

Abstract
A 56-year-old man was admitted to our hospital for renal dysfunction and symmetrical swelling of submandibular glands. Laboratory and imaging findings were consistent with immunoglobulin G4-related disease (IgG4RD). Histological findings of the submandibular gland and the kidney were also consistent with IgG4RD. However, the patient did not have elevated serum or tissue IgG4 levels. Oral prednisolone therapy, initially 50 mg/day and gradually tapered over 12 months, improved his laboratory abnormalities and the swelling of his affected organs. These findings prompted our final diagnosis of IgG4RD. IgG4RD is a newly recognised disease with an unknown aetiology. This case suggests that IgG4 antibodies do not play a primary role in the aetiology of IgG4RD. Furthermore, clinicians should not exclude the diagnosis of IgG4RD in patients lacking elevated IgG4 levels in their affected tissues, particularly if they have other features of IgG4RD. Steroid therapy should be considered for such patients.
Background
Immunoglobulin G4-related disease (IgG4RD) is a disease with an unknown aetiology that is characterised by marked lymphoplasmacytic infiltration of IgG4-positive plasma cells into affected tissues.1–4 However, it is unknown whether IgG4 plays primary or secondary roles in its aetiology.
Case presentation
A 56-year-old Japanese man with a 3-month history of palpable non-tender masses under his jaw and general fatigue without fever, night sweats, weight loss or other symptoms of infection was referred to our institution. He was diagnosed with benign prostatic hyperplasia 10 months ago and was treated with 8 mg/day silodosin. He had no history of asthma or sinusitis. He was only treated with silodosin. He had not recently used any over-the-counter drugs and he denied taking any herbal medicines or banned substances. Our examination revealed symmetrical swelling of his submandibular glands, which felt like hard elastic on palpation, and were not fixed to the adjacent tissue. We also found three enlarged and movable lymph nodes, with diameters of 2 cm for one node and 1 cm for two nodes. Other superficial lymph nodes were not palpable.
Laboratory tests revealed the following values: white cell count, 8.6×103/μl with eosinophilia (eosinophil, 782/μL); red blood cell count, 3.51×106/μL, haemoglobin, 10.7 g/dL; haematocrit, 31.4%; platelet count, 224×103/μL; serum creatine, 2.75 mg/dL; C reactive protein, 0.3 mg/dL; amylase, 83 U/L (normal range, 0–70 U/L); lipase, 211 U/L (0–49 U/L); glucose, 88 mg/dL; lactate dehydrogenase, 183 U/L (80–230 U/L); β2 microglobulin, 7.5 mg/L (1.0–1.9 mg/L); IgG, 4 193 mg/dL (870–1700 mg/dL); IgE, 547 IU/mL (0–173 IU/mL); complement (C) 3, 25 mg/dL; C4, 1 mg/dL; C1q immune complexes, 26.2 μg/mL (0.0–0.3 μg/mL); and ferritin, 238 ng/mL (39.4–340 ng/mL). Urinalysis revealed protein (2+), blood (2+) and 1–4 erythrocytes and 1–4 white blood cells per high-power field without casts. Urinary protein excretion was 2.8 g/day. His urinary β2 microglobulin level was 10 900 μg/l (0–230 μg/L). Immunological studies revealed the following: antinuclear antibody titre, 1:160 (mixed homogeneous and speckled pattern); anti-dsDNA IgG titre, 16 IU/mL (0–12 IU/mL); and anti-ssDNA IgG titre, 38 AU/mL (0–25 AU/mL). The tests for anti-SSA/Ro and anti-SSB/La antibodies were negative. M-protein and Bence-Jones protein were not detected in serum or urine. We did not perform serological tests for HIV.
Contrast-enhanced cervical-thoraco-abdominal pelvic CT revealed swelling of his bilateral submandibular glands, multiple lymph nodes in the neck and mediastinum, a diffusely enlarged pancreas with delayed enhancement, diffusely enlarged kidneys with multiple low-density lesions and a soft tissue mantle surrounding his abdominal aorta (figure 1).
Figure 1.

Contrast-enhanced cervical–thoraco–abdominal pelvic CT images taken before and 18 months after treatment. (A–C), Images taken before treatment show swelling of the bilateral submandibular glands and adjacent lymph nodes (A), a diffusely enlarged pancreas (B), diffusely enlarged kidneys with multiple low-density lesions and his abdominal aorta is surrounded by a soft tissue mantle (arrow) (C), (D–F Images taken 18 months after treatment show that the submandibular glands and lymph nodes (D), pancreas (E) and kidneys (F) have normalised in size. The soft tissue mantle has also disappeared (bottom).
