

Abstract
Aim
Cannabinoid receptor type 1 (CB1) antagonists have been developed for the treatment of obesity and associated risk factors. Surinabant is a high affinity CB1 antagonist in vitro. The aim of this study was to assess the magnitude of inhibition by surinabant of CNS effects and heart rate induced by Δ9-tetrahydrocannabinol (THC) in humans.
Methods
This was a double-blind, placebo-controlled, randomized, four period six sequence crossover study. Thirty healthy young male occasional cannabis users (<1 per week) were included. A single oral dose of surinabant (5, 20 or 60 mg) or placebo was administered followed 1.5 h later by four intrapulmonary THC doses (2, 4, 6 and 6 mg) or vehicle, administered at 1 h intervals. The wash-out period was 14–21 days. Subjective and objective pharmacodynamic (PD) measurements were performed. A population PK–PD model for THC and surinabant quantified PK and PD effects.
Results
Surinabant 20 and 60 mg inhibited all THC-induced PD effects in a similar range for both doses with inhibition ratios ranging from 68.3% (95% CI = 32.5, 104.2; heart rate) to 91.1% (95% CI = 30.3, 151.8; body sway). IC50 ranged from 22.0 ng ml−1 [relative standard error (RSE) = 45.2%; body sway] to 58.8 ng ml−1 (RSE = 44.2%; internal perception). Surinabant 5 mg demonstrated no significant effects.
Conclusions
The dose-related inhibition by surinabant, without any effect of its own, suggests that this compound behaves as a CB1 receptor antagonist in humans at these concentrations. A single surinabant dose between 5 to 20 mg and above was able to antagonize THC-induced effects in humans.
Keywords: CB1 receptor antagonist, pharmacodynamics, pharmacokinetics, PK-PD modelling, SR147778, surinabant
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Rimonabant was the first cannabinoid receptor type 1 (CB1) inverse agonist that reached the market. However, with the doses used in patients, it caused severe side effects. We wanted to investigate the dose–effect profile and the effective dose range of surinabant, a new CB1 antagonist in healthy subjects.
WHAT THIS STUDY ADDS
This study provides insight into the effective surinabant dose range in healthy subjects using a Δ9-tetrahydrocannabinol (THC) challenge test. Surinabant is able to inhibit various tests, such as heart rate, body sway and feeling high. In our model, surinabant is already maximally effective at single doses of 20 mg, and possibly even below.
Introduction
Research on the cannabinoid system started several decades ago with the isolation of Δ9-tetrahydrocannabidiol (THC) from the plant Cannabis sativa 1. Since the 1990s, when cannabinoid receptors type 1 (CB1) and type 2 (CB2) 2 were cloned, the number of studies on the cannabinoid system and its application to medical practice has increased rapidly 3, 4. Modulators of CB1 receptors became of special interest for medical indications. CB1 receptors are mainly located in brain areas such as the cortex, basal ganglia, hippocampus, hypothalamus and cerebellum, in the spinal cord, and in peripheral tissues such as adipose tissue, the heart and intestines 5. THC is the most well-known agonist of the CB1 receptor and induces a wide range of effects corresponding to the widespread location of CB1 receptors. These effects include involvement in feeding behaviour and pain 5–8.
In the 1990s, the alimentary effects led to the theory that if appetite enhancement is regulated by CB1 receptors, then antagonism of these receptors would suppress appetite, resulting in weight loss. With the increasing global problem of obesity and related factors, this topic became of special interest for pharmaceutical companies. From 1994, the first CB1 inverse agonist rimonabant (at that time believed to be an antagonist) was discovered and developed by Sanofi 9. Besides efficacy in obesity and associated risk factors 6, 7, results from pre-clinical and clinical research also showed the beneficial effects of CB1 antagonists on alcohol and nicotine abuse 10–13. In 2006, the European Commission granted a marketing authorization for rimonabant as an adjunct to diet and exercise for the treatment of obese patients, or overweight patients with associated risk factors such as dyslipidaemia, diabetes mellitus type 2 or cardiovascular risk factors 14.
However, after a recommendation of suspension of rimonabant's marketing authorization by the European Medicines Agency (EMEA) in 2008, rimonabant was withdrawn from the market 15. The EMEA had drawn the conclusion that the weight loss did not outweigh the psychiatric side effects, especially depression 16. At around the same time, Merck announced the withdrawal of their CB1-antagonist taranabant from phase II and III studies for the indications of smoking cessation and obesity, also due to psychiatric side effects including depression, irritability, anxiety and suicidality 17–21. The results of clinical studies on rimonabant and taranabant showed that both the desired and undesired effects were dose related, with greater efficacy and more adverse events at the highest doses 7, 17–19. While the significant weight loss effects can only be measured after a few weeks, Morrison et al. reported that with taranabant, the largest percentage of psychiatric adverse events occurred within the first 4 days of treatment 21.
These clinical findings with CB1 antagonists do not invalidate attempts to address obesity treatment or smoking cessation via antagonism of the CB1 receptor, although clearly careful attention should be paid to potentially harmful effects 22. The clinically effective level might be found in a lower dose range of the CB1 antagonist compared with doses that cause psychiatric side effects 23. In the available literature on CB1 antagonists, there is a lack of information on different dose or plasma concentration ranges, and the relation between the various pharmacodynamic parameters, i.e. efficacy parameters and safety profile. Therefore, for future CB1 antagonist studies a possible safety window between clinically effective dose levels and doses with undesirable effects should be examined carefully.
For example, rimonabant 20 mg demonstrated a reduction of both weight gain and smoking cessation in humans, whereas Tonstad & Aubin found, that CB1 antagonist surinabant 5 mg did not improve smoking cessation, but had a small effect on reducing weight gain 24, 25.
Acute administration of CB1 antagonists does not give measurable effects in healthy volunteers, which hampers the accurate determination of dose–response relationships and prediction of minimal pharmacological effect levels in early drug development. Therefore, we previously developed the THC challenge test 26, 27. This test is used to quantify the displacement of the concentration–effect curve of the CB1 agonist THC, by different doses of a CB1 antagonist for various pharmacodynamic parameters. The THC-challenge test showed clear dose-related effects of the CB1 antagonist drinabant (AVE1625) in a previous study, after single doses that did not cause any detectable effect of their own, and which were lower than predicted from preclinical experiments 26. As a consequence, the dose range for subsequent phase II trials was reduced. A very recent study on smoking cessation found that another CB1 antagonist, surinabant, had a small effect on weight gain, whereas it had no effect on smoking cessation 24.
After repeated dose oral administration for 14 days in young subjects, surinabant was rapidly absorbed with a median tmax of ∼2 h. After a single dose administration of 20 to 80 mg, Cmax and AUC increased less than dose proportionally (2.0-and 2.9-fold respectively). The 4-fold dose increase in a repeated dosing study had a 2.1- and 2.1-fold increase of Cmax and AUC(0,24h). The terminal half-life was not dose proportional and ranged between 161 and 183 h for 14 day multiple doses (20 to 80 mg day−1). Steady-state was achieved by day 13 and the mean accumulation ratios were 1.3 (Cmax) and <2.6 (AUC(0,24h)) 28. A previous pharmacokinetic trial in humans found that surinabant elimination took place primarily through the faeces 29. An in vitro study identified CYP3A4 as the major CYP isoform involved in the metabolism of surinabant 30.
