

Abstract
Histone deacetylases (HDACs) emerged as important drug targets in epigenetics. The most common HDAC inhibitors use hydroxamic acids as zinc binding groups despite unfavorable pharmacokinetic properties. A two-stage protocol of M05-2X calculations of a library of 48 fragments in a small model active site, followed by QM/MM hybrid calculations of the full enzyme with selected binders is used to prospectively select potential bidentate zinc binders. The energetics and interaction patterns of several zinc binders not previously used for the inhibition of HDACs are discussed.
The acetylation state of lysine residues on the surface of histones, which plays a key role during epigenetic regulation, is controlled by histone deacetylases (HDACs). Three HDAC classes (class I, II, IV) are zinc dependent, while class III HDACs are NAD+ dependent sirtuins.1 Inhibition of zinc-dependent HDACs shows great potential in cancer therapy2,3 as well as in a wide range of non-cancer disorders.4 Two HDAC inhibitors, suberoylanilide hydroxamic acid (SAHA) and FK228 have been approved for human use by the FDA and more than 20 others are in different stages of clinical trials.
All current inhibitors of HDACs share a common pharmacophore, shown in Figure 1 for SAHA, consisting of a zinc binding group (ZBG) or warhead to chelate the catalytic zinc ion, a capping group binding on the surface of the active site pocket, and a linker between the ZBG and the cap. Although the effects of the cap group5,6 and the linker7 have been explored by this group and others, it is generally thought that the ZBG is to a significant extent responsible for the potency and sometimes the isoform selectivity8 of HDAC inhibitors. Hydroxamates are among the most effective zinc binding group known in both natural9 and non-natural HDAC inhibitors,10 but poor pharmacokinetics11 due to rapid clearance and severe toxicity12 due to non-specific metal binding by hydroxamates have motivated the search for non-hydroxamate ZBGs.13,14,15 Most non-hydroxamate ZBGs are much less potent than their hydroxamic acid counterparts.
Figure 1.
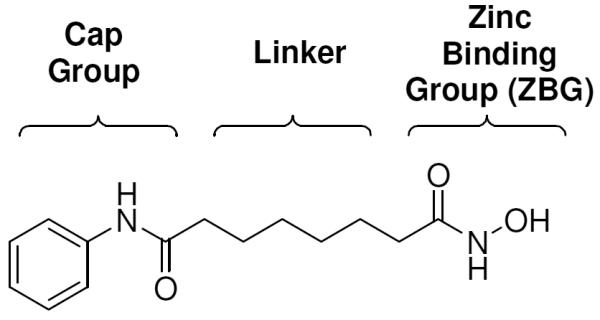
Common pharmacophore for HDAC inhibitors
For the case of matrix metalloproteases (MMPs), another zinc-dependent enzyme family, Cohen and co-workers studied a fragment library.16,17 However, the fragments identified for MMPs are not directly transferrable to HDACs due to the differences in the active site charge and geometry. A similar experimental fragment screening study of ZBGs in HDACs has to the best of our knowledge not yet been performed, presumably due to the nature of the HDAC assay that requires a significant pre-incubation period.10
There have been several computational studies of ZBGs for HDACs. Vanommeslaeghe et al. ranked a series of ZBGs using computational studies of a binding site model and proposed a general framework for chemical groups binding to HDAC.18,19 We used a smaller model to estimate the binding ability of selected ligands and uncovered the importance of the correct pKa of the ZBG.20 McCarren et al. explored the substitution pattern of α-aminocarbonyl groups by tuning electronic effects.21 All of these studies studied relatively small libraries in a retrospective fashion. Here, we present a computational study of ~50 ZBGs in a prospective fashion. We envisioned a two-stage protocol where an initial rapid screen of a small model focuses on the metal binding interactions themselves, followed by a secondary screen of selected binders that for the first time considers a complete model including the secondary interactions in the binding site. The goal of the study is to suggest novel ZBGs similar in potency to the currently studied hydroxamates and other known ZBGs.
