

Abstract
Patients undergoing glucocorticoid therapy for a variety of disorders, including autoimmune diseases and hematological malignancies, are at risk of developing osteonecrosis. Despite extensive research in both patients and animal models, the underlying pathogenesis remains unclear. Proposed inciting mechanisms include intravascular thrombotic occlusion, marrow fat hypertrophy, osteocyte and/or endothelial cell apoptosis, hypercoagulability, and vasoconstriction of specific arteries and arterioles supplying bone. Our laboratory has developed a model of steroid-induced osteonecrosis in BALBcJ mice which reflects clinically relevant exposures to glucocorticoids in which treated mice develop osteonecrosis of the distal femoral epiphysis when administered 4 to 8 mg/L dexamethasone in drinking water for 6 weeks. We identified lesions in arterioles supplying this area, with the mildest occurring in knees without any evidence of osteonecrosis. However, arteriopathy was more common among mice that did versus did not develop osteonecrosis (P < 0.0001); in mice with osteonecrosis, the associated vessels showed transmural necrosis and thickening of the vessel wall progressing to the point of luminal obstruction. In the most severe cases of osteonecrosis, end-stage lesions consisted of fully occluded vessels with marrow and bone necrosis involving the entire epiphysis. We propose that a primary arteriopathy is the initiating event in the genesis of steroid-induced osteonecrosis and provides a basis for future investigation of this disease process.
The association between corticosteroid use and osteonecrosis (ON) was first described in patients receiving renal transplant who were undergoing immunoablation or immunosuppression as part of their transplantation regimen.1 Glucocorticoids are used extensively to treat diverse diseases such as rheumatoid arthritis, asthma, and lymphoid malignancies. The incidence of ON is as high as 37% in patients with systemic lupus erythematosus,2 and a recent prospective magnetic resonance image screening of children being treated for acute lymphoblastic leukemia showed an 18% incidence of symptomatic ON and a 72% incidence of magnetic resonance image-detectable ON.3
No consensus is widely held as to the pathogenesis of corticosteroid-induced ON. It is generally agreed that the end point is the interruption of blood supply to the bone, leading to death of osteocytes and adipocytes with resultant bone marrow edema and, ultimately, destruction of the architecture of the bone.4 Proposed inciting mechanisms include intravascular thrombotic occlusion, extravascular adipogenesis/fat hypertrophy, osteocyte and/or endothelial cell apoptosis, hypercoagulability, and vasoconstriction of arteries and arterioles that supply the bone.5
Our laboratory has developed a model of steroid-induced ON in BALBcJ mice which reflects clinically relevant exposures to glucocorticoids.6 The administration of dexamethasone (DEX) in the drinking water at 4 mg/L in the first week of treatment and 2 mg/L for the remainder of a 12-week treatment resulted in a 40% incidence of ON of the distal femoral epiphysis. This mouse model was developed to test the influence of various chemotherapeutic drug combinations and treatment schedules, as well as specific gene-related effects, on the development of ON.
In the present study, we investigated the pathogenesis of DEX-induced ON in BALB/c mice by careful examination and comparison of sequential histological sections of affected and unaffected bones showing various stages of lesion development.
Materials and Methods
Chemicals
DEX sodium phosphate solution was purchased from American Pharmaceutical Partners, Inc. (Schaumburg, IL). Escherichia coli-asparaginase was obtained from Ovation Pharmaceuticals (Deerfield, IL); 6-mercaptopurine was obtained from Acros Organics (Fair Lawn, NJ). Sulfamethoxazole and trimethoprim oral suspension was obtained from Hi-Tech Pharmacal Co., Inc. (Amityville, NY), and tetracycline was purchased from Sigma (St. Louis, MO). The folic acid–deficient diet for mice was obtained from TestDiet (Richmond, IN).
Treatment Regimens
The tissues evaluated for this study were obtained from mice used in a variety of pilot experiments that were performed to explore the effects of possible risk factors for DEX-induced ON, such as age, duration, and dose of treatment with DEX, and the effect of various chemotherapeutic agents used in combination with DEX. Male BALB/c mice were used in all experiments because previous studies showed this strain to be most susceptible to DEX-induced ON, with male mice showing a higher incidence than female mice.6 Herein, we used two substrains of BALB/c mice: BALBcJ (Jackson Laboratories, Sacramento, CA) and BALBcAnN (Harlan Laboratories, Houston, TX). These two substrains have been reported to have different susceptibility to DEX-induced adverse effects.7 Treatment started at postnatal days 24 to 28 and lasted for 6 weeks. DEX (4 mg/L or 8 mg/L) and 6-mercaptopurine (2 mg/L or 4 mg/L) were administered in the drinking water, whereas asparaginase was administered by intraperitoneal injections at 7500 IU/kg once or twice weekly. To prevent opportunistic infections, 1000 mg/L tetracycline, 7 days a week, and 600 mg/L and 120 mg/L, respectively, sulfamethoxazole and trimethoprim oral suspension 3.5 days a week were also added to drinking water. Mice were fed a folic acid–deficient diet that contained <0.05 ppm folic acid during treatment; this diet was used previously in developing the murine model of glucocorticoid-induced ON.6 All experiments involving the production and use of mice were approved by the Institutional Animal Care and Use Committee of St. Jude Children’s Research Hospital. The treatment protocols and number of mice evaluated after treatment with DEX with or without asparaginase or 6-mercaptopurine are outlined in Table 1.