The aim of this study was to investigate the pharmacodynamic/pharmacokinetic relationships of surinabant using the THC challenge test in healthy volunteers.
Methods
Study design
This was a single centre double-blind, placebo-controlled, randomized, six treatment, four period, six sequence incomplete balanced crossover study with a wash-out period of at least 2 weeks.
Subjects and power calculation
Healthy male volunteers aged 18 to 45 years were included in the study. Subjects had to be cannabis users for at least 1 year with a frequency of use of no more than once a week to minimize the risk on adverse effects from naive subjects, as well as to avoid tolerance. Subjects had to be able to refrain from using cannabinoids from at least 3 weeks prior to the first treatment period up to the end of the study.
Thirty-six subjects were planned to be randomized and treated in order to obtain at least 24 subjects completing the four periods (four subjects per sequence, each treatment given to a total of 16 subjects). A sample size of 16 subjects per treatment group was to provide a power of at least 90% to demonstrate a 50% inhibition of THC-induced effect on body sway, alertness and feeling high, using a two-sided paired t-test at the 5% alpha level. These parameters gave consistent and robust THC effects in previous studies, and were therefore chosen for the power calculation 26, 27, 31. Calculations were based on CB1 antagonist placebo + THC effects and within-subject standard deviations as demonstrated in a previous study 26.
Procedure
Subjects gave written informed consent after full explanation of what was involved, and before any study specific procedure was performed. Eligible subjects were enrolled in the study after a general health screen within 3 weeks before the first study day. Subjects were acquainted with the experimental methods and conditions in a training session including the inhalation procedure using THC vehicle. An alcohol breath test and urine drug screen had to be negative on each study day. Pharmacodynamic (PD) and pharmacokinetic (PK) measurements were frequently performed on all study days. A follow-up visit was scheduled between 12 and 18 days after the last study day. The study protocol was approved by the Medical Ethics Review Board of Leiden University Medical Center and complied with the principles of ICH-GCP, the Helsinki declaration and Dutch laws and regulations.
Treatments
Subjects received randomized administration of four out of the following six treatments: surinabant 5 mg or 20 mg or 60 mg + THC, surinabant 60 mg + placebo THC, placebo surinabant + THC, and placebo surinabant + placebo THC. Starting from the expected tmax of surinabant at 1.5 h, four doses of inhaled THC (2, 4, 6 and 6 mg) or placebo were administered at 1 h intervals.
Surinabant was administered as oral capsules (Sanofi-Synthélabo Recherche, Toulouse, France). The soft gelatin capsules contained 5 mg, 10 mg or 20 mg surinabant or placebo and the following excipients: polyoxyl 40 hydrogenated castor oil, propylene glycol monolaurate type II, triglycerides medium-chain (caprylic-capric acid 60-40), caprylocaproyl macrogolglycerides type 400, gelatin, glycerol, titanium dioxide and purified water.
THC 2, 4, and 6 mg was diluted in 200 μl 100% ethanol (Farmalyse b.v., Zaandam, the Netherlands) or THC vehicle, which consisted of ethanol only. This amount of ethanol was considered too small to cause any effects that would interfere with THC effects. The THC was vaporized into a balloon using a Volcano vaporizer® (Storz & Bickel GmbH & Co. KG, Tuttlingen, Germany). Subjects inhaled the full contents of the balloon within 2 min using a standard paced puffing protocol as previously described by Zuurman et al. 27.
Surinabant dosages were selected in order to obtain sub-effective and effective plasma concentrations, based on phase 2 efficacy results in obesity and on PK data from a phase 1 study (study numbers DRI5029 and TDR 5736, data on file). THC dosages were selected in order to reach and maintain clear, sub-maximal central nervous system effects, based on PK–PD model simulations that were based on a previous study 31. Procedures to evaporate the solution and inhalation of the vapour were done according to a method previously described by Zuurman et al. 27.
Outcome measures
Pharmacokinetic measurements
For surinabant and THC PK analysis, venous blood samples were taken via a cannula that was inserted at the start of the study day 30 min after arrival, before any measurements were performed. Surinabant samples were drawn pre-dose and at fixed time points after dosing from t = 45 min up to t = 24 h. THC samples were taken pre-dose and three times after each of the first three THC administrations, and four times after the fourth THC administration.
Pharmacodynamic assessments
The choice of the PD endpoints was based on a previous review and previous studies by Zuurman et al. 26, 27, 32. The PD measurements were performed twice pre-dose, twice after surinabant administration before the first THC inhalation, three times after each of the first three THC inhalations and nine times after the fourth THC inhalation up to t = 9 h 16 min. Vital signs (heart rate and blood pressure) were measured ten times per study day of which two were pre-dose.
Body sway
The body sway meter (André Ibelings, TNO/ICT, Delft, the Netherlands) is an objective assessment of antero-postural sway in mm per 2 min. The antero-postural sway is regulated by different factors, such as attention and motor coordination, involving the central and peripheral nervous system and vestibular processes. Visual feedback was eliminated by closing the eyes. Measurements were performed according to a procedure previously described 27.
Visual analogue scales (VAS)
VAS by Bond & Lader is a 16-item assessment of subjective effect on alertness (composition of items alert/drowsy, strong/feeble, muzzy/clear-handed, well coordinated/clumsy, lethargic/energetic, mentally slow/quick-witted, attentive/dreamy, incompetent/proficient and interested/bored), on mood (composition of items contended/discontended, troubled/tranquil, happy/sad, antagonistic/amicable, and withdrawn/gregarious) and calmness (composition of items calm/excited and tense/relaxed) 33. The adapted version of VAS by Bowdle 34 is a 13-item assessment of subjective effects on feeling high and on factors of internal and external perception, which are both compositions of items that are affected differently by THC as previously described 27.
Heart rate and blood pressure
Heart rate and blood pressure were measured using the Nihon-Koden (Lifescope EC, Tokyo, Japan) blood pressure apparatus. All heart rate measurements were used for PD analysis.
Adverse events and concomitant medication were continuously recorded from screening until the follow-up period.
Bioanalyses
Surinabant samples
Venous blood was collected in 4.5 ml EDTA tubes. The blood samples were kept on ice and centrifuged within 30 min of collection at 2000 g at 4°C for 10 min. The plasma was transferred into 2 ml Sarstedt polypropylene tubes and stored at −20°C. Samples were analyzed by the Global Metabolism and Pharmacokinetics department of Sanofi (Malvern, PA, USA) using a liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) method with a lower limit of quantification (LLOQ) of 1.0 ng ml−1.