Using the approach discussed in the methods section, we calculated a library of putative bi-dentate ligands bound to a small model active site using the M05-2X method. Previous studies22,23 suggest a 5-coordinated zinc in the active site. A library of minimal fragments that allow bidentate binding through any two of oxygen, nitrogen or sulfurs in 1–4, 1–5, or 1–6 positions was thus constructed. Both neutral and anionic ligands were considered, even though ions would be subject to a significant desolvation penalty. Experimental and computational work on zinc enzymes suggest that binding to the zinc ion lowers the pKa of the bound ligands.20,24,25 Weak acids are thus likely to bind the zinc ion in the deprotonated form with the acidic proton transferred to a neighbor His145-Asp179 dyad (residue numbering from HDAC226). It has been proposed that one reason for the high potency of the hydroxamic acid ligand is the fact that, with a pKa of 9.4, it is neutral in solution and therefore not subject to a desolvation penalty, but negatively charged upon coordination to the zinc ion.20 The balance between the energy penalty of the deprotonation process and energy gain from the favorable electrostatic interaction of two new formed opposite charge centers requires the pKa of a ligand to be in a very narrow window of ~9–10. However, the deprotonation process itself beyond the selection of appropriate groups was not considered in this study. Hydroxamic acids, phenols, and sulfhydryl group are therefore considered to be good building blocks for the fragment design. It should be noted, this proposal has been challenged based on QM/MM simulations.27
The final scaffold library contained 48 compounds and is shown in Figure 2 with the coordinating atoms colored in blue. Most of these compounds in the library are known and are commercially available except for 3c, 4c, 5b and 3f, which were not previously described, and 3a and 3b which are not commercially available. In the following discussion, deprotonated and protonated fragments are discussed separately in the sake of clarity. Only promising ZBGs will be discussed in more detail.
Figure 2.

Fragment library with the interaction energy with the fixed small model is showed in red, and atoms designed to coordinate with zinc ion in blue.
Focusing first on the negative fragments, hydroxamates 1a–5a are consistently predicted to be strong zinc binders, in agreement with experiment.17,282a (N-methyl-N'-hydroxyurea) was predicted to be an even stronger binder than 1a, also in agreement with the previous results.18 Although there are small differences to the geometry described there, probably due to the differences in the active site models used, it is gratifying to note that the minimal model is able to rapidly provide rankings in agreement with the much bigger model used previously.18–20 Similarly, compounds 1a, 1b, and a close analog of 1e have been discussed previously for the case of a more flexible model active site, which significantly increases the computational requirements. Correcting for the different reference values of the models used, the binding energy of 1a, 1b and 1e would be −3.0, −1.3 and 5.4 kcal/mol, compared to the binding free energy in Wang's study were −6.7, −4.6, and 8.9 kcal/mol, respectively.20 Although different models and different levels of theory were used, the same rank ordering and similar relative energies are obtained, suggesting that the approximations made in our model are reasonable. To the best of our knowledge, the cyclic hydroxamates 3a and 4a as well as the pyridine-N-oxide 5a, which are likely to be more stable towards hydrolysis, have not yet been studied as ZBGs in HDACs. Based on these results, they are promising candidates for experimental studies.
Besides the hydroxamates 1a–5a, the binding energy comparison predicts hydroxypyrone (1b) and hydroxypyridinones(2b, 4b) to also be favorable binders. Guaiacolate (4d) is reported to form a 1:2 complex with Zn(II) ion by IR spectroscopy analysis.29 With a predicted binding energy similar to hydroxamic acid it is of interest but its pKa is with 9.930 relatively high. Although the transferability of results from MMPs to HDACs is unclear, 8-hydroxyquinoline (5d) which was previously identified as a promising fragment for MMP inhibitors,17 is also predicted to be favorable in the model site discussed here. Interestingly, the sulfur-containing groups, 2c, 3c, 4c, 3b, 5b, and 3d, which were found to be good ZBGs in MMPs17 are predicted to have lower interaction energies than the corresponding mono-dentate fragment, methylthiolate (calculated to be −20.8 kcal/mol), the metal binding group in naturally occurring HDAC inhibitors.5,6 When comparing pairs of compounds where the heteroatom is part of a ring system (e.g. 1b and 6b, 2b and 4b, 4c and 3c, 5c and 6c, it is interesting to note that the isomer with the heteroatoms in the ring closer to the metal binding substitutions consistently have a lower binding energy than those with the heteroatom further away. This can be rationalized by the fact that heteroatoms in these ring systems have same resonance effect to contribute a pair of electrons to form an aromatic system, but the inductive effect in the nearby position is stronger.
