

Abstract
Aim
The aim of this study was to develop a PK/PD model to assess drug–drug interactions between dabigatran and P-gp modulators, using the example of clarithromycin, a strong inhibitor of P-gp.
Methods
Ten healthy male volunteers were randomized to receive in the first treatment period a single 300 mg dose of dabigatran etexilate (DE) and in the second treatment period 500 mg clarithromycin twice daily during 3 days and then 300 mg DE plus 500 mg clarithromycin on the fourth day, or the same treatments in the reverse sequence. Dabigatran plasma concentration and ecarin clotting time (ECT) were measured on 11 blood samples. Models were built using a non-linear mixed effect modelling approach.
Results
The best PK model was based on an inverse Gaussian absorption process with two compartments. The relationship between dabigatran concentration and ECT was implemented as a linear function. No continuous covariate was associated with a significant decrease in the objective function. The concomitant administration of clarithromycin induced a significant change only in DE bioavailability, which increased from 6.5% to 10.1% in the presence of clarithromycin. Clarithromycin increased peak concentration and AUC by 60.2% and 49.1% respectively.
Conclusion
The model proposed effectively describes the complex PK of dabigatran and takes into account drug–drug interactions with P-gp activity modulators, such as clarithromycin.
Keywords: bioavailability, clarithromycin, dabigatran, drug–drug interaction, P-glycoprotein inhibition, population pharmacokinetics and pharmacodynamics
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Dabigatran etexilate has a bioavailability of 6.5% due to a complex absorption process.
Dabigatran etexilate is a substrate for P-gp and its reflux can be modulated by other drugs.
P-gp inhibitors increase the AUC of dabigatran from about 50% to over 200%.
WHAT THIS STUDY ADDS
The pharmacokinetics and pharmacodynamics of dabigatran were described by a two compartment model with an absorption following an inverse Gaussian law, associated with a linear effect model.
We showed that this phenomenon is explained solely by an increase in bioavailability from 6.5 to 10%.
Exposure to dabigatran is increased by 50% in the presence of clarithromycin and is characterized by substantial variability.
Introduction
Dabigatran etexilate (DE) is an anticoagulant drug used for the prevention of venous thromboembolism after orthopaedic surgery and for the long term prevention of stroke in patients with atrial fibrillation 1, 2. DE is a synthetic, orally administered prodrug that is rapidly converted into the active drug, dabigatran, by a serum esterase. It exerts its activity by direct competitive inhibition of thrombin. DE has a bioavailability of 6.5% due to a complex absorption process 3, necessitating first, an acid environment for dissolution and second, active transport by P-glycoprotein (P-gp) 3, 4. P-gp-mediated transport through the enterocyte, from the basolateral to the apical side, could be involved in the excretion of DE in the gut. Dabigatran is not metabolized by the cytochrome P450 enzyme system and 80% of the dose administered is directly excreted by the kidneys 3, 5. In clinical practice, dabigatran can be given as a fixed dose without monitoring.
Compared with other anticoagulant drugs, such as vitamin K antagonists, dabigatran has a low potential for drug–drug interactions. However, it is a substrate for P-gp and its reflux can be modulated by other drugs. P-gp inducers and inhibitors can, respectively, decrease or increase its bioavailability. An increase in dabigatran area under the concentration vs. time curve (AUC), ranging from near 50% to over 200%, has been reported after co-administration of amiodarone, ketoconazole, quinidine or verapamil 5. Some of these drug–drug interactions are strong enough to warrant contraindications or precautions for use. Other interactions involving P-gp modulators might also be clinically relevant and necessitate dose adjustments, possibly based on drug monitoring.
The aim of this study was to develop a PK/PD model to assess drug–drug interactions between dabigatran and P-gp modulators, using clarithromycin, a strong inhibitor of P-gp, as an example.
Methods
Study design
This was a single centre, randomized, open label study with a two-way crossover design (clinical trial.org registration no.: NCT01385683). The protocol complied with the principles of the declaration of Helsinki (2002 version) and those of Good Clinical Practice established by the International Conference on Harmonization. It was approved by the French regulatory authority (AFSSAPS) and the local Ethics Committee.
Participants
Ten healthy male Caucasian volunteers aged from 18 to 35 years, were included in the study after a complete physical examination and laboratory tests. To be included, subjects had to have normal coagulation test results with respect to the following parameters: platelet count, international normalized ratio, activated partial thromboplastin time (aPPT) and fibrinogen level. Volunteers with renal insufficiency, hepatic insufficiency or a peptic ulcer, and those who presented a lesion at risk of bleeding or had undergone surgery during the month preceding the study, were excluded. Subjects with known hypersensitivity to DE or clarithromycin, or who had taken any medicine during the week before the study were also excluded. Written informed consent was obtained from each volunteer before inclusion in the study and genetic analysis.