Because we strongly suspected IgG4RD, the patient's serum IgG subclasses were analysed, which gave the following values: IgG1, 2520 mg/dL (normal range, 320–740 mg/dL); IgG2, 298 mg/dL (208–754 mg/dL); IgG3, 399 mg/dL (6.6–88 mg/dL); and IgG4, 7.5 mg/dL (4.8–105 mg/dL).
A needle biopsy specimen taken from the submandibular gland showed diffuse infiltration of lymphocytes and plasma cells, together with periductal fibrosis, similar to sclerosing sialadenitis (figure 2A). Obliterative phlebitis was not be found in the specimen. Pathological analysis of the lymph node also revealed marked lymphoplasmacytic infiltration. The kidney biopsy specimen showed diffuse infiltration of lymphoplasmacytes and eosinophils, with marked interstitial fibrosis (figure 2B). Immunohistochemical staining of these three tissues for CD3 and CD20 revealed that these lymphocytes were polyclonal and largely consisted of CD3-positive small T cells. Most of the glomeruli within the specimen showed mild thickening of the capillary walls without spike formation. There was no evidence of crescent formation, endocapillary proliferation, fibrinoid necrosis or thrombosis. Immunofluorescence revealed diffuse granular staining for IgG and C3 in the glomerular basement membrane, but staining for IgA, IgM, C1q, and C4 was negative (figure 2C). Electron microscopy showed subepithelial electron-dense deposits (figure 2D). From these findings, we reached the diagnosis of tubulointerstitial nephritis with membranous nephropathy, a common complication of IgG4RD.4 5 Although immunohistochemical staining for IgG and IgG4 using a mouse monoclonal antibody to human IgG4 (clone HP6025; Invitrogen, Carlsbad, California, USA) revealed numerous IgG-positive cells, there were few IgG4-positive cells. The IgG4-positive to IgG-positive plasma cell ratio was <2% (figure 3).
Figure 2.

Light microscopy, immunofluorescence and electron microscopy findings. (A) Light microscopy of the right submandibular gland shows massive lymphoplasmacytic infiltration with fibrosis (H&E; original magnification, ×200). (B) Light microscopy of the right kidney shows diffuse lymphoplasmacytic infiltration, with marked interstitial fibrosis (H&E; original magnification, ×100). Angitis, granulomatous lesions, neutrophil infiltration and advanced tubulitis were not found. (C) Immunofluorescence of the glomerulus showing diffuse granular staining for IgG in the glomerular basement membrane. (D) Electron microscopy of the glomerulus showing focal effacement of podocyte foot processes, with scattered subepithelial electron-dense deposits.
Figure 3.
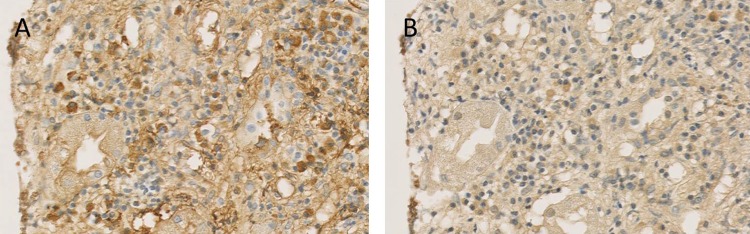
Immunohistochemical staining of the kidney. Immunohistochemical staining of the kidney for IgG (A), and IgG4 (B), shows numerous IgG-positive cells with scant IgG4-positive cells. His IgG4-positive to IgG-positive plasma cell ratio was <2% (original magnification, ×200).
Treatment
Oral prednisolone therapy (0.8 mg/kg/day) was started on day 10 and was continued at this dose for 2 weeks. The dose was then gradually tapered over 12 months.
Outcome and follow-up
Oral prednisolone therapy improved his laboratory abnormalities and the swelling of his affected organs (figure 1). Eosinophilia disappeared shortly after starting treatment while the serum levels of IgG and IgE normalised within 2 months, and the serum complement levels normalised within 3 months. Antibodies against dsDNA and ssDNA, and circulating C1q-immune complexes had disappeared by 3 months. His serum creatine decreased gradually to 1.46 mg/dl at 4 months and was stable thereafter. Urine occult blood had disappeared at 5 months, and urinary protein disappeared by 1 year. The patient has remained stable for >1 year since discontinuing steroid therapy.