Table 1.
Incidence of ON and Arteriopathy in Mice Treated with DEX with or without ASP and/or 6MP
| Strain | No. (%) | Evaluated for arteriopathy, no. | Arteriopathy positive, no. (%) | Arteriopathy negative, no. (%) | P value∗ |
|---|---|---|---|---|---|
| BALBcAnN | |||||
| DEX 4 mg (n = 56) | 0.21 | ||||
| Positive ON | 3 (5.4) | 1 | 1 (100) | 0 (0) | |
| Negative ON | 53 (94.6) | 13 | 2 (15.4) | 11 (84.6) | |
| DEX 4 mg + ASP (n = 19) | 0.17 | ||||
| Positive ON | 2 (10.5) | 2 | 2 (100) | 0 (0) | |
| Negative ON | 17 (89.5) | 2 | 0 (0) | 2 (100) | |
| DEX 8 mg (n = 54) | 0.0002 | ||||
| Positive ON | 17 (31.5) | 12 | 12 (100) | 0 (0) | |
| Negative ON | 37 (68.5) | 16 | 5 (31.3) | 11 (68.8) | |
| DEX 8 mg + ASP (n = 59) | 0.02 | ||||
| Positive ON | 8 (13.6) | 8 | 7 (87.5) | 1 (12.5) | |
| Negative ON | 51 (86.4) | 8 | 2 (25.0) | 6 (75.0) | |
| BALBcAnN total (n = 188) | <0.0001 | ||||
| Positive ON | 30 (16.0) | 23 | 22 (95.7) | 1 (4.3) | |
| Negative ON | 158 (84.0) | 39 | 9 (23.1) | 30 (76.9) | |
| BALBcJ | |||||
| DEX 4 mg (n = 137) | <0.0001 | ||||
| Positive ON | 43 (31.4) | 29 | 29 (100) | 0 (0) | |
| Negative ON | 94 (68.6) | 21 | 11 (52.4) | 10 (47.6) | |
| DEX + 6MP (n = 99) | <0.0001 | ||||
| Positive ON | 38 (38.4) | 35 | 35 (100) | 0 (0) | |
| Negative ON | 61 (61.6) | 25 | 13 (52.0) | 12 (48.0) | |
| DEX 4 mg + ASP (n = 21) | 0.14 | ||||
| Positive ON | 7 (33.3) | 4 | 4 (100) | 0 (0) | |
| Negative ON | 14 (66.7) | 3 | 1 (33.3) | 2 (66.7) | |
| BALB/cJ total (n = 257) | <0.0001 | ||||
| Positive ON | 88 (34.2) | 68 | 68 (100) | 0 (0) | |
| Negative ON | 169 (65.8) | 49 | 25 (51.0) | 24 (49.0) | |
| All Mice | |||||
| (n = 445) | <0.0001 | ||||
| Positive ON | 118 (26.5) | 91 | 90 (98.9) | 1 (1.1) | |
| Negative ON | 327 (73.5) | 88 | 34 (38.6) | 54 (61.4) | |
ASP, asparaginase; 6-MP, 6-mercaptopurine.
P values for comparing the incidence of arteriopathy in ON-positive and ON-negative cases, using χ2 test (if the expected number of cases in each category is ≥5) or Fisher’s exact test (if the expected number of cases in any category is <5).
Histopathology
At necropsy, both hind limbs were collected, fixed in 10% formalin, decalcified in 10% formic acid, routinely processed, sagittally sectioned, and stained with H&E. Sections were evaluated for the presence of ON. Also documented at that time was the presence or absence of evaluable arteriolar branches of the medial genicular artery located along the surface of the distal femoral condyles. Mice with ON or arteriolar lesions in one or both legs were classified as ON positive or arteriopathy positive, respectively. Elastic stains (kit AR163; Dako, Carpinteria, CA) were performed on sections previously stained with H&E after removal of the coverslip and de-staining.
Immunohistochemistry
For immunohistochemistry, 4- to 6-μm serial sections were first deparaffinized in xylene and then rehydrated through graded alcohols. Endogenous peroxidases were blocked by incubating slides for 5 minutes with 3% aqueous hydrogen peroxide. Primary antibodies used in this study included CD31 (catalog #553370; Pharmingen, San Diego, CA) and α-smooth muscle actin (α-SMA; catalog #M0851; Dako). For labeling of CD31, all of the following steps were performed on an autostainer at room temperature with Tris-buffered saline and Tween 20 (catalog #TA-999TT; ThermoShandon, Freemont, CA) buffer rinses between steps. Enzyme-induced epitope retrieval was done with proteinase K (10 minutes; catalog #S3020; Dako). Sections were incubated with anti-CD31 antibody at 1:20 for 60 minutes and then with the secondary biotinylated rabbit anti-rat antibody (Vector; catalog #BA-4001; Burlingame, CA) at 1:200 for 10 minutes. For the labeling of α-SMA, the primary antibody was first biotinylated with the ARK kit (catalog #K3954; Dako) according to the manufacturer’s instructions. Heat-induced epitope retrieval was performed in a decloaking chamber with citrate buffer, pH 6, for 30 minutes at 98°C (Invitrogen, Carlsbad, CA; catalog #00-5000). Sections were incubated with the biotinylated primary antibody for 30 minutes. Bound antibodies were detected with the horseradish peroxidase-labeled streptavidin method (catalog #TS-125-HR; ThermoShandon), with 3,3′-diaminobenzidine (catalog #TA-125-HDX; ThermoShandon) as chromogenic substrate (DAB; catalog #TA-125-HDX; ThermoShandon). Sections were counterstained with Mayer’s hematoxylin (catalog #TA-125-MH; ThermoShandon).