THC samples
For determination of the concentration of plasma THC and its metabolites 11-hydroxy-THC (11-OH-THC) and 11-nor-9-carboxy-THC (THC-COOH) venous blood was collected in 4 ml EDTA tubes. As cannabinoids are photosensitive compounds, samples were protected from light at all times. The tubes were kept on ice and centrifuged for 10 min at 2000 g at 4°C. The plasma was transferred into 2 ml brown Sarstedt polypropylane tubes and stored at −20°C. Plasma samples were analyzed by PRA International (Zuidlaren, the Netherlands). Plasma THC as well as metabolite concentrations (11-OH-THC and THC-COOH) were determined using a LC-MS/MS method with a LLOQ of 0.5 ng ml−1.
Statistical analyses
Adverse effects
Evaluation of the safety data was based on the review of individual values and descriptive statistics. Vital signs (heart rate and blood pressure) were analyzed using descriptive statistics. Adverse events were coded according to the Medical Dictionary for Regulatory Activities (MedDRA version 9.0).
Non-compartmental pharmacokinetics
PK parameters of surinabant, THC, 11-OH-THC and THC-COOH were determined for each period by non-compartmental analysis of plasma concentrations and real time values using PKDMS Version 1.3 with WinNonlin Professional Version 4.01.
Pharmacodynamics
PD parameters were analyzed using a linear mixed effect model with treatment, period, time and treatment by time as fixed effects, subject and subject by treatment as random effects and with the baseline value as covariate. The baseline value was defined as the calculated mean of pre-dose assessments for each occasion. From this model, pairwise differences and corresponding 95% confidence intervals (CI) were estimated to verify the effects of THC and to assess the intrinsic and inhibitory activity of surinabant. This analysis was conducted on data measured from the third THC inhalation up to 3 h after the fourth inhalation to measure at maximum THC effects. The model was fitted by estimated generalized least squares using SAS PROC MIXED.
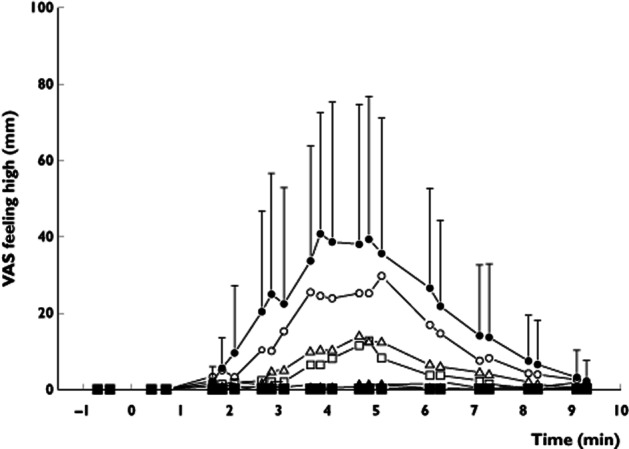
Inhibition ratios as defined in percentages were estimated (with 95% CI) within the mixed model framework for each surinabant dose separately using the following formula below. Each parameter in the formula represents the effect that was measured at a certain time point for the indicated treatment:
 |
Body sway data and item ‘feeling high’ on the VAS Bowdle were analyzed after log (VAS score + 2) transformation.
Population PK–PD modelling
Population PK and PK–PD modelling was performed using the non-linear mixed effect modelling package nonmem (version 5, ICON Development Solutions, Ellicott City, MD, USA) 35 running on a Linux cluster 36. Model development was guided by visual (goodness of fit plots) and statistical criteria based on minimization of the objective function value, uncertainty of parameter estimates and biologically plausible values. The first order conditional estimation method with interaction (FOCE-I) was used throughout the analysis.
Population PK models were developed to describe the time course of surinabant and THC concentrations. Subsequently, PK–PD models were developed for the separate PD measures that quantify the relationship between the plasma concentrations of surinabant and THC and the observed effects, using an agonist-antagonist interaction model, as shown in Equation 1:
 |
(1) |
| (2) |
Equation 2 models the effect at a specific time point and occasion. The empirical Bayes estimates of the individual PK parameters were used to develop separate PK–PD models for the evaluated PD parameters.
The PK–PD relationship for THC was described using an effect compartment model in which the effect compartment rate constant (Keo) accounts for the delay between PK and PD (i.e. hysteresis). This parameter can also be expressed as the effect compartment equilibrium half-life (t50), which was calculated by the following equation:
| (3) |
The relation between the effect compartment concentration and the observed effect was initially modelled using a maximal effect model, in terms of baseline, EC50 and Emax. When the data showed no maximal effect relationship, a linear slope function was estimated.
As all subjects had PK sampling on more than one occasion for THC, interoccasion variability (IOV) was evaluated for the relative bioavailability. A THC dose was defined as an occasion. Interindividual variability (IIV) and IOV in a PK parameter, P, were included in the model and assumed to be log-normally distributed, according to Equation 4:
| (4) |
where Pjk is an individual PK parameter for the jth individual and the kth occasion, TVP is the typical value of the PK parameter, and ηj and τk are the independent and normally distributed between- and within-subject random variability with mean of zero and variance ΩP and ΠP, respectively. Different combinations of η correlation (ω-block) and η fixed at zero were evaluated. The selection of an ω-block, if any, was made on the basis of the decrease of the objective function value (OFV). The residual variability was evaluated using a proportional error model for the population PK analysis and using an additive error model for the population PK–PD analysis according to equations 5 and 6, respectively:
| (5) |
| (6) |
where Cobs was the observed concentration or effect, Cpred was the corresponding model predicted concentration or effect and ε was the departure of the observed from the predicted concentration or effect, which was assumed to follow a random normal distribution with a mean of 0 and variance, Σ.
Results
Subject demographics
Thirty healthy young males were randomized and treated and 28 subjects completed four occasions. One subject discontinued from the study after the first study occasion (surinabant 5 mg + THC) due to personal reasons and one subject discontinued due to an adverse event during the second visit (placebo surinabant + THC). Thirty subjects were evaluated for pharmacodynamic and pharmacokinetic analysis. Subject demographics were balanced for all treatment arms (mean age = 23.2 years, SD = 5.3; weight = 78.94 kg, SD = 8.23; height = 187.7 cm, SD = 6.7; BMI = 22.39 kg m−2, SD = 1.94). All subjects were of Caucasian ethnicity (one subject was of half Asian, half Caucasian origin).
Adverse effects
Adverse events were of mild to moderate intensity and transitory in nature, and no serious adverse events were reported during the study. One subject discontinued his second occasion with placebo + THC challenge treatment due to vasovagal syncope, which occurred 8 min after the second THC inhalation (4 mg). The safety profile of adverse events was similar in the surinabant 60 mg group (10 out of 18; 56% of the subjects had adverse events) compared with the placebo group (8 out of 19; 42%). The most frequent adverse events in the surinabant 60 mg + THC vehicle group were headache (28%), somnolence (17%) and nausea (17%). A higher incidence of psychiatric, nervous system and gastrointestinal disorders was observed during THC treatment (95%), which were dose dependently decreased by surinabant co-treatment (90% in the surinabant 5 mg group, 82% in the surinabant 20 mg group, and 63% in surinabant 60 mg + THC treatment group). These adverse events include euphoric mood (feeling high, collected after spontaneous reporting independent from the VAS feeling high scores, 45%), dizziness (50%), somnolence (45%), headache (30%), dry mouth (20%) and nausea (15%).