Differential diagnosis
IgG4RD is a newly recognised fibroinflammatory condition.1 It affects various organs, including the pancreas, bile ducts, lacrimal gland, salivary gland, central nervous system, thyroid, lung, liver, gastrointestinal tract, kidney, prostate, retroperitoneum, arteries, lymph nodes, skin and breast. Many patients with IgG4RD have synchronous or metachronous lesions in several organs.1–3 Pathologically, IgG4RD is characterised by tumefactive lesions, a dense lymphoplasmacytic infiltrate rich in IgG4-positive plasma cells, storiform fibrosis and eosinophilic infiltration.2 Although the clinical symptoms vary depending on the affected organs, most patients with IgG4RD present with polyclonal γ-globulinaemia, elevated IgE levels, hypocomplementaemia, swelling of the affected organs and delayed enhancement of the pancreas and multiple low-density lesions of the kidney on contrast-enhanced CT. Patients also respond well to steroid therapy.1–4 Based on these typical features, the patient's clinical presentation, laboratory results, imaging findings, histological findings and the response to steroid therapy were consistent with IgG4RD, except there was scant IgG4 in the serum and affected tissues. Although obliterative phlebitis was absent on the biopsy specimen, it is sometimes difficult to obtain a comprehensive histological assessment using needle biopsy samples alone.2 Hence, we searched for possible explanations for the scarcity of IgG4 in this patient. We first thought that the patient had another condition. Malignancies, Sjögren's syndrome, Castleman's disease, granulomatous polyangiitis, sarcoidosis and Churg-Strauss syndrome, must be ruled out in the diagnosis of IgG4RD.2 3 However, these diseases were unlikely in our patient based on the serological, histological and imaging findings, as well as the clinical course. Although serum anti-dsDNA IgG and anti-ssDNA IgG positivity may prompt the diagnosis of systemic lupus erythematosus (SLE), our patient did not fulfil the diagnostic criteria for SLE, nor did he have features suggestive of lupus nephritis in the biopsy specimen.
Another potential explanation for the scarcity of IgG4 in this patient is that the disease had progressed to a fibrotic stage in which fewer plasma cells are present and fibrosis may predominate in the involved tissue.2 However, the dense lymphoplasmacytic infiltrate in the biopsy specimen suggested that the patient was in an active disease stage. Therefore, we finally concluded that the patient had IgG4RD without IgG4 antibodies, or ‘IgG4-negative IgG4RD’.
Discussion
Although approximately 30% of patients with IgG4RD have normal serum IgG4 levels,6 to the best of our knowledge, this is the first case report of a patient who was strongly suspected of having IgG4RD associated with scant IgG4-positive cells in the affected tissues.
Although IgG4RD is so named because it reflects the ubiquity of IgG4 within the affected organs,7 much remains unknown about the role of IgG4 in the aetiology of the disease. Its inability to bind to the C1q complement, its tendency to promote anti-inflammatory processes and the fact that most tissue-infiltrating and circulating IgG4-positive cells are polyclonal suggest that IgG4 antibodies are involved in a response to an inflammatory stimulus, rather than acting as tissue-destructive immunoglobulins.1
This patient supports the hypothesis that IgG4 antibodies do not play a primary role in the aetiology of IgG4RD. This patient also suggests that clinicians should not exclude the diagnosis of IgG4RD in patients lacking elevated IgG4 levels in their affected tissues, particularly if they show other features of IgG4RD. Clinicians should consider steroid therapy in such patients.
Learning points.
Immunoglobulin G4-related disease (IgG4RD) is a disease with an unknown aetiology that is characterised by marked lymphoplasmacytic infiltration of IgG4-positive plasma cells into affected tissues.1–3 However, it is unknown whether IgG4 plays primary or secondary roles in its aetiology.
The findings in this patient suggest that IgG4RD develops without IgG4 antibodies in some patients.
Clinicians should not exclude the diagnosis of IgG4RD in patients lacking elevated IgG4 levels in their affected tissues, particularly if they show other features of IgG4RD. Steroid therapy should be considered in such patients.
Footnotes
Contributors: TM contributed to the creation of the concept of this article. TS contributed to the interpretation and analysis of the pathological points. TM, TS, NH and SM equally contributed to drafting the article and revising it critically for important intellectual content and finally approved the version to be published.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med 2012;2013:539–51 [DOI] [PubMed] [Google Scholar]
- 2.Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol 2012;2013:1181–92 [DOI] [PubMed] [Google Scholar]
- 3.Umehara H, Okazaki K, Masaki Y, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol 2012;2013:21–30 [DOI] [PubMed] [Google Scholar]
- 4.Raissian Y, Nasr SH, Larsen CP, et al. Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol 2011;2013:1343–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexander MP, Larsen CP, Gibson IW, et al. Membranous glomerulonephritis is a manifestation of IgG4-related disease. Kidney Int 2013;2013:455–62 [DOI] [PubMed] [Google Scholar]
- 6.Sah RP, Chari ST. Serologic issues in IgG4-related systemic disease and autoimmune pancreatitis. Curr Opin Rheumatol 2011;2013:108–13 [DOI] [PubMed] [Google Scholar]
- 7.Stone JH, Khosroshahi A, Deshpande V, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum 2012;2013:3061–7 [DOI] [PMC free article] [PubMed] [Google Scholar]