Among the neutral fragments, 1f (4-hydroxymethylimidazole) is ranked to have the most favorable interactions, significantly stronger than the benzamidite 1e that acts as the ZBG in widely studied HDAC inhibitors such as MS275. The 3'-N of the imidazole has a favorable interaction with the zinc ion and the hydroxyl group forms a hydrogen bond with the carboxyl group in the active site. Fragments related to 1f such as 1h, 2f, 2i, 2h and 2e are calculated to have similar binding energies and contain an amino group. Amino groups have been used as ZBGs in neutral HDAC inhibitors in other contexts (e.g. α-amino carbonyls in PDB 3MAX and 3SFF). In addition to acting as a Lewis base, the amino group can form two additional hydrogen bonds with His145 and His146 in the active site. As will be discussed in more detail below, an investigation of these interactions requires a more complete model. Although the hydroxypyrones and hydroxypyridinone anions discussed earlier chelate the zinc ion well, the same is not found for the analogous neutral aminopyrone and aminopyridinone which cannot be deprotonated in the active site and which is a weaker Lewis base due to the conjugation of the free electron pair to the aromatic system. Nevertheless, the aliphatic amines are reasonable Lewis acids while the corresponding amides are predicted to not bind well, as indicated by the comparison of 2f with 5f and 2i with 4i. The cycloalkane-1,2-diol compounds, 1g–5g all have a similar binding affinity similar to 1e. The conformation of these cycloalkane systems differs significantly, but has only a small effect on the calculated binding affinity. This could potentially be exploited in a model structure to display other substituents at well-defined positions in the pocket without changing the binding to the zinc significantly. Recently, trifluoromethyloxadiazole (TFMO) was introduced as a novel ZBG for class IIa HDACs.31 The binding energy of TMFO was calculated to be the weakest among the ligands studied here, in line with the authors assertion that it is a non-chelating group.
Although all these fragments were designed to coordinate the zinc ion in a bidendate fashion, about half of the fragments showed significant differences in the bond lengths between the zinc and the heteroatoms (for detailed information, see Tables S1 and S2 in the Supporting Information), making the effectively mono-dentate binders modes. This is due to the localization of the negative charge on one of the heteroatoms, and could be more pronounced interaction of the heteroatoms with other atoms. In the actual active site, this could include the side chains of Tyr308, His145 and His146 which are positioned to possibly form important hydrogen bonds with the ligand and which are not included in the minimal model. The pharmacophore model proposed in Vanommeslaeghe et al.18,19 includes some of these interactions, but even this model is necessarily limited in size and it is not clear if all relevant interactions are captured.
To circumvent this problem and to investigate whether these fragments could form either favorable or unfavorable interaction with residues close to the active site, we performed QM/MM hybrid calculations of the complete system based on the crystal structure of HDAC2 (PDB 3MAX26) with selected fragments. The top ranking fragments from Figure 2 were superimposed back to the model atom in the crystal structure. 8-hydroxyquinoline (5d, shown in orange in Figure 3 left), despite its favorable binding energy with the model active site, has a number of unfavorable steric interactions in the small binding site and was therefore not pursued further. In contrast, fragments 1a, 2a, 3a, 4a, 6d, 1e, 2e, 1h, 2h, and 3h, which have good binding energies, fit well in the pocket, and form good interactions with neighboring residues in the active site pocket that could potentially be extended by adding substituents as part of further hits-to-lead development. 11 neutral and 12 anionic fragments were studied by QM/MM optimizations. 4a and 1h (Figure 3 middle and right) are discussed as representative examples while the structures of the remaining ligands can be found in the Supporting Information.