Results
Overall, 34.2% (88 of 257) of BALBcJ and 16.0% (30 of 188; P < 0.0001) of BALBcAnN mice developed ON of the distal femoral epiphysis, characterized by necrotic bone marrow adipose tissue and hematopoietic cells, ghost osteocytes, and empty osteocyte lacunae within trabecular bone [Figure 1, B and D (compare with control in Figure 1, A and C)]; we have previously reported on the substrain difference in occurrence of ON.7 An arteriolar branch of the medial genicular artery coursing along the dorsal surface of the distal femur was detected in 40.2% (179 of 445) of the mice. Lesions were present in the vessels of 98.9% (90 of 91) of mice with ON and in 38.6% (34 of 88; P < 0.0001) of mice without ON [Figure 1F (compare with control in Figure 1E) and Table 1], indicating that arteriopathy was associated with the occurrence of ON and that vascular lesions could be detected in mice who had no signs of ON. In the one mouse with ON that was negative for vascular lesions, the area of necrosis was <10% of the epiphysis, and it is possible that, although an unaffected vessel was present in the section, the unaffected vessel was in a different plane of section.
Figure 1.
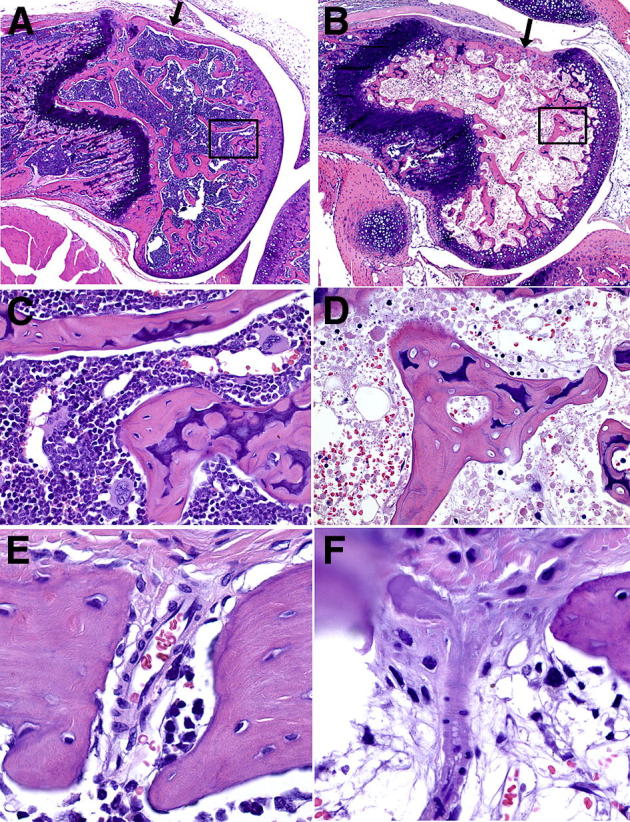
H&E-stained sagittal sections of the distal femoral epiphysis, physis, and metaphysis in control (A, C, and E) and ON (B, D, and F) mice. Location of vessels supplying the epiphysis is indicated in A and B by an arrow. A: Normal marrow and trabecular bone. B: Steroid-induced severe ON, diffusely necrotic epiphyseal marrow, and trabecular bone. C: Magnification of boxed area in A shows healthy osteocytes within lacunae and normal hematopoietic cells. D: Magnification of boxed area in B shows loss of osteocytes within lacunae and necrotic bone marrow. E: Normal transcortical arteriole. F: Degeneration, necrosis, and luminal occlusion of transcortical arteriole in mouse with ON. Original magnification: ×40 (A and B); ×400 (C and D); ×600 (E and F).
The mildest vascular lesions were observed in eight mice without ON and included loss of endothelium, necrosis of vascular smooth muscle cells, and thickening and basophilia of the vessel wall (Figure 2A). When stained with trichrome (Figure 2B), loss of trichrome-positive staining was observed in the thickened area of the vascular wall, perhaps indicating loss of protein because of necrosis of smooth muscle cells. Supporting this conclusion was the loss of α-SMA (Figure 2C), and loss of endothelial CD31 labeling (Figure 2D) in the affected areas. It should be noted that at this stage these changes in the vessel wall were present without evidence of luminal obstruction (Figure 3A). In some mice, these lesions progressed to transmural necrosis and thickening of the vessel wall, fragmentation of the internal elastic lamina, and mural thickening to the point of luminal occlusion (Figure 3B). Elastin histochemistry clearly showed the fragmentation of the internal elastic lamina in the involved sites (Figure 3, C and D). At this stage, there was generally only localized necrosis of the bone marrow in the femoral epiphysis and only rare empty osteocyte lacunae. In some vessels, thrombi developed proximal to areas of vascular occlusion (Figure 3, E and F); at this stage often more extensive marrow necrosis was accompanied by occasional empty osteocyte lacunae. In the most severe and advanced cases of ON, end-stage lesions consisted of fully occluded vessels with marrow and bone necrosis that involved the entire epiphysis (Figure 3, G and H).