Treatment procedure
Following screening, volunteers were randomized 1:1 to receive either first DE alone and then a combination of DE and clarithromycin, or first the combination treatment and then DE alone. The two treatment periods were separated by a 6 day washout period. In the DE treatment period, each volunteer received, at time zero (t0: 07.30 h), a 300 mg oral dose of DE (two Pradaxa® 150 mg capsules; Boehringer Ingelheim, Ingelheim, Germany). During the DE plus clarithromycin treatment period, volunteers received on the first 3 days 500 mg clarithromycin (Zeclar®, 500 mg tablet, Abott France, France) twice daily (at 09.00 h and 18.00 h). On the 4th day they received 300 mg of DE plus 500 mg of clarithromycin at t0 (07.30 h). The clarithromycin administration regimen was chosen on the grounds of its relevance to clinical practice. DE was administered with 300 ml water (150 ml immediately, then 150 ml 30 min later) to subjects in a semi-supine position after an overnight fast of at least 8 h. Standardized meals were served at 12.00 h and 18.00 h.
Sample collection and analysis
For pharmacokinetic (PK) and pharmacodynamic (PD) analysis, 5 ml blood samples were drawn in tubes at 0, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10 and 23.5 h after DE administration for measurement of plasma concentrations of dabigatran. All analyses were performed on venous blood samples collected by venipuncture, using whole blood collected in citrate tubes for PD and heparinized tubes for PK analysis.
Dabigatran plasma concentrations were determined using the validated liquid chromatography-tandem mass spectrometry method 6. The intra- and inter-day precision values were below 11.3% and accuracy was within 93.8% and 108.8%. The lower limit of quantification was 2 μg l−1.
The anticoagulant effect of dabigatran was measured by an ecarin clotting time (ECT) assay, as described by Nowak 7. The intra- and inter-day precision values were below 2.5%. ECT is the reference coagulation test for analysis of dabigatran's anticoagulant effect 8.
Model development and evaluation
Data analysis was performed using MONOLIX non-linear mixed-effects modelling software (version 4.12, release 2; INRIA – Institut National de Recherche en Informatique et en Automatique, France) 9. The stochastic approximation expectation maximization algorithm combined with the Markov chain Monte Carlo simulation procedure was used to estimate the maximum likelihood of the model. The parameters of the model were assumed to be log-normally distributed in the population studied.
We fitted the PK and PD data to the model to try to identify the mechanism of the interaction. Modelling was performed in several steps. The first step consisted in identifying the structural PK model for dabigatran without clarithromycin. The second step comprised the estimation of PK/PD parameters and relevant covariates. The third step consisted in testing the inter-occasional variability (IOV) on the full data set (with and without clarithromycin). Finally, drug–drug interaction was investigated applying the categorical covariate ‘concomitant treatment’ to model parameters. In our study, we set the population mean value of DE bioavailability when administered alone at 6.5%, as we had no pharmacokinetic data for DE injected intravenously 3.
Pharmacokinetic model
As drug–drug interaction certainly occurs at the absorption level, the main issue in model development was to determine the most relevant absorption process. Zero order, first order and inverse Gaussian absorptions were tested. The inverse Gaussian absorption process was implemented as follows:
where fA(t) represents the quantity of DE that passes into the central compartment during the time interval dt, D is the dose administered at time t, F is the bioavailability, MAT is the mean absorption time and CV2 is the relative dispersion of absorption time 10, 11. This type of absorption process is well suited to test interactions based on a change in bioavailability and has already been used to model the mechanism of P-gp inhibition 12.
One and two compartment models were tested for the structural component of the model.
Pharmacodynamic model
A linear relationship, previously described in population model analysis, and an Emax model were evaluated to describe the relationship between the concentration of dabigatran and its effect 8.
