Decorin binds to a narrow region of the epidermal growth factor (EGF) receptor, partially overlapping but distinct from the EGF-binding epitope. J Biol Chem. 2002;277:35671–35681. doi: 10.1074/jbc.M205317200. [DOI] [PubMed] [Google Scholar]
- 17.Cabello-Verrugio C., Santander C., Cofré C., Acuña M.J., Melo F., Brandan E. The internal region leucine-rich repeat 6 of decorin interacts with low density lipoprotein receptor-related protein-1, modulates transforming growth factor (TGF)-β-dependent signaling, and inhibits TGF-β-dependent fibrotic response in skeletal muscles. J Biol Chem. 2012;287:6773–6787. doi: 10.1074/jbc.M111.312488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vial C., Gutiérrez J., Santander C., Cabrera D., Brandan E. Decorin interacts with connective tissue growth factor (CTGF)/CCN2 by LRR12 inhibiting its biological activity. J Biol Chem. 2011;286:24242–24252. doi: 10.1074/jbc.M110.189365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang G., Chen S., Goldoni S., Calder B.W., Simpson H.C., Owens R.T., McQuillan D.J., Young M.F., Iozzo R.V., Birk D.E. Genetic evidence for the coordinated regulation of collagen fibrillogenesis in the cornea by decorin and biglycan. J Biol Chem. 2009;284:8888–8897. doi: 10.1074/jbc.M806590200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McEwan P.A., Scott P.G., Bishop P.N., Bella J. Structural correlations in the family of small leucine-rich repeat proteins and proteoglycans. J Struct Biol. 2006;155:294–305. doi: 10.1016/j.jsb.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 21.Danielson K.G., Baribault H., Holmes D.F., Graham H., Kadler K.E., Iozzo R.V. Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J Cell Biol. 1997;136:729–743. doi: 10.1083/jcb.136.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen S., Oldberg A., Chakravarti S., Birk D.E. Fibromodulin regulates collagen fibrillogenesis during peripheral corneal development. Dev Dyn. 2010;239:844–854. doi: 10.1002/dvdy.22216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Birk D.E., Fitch J.M., Babiarz J.P., Linsenmayer T.F. Collagen type I and type V are present in the same fibril in the avian corneal stroma. J Cell Biol. 1988;106:999–1008. doi: 10.1083/jcb.106.3.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scholzen T., Solursh M., Suzuki S., Reiter R., Morgan J.L., Buchberg A.M., Siracusa L.D., Iozzo R.V. The murine decorin: complete cDNA cloning, genomic organization, chromosomal assignment, and expression during organogenesis and tissue differentiation. J Biol Chem. 1994;269:28270–28281. [PubMed] [Google Scholar]
- 25.Fisher L.W., Stubbs J.T., 3rd, Young M.F. Antisera and cDNA probes to human and certain animal model bone matrix noncollagenous proteins. Acta Orthop Scand Suppl. 1995;266:61–65. [PubMed] [Google Scholar]
- 26.Hausser H., Schönherr E., Müller M., Liszio C., Bin Z., Fisher L.W., Kresse H. Receptor-mediated endocytosis of decorin: involvement of leucine-rich repeat structures. Arch Biochem Biophys. 1998;349:363–370. doi: 10.1006/abbi.1997.0471. [DOI] [PubMed] [Google Scholar]
- 27.Brandan E., Retamal C., Cabello-Verrugio C., Marzolo M.P. The low density lipoprotein receptor-related protein functions as an endocytic receptor for decorin. J Biol Chem. 2006;281:31562–31571. doi: 10.1074/jbc.M602919200. [DOI] [PubMed] [Google Scholar]
- 28.Walter P., Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 29.Tsang K.Y., Chan D., Bateman J.F., Cheah K.S. In vivo cellular adaptation to ER stress: survival strategies with double-edged consequences. J Cell Sci. 2010;123:2145–2154. doi: 10.1242/jcs.068833. [DOI] [PubMed] [Google Scholar]
- 30.Fisher L.W., Termine J.D., Young M.F. Deduced protein sequence of bone small proteoglycan I (biglycan) shows homology with proteoglycan II (decorin) and several nonconnective tissue proteins in a variety of species. J Biol Chem. 1989;264:4571–4576. [PubMed] [Google Scholar]