Figure 2.
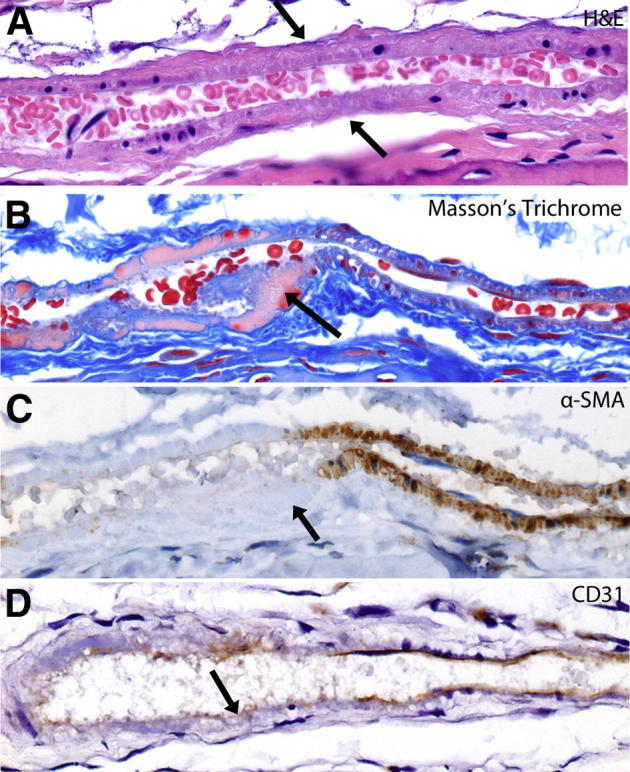
Mildest visible lesion in arterioles supplying the distal femoral epiphysis in steroid-treated mice. A: H&E staining shows segmental mild thickening of vessel wall, increased basophilia, and loss of smooth muscle cell and endothelial cell nuclei in area indicated by arrow. B: Masson’s trichrome, segmental loss of trichrome-positive staining in area of vascular smooth muscle cells. Segment indicated by arrow. C: α-SMA immunohistochemistry shows loss of α-SMA in area of smooth muscle necrosis. Segment indicated by arrow. D: CD31 immunohistochemistry shows segmental loss of endothelial cells. Area indicated by the arrow. Original magnification: ×600 (A–D).
Figure 3.
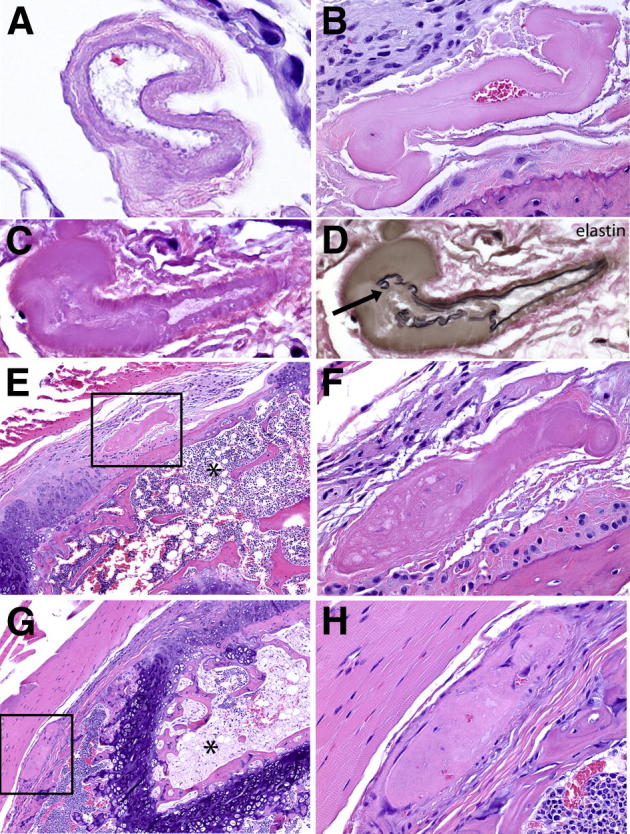
Vascular lesions in arterioles supplying the distal femoral epiphysis. A: H&E staining of cross-section of arteriole shows transmural necrosis and the absence of intraluminal obstruction. B: H&E staining of cross-section of arteriole shows transmural necrosis, thickening of the vessel wall, and partial luminal obstruction. C: H&E staining shows diffuse transmural necrosis with segmental thickening of the vessel wall and partial loss of the internal elastic lamina. D: Elastin histochemistry performed after removal of coverslip and de-staining of section in C, showing segmental loss and discontinuity of the internal elastic lamina, indicated by arrow. E: H&E staining of sagittal section of distal femoral epiphysis shows complete occlusion of vascular lumen by a thrombus proximal to the area of mural thickening (boxed area) and diffuse, extensive bone marrow necrosis (asterisk) and focal, partial necrosis of trabecular bone. F: Magnification of boxed area in E. G: H&E staining of sagittal section of distal femoral epiphysis, physis, and metaphysis shows thrombosis of segment of vessel located proximal to the vessel shown in boxed area of E, concurrent with diffuse, extensive bone marrow and trabecular bone necrosis (asterisk). H: Magnification of boxed area in G. Original magnification: ×600 (A); ×400 (B–D, F, and H); ×100 (E and G).