Covariate model
Body mass index, weight, lean body weight, size, age and creatinine clearance [calculated using the Modification of Diet in Renal Disease (MDRD) and Cockcroft–Gault equations] were tested as continuous covariates on absorption and structural parameters. The concomitant administration of clarithromycin, was included as a categorical covariate in the absorption model in order to test the presence of an interaction between dabigatran and clarithromycin:
where θi is one of the population parameters, 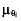 is the typical value of the parameter θi,
is the typical value of the parameter θi, 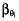 is the covariate effect, Clari is equal to 1 during the clarithromycin plus DE treatment period and 0 during the DE alone period,
is the covariate effect, Clari is equal to 1 during the clarithromycin plus DE treatment period and 0 during the DE alone period, 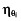 is the inter-patient variability in the absence of clarithromycin, and
is the inter-patient variability in the absence of clarithromycin, and 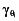 is the increase in inter-patient variability in the presence of clarithromycin.
is the increase in inter-patient variability in the presence of clarithromycin.
The covariates were added to the model in accordance with the procedure described elsewhere 13. This involved a forward inclusion step and a backward elimination step. A decrease in the objective function of at least 3.84 was required to identify a significant covariate.
Model evaluation
Model evaluation and selection was based on visual inspection of goodness of fit plots, the precision of parameter estimates and a decrease in objective function. The goodness of fit was established by plotting the population predictions of the model vs. observations, the individual predictions vs. observations, and the normalized prediction distribution errors (NPDE) vs. time 14. The visual predictive check (VPC) was generated by simulating 1000 times the parameters of the 10 subjects. The ability of the model to describe the observations was evaluated by inspection of the distribution of the simulated concentrations. Finally, the median parameter values and the 90% prediction interval of the VPC replicates were compared with the observations comprising the original dataset.
Results
Population description
All the subjects completed the study and were included in the PK and PD analyses. The median age (range) was 22 (18–33) years, median height 180 (175–188) cm and median weight 75 (64–82) kg. With regard to laboratory data, median aPPT (range) was 1.04 (0.9–1.15) s, median plasma fibrinogen concentration 2.25 (1.6–3) g l−1 and median plasma creatinine concentration 77 (72–95) μmol l−1.
Population PK model
The best absorption model was that based on an inverse Gaussian absorption process. This was associated with the best objective function value (758.6), first order and zero order absorption processes being associated with objective function values of 959.5 and 946.7, respectively (on the basis of a two compartment structure).
The parameters defining the two compartment structure of the model were the clearance (CL), the inter-compartmental clearance (Q), and the central and peripheral compartments (Vc and Vp respectively). Random effect was tested only on Vc and absorption parameters (F, CV2 and MAT). No covariance was found between the parameters CL and Vc. Inter-individual variability was estimated for MAT, CV2, F and Vc. Residual variability was best described using a combined (additive plus proportional) error model. The values of these parameters are shown in Table 1.
Table 1.
Values of pharmacokinetic and pharmacodynamic parameters
| Population mean (% RSE) | Interpatient SD (% RSE) | |
|---|---|---|
| Absorption model parameters | ||
| MAT | 1.65 (4) | 0.0722 (16) |
| CV | 0.622 (5) | 0.152 (5) |
| F | 0.065 (fixed) | 0.386 (13) |
| F* | 0.101 (1.4) | 0.725 (12.7) |
| Disposition model parameters | ||
| VC | 48.3 (12) | 0.105 (41) |
| VP | 68.7 (6) | – |
| Q | 20.6 (8) | – |
| CL | 14.8 (7) | – |
| Effect model parameters | ||
| ECT0 | 31.3 (1) | – |
| K | 0.435 (6) | 0.178 (6) |
| Error model | ||
| σconcentration,additive | 4.65 (16) | – |
| σconcentrationproportional | 0.105 (11) | – |
| σECT,proportional | 0.078 (5) | – |
Bioavailability in presence of clarithromycin. RSE, relative standard error.
None of the continuous covariates tested (weight, body mass index, body surface area, creatinine clearance) was associated with a significant decrease in the objective function.
Population PK/PD model
The best PK/PD model corresponded to a linear relationship between dabigatran concentration and its anticoagulant effect, based on the ECT, implemented as follows:
where K is a proportionality coefficient, ECT0 is the baseline ECT value and CDabi the plasma concentration of dabigatran. Random effect was tested only on K. The error model was proportional. The values of these parameters are presented in Table 1.
No relevant IOV was estimated on the PK and PD model parameters.
Testing the interaction
The administration of clarithromycin induced a significant change only in the bioavailability of DE. The population mean value of F increased from 6.5% (pre-set value; range 2.8–12.1) to 10.1% (4.1–26.9) in the presence of clarithromycin, the inter-individual variability of F increasing from 0.386 to 0.725 (CV%). The modification of the absorption rate of dabigatran and its variability in the presence of clarithromycin is illustrated in Figure 1. The impact of clarithromycin on peak concentration was an increase of 60.2% from (median, min–max) 174.4 μg l−1 (92.3–310.0) without clarithromycin to 279.4 μg l−1 (71.6–782.1) with clarithromycin. On AUC, an increase of 49.1% was observed from 1220.5 μg l−1 h (586.5–2227.2) without clarithromycin to 1820.4 μg l−1 h (521.0–5000.2) with clarithromycin.