No clinically relevant changes were found for blood pressure, haematology, biochemistry, urinalysis or any of the ECG intervals. Heart rate changes were analyzed as PD parameters.
PK analysis
Surinabant
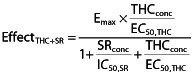
Mean surinabant plasma concentration–time profiles are shown in Figure 1 and an overview of surinabant PK parameters is given in Table 1. Mean surinabant exposure was generally similar with or without THC challenge after surinabant 60 mg (Figure 1). Median tmax was 1.58 h for all surinabant dosages, corresponding to the start time of the THC challenge. Surinabant exposure increased in a less than dose proportional manner. A 12-fold increase in surinabant dose (from 5 mg to 60 mg) gave a 6.91-fold increase of Cmax (P < 0.0001) and an 8.08-fold increase of AUC(0,24 h) (P < 0.0001).
Figure 1.
Mean and predicted plasma concentration–time curve of surinabant with SDs. Surinabant was administered at time point zero and the first blood sample for bio-analysis was taken pre-dose. The open circles are surinabant concentrations after surinabant 5 mg + THC, the open triangles are surinabant 20 mg + THC, the open squares are surinabant 60 mg + THC treatment and the closed squares are after surinabant 60 mg + placebo THC treatment. The dotted lines with plus signs represent the predicted surinabant plasma concentration−time curves
Table 1.
Non-compartmental PK parameters for surinabant (5, 20 and 60 mg), Mean (CV%) ± SD of surinabant PK parameters
| Surinabant 5 mg + THC challenge (n = 20) | Surinabant 20 mg + THC challenge (n = 19) | Surinabant 60 mg + THC challenge (n = 19) | Surinabant 60 mg + THC vehicle (n = 18) | |
|---|---|---|---|---|
| Cmax (ng ml−1) | 104 (31) ± 32.6 | 334 (24) ± 79.0 | 719 (26) ± 190 | 749 (21) ± 157 |
| tmax*(h) | 1.58 (0.750, 1.58) | 1.58 (0.750, 2.58) | 1.58 (0.750, 2.58) | 1.58 (0.750, 2.58) |
| AUC(0,24 h) (ng ml−1 h) | 543 (50) ± 271 | 1860 (30) ± 557 | 4390 (32) ± 1420 | 4870 (28) ± 1380 |
Median (minimum, maximum).
Population PK analysis showed that surinabant PK were best described with a two-compartment model with first-order elimination and first-order absorption with a lag time. Population PK parameters were estimated with good precision (relative standard error (RSE) < 22.0). Population PK parameter estimates are given in Table 2.
Table 2.
Population PK parameters for surinabant and THC.
| Parameter | Surinabant | THC | ||
|---|---|---|---|---|
| Estimate (RSE) | IIV (RSE) | Estimate (RSE) | IIV (RSE) | |
| Clearance/F (l h−1) | 4.69 (13.0) | 72.1 (27.7) | 293 (7.58) | 11.8 (25.0) |
| Central volume of distribution/F (l) | 3.74 (22.0) | 74.8 (34.9) | 43.6 (8.03) | 15.2 (36.0) |
| Peripheral volume of distribution/F (l) | 491 (6.27) | 30.6 (23.9) | 136 (8.97) | – |
| Intercompartmental clearance/F (l h−1) | 15.3 (3.70) | 16.3 (30.8) | 166 (8.01) | – |
| Absorption rate constant (ka, h−1) | 0.406 (3.18) | 6.40 (115) | – | – |
| Lag time (h) | 0.591 (5.91) | – | – | – |
| Dose effect on ka* | −0.00164 (16.4) | – | – | – |
| Interoccasion variability on relative bioavailability (CV%) | – | – | 55.8 (12.6) | – |
| Proportional residual error (CV%) | 18.2 (10.0) | – | 15.9 (14.6) | – |
CV, coefficient of variation (%); F, bioavailability; IIV, inter-individual variability (%); RSE, relative standard error (%).
Dose effect on ka (α): ka (dose) = ka (5mg) + α × (dose − 5).
THC
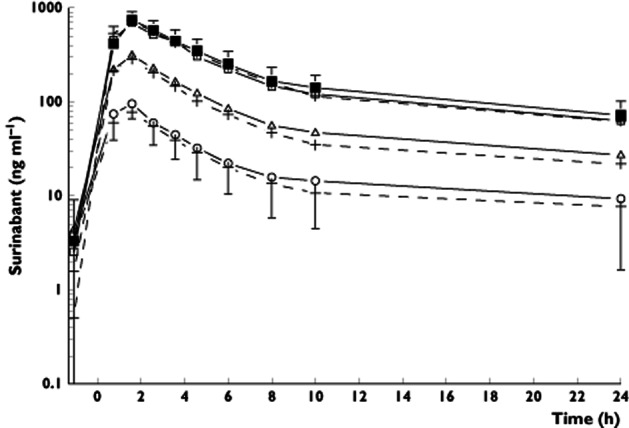
Mean THC plasma concentration–time profiles are shown in Figure 2. THC peak plasma concentration increased for the fourth inhalation, as co-administration of surinabant increased (Figure 2, P = 0.0006). A similar increase was observed for 11-OH-THC, and to a lesser extent for THC-COOH (data not shown).
Figure 2.
Mean plasma concentration-time curve of THC with SDs. The arrows indicate the time points of THC administration. The closed circles are the THC concentrations after placebo surinabant + THC treatment, the open circles are surinabant 5 mg + THC, the triangles are surinabant 20 mg + THC, and the squares are surinabant 60 mg + THC treatment. The graph shows a rather repetitive pattern after each THC administration: the blood samples were taken at 5, 30 and 57 min after the first, second and third inhalation, and at 5, 20, 89 and 130 min after the fourth THC inhalation
A two-compartmental model with linear elimination best described the THC PK data. A model with Michaelis-Menten elimination, as was used in a previous study 31, did not significantly improve the model (data not shown). PK parameter estimations were relatively good, with a RSE up to 14.6%. Relative bioavailability fractions were implemented for each dose within an individual allowing the estimation of intra-individual variability in absorption. Inter-occasion variability of bioavailaibility was shown to significantly improve the model, and was estimated to be 55.8%. Inter-individual variability was estimated for central clearance and central volume of distribution. An overview of population pharmacokinetic parameters is given in Table 2.