Discussion
We observed that an arteriopathy involving branches of the medial genicular artery supplying the distal femoral epiphysis preceded the development of ON. The mildest vascular lesions are characterized by loss of endothelium and necrosis of arterial smooth muscle, and more severe lesions include transmural necrosis of the vessel wall, luminal occlusion, and proximal thrombosis. It is our hypothesis that these mild, moderate, and severe lesions indicate progressive stages in the generation of ON. Our findings clearly indicate that these vascular lesions can exist without the characteristic bone lesions of ON in the epiphyseal marrow and trabecular bone, which include marrow necrosis, edema, and loss of osteocytes. Because of the small size of the branches of the medial genicular artery and the method of sectioning, the blood vessels were not present in all sections analyzed, but it is presumed that, given more extensive sampling of the bones, all of those with ON would have concomitant vascular lesions.
The pattern and extent of lesions present in the blood vessels, marrow, and bone suggest that occlusion of the vasculature supplying one portion of the epiphysis, resulting in marrow with or without bone necrosis in a focal area, initiates a cascade of events within the epiphysis that results in complete necrosis. It is likely that focal necrosis in one part of the femoral epiphysis can initiate edema and inflammation in the confined space of epiphyseal marrow that results in a compartment syndrome that compromises local blood flow. The hypothesis that increased intramedullary pressure leads to ON was first proposed by Arlet et al8 in 1972, although the mechanism and sequence of events they put forth differs from what we are suggesting here. Arlet et al8 proposed that the process of ON begins with compression of the intraosseous sinusoidal system, which leads to venous stasis and eventually to arterial obstruction and subsequent bone injury. In contrast, our findings indicate that the initiating event is within the supplying arteries and arterioles and that the subsequent inflammation, edema, and necrosis in the epiphysis are secondary, self-propagating events.
Interestingly, similar lesions in the arteries and arterioles supplying the femoral head have been reported in humans. For example, thickening of the intima and various degrees of intimal damage were seen in hip capsule arteries in patients who received a renal transplant who were treated with high-dose steroids but showed no overt symptoms of hip disease before death.9 In another study, Saito et al10 examined core biopsy specimens from femoral heads with silent ON of the femoral head, advanced ON of the femoral head, and osteoarthrosis controls. The most consistent finding in patients with ON of the femoral head was what was described by the investigators as an “arteriopathy which comprised the structural damage to the vascular wall of arterioles and the residue of broken-down blood vessels.”10,p102 The tunica media showed various combinations of degeneration, loss of elastin and collagen fibers, disruption of the internal elastic lamina, and smooth muscle cell necrosis and loss. Only mild changes in the tunica intima were observed.10
In mice, structural changes similar to those described here were observed in response to local corticosteroid treatment. Delivery of DEX to the femoral artery with the use of a drug-eluting polymer cuff induced dose-dependent medial atrophy, a reduction in vascular smooth muscle cells and collagen content, and disruption of the internal elastic lamina.11 Local DEX delivery to arteries has been tested in other animal models in an attempt to inhibit restenosis, with inconsistent effects on neointimal hyperplasia; however, no mention of adverse changes to the medial tissue was reported.12–14 This may have been due to different concentrations of DEX or to species differences in the vascular delivery of or reactivity to the drug.
In comparing several different strains of mice, we previously found BALB/c mice to be the most susceptible to DEX-induced ON.6 The reason for the increased susceptibility in this strain of mice has not been determined, but several studies have reported that BALB/c mice have fewer native collateral blood vessels in the hind limb than do mice of other strains.15–17 In models of hind limb ischemia induced with femoral artery ligation, the reduced collateral circulation in BALB/c results in greater reduction in perfusion immediately after femoral ligation, slower recovery of perfusion, greater impairment of hind limb use, and more severe ischemia.16,17 The reduced collateral circulation in BALB/c mice may explain why they are more prone to steroid-induced ON. It is possible that other strains of mice experience similar damage to the supplying arteries and arterioles but have sufficient collateral circulation to maintain perfusion at a high enough level to prevent marrow or bone necrosis.
Our previous studies showed development of ON in the distal femur (femoral condyles) but not in the femoral head.6 Most studies that use other animal models of ON, including those in rats, rabbits, and pigs, have focused on the femoral head and/or the vessels feeding the femoral head, that is, the lateral epiphyseal arteries.18,19 It is unknown whether ON occurs in the femoral condyles in these other animal models, because these sites do not appear to have been examined. Importantly, steroid-induced ON in humans results in necrosis in the distal femur as frequently as in the femoral head. Sakamoto et al20 reported the incidence of steroid-induced ON to be 63% in the hip and 51% in the knee, when diagnosed by magnetic resonance image screening. A more recent screening study found an overall incidence of ON of 21.5% in patients before bone marrow transplantation, with 11.7% involving the hip and 23.5% the knee.21 What is common to the skeletal areas more frequently developing ON, whether at the hip or knee joint, is the vulnerability of these sites to vascular lesions as a result of the unique vascular anatomy in those locations; their nutrient vessels are tortuous and have limited anastomoses.22 Analysis of the arterial supply to the distal femur in humans has shown that the intraosseous supply to the medial condyle appears to consist of a single nutrient vessel supplying the subchondral bone with an apparent watershed area of limited supply.22 In addition, it is likely that differences in mechanical stress placed on individual vessels due to variations in areas of greatest weight-bearing influence which bones ultimately undergo ON.