Figure 3.

left: Optimized Structure of 5d (orange), 4a (yellow) and 1h (green) in the HDAC2 pocket: the optimized structure of fragment and model complex is superposed into the crystal structure of HDAC2 (PDB 3MAX). The binding conformation of 4a (middle) and 1h (right) in HDAC2 binding site after QM/MM optimization
Hydroxamate 4a was studied in the deprotonated form with a proton transferred to His145. After QM/MM optimization, the conformation obtained is similar to the one observed to a hydroxamate inhibitor bound to HDAC8 (PDB 2V5X), except for the hydrogen bond with His146, presumably because the six membered ring prevents the reorientation necessary for the formation of this bond. It is interesting to note that in agreement with the proton shuttle mechanism proposed by Finnin et al. and reminiscent of the catalytic triade of serine proteases,32 the proton is shared between His145 and Asp179. Figure 3, middle, also shows the extensive hydrogen bonding network 4a engages in, illustrating the value of studies of selected ZBGs in the full model.
This hydrogen bonding network is even more important for neutral fragments such as 1h, shown in Figure 3 on the right, where the interaction with Zn2+ is weaker as discussed earlier. The hydroxyl group is hydrogen bonded to Tyr308 as well as to Asp269. The amine group forms two hydrogen bonds with His145 and His146 in addition to the complexation to Zn2+. Both compounds are well accommodated in the pocket, and could be promising ZBGs in HDAC inhibitors.
In summary, a new, two-step strategy to identify potential zinc binding fragments for HDAC inhibitors was used. Electronic structure calculations were used to rapidly screen a fragment library to identify ZBGs with good binding affinity in a small active site model. Promising candidates were studied in the protein pocket, first by simple positions to check for steric interactions, followed by QM/MM optimization of the fragments in the real protein environment. The QM/MM calculations allow a comparison of the results from the small model with the full system and emphasize the importance of the secondary interactions in the active site, especially for neutral ligands. The results are in good agreement with the available experimental data for the known ZBGs and predict additional ZBGs such as α-aminoalcohols, guaiacolate, and 2-(aminomethyl)pyridines that are promising candidates for experimental studies.
Methods
Model building
A fixed small active site model, shown in Scheme 1 consisting of one 4-methylimidazole and two acetates, representing the first coordination shell of zinc by Asp181, His183, and Asp269 was used. The coordinates of the model were extracted from the human HDAC2 (PDB 3MAX) of 2.05 Å resolution, with hydrogens added. As outlined by Vanommeslaeghe et al.18 and McCarren et al.,21 a fixed model is able to avoid the problem of changes in the side chain conformations in the model that are unlikely to occur in the enzyme. In addition, a structural comparison of the first coordination shell in the available human HDAC PDB structures23 shows that the Zn ion and the first shell residues are rigid. (See Supporting Information, Figure S1) Different conformations and protonation states were considered where appropriate, and only the most stable structure is discussed here.
Scheme 1.
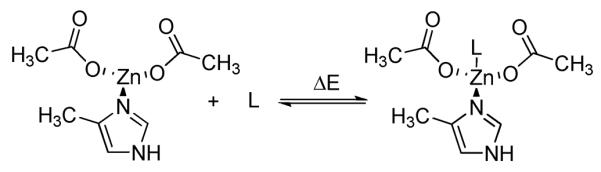
Reaction for calculating the binding energy of zinc binding group L
To ensure the steric compatibility with the deeply burried binding site of HDACs, each ligand was manually docked into the pocket of HDAC2 (3MAX) to confirm a bidentate mode and avoid a possible steric repulsion between the ligand and the neighboring residues in the pocket. The coordinates of the ligand and small active site model were then extracted from the complex structure with hydrogen added appropriately. The ligands were optimized in the presence of the fixed active site model where the heavy atoms in the model are fixed. After optimization, the complex with the fixed model and ligand was superimposed with the model in the protein (3MAX) to visualize the conformation of the ligand in the real pocket environment. If there were several docked conformations for a ligand, the conformation which with the lowest binding energy and without apparent repulsion with the protein in the optimized structure were used for comparison with other ligands.