Pharmacodynamics
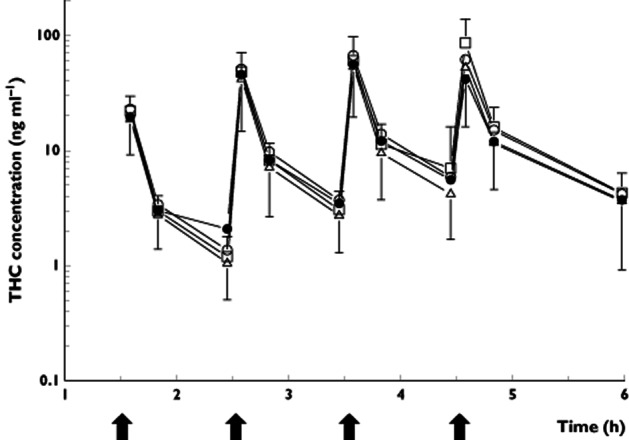
THC-induced significant effects on all pharmacodynamic measurements, except for VAS calmness, compared with the placebo surinabant + placebo THC condition. Surinabant 20 and 60 mg were able to reduce significantly all THC-induced effects on the central nervous system and heart rate compared with surinabant placebo + THC challenge. The inhibition ratios for surinabant 20 mg and 60 mg did not differ significantly. Surinabant completely or almost completely (>80% inhibition) inhibited THC-induced effects, except on heart rate and feeling high where submaximal inhibition was observed (Table 3). Surinabant 5 mg was not able to inhibit any of the THC-induced effects significantly. By itself, 60 mg surinabant did not induce any significant effect on the central nervous system parameters nor on heart rate, compared with surinabant placebo + THC placebo treatment. A graph with the observed effects of feeling high can be found in Figure 3.
Table 3.
Ratios and 95% confidence intervals (CIs) of inhibition by surinabant (5, 20 and 60 mg) on THC-induced effects, measured from the third THC inhalation until 3 h after the fourth inhalation
| PD assessment | Surinabant dose (mg) | % Inhibition (estimate) | 95% CI |
|---|---|---|---|
| Body sway | 5 | 13.6 | (−32.6, 59.7) |
| 20 | 93.1 | (31.9, 154.3) | |
| 60 | 91.1 | (30.3, 151.8) | |
| VAS alertness | 5 | −8.9 | (−54.9, 37.0) |
| 20 | 72.5 | (18.3, 126.7) | |
| 60 | 82.5 | (25.7, 139.4) | |
| VAS feeling high | 5 | 10.0 | (−20.9, 40.9) |
| 20 | 68.0 | (31.6, 104.4) | |
| 60 | 70.0 | (33.2, 106.9) | |
| VAS external perception | 5 | 17.1 | (−18.3, 52.6) |
| 20 | 88.7 | (43.2, 134.3) | |
| 60 | 89.0 | (43.3, 134.7) | |
| VAS internal perception | 5 | 37.9 | (−5.1, 80.9) |
| 20 | 89.9 | (37.0, 142.8) | |
| 60 | 91.6 | (38.3, 145.0) | |
| Heart rate | 5 | 17.6 | (−13.0, 48.1) |
| 20 | 75.4 | (38.4, 112.3) | |
| 60 | 68.3 | (32.5, 104.2) |
Figure 3.
Graph with observed feeling high effects and SDs. Two baseline measurements were recorded before surinabant administration. The closed triangles are feeling high scores after placebo surinabant + placebo THC administration, the closed circles are after placebo surinabant + THC treatment, the open circles are surinabant 5 mg + THC, the open triangles are surinabant 20 mg + THC, the open squares are surinabant 60 mg + THC treatment and the closed squares are after surinabant 60 mg + placebo THC treatment
Population PK–PD
A schematic representation of the basic structure of the PK–PD model is visualized in Figure 4. The effect of THC on body sway and feeling high were best described by maximum effect models, relating the effect to the concentration in the effect compartment 37. These models included inter-individual variability on the baseline value, Emax and Keo (Table 4). The effect of surinabant on THC-induced feeling high was best described using a partial antagonism model. Internal and external perception and alertness were best described by a linear response model, relating the effect to the concentration in the effect compartment. These models included variability on the baseline value along with inter-individual variability on baseline, slope and Keo (Table 4). As some subjects appeared not to show any changes in internal perception following the THC challenge, a model excluding non-responders was evaluated, but no improvement was seen. The effect compartment equilibrium half-lives for alertness (120 min) and body sway (89 min) were larger compared with feeling high (40 min), internal (44 min) and external perception (48 min). This means that THC-induced effects on alertness and body sway have a later onset than effects on feeling high, internal and external perception and that they last longer. For heart rate, no PK–PD model was developed. In the placebo group, the sampling scheme during the post-prandial period in which heart rate increase was observed was too sparse for accurate PK–PD modelling.
Figure 4.
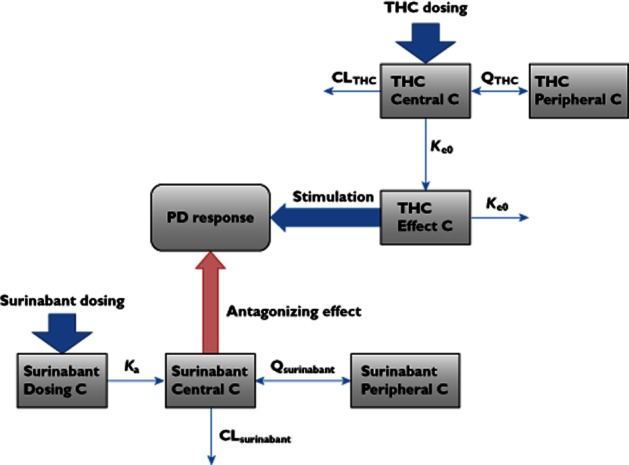
Schematic overview of PK–PD model (for detailed background information, see 41). The central compartment refers to the central circulation. C, compartment; CL, clearance; ka, absorption rate constant; Ke0, effect compartment equilibration rate constant; Q, intercompartmental clearance
Table 4.
Population PK–PD parameter estimates for body sway, VAS feeling high, alertness, external perception and internal perception
| PD parameter | Population parameter estimate (RSE%) | Inter-individual variability CV% (RSE%) | Inter-occasion variability CV% (RSE%) |
|---|---|---|---|
| Body sway | |||
| Baseline (ln mm) | 5.46 (1.26) | 6.66 (24.3) | 3.00 (32.2) |
| Emax (log mm) | 0.829 (24.5) | 68.8 (40.2) | – |
| EC50*(ng ml−1) | 7.24 (42.8) | – | – |
| Ke0 (h−1) | 0.466 (17.9) | 73.4 (33.6) | – |
| IC50 (ng ml−1) | 22.0 (45.2) | – | – |
| Residual variability (SD of additive error) | 0.212 (10.5) | – | – |
| Feeling high | |||
| Baseline (log mm) | 0.321 (3.96) | 21.6 (38.5) | – |
| Emax (log mm) | 0.713 (31.6) | 124 (39.6) | – |
| EC50* (ng ml−1) | 6.98 (33.5) | – | – |
| Ke0 (h−1) | 1.04 (17.4) | 71.6 (32.4) | – |
| IC50 (ng ml−1) | 30.5 (61.6) | – | – |
| Maximum inhibition | 0.751 (20.6) | – | – |
| Residual variability (SD of additive error) | 0.254 (19.1) | – | – |
| Alertness | |||
| Baseline (mm) | 49.4 (1.10) | 5.13 (47.9) | 180 (37.0) |
| Slope (ng−1 ml−1) | 0.547 (45.2) | 98.1 (53.5) | – |
| Ke0 (h−1) | 0.347 (33.7) | 4.64 (26.0) | – |
| IC50 (ng ml−1) | 33.6 (45.8) | – | – |
| Residual variability (SD of additive error) | 3.30 (18.3) | – | – |
| External perception | |||
| Baseline (log mm) | 0.367 (0.529) | – | 3.86 (46.1) |
| Slope (ng−1 ml−1) | 0.00258 (41.9) | 154 (29.4) | – |
| Ke0 (h−1) | 0.868 (16.9) | 69.9 (30.1) | – |
| IC50 (ng ml−1) | 37.1 (59.6) | – | – |
| Residual variability (SD of additive error) | 0.0182 (19.1) | – | – |
| Internal perception | |||
| Baseline (log mm) | 0.366 (0.508) | 2.68 (68.2) | 1.46 (36.9) |
| Slope (ng−1 ml−1) | 0.000869 (38.2) | 151 (35.1) | – |
| Ke0 (h−1) | 0.955 (20.1) | 71.4 (45.5) | – |
| IC50 (ng ml−1) | 58.8 (44.2) | – | – |
| Residual variability (SD of additive error) | 0.0123 (22.8) | – | – |
EC50 of THC effect. CV, coefficient of variation (%); EC50, concentration producing 50% of Emax; Emax, maximal effect; IC50, concentration producing 50% of inhibition of THC Emax; Ke0, effect compartment equilibration rate constant; RSE, relative standard error (%); SD, standard deviation.