Other animal models also support a primary role for vascular dysfunction in the pathogenesis of ON. Spontaneously hypertensive rats (SHR), a strain obtained from Wistar Kyoto rats23 are used as animal models for hypertension and cerebrovascular disorders, and approximately 50% develop spontaneous necrosis of the femoral head from 10 to 20 weeks of age.24 Stroke-prone SHR (SHRSP) are rats obtained by selective mating of SHR25; they have a reported ON incidence of 70% at 10 to 14 weeks of age.26 Notably, steroid treatment in SHRSP rats potentiates the development of ON; 2 weeks after the administration of a 20-mg/kg intramuscular injection of methylprednisolone in 17-week-old rats, 95.2% of treated rats exhibited ON compared with 51.2% of rats in the control group.27 Although the molecular mechanism underlying the susceptibility of SHR and SHRSP rats to ON is unknown, these findings suggest that it is potentiated by corticosteroids.
Both direct and indirect effects of glucocorticoids have been proposed to be responsible for vascular damage leading to ON.5,28 Examinations of the direct pharmacologic effects of corticosteroids on blood vessels supplying the femoral head have been performed in ON models in pigs and rabbits. Corticosteroids reduce femoral head blood flow in pigs,29 have a vasoconstrictive effect on lateral epiphyseal arteries of the femoral head,30,31 and potentiate the vasoconstriction elicited by endothelin-1.31 ON of the femur and humerus was prevented in a rabbit model when a nitrate skin patch was administered at the same time as intramuscular administration of methylprednisolone at 20 mg/kg of body weight.32 Glucocorticoid receptors have been shown in the media of the canine aorta,33 and numerous in vitro studies have provided evidence that the glucocorticoid receptor is present in both the vascular smooth muscle34–36 and the vascular endothelium.35,37,38 In addition to the expected effect of changes in gene expression, studies have indicated that they may mediate the effect of glucocorticoids on the influx of Na+ or Ca2+ into cells.35,39 It is possible that the same mechanisms regulating the vasoconstrictive actions of glucocorticoids, and thereby reduced blood flow, also lead to eventual degeneration and necrosis of vascular smooth muscle cells and endothelium. However, the pharmacologic regulation of blood flow to bone has been shown to differ from that to other organs40; therefore, the specific mechanism leading to necrosis of blood vessels supplying the epiphysis as observed in this study may be unique.
Our finding that progressive lesions develop in the arterioles supplying the distal femoral epiphyses before any abnormalities become evident in the bone or bone marrow indicates that a primary arteriopathy is the underlying cause of steroid-induced ON. A better understanding of the origin and pathogenesis of steroid-induced ON should prove useful in the development of improved diagnostic and therapeutic approaches in the prevention and treatment of this disease process.
Acknowledgments
We thank David Jenkins, Natalie Mannon, Jean Cai, and members of the Animal Resource Center for their assistance with the drug administration experiments and Pamela Johnson, Hermitta McLaurine, and other members of the St. Jude Veterinary Pathology Core laboratory for their assistance in preparing the tissues and histology slides for analysis of osteonecrosis.
Footnotes
Supported by the National Cancer Institute grants CA 142665 and CA 21765 and by the American Lebanese Syrian Associated Charities (ALSAC).
Current address of J.K., Department of Pharmacy Pharmacology Research, University of Texas MD Anderson Cancer Center, Houston, TX.
References
- 1.Arfi S., Moreau F., Heuclin C., Kreis H., Paolaggi J.B., Auguier L. [Aseptic osteonecrosis in renal transplantation; apropos of 29 cases] Rev Rhum Mal Osteoartic. 1975;42:162–176. French. [PubMed] [Google Scholar]
- 2.Shigemura T., Nakamura J., Kishida S., Harada Y., Ohtori S., Kamikawa K., Ochiai N., Takahashi K. Incidence of osteonecrosis associated with corticosteroid therapy among different underlying diseases: prospective MRI study. Rheumatology. 2011;50:2023–2028. doi: 10.1093/rheumatology/ker277. [DOI] [PubMed] [Google Scholar]
- 3.Kawedia J.D., Kaste S.C., Pei D., Panetta J.C., Cai X., Cheng C., Neale G., Howard S.C., Evans W.E., Pui C.H., Relling M.V. Pharmacokinetic, pharmacodynamic, and pharmacogenetic determinants of osteonecrosis in children with acute lymphoblastic leukemia. Blood. 2011;117:2340–2347. doi: 10.1182/blood-2010-10-311969. quiz 2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Powell C., Chang C., Gershwin M.E. Current concepts on the pathogenesis and natural history of steroid-induced osteonecrosis. Clin Rev Allergy Immunol. 2011;41:102–113. doi: 10.1007/s12016-010-8217-z. [DOI] [PubMed] [Google Scholar]