The initial structure of the protein for the QM/MM calculations was prepared from the crystal structure of the complex of HDAC2 with N-(4-aminobiphenyl-3-yl)benzamide (3MAX). Waters and ions, except for the catalytic zinc ion, as well as alternate positions were excluded using SwissPdb Viewer.33 Hydrogen atoms were added and optimized using Protein Local Optimization Program (PLOP).34 The protonation states of the charged residues were checked manually, with all Asp and Glu in a negative charge state, and all Arg and Lys in a positive charge state. In analogy to our earlier MD simulations,5 His146 and His286 were treated as singly protonated on the δ site, His33, His38, His44, His62, His73, His172, His183, His184, His204, and His349 as singly protonated on the ε site. Consistent with our earlier work,35,36 His145 was treated as doubly protonated when bound with a negative ligand (e.g. 4a), but as singly protonated on the δ site when bound with a neutral ligand, 1h.
DFT Calculations
All DFT calculations were carried out using G09.37 Based on the benchmark studies of Sorkin et al.,38 the M05-2X39 functional together with a 6–31+g* basis set on all main group elements and 6–311g* on Zn2+ were used. The SMD40 solvent model for water was used in all the optimizations to represent the polar environment in the active site of HDAC. The structures for the small model without ligand (fixed heavy atoms), the ligand, and the small model with the ligand (with all the heavy atoms in the model fixed) were optimized with the solvent effect of water modeled by SMD, and the binding energy was calculated as shown in Scheme 1.
QM/MM Calculation
For the QM and MM partition, the QM subsystem included the side chains of His145, His146, Asp179, Asp181, His183, and Asp269, the phenol group of Tyr308, the ligand, and the catalytic zinc ion. Hydrogen atoms were added to saturate the dangling bond at the QM/MM boundary. The ligand conformations (minimally rotated or translated if necessary) were taken from the DFT optimization with the fixed small active site model. The QM part was treated by the M05-2X functional with 6–311g* for zinc atom and 6–31+g* for all other QM atoms. The MM level was described by the Amber force field (parm99). TAO41 was used for the preparation and analysis of the ONIOM calculations. The zinc parameters developed by Merz and coworkers were used.42 Atoms within a 20 Å distance from the zinc ion were allowed to move. Other atoms in the system were fixed to decrease the energy fluctuation and save CPU time. An electronic embedding scheme was used for the geometry optimization of 4a and 1h to consider the electronic interaction between QM and MM parts in detail, while a mechanical embedding scheme was used for the other examples listed in the Supporting Information
Supplementary Material
Acknowledgments
We gratefully acknowledge the support of this work by the National Institutes of Health (RO1 CA152314), the National Science Foundation of China (21133002), and the Shenzhen Peacock Program (KQTD201103). We also thank Brandon Haines at the University of Notre Dame for help with the ONIOM calculations in G09 and Dr. Xinhao Zhang and Prof. Yun-Dong Wu (PKUSZ) for helpful discussions.