Figure 1.
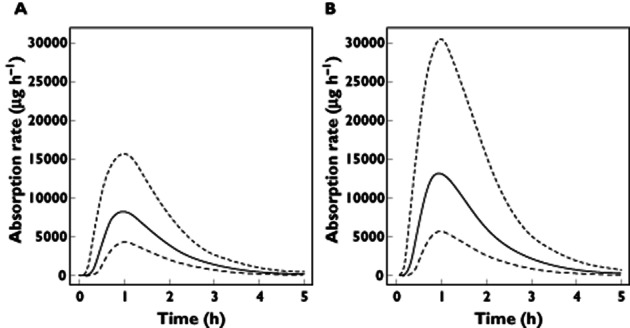
Absorption rate of dabigatran with and without clarithromycin. The solid black line represents the median absorption rate of dabigatran without clarithromycin (A) and with clarithromycin (B). The dashed lines indicate the 5% and 95% percentiles
Model evaluation
The goodness of fit plots for the model (Supplementary Figure S1) show no apparent bias in model prediction except for high concentration and ECT values. The NPDE was not biased. According to the VPC (Figure 2), average observed values were well predicted and only extreme profiles were outside 90% of the simulated values, showing the good predictive properties of the model.
Figure 2.
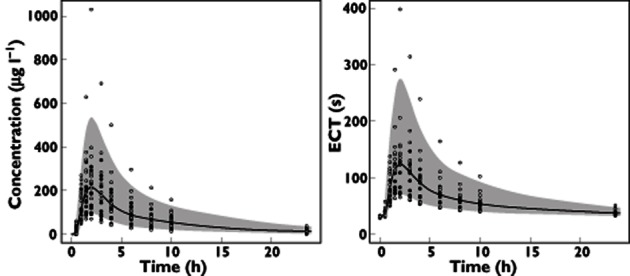
Visual predictive check. The envelope indicates the 90% confidence interval of simulations. The grey circles represent data obtained without clarithromycin. The white circles represent data obtained with clarithromycin
Discussion
Our study showed that the best model to describe the pharmacokinetics and pharmacodynamics of dabigatran in healthy volunteers was a two compartment model with an absorption following an inverse Gaussian law, associated with a linear effect model. To the best of our knowledge, this is the first population model developed on the basis of rich pharmacokinetic and pharmacodynamic data allowing precise description of the complex mechanism of absorption of DE. Other population models published were developed on the basis of patient cohorts receiving thromboembolism prophylaxis after orthopaedic surgery or in the context of atrial fibrillation. These models, based on sparse blood sampling did not allow relevant characterization of the complex absorption of dabigatran DE 8, 15, 16.
Concerning methodology, we employed an original and powerful statistical approach to test for drug–drug interaction. In contrast to standard bioequivalence approaches, population models are little used, despite their demonstrated superiority for the evaluation of drug–drug interactions in terms of statistical power to demonstrate such interactions 17, 18. These models also permit elucidation of the mechanism underlying the interaction. In our case, recourse to an inverse Gaussian model allowed identification of the different stages of the absorption phase and consequently precise evaluation of the drug–drug interaction. This approach has already been successfully used to model P-gp inhibition and its effect on the absorption of digoxin, another P-gp substrate 12.
In our study, clarithromycin increased exposure to dabigatran (AUC) by more than 50%. This increase resulted in a proportional prolongation of coagulation time. We showed that this phenomenon is explained solely by an increase in bioavailability from 6.5 to 10%. This result illustrates perfectly the mechanism involved in the inhibition of P-gp by clarithromycin.
In another study, evaluating the same interaction, the authors showed 19 and 20% increases in AUC and Cmax, respectively 5. This difference may be explained by the greater interindividual variability in DE bioavailability observed in the presence of clarithromycin (38.6 vs. 72.5%) in our study, the effect of clarithromycin differing substantially from one volunteer to another (Figure 3). This variability could be explained by genetic factors, such as polymorphism of the gene coding for P-gp and CES1, which limits the biotransformation of the drug's oral etexilate form to active dabigatran. In a recent follow-up subanalysis of the RE-LY study (atrial fibrillation treated with dabigatran etexilate), the authors identified a common gene variant that seems to influence the bleeding risk associated with the drug 2, 19. Their conclusion is that a genetic polymorphism of ABCB1 (rs4148738) and CES1 (rs8192935) were significantly associated with higher dabigatran peak concentrations.