- 5.Kerachian M.A., Seguin C., Harvey E.J. Glucocorticoids in osteonecrosis of the femoral head: a new understanding of the mechanisms of action. J Steroid Biochem Mol Biol. 2009;114:121–128. doi: 10.1016/j.jsbmb.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang L., Boyd K., Kaste S.C., Kamdem Kamdem L., Rahija R.J., Relling M.V. A mouse model for glucocorticoid-induced osteonecrosis: effect of a steroid holiday. J Orthop Res. 2009;27:169–175. doi: 10.1002/jor.20733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawedia J.D., Janke L., Funk A.J., Ramsey L.B., Liu C., Jenkins D., Boyd K.L., Relling M.V. Substrain-specific differences in survival and osteonecrosis incidence in a mouse model of osteonecrosis. Comp Med. 2012;62:466–471. [PMC free article] [PubMed] [Google Scholar]
- 8.Arlet J., Ficat P., Lartigue G., Tran M.A. [Clinical research on intraosseous pressure in the upper femoral metaphysis and epiphysis in humans. Application to the diagnosis of ischemia and necrosis] Rev Rhum Mal Osteoartic. 1972;39:717–723. French. [PubMed] [Google Scholar]
- 9.Spencer J.D., Brookes M. Avascular necrosis and the blood supply of the femoral head. Clin Orthop Relat Res. 1988;235:127–140. [PubMed] [Google Scholar]
- 10.Saito S., Ohzono K., Ono K. Early arteriopathy and postulated pathogenesis of osteonecrosis of the femoral head. The intracapital arterioles. Clin Orthop Relat Res. 1992;(277):98–110. [PubMed] [Google Scholar]
- 11.Pires N.M., Schepers A., van der Hoeven B.L., de Vries M.R., Boesten L.S., Jukema J.W., Quax P.H. Histopathologic alterations following local delivery of dexamethasone to inhibit restenosis in murine arteries. Cardiovasc Res. 2005;68:415–424. doi: 10.1016/j.cardiores.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 12.Villa A.E., Guzman L.A., Chen W., Golomb G., Levy R.J., Topol E.J. Local delivery of dexamethasone for prevention of neointimal proliferation in a rat model of balloon angioplasty. J Clin Investig. 1994;93:1243–1249. doi: 10.1172/JCI117078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lincoff A.M., Furst J.G., Ellis S.G., Tuch R.J., Topol E.J. Sustained local delivery of dexamethasone by a novel intravascular eluting stent to prevent restenosis in the porcine coronary injury model. J Am Coll Cardiol. 1997;29:808–816. doi: 10.1016/s0735-1097(96)00584-0. [DOI] [PubMed] [Google Scholar]
- 14.Strecker E.P., Gabelmann A., Boos I., Lucas C., Xu Z., Haberstroh J., Freudenberg N., Stricker H., Langer M., Betz E. Effect on intimal hyperplasia of dexamethasone released from coated metal stents compared with non-coated stents in canine femoral arteries. Cardiovasc Intervent Radiol. 1998;21:487–496. doi: 10.1007/s002709900309. [DOI] [PubMed] [Google Scholar]
- 15.Zbinden S., Clavijo L.C., Kantor B., Morsli H., Cortes G.A., Andrews J.A., Jang G.J., Burnett M.S., Epstein S.E. Interanimal variability in preexisting collaterals is a major factor determining outcome in experimental angiogenesis trials. Am J Physiol Heart Circ Physiol. 2007;292:H1891–1897. doi: 10.1152/ajpheart.00537.2006. [DOI] [PubMed] [Google Scholar]
- 16.Chalothorn D., Faber J.E. Strain-dependent variation in collateral circulatory function in mouse hindlimb. Physiol Genomics. 2010;42:469–479. doi: 10.1152/physiolgenomics.00070.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Helisch A., Wagner S., Khan N., Drinane M., Wolfram S., Heil M., Ziegelhoeffer T., Brandt U., Pearlman J.D., Swartz H.M., Schaper W. Impact of mouse strain differences in innate hindlimb collateral vasculature. Arterioscler Thromb Vasc Biol. 2006;26:520–526. doi: 10.1161/01.ATV.0000202677.55012.a0. [DOI] [PubMed] [Google Scholar]
- 18.Jones L.C., Allen M.R. Animal models of osteonecrosis. Clin Rev Bone Miner Metab. 2011;9:63–80. [Google Scholar]
- 19.Kerachian M.A., Harvey E.J., Cournoyer D., Chow T.Y., Nahal A., Seguin C. A rat model of early stage osteonecrosis induced by glucocorticoids. J Orthop Surg Res. 2011;6:62. doi: 10.1186/1749-799X-6-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakamoto M. [A prospective study of steroid-induced osteonecrosis by MRI screening] Nihon Seikeigeka Gakkai Zasshi. 1994;68:367–378. Japanese. [PubMed] [Google Scholar]
- 21.Sharma S., Yang S., Rochester R., Britton L., Leung W.H., Yang J., Neel M.D., Ness K.K., Kaste S.C. Prevalence of osteonecrosis and associated risk factors in children before allogeneic BMT. Bone Marrow Transplant. 2011;46:813–819. doi: 10.1038/bmt.2010.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reddy A.S., Frederick R.W. Evaluation of the intraosseous and extraosseous blood supply to the distal femoral condyles. Am J Sports Med. 1998;26:415–419. doi: 10.1177/03635465980260031201. [DOI] [PubMed] [Google Scholar]