Footnotes
Supporting Information Cartesian coordinates and energies of all species discussed as well as pdb files for the QM/MM calculations. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- (1).Gregoretti IV, Lee Y-M, Goodson HV. J. Mol. Biol. 2004;17 doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- (2).Pontiki E, Hadjipavlou-Litina D. Med. Res. Rev. 2012;32:1. doi: 10.1002/med.20200. [DOI] [PubMed] [Google Scholar]
- (3).Bertrand P. Eur. J. Med. Chem. 2010;45:2095. doi: 10.1016/j.ejmech.2010.02.030. [DOI] [PubMed] [Google Scholar]
- (4).Wiech NL, Fischer JF, Helquist P, Wiest O. Curr. Top. Med. Chem. 2009;9:257. doi: 10.2174/156802609788085241. [DOI] [PubMed] [Google Scholar]
- (5).Bowers AA, Greshock TJ, West N, Estiu G, Schreiber SL, Wiest O, Williams RM, Bradner JE. J. Am. Chem. Soc. 2009;131:2900. doi: 10.1021/ja807772w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bowers AA, West N, Newkirk TL, Troutman-Youngman AE, Schreiber SL, Wiest O, Bradner JE, Williams RM. Org. Lett. 2009;11:1301. doi: 10.1021/ol900078k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Weerasinghe SVW, Estiu G, Wiest O, Pflum MK. J. Med. Chem. 2008;51:5543. doi: 10.1021/jm800081j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Methot JL, Chakravarty PK, Chenard M, Close J, Cruz JC, Dahlberg WK, Fleming J, Hamblett CL, Hamill JE, Harrington P, Harsch A, Heidebrecht R, Hughes B, Jung J, Kenific CM, Kral AM, Meinke PT, Middleton RE, Ozerova N, Sloman DL, Stanton MG, Szewczak AA, Tyagarajan S, Witter DJ, Secrist JP, Miller TA. Bioorg. Med. Chem. Lett. 2008;18:973. doi: 10.1016/j.bmcl.2007.12.031. [DOI] [PubMed] [Google Scholar]
- (9).Fennell KA, Moellmann U, Miller MJ. J. Org. Chem. 2008;73:1018. doi: 10.1021/jo7020532. [DOI] [PubMed] [Google Scholar]
- (10).Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R. Nat. Chem. Biol. 2010;6:238. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Summers JB, Gunn BP, Mazdiyasni H, Goetze AM, Young PR, Bouska JB, Dyer RD, Brooks DW, Carter GW. J. Med. Chem. 1987;30:2121. doi: 10.1021/jm00394a032. [DOI] [PubMed] [Google Scholar]
- (12).Kelly WK, Richon VM, O'Connor O, Curley T, MacGregor-Curtelli B, Tong W, Klang M, Schwartz L, Richardson S, Rosa E, Drobnjak M, Cordon-Cordo C, Chiao JH, Rifkind R, Marks PA, Scher H. Clin. Cancer Res. 2003;9:3578. [PubMed] [Google Scholar]
- (13).Suzuki T, Matsuura A, Kouketsu A, Nakagawa H, Miyata N. Bioorg. Med. Chem. Lett. 2005;15:331. doi: 10.1016/j.bmcl.2004.10.074. [DOI] [PubMed] [Google Scholar]
- (14).Suzuki T, Miyata N. Mini-Rev. Med. Chem. 2006;6:515. doi: 10.2174/138955706776876186. [DOI] [PubMed] [Google Scholar]
- (15).Suzuki T, Miyata N. Curr. Med. Chem. 2005;12:2867. doi: 10.2174/092986705774454706. [DOI] [PubMed] [Google Scholar]
- (16).Agrawal A, Johnson SL, Jacobsen JA, Miller MT, Chen L-H, Pellecchia M, Cohen SM. ChemMedChem. 2010;5:195. doi: 10.1002/cmdc.200900516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Jacobsen JA, Fullagar JL, Miller MT, Cohen SM. J. Med. Chem. 2011;54:591. doi: 10.1021/jm101266s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Vanommeslaeghe K, Loverix S, Geerlings P, Tourwé D. Bioorg. Med. Chem. 2005;13:6070. doi: 10.1016/j.bmc.2005.06.009. [DOI] [PubMed] [Google Scholar]
- (19).Vanommeslaeghe K, Proft FD, Loverix S, Tourwé D, Geerlings P. Bioorg. Med. Chem. 2005;13:3987. doi: 10.1016/j.bmc.2005.04.001. [DOI] [PubMed] [Google Scholar]