The EC50 of THC for body sway was similar to that of feeling high (7.24 ng ml−1 and 6.98 ng ml−1, respectively). No EC50 could be calculated for the other PD parameters, as a linear model best described these parameters. The IC50 of surinabant for body sway was approximately half of the IC50 value for internal perception (22.0 ng ml−1 vs. 58.8 ng ml−1). This means that 50% inhibition of THC-induced body sway increase is established with a surinabant concentration that is approximately half of the concentration needed to reduce the effects on internal perception by half. IC50 values for feeling high, alertness and external perception were similar (30.5 ng ml−1, 33.6 ng ml−1 and 37.1 ng ml−1, respectively). A summary of population PK–PD model parameters can be found in Table 4.
Discussion
The objective of this study was to investigate the interaction of oral surinabant and inhaled THC on central nervous system effects and heart rate in healthy subjects. We have demonstrated that doses of 20 and 60 mg surinabant are able to inhibit THC-induced effects on central nervous system parameters and heart rate by 68.0% to 91.6%, whereas surinabant 5 mg was unable to antagonize any THC effect. Surinabant 60 mg alone had no acute effects, particularly not on mood.
Pharmacokinetics
With increasing doses of surinabant, maximum plasma concentration (Cmax) and AUC(0,24 h) increased in a less than dose-proportional manner. This was also found in the population PK model. A negative dose effect on the absorption rate constant improved the model. Physiologically, this could be explained by saturation of absorption of surinabant, poor dissolution or an increase of transit time from the blood. The exact mechanism is unknown.
THC peak plasma concentration increased as co-administration of surinabant increased, which was represented in the population PK model by a relatively high inter-occasion variability on bioavailability of 55.8%. Rather than representing a PK interaction, this could be due to a pharmacodynamic compensation in this group of experienced cannabis users. Subjects who received surinabant in combination with THC experienced less of their familiar subjective effects while inhaling THC. Consequently, they may have tried to inhale THC maximally during concomitant surinabant treatment. On the other hand, less THC was required to induce the desired high feelings, while on surinabant placebo. The standardized paced puffing inhalation protocol should have prevented this type of variability. However, it is possible that some subjects were able to regulate the amount of THC by breathing out through the nose. Therefore, the inhalation protocol has since been adapted by adding the use of a nose clamp for future studies.
Pharmacodynamics
In contrast to a paced puffing protocol, complete self-regulation of THC administration would allow subjects to titrate for the expected or desired PD effects. This would lead to inaccurate estimations of the antagonistic effects, which could explain the differences in the effect size between our study and a previous study by Huestis et al. In the latter study in which a cannabis challenge was applied, rimonabant doses up to 90 mg gave 43% inhibition on subjective feeling high and 59% inhibition on heart rate increase 38, 39, whereas for surinabant, reductions were 70% and 75%, respectively. The rimonabant doses produced plasma concentrations in the upper range of the therapeutic window, suggesting that the levels of inhibition that were found in the current study could be over the therapeutic range. Although this cannot be excluded without a comparison with the results of clinical studies, it is perhaps more likely that the disparate estimates are related to differences in inhalation methodology. In Huestis et al.'s study, subjects inhaled THC from cannabis cigarettes, which allows a certain freedom to self-regulate the amount of inhaled THC by the deepness and the number of the inhalations. THC Cmax was 130 ng ml−1 in the study by Huestis et al. and 83.48 ng ml−1 in the current study. With self-regulated titration for PD effects, subjects compensate for a certain amount of effect inhibition, leading to an underestimation of rimonabant's antagonistic potency. This is more difficult if THC is administered with an evaporation device and subjects are instructed to inhale the full contents of the balloon. In view of these differences, it seems more likely that the suppression caused by suribanant is in the same range as the effects of rimonabant. Furthermore, the variety of active compounds from cannabis could interfere with the THC and antagonist effects. The time period from which the inhibition ratios were calculated was different for both studies (1 h vs. 4.5 h).
Another study using the CB1 antagonist drinabant (AVE1625) had a similar design to the current study 26. Drinabant 20 mg and 60 mg induced maximal inhibition on heart rate, VAS feeling high, internal and external perception, but not on body sway and VAS alertness. Surinabant caused suppression of all these THC responses, including near complete inhibition of body sway and VAS alertness, but it had sub-maximal effects on heart rate and high feeling. This indicates possible differences in clinical efficacy between surinabant and drinabant. We have argued that THC-induced tachycardia is (primarily) mediated peripherally, based on a previous PK–PD study in which the equilibration half-life of heart rate was significantly shorter compared with the other centrally mediated effect parameters 31. In line with this conjecture, pre-clinical studies also suggest that surinabant and drinabant have different central and peripheral mediated effects. Conversely, effects on food intake, which could be peripherally mediated 40, are found at 0.3 mg kg−1 oral drinabant, while the effective dose of oral surinabant was 3.0 kg mg−1 (unpublished data). No plasma concentrations or PK–PD-relations were determined in these preclinical experiments. These findings could be explained by a larger or faster brain penetration for surinabant compared with drinabant, whereas drinabant appears to have a relatively larger peripheral effect. If so, the effect of surinabant on feeling high seems small (around 70%) compared with drinabant (up to 101%), but the reliability of this inhibition ratio may have been diminished by a fairly large intra-individual variability (124%).
Surinabant 5 mg was unable to inhibit significantly any of the THC-induced central nervous system effects and heart rate, which were suppressed by surinabant 20 mg and 60 mg. This implies that surinabant effects are dose-dependent. Inhibition ratios of surinabant 20 mg were similar to 60 mg, indicating that 20 mg is able to induce maximal effects.