- 31.Hassell J.R., Birk D.E. The molecular basis of corneal transparency. Exp Eye Res. 2010;91:326–335. doi: 10.1016/j.exer.2010.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu C.Y., Birk D.E., Hassell J.R., Kane B., Kao W.W. Keratocan-deficient mice display alterations in corneal structure. J Biol Chem. 2003;278:21672–21677. doi: 10.1074/jbc.M301169200. [DOI] [PubMed] [Google Scholar]
- 33.Chakravarti S., Zhang G., Chervoneva I., Roberts L., Birk D.E. Collagen fibril assembly during postnatal development and dysfunctional regulation in the lumican-deficient murine cornea. Dev Dyn. 2006;235:2493–2506. doi: 10.1002/dvdy.20868. [DOI] [PubMed] [Google Scholar]
- 34.Chen S., Birk D.E. The regulatory roles of small leucine-rich proteoglycans in extracellular matrix assembly. FEBS J. 2013;280:2120–2137. doi: 10.1111/febs.12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamandé S.R., Bateman J.F. Procollagen folding and assembly: the role of endoplasmic reticulum enzymes and molecular chaperones. Semin Cell Dev Biol. 1999;10:455–464. doi: 10.1006/scdb.1999.0317. [DOI] [PubMed] [Google Scholar]
- 36.Makareeva E., Leikin S. Procollagen triple helix assembly: an unconventional chaperone-assisted folding paradigm. PLoS One. 2007;2:e1029. doi: 10.1371/journal.pone.0001029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cawthorn T.R., Moreno J.C., Dharsee M., Tran-Thanh D., Ackloo S., Zhu P.H., Sardana G., Chen J., Kupchak P., Jacks L.M., Miller N.A., Youngson B.J., Iakovlev V., Guidos C.J., Vallis K.A., Evans K.R., McCready D., Leong W.L., Done S.J. Proteomic analyses reveal high expression of decorin and endoplasmin (HSP90B1) are associated with breast cancer metastasis and decreased survival. PLoS One. 2012;7:e30992. doi: 10.1371/journal.pone.0030992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Austin B.A., Coulon C., Liu C.Y., Kao W.W., Rada J.A. Altered collagen fibril formation in the sclera of lumican-deficient mice. Invest Ophthalmol Vis Sci. 2002;43:1695–1701. [PubMed] [Google Scholar]
- 39.Chakravarti S., Petroll W.M., Hassell J.R., Jester J.V., Lass J.H., Paul J., Birk D.E. Corneal opacity in lumican-null mice: defects in collagen fibril structure and packing in the posterior stroma. Invest Ophthalmol Vis Sci. 2000;41:3365–3373. [PMC free article] [PubMed] [Google Scholar]
- 40.Oldberg A., Antonsson P., Moses J., Fransson L.A. Amino-terminal deletions in the decorin core protein leads to the biosynthesis of proteoglycans with shorter glycosaminoglycan chains. FEBS Lett. 1996;386:29–32. doi: 10.1016/0014-5793(96)00407-3. [DOI] [PubMed] [Google Scholar]
- 41.Zhou Y.B., Liu F., Zhu Z.D., Zhu H., Zhang X., Wang Z.Q., Liu J.H., Han Z.G. N-glycosylation is required for efficient secretion of a novel human secreted glycoprotein, hPAP21. FEBS Lett. 2004;576:401–407. doi: 10.1016/j.febslet.2004.09.039. [DOI] [PubMed] [Google Scholar]
- 42.Lee J.H., Ki C.S., Chung E.S., Chung T.Y. A novel decorin gene mutation in congenital hereditary stromal dystrophy: a Korean family. Korean J Ophthalmol. 2012;26:301–305. doi: 10.3341/kjo.2012.26.4.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mutant decorin is not degraded through the endosome-lysosome pathway. HEK293T cells were transfected with either WT or mutant decorin cDNA. At 36 hours after transfection, the cells were treated with 20 mmol/L NH4Cl to inhibit lysosomal digestion or with 2.5 μmol/L methyl-β-cyclodextrin to inhibit caveolin-dependent endocytosis. Cells were trypsinized, washed with PBS, and collected at 24 hours after treatment. Immunoblots show that intracellular decorin levels did not change after either treatment.