- (20).Wang D, Helquist P, Wiest O. J. Org. Chem. 2007;72:5446. doi: 10.1021/jo070739s. [DOI] [PubMed] [Google Scholar]
- (21).McCarren P, Hall ML, Whitehead L. Chem. Biol. Drug. Des. 2012;80:203. doi: 10.1111/j.1747-0285.2012.01382.x. [DOI] [PubMed] [Google Scholar]
- (22).Wu R, Hu P, Wang S, Cao Z, Zhang Y. J. Chem. Theory Comput. 2009;6:337. doi: 10.1021/ct9005322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ficner R. Curr. Top. Med. Chem. 2009;9:235. doi: 10.2174/156802609788085304. [DOI] [PubMed] [Google Scholar]
- (24).Cross JB, Duca JS, Kaminski JJ, Madison VS. J. Am. Chem. Soc. 2002;124:11004. doi: 10.1021/ja0201810. [DOI] [PubMed] [Google Scholar]
- (25).Hightower KE, Huang C.-c., Casey PJ, Fierke CA. Biochemistry. 1998;37:15555. doi: 10.1021/bi981525v. [DOI] [PubMed] [Google Scholar]
- (26).Bressi JC, Jennings AJ, Skene R, Wu Y, Melkus R, Jong RD, O'Connell S, Grimshaw CE, Navre M, Gangloff AR. Bioorg. Med. Chem. Lett. 2010;20:3142. doi: 10.1016/j.bmcl.2010.03.091. [DOI] [PubMed] [Google Scholar]
- (27).Wu R, Lu Z, Cao Z, Zhang Y. J. Am. Chem. Soc. 2011;133:6110. doi: 10.1021/ja111104p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Puerta DT, Mongan J, Tran BL, McCammon JA, Cohen SM. J. Am. Chem. Soc. 2005;127:14148. doi: 10.1021/ja054558o. [DOI] [PubMed] [Google Scholar]
- (29).Omura Y. Spectrochim. Acta Part A. 1998;54:507. [Google Scholar]
- (30).Serjeant EP, Dempsey B. Ionisation Constants of Organic Acids in Aqueous Solutions. Pergamon; New York: 1979. [Google Scholar]
- (31).Lobera MM, K. P., Pohlhaus DT, Wright QG, Trocha M, Schmidt DR, Baloglu E, Trump RP, Head MS, Hofmann GA, Murray-Thompson M, Schwartz B, Chakravorty S, Wu Z, Mander PK, Kruidenier L, Reid RA, ; Burkhart WT,BJ, Rong JX, Wagner C, Moyer MB, Wells C, Hong X, Moore JT, Williams JD, Soler D, Ghosh S, Nolan MA. Nature Chem. Biol. 2013 doi: 10.1038/nchembio.1223. advance online publication doi:10.1038/nchembio.1223. [DOI] [PubMed] [Google Scholar]
- (32).Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Nature. 1999;401:188. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- (33).Guex N, Peitsch MC. Electrophoresis. 1997;18:2714. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- (34).Jacobson MP, Pincus DL, Rapp CS, Day TJF, Honig B, Shaw DE, Friesner RA. Proteins: Struct., Funct., Bioinform. 2004;55:351. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- (35).Estiu G, Greenberg E, Harrison CB, Mazitschek R, Bradner JE, Wiest O. J. Med. Chem. 2008;51:2898. doi: 10.1021/jm7015254. [DOI] [PubMed] [Google Scholar]
- (36).Estiu G, Greenberg E, Mazitschek R, Bradner JE, Wiest O. Bioorg. Med. Chem. 2010;18:4103. doi: 10.1016/j.bmc.2010.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery J,JA, ; Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian, Inc.; Wallingford CT: 2009. [Google Scholar]
- (38).Sorkin A, Truhlar DG, Amin EA. J. Chem. Theory Comput. 2009;5:1254. doi: 10.1021/ct900038m. [DOI] [PubMed] [Google Scholar]
- (39).Zhao Y, Schultz NE, Truhlar DG. J. Chem. Theory Comput. 2006;2:364. doi: 10.1021/ct0502763. [DOI] [PubMed] [Google Scholar]
- (40).Marenich AV, Cramer CJ, Truhlar DG. J. Phys. Chem. B. 2009;113:6378. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- (41).Tao P, Schlegel HB. J. Comput. Chem. 2010;31:2363. doi: 10.1002/jcc.21524. [DOI] [PubMed] [Google Scholar]
- (42).Hoops SC, Anderson KW, Merz KM. J. Am. Chem. Soc. 1991;114:8262. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.