- 23.Okamoto K., Aoki K. Development of a strain of spontaneously hypertensive rats. Jpn Circ J. 1963;27:282–293. doi: 10.1253/jcj.27.282. [DOI] [PubMed] [Google Scholar]
- 24.Hirano T., Iwasaki K., Yamane Y. Osteonecrosis of the femoral head of growing, spontaneously hypertensive rats. Acta Orthop Scand. 1988;59:530–535. doi: 10.3109/17453678809148778. [DOI] [PubMed] [Google Scholar]
- 25.Okamoto K., Yamori Y., Nagaoka A. Establishment of stroke-prone spontaneously hypertensive rat (Shr) Circ Res. 1974;34:I143–I153. [Google Scholar]
- 26.Naito S., Ito M., Sekine I., Hirano T., Iwasaki K., Niwa M. Femoral head necrosis and osteopenia in stroke-prone spontaneously hypertensive rats (SHRSPs) Bone. 1993;14:745–753. doi: 10.1016/8756-3282(93)90206-p. [DOI] [PubMed] [Google Scholar]
- 27.Murata M., Kumagai K., Miyata N., Osaki M., Shindo H. Osteonecrosis in stroke-prone spontaneously hypertensive rats: effect of glucocorticoid. J Orthop Sci. 2007;12:289–295. doi: 10.1007/s00776-007-1129-y. [DOI] [PubMed] [Google Scholar]
- 28.Kerachian M.A., Harvey E.J., Cournoyer D., Chow T.Y., Seguin C. Avascular necrosis of the femoral head: vascular hypotheses. Endothelium. 2006;13:237–244. doi: 10.1080/10623320600904211. [DOI] [PubMed] [Google Scholar]
- 29.Drescher W., Schneider T., Becker C., Hobolth J., Ruther W., Hansen E.S., Bunger C. Selective reduction of bone blood flow by short-term treatment with high-dose methylprednisolone. An experimental study in pigs. J Bone Joint Surg Br. 2001;83:274–277. doi: 10.1302/0301-620x.83b2.10323. [DOI] [PubMed] [Google Scholar]
- 30.Drescher W., Bunger M.H., Weigert K., Bunger C., Hansen E.S. Methylprednisolone enhances contraction of porcine femoral head epiphyseal arteries. Clin Orthop Relat Res. 2004:112–117. doi: 10.1097/01.blo.0000127587.92303.b4. [DOI] [PubMed] [Google Scholar]
- 31.Drescher W., Li H., Lundgaard A., Bunger C., Hansen E.S. Endothelin-1-induced femoral head epiphyseal artery constriction is enhanced by long-term corticosteroid treatment. J Bone Joint Surg Am. 2006;88(Suppl 3):173–179. doi: 10.2106/JBJS.F.00773. [DOI] [PubMed] [Google Scholar]
- 32.Drescher W., Beckmann R., Kasch R., Pufe M., Knobe M., Kweider N., Hassenpflug J., Tingart M., Pufe T., Kadyrov M. Nitrate patch prevents steroid-related bone necrosis. J Orthop Res. 2011;29:1517–1520. doi: 10.1002/jor.21420. [DOI] [PubMed] [Google Scholar]
- 33.Horwitz K.B., Horwitz L.D. Canine vascular tissues are targets for androgens, estrogens, progestins, and glucocorticoids. J Clin Investig. 1982;69:750–758. doi: 10.1172/JCI110513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Provencher P.H., Saltis J., Funder J.W. Glucocorticoids but not mineralocorticoids modulate endothelin-1 and angiotensin II binding in SHR vascular smooth muscle cells. J Steroid Biochem Mol Biol. 1995;52:219–225. doi: 10.1016/0960-0760(94)00168-l. [DOI] [PubMed] [Google Scholar]
- 35.Kornel L., Nelson W.A., Manisundaram B., Chigurupati R., Hayashi T. Mechanism of the effects of glucocorticoids and mineralocorticoids on vascular smooth muscle contractility. Steroids. 1993;58:580–587. doi: 10.1016/0039-128x(93)90099-9. [DOI] [PubMed] [Google Scholar]
- 36.Tsugita M., Iwasaki Y., Nishiyama M., Taguchi T., Shinahara M., Taniguchi Y., Kambayashi M., Terada Y., Hashimoto K. Differential regulation of 11beta-hydroxysteroid dehydrogenase type-1 and -2 gene transcription by proinflammatory cytokines in vascular smooth muscle cells. Life Sci. 2008;83:426–432. doi: 10.1016/j.lfs.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 37.Wallerath T., Witte K., Schafer S.C., Schwarz P.M., Prellwitz W., Wohlfart P., Kleinert H., Lehr H.A., Lemmer B., Forstermann U. Down-regulation of the expression of endothelial NO synthase is likely to contribute to glucocorticoid-mediated hypertension. Proc Natl Acad Sci U S A. 1999;96:13357–13362. doi: 10.1073/pnas.96.23.13357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ray K.P., Searle N. Glucocorticoid inhibition of cytokine-induced E-selectin promoter activation. Biochem Soc Trans. 1997;25:189S. doi: 10.1042/bst025189s. [DOI] [PubMed] [Google Scholar]
- 39.Kornel L., Prancan A.V., Kanamarlapudi N., Hynes J., Kuzianik E. Study on the mechanisms of glucocorticoid-induced hypertension: glucocorticoids increase transmembrane Ca2+ influx in vascular smooth muscle in vivo. Endocr Res. 1995;21:203–210. doi: 10.3109/07435809509030436. [DOI] [PubMed] [Google Scholar]
- 40.Brinker M.R., Lippton H.L., Cook S.D., Hyman A.L. Pharmacological regulation of the circulation of bone. J Bone Joint Surg Am. 1990;72:964–975. [PubMed] [Google Scholar]