PK–PD
The PK–PD models adequately described the time course of PK and PD effects of THC and the antagonism of these PD effects by surinabant. The THC models of body sway, feeling high and alertness are generally comparable with the THC model that was constructed in a previous study by Strougo et al. 31. The maximal effect of THC on feeling high was smaller in the current study compared with the previous study by Strougo et al. (0.713 log mm vs. 1.68 log mm). A linear response model best fitted the external perception data in this study, while Strougo et al. found a maximal effect model best described their data. The difference observed in this study might be explained by the THC dose range, which could have been insufficient for detecting a maximal effect.
For surinabant, various IC50 values were found for central nervous system parameters, with a smaller IC50 value for body sway, which may be regulated by central as well as peripheral processes, compared with the purely centrally mediated measures. This variability of PK–PD parameters implies that surinabant has a variety of effect compartments, even within the CNS, which could be functional or kinetic. Also, the effect compartment equilibrium rate constant, or Keo, showed differences among the various PD measures, which means that some effects have a later onset and longer duration than other effects. This could be caused by several factors that could not be determined in this study, such as a difference in penetration rate between the different effect compartments. These findings also support the hypothesis that the clinically effective level of surinabant might be found at different concentrations compared with the levels that are needed to induce adverse side effects.
This agonist-antagonist PK–PD interaction model can be used for prediction of surinabant concentration–effect profiles in future studies, even if these studies have a different design or dosing regimen. As surinabant and rimonabant are very similar in structure and action, the population PK–PD model of surinabant could also be used to estimate concentration-effect profiles of rimonabant to a certain extent. Conversely, as rimonabant has been used extensively in patient studies, a patient population PK–PD model could theoretically be used to predict concentration-effect profiles for surinabant in patients, with the aim of finding an optimal therapeutic window, ranging between the dose-dependent desired and undesired effects. Currently, however, such quantitative predictions are hampered by the as yet unknown relationships between the PD (central and peripheral) biomarkers and the clinical (metabolic and psychiatric) endpoints. At any rate, surinabant was found to be a potent CB1-antagonist, at single doses that did not cause any adverse systemic or CNS effects in healthy subjects. However, this information is insufficient to draw conclusions on the effects after a multiple dose regimen. Therefore, future studies should investigate the optimal surinabant dose and its effects after long term use, with a particular focus on the occurrence of psychiatric side effects.
Acknowledgments
We thank Rik Schoemaker for his advice on the study design.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare this study was financially supported by Sanofi; Christine Roy, Geraldine Ferron, Sandrine Turpault, Franck Poitiers and Jean-Louis Pinquier were employed by Sanofi in the previous 3 years and were shareholders. There are no other relationships or activities that could appear have influenced the submitted work.
References
- 1.Mechoulam R, Gaoni Y. A total synthesis of dl-delta-1-tetrahydrocannabinol, the active constituent of hashish. J Am Chem Soc. 1965;87:3273–3275. doi: 10.1021/ja01092a065. [DOI] [PubMed] [Google Scholar]
- 2.Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (3rd edn.) 2008;153(Suppl. 2):S1–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 4.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 5.Bermudez-Silva FJ, Viveros MP, McPartland JM, Rodriguez FF. The endocannabinoid system, eating behavior and energy homeostasis: the end or a new beginning? Pharmacol Biochem Behav. 2010;95:375–382. doi: 10.1016/j.pbb.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 6.Ravinet TC, Arnone M, Delgorge C, Gonalons N, Keane P, Maffrand JP, Soubrie P. Anti-obesity effect of SR141716, a CB1 receptor antagonist, in diet-induced obese mice. Am J Physiol Regul Integr Comp Physiol. 2003;284:R345–R353. doi: 10.1152/ajpregu.00545.2002. [DOI] [PubMed] [Google Scholar]
- 7.Van Gaal LF, Rissanen AM, Scheen AJ, Ziegler O, Rossner S. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet. 2005;365:1389–1397. doi: 10.1016/S0140-6736(05)66374-X. [DOI] [PubMed] [Google Scholar]
- 8.Pertwee RG. Emerging strategies for exploiting cannabinoid receptor agonists as medicines. Br J Pharmacol. 2009;156:397–411. doi: 10.1111/j.1476-5381.2008.00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rinaldi-Carmona M, Barth F, Heaulme M, Shire D, Calandra B, Congy C, Martinez S, Maruani J, Neliat G, Caput D. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994;350:240–244. doi: 10.1016/0014-5793(94)00773-x. [DOI] [PubMed] [Google Scholar]
- 10.de Rodriguez FF, Roberts AJ, Bilbao A, Koob GF, Navarro M. Cannabinoid receptor antagonist SR141716A decreases operant ethanol self administration in rats exposed to ethanol-vapor chambers. Zhongguo Yao Li Xue Bao. 1999;20:1109–1114. [PubMed] [Google Scholar]
- 11.Centre for Reviews and Dissemination. Health Information Resources. 3-12-2004. Ref Type: Online Source. 2004.
- 12.Cohen C, Perrault G, Griebel G, Soubrie P. Nicotine-associated cues maintain nicotine-seeking behavior in rats several weeks after nicotine withdrawal: reversal by the cannabinoid (CB1) receptor antagonist, rimonabant (SR141716) Neuropsychopharmacology. 2005;30:145–155. doi: 10.1038/sj.npp.1300541. [DOI] [PubMed] [Google Scholar]
- 13.Cohen C, Perrault G, Voltz C, Steinberg R, Soubrie P. SR141716, a central cannabinoid (CB(1)) receptor antagonist, blocks the motivational and dopamine-releasing effects of nicotine in rats. Behav Pharmacol. 2002;13:451–463. doi: 10.1097/00008877-200209000-00018. [DOI] [PubMed] [Google Scholar]
- 14.Wathion N. Public statement on Acomplia (rimonabant): withdrawal of the marketing authorisation in the European Union. 2009. EMEA/39457/2009. 1-30-2009. Ref Type: Internet Communication.
- 15.The European Medicines Agency (EMEA) The European Medicines Agency recommends suspension of the marketing authorisation of Acomplia. Doc.Ref.EMEA/CHMP/537777/2008. 10-23-2008. Ref Type: Internet Communication. 2008.
- 16.The European Medicines Agency (EMEA) Questions and answers on the recommendation to suspend the marketing authorisation of Acomplia (rimonabant). Doc.Ref.EMEA/537153/2008. 10-23-2008. Ref Type: Internet Communication. 2008.
- 17.Merck & Co., Inc. Merck discontinues development of investigational medicine taranabant for obesity. 10-2-2008. Ref Type: Internet Communication. 2008.
- 18.Aronne LJ, Tonstad S, Moreno M, Gantz I, Erondu N, Suryawanshi S, Molony C, Sieberts S, Nayee J, Meehan AG, Shapiro D, Heymsfield SB, Kaufman KD, Amatruda JM. A clinical trial assessing the safety and efficacy of taranabant, a CB1R inverse agonist, in obese and overweight patients: a high-dose study. Int J Obes (Lond) 2010;34:919–935. doi: 10.1038/ijo.2010.21. [DOI] [PubMed] [Google Scholar]
- 19.Kipnes MS, Hollander P, Fujioka K, Gantz I, Seck T, Erondu N, Shentu Y, Lu K, Suryawanshi S, Chou M, Johnson-Levonas AO, Heymsfield SB, Shapiro D, Kaufman KD, Amatruda JM. A one-year study to assess the safety and efficacy of the CB1R inverse agonist taranabant in overweight and obese patients with type 2 diabetes. Diabetes Obes Metab. 2010;12:517–531. doi: 10.1111/j.1463-1326.2009.01188.x. [DOI] [PubMed] [Google Scholar]
- 20.Proietto J, Rissanen A, Harp JB, Erondu N, Yu Q, Suryawanshi S, Jones ME, Johnson-Levonas AO, Heymsfield SB, Kaufman KD, Amatruda JM. A clinical trial assessing the safety and efficacy of the CB1R inverse agonist taranabant in obese and overweight patients: low-dose study. Int J Obes (Lond) 2010;34:1243–1254. doi: 10.1038/ijo.2010.38. [DOI] [PubMed] [Google Scholar]
- 21.Morrison MF, Ceesay P, Gantz I, Kaufman KD, Lines CR. Randomized, controlled, double-blind trial of taranabant for smoking cessation. Psychopharmacology (Berl) 2010;209:245–253. doi: 10.1007/s00213-010-1790-2. [DOI] [PubMed] [Google Scholar]
- 22.Kirilly E, Gonda X, Bagdy G. CB1 receptor antagonists: new discoveries leading to new perspectives. Acta Physiol (Oxf) 2012;205:41–60. doi: 10.1111/j.1748-1716.2012.02402.x. [DOI] [PubMed] [Google Scholar]
- 23.Cohen AF. Developing drug prototypes: pharmacology replaces safety and tolerability? Nat Rev Drug Discov. 2010;9:856–865. doi: 10.1038/nrd3227. [DOI] [PubMed] [Google Scholar]
- 24.Tonstad S, Aubin HJ. Efficacy of a dose range of surinabant, a cannabinoid receptor blocker, for smoking cessation: a randomized controlled clinical trial. J Psychopharmacol. 2012;26:1003–1009. doi: 10.1177/0269881111431623. [DOI] [PubMed] [Google Scholar]
- 25.Cahill K, Ussher MH. Cannabinoid type 1 receptor antagonists for smoking cessation. Cochrane Database Syst Rev. 2011;(3) doi: 10.1002/14651858.CD005353.pub4. CD005353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zuurman L, Roy C, Schoemaker R, Amatsaleh A, Guimaeres L, Pinquier J, Cohen A, van Gerven J. Inhibition of THC-induced effects on the central nervous system and heart rate by a novel CB1 receptor antagonist AVE1625. J Psychopharmacol. 2010;24:363–371. doi: 10.1177/0269881108096509. [DOI] [PubMed] [Google Scholar]
- 27.Zuurman L, Roy C, Schoemaker RC, Hazekamp A, Den Hartigh J, Bender JC, Verpoorte R, Pinquier JL, Cohen AF, van Gerven JM. Effect of intrapulmonary tetrahydrocannabinol administration in humans. J Psychopharmacol. 2008;22:707–716. doi: 10.1177/0269881108089581. [DOI] [PubMed] [Google Scholar]
- 28.Sanofi-aventis. A clinical study to investigate the tolerability and pharmacokinetic parameters of SR147778 after repeated oral administrations (soft gelatin capsules) in young healthy male subjects; CSRCO-TDR5736-EN-E01 – Data on file. 2006. Ref Type: Unpublished Work.
- 29.Sanofi-Aventis. Excretion balance, pharmacokinetics, metabolites profiling and identification after single oral administration of SR147778 using [14C]-labeled compound in healthy male subjects. Open, non-randomized and single center study, CSRCO-BEX4499-EN-E01 – Data on file. 2006. Ref Type: Unpublished Work.
- 30.Sanofi-Aventis. Determination of the cytochrome P450 (CYP) isoforms involved in the oxidative metabolism of SR147778 in vitro, CMPKCO-MIH0135-EN-E01 – Data on file. 2006. Ref Type: Unpublished Work.
- 31.Strougo A, Zuurman L, Roy C, Pinquier JL, van Gerven JM, Cohen AF, Schoemaker RC. Modelling of the concentration–effect relationship of THC on central nervous system parameters and heart rate – insight into its mechanisms of action and a tool for clinical research and development of cannabinoids. J Psychopharmacol. 2008;22:717–726. doi: 10.1177/0269881108089870. [DOI] [PubMed] [Google Scholar]
- 32.Zuurman L, Ippel AE, Moin E, van Gerven JM. Biomarkers for the effects of cannabis and THC in healthy volunteers. Br J Clin Pharmacol. 2009;67:5–21. doi: 10.1111/j.1365-2125.2008.03329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bond A, Lader M. The use of analogue scales in rating subjective feelings. Br J Med Psychol. 1974;47:211–218. [Google Scholar]
- 34.Bowdle TA, Radant AD, Cowley DS, Kharasch ED, Strassman RJ, Roy-Byrne PP. Psychedelic effects of ketamine in healthy volunteers: relationship to steady-state plasma concentrations. Anesthesiology. 1998;88:82–88. doi: 10.1097/00000542-199801000-00015. [DOI] [PubMed] [Google Scholar]
- 35.NONMEM project group. NONMEM users guide. 1992. Ref Type: Computer Program.
- 36.Speth H. A Linux cluster for population pharmacokinetic analyses. Int J Clin Pharmacol Ther. 2004;42:189–190. [PubMed] [Google Scholar]
- 37.Ferron G, Klumpers LE, van Gerven JMA, Roy C. PK and PK/PD modeling of CB1 blocker antagonism of THC induced CNS and heart rate effect. PAGE, Abstracts 17. 2008. Ref Type: Abstract.
- 38.Huestis MA, Gorelick DA, Heishman SJ, Preston KL, Nelson RA, Moolchan ET, Frank RA. Blockade of effects of smoked marijuana by the CB1-selective cannabinoid receptor antagonist SR141716. Arch Gen Psychiatry. 2001;58:322–328. doi: 10.1001/archpsyc.58.4.322. [DOI] [PubMed] [Google Scholar]
- 39.Huestis MA, Boyd SJ, Heishman SJ, Preston KL, Bonnet D, Le FG, Gorelick DA. Single and multiple doses of rimonabant antagonize acute effects of smoked cannabis in male cannabis users. Psychopharmacology (Berl) 2007;194:505–515. doi: 10.1007/s00213-007-0861-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gomez R, Navarro M, Ferrer B, Trigo JM, Bilbao A, Del AI, Cippitelli A, Nava F, Piomelli D, de Rodriguez FF. A peripheral mechanism for CB1 cannabinoid receptor-dependent modulation of feeding. J Neurosci. 2002;22:9612–9617. doi: 10.1523/JNEUROSCI.22-21-09612.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mager DE, Wyska E, Jusko WJ. Diversity of mechanism-based pharmacodynamic models. Drug Metab Dispos. 2003;31:510–518. doi: 10.1124/dmd.31.5.510. [DOI] [PubMed] [Google Scholar]