Figure 3.
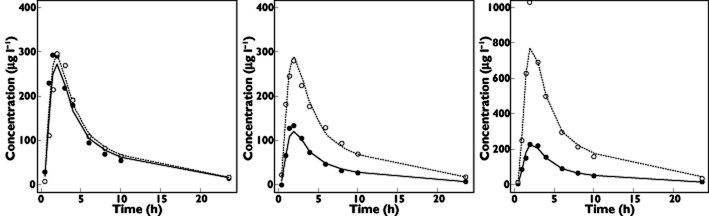
Pharmacokinetic profiles of three volunteers. Fit of dabigatran plasma concentration with (solid line) and without (dashed line) clarithromycin. Grey circles represent data obtained without clarithromycin. White circles represent data obtained with clarithromycin
In conclusion, the proposed model effectively describes the complex pharmacokinetics of dabigatran, allowing drug–drug interactions related to P-gp modulation to be taken into account. Using this model, we showed that exposure to dabigatran is increased by 50% in the presence of clarithromycin and is characterized by substantial variability. In contrast to non-compartmental analysis, the population analysis approach is an efficient tool to investigate other drug–drug interactions with dabigatran concerning P-gp inhibitors, inductors or substrates.
Acknowledgments
This research has received funding support from the University Hospital of Saint-Etienne, and was promoted by the University Hospital of Saint-Etienne.
Competing Interests
All authors have completed the Unified Competing Interest from http://www.icmje.org/coi_disclorure.pdf. and declare there is no support from any organization for the submitted work, no financial relationships with any organization that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1
Goodness of fit plots. The grey circles represent data obtained without clarithromycin. The white circles represent data obtained with clarithromycin.The solid line indicates the line of identity. The dashed line indicates the regression line
References
- 1.Eriksson BI, Dahl OE, Büller HR, Hettiarachchi R, Rosencher N, Bravo M-L, Ahnfelt L, Piovela F, Stangier J, Lebo KA, Reilly P. A new oral direct thrombin inhibitor, dabigatran etexilate, compared with enoxaparin for prevention of thromboembolic events following total hip or knee replacement: the BISTRO II randomized trial. J Thromb Haemost. 2005;3:103–111. doi: 10.1111/j.1538-7836.2004.01100.x. [DOI] [PubMed] [Google Scholar]
- 2.Connolly SJ, Ezekowitz MD, Yusuf S, Eikelboom J, Oldgren J, Parekh A, Pogue J, Reilly PA, Themeles E, Varrone J, Wang S, Alings M, Xavier D, Zhu J, Diaz R, Lewis BS, Darius H, Diener HC, Joyner CD, Wallentin L RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361:1139–1151. doi: 10.1056/NEJMoa0905561. [DOI] [PubMed] [Google Scholar]
- 3.Blech S, Ebner T, Ludwig-Schwellinger E, Stangier J, Roth W. The metabolism and disposition of the oral direct thrombin inhibitor, dabigatran, in humans. Drug Metab Dispos. 2008;36:386–399. doi: 10.1124/dmd.107.019083. [DOI] [PubMed] [Google Scholar]
- 4.Stangier J, Eriksson BI, Dahl OE, Ahnfelt L, Nehmiz G, Stähle H, Rathgen K, Svärd R. Pharmacokinetic profile of the oral direct thrombin inhibitor dabigatran etexilate in healthy volunteers and patients undergoing total hip replacement. J Clin Pharmacol. 2005;45:555–563. doi: 10.1177/0091270005274550. [DOI] [PubMed] [Google Scholar]
- 5.CHMP. Pradaxa, INN- dabigatran etexilate CHMP assessment report for radaxa. EMEA, editor. 2008 Apr 10; 1–36. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000829/WC500041062.pdf (last accessed 30 September 2012)
- 6.Delavenne X, Moracchini J, Laporte S, Mismetti P, Basset T. UPLC MS/MS assay for routine quantification of dabigatran – a direct thrombin inhibitor – in human plasma. J Pharm Biomed Anal. 2012;58:152–156. doi: 10.1016/j.jpba.2011.09.018. [DOI] [PubMed] [Google Scholar]
- 7.Nowak G. The ecarin clotting time, a universal method to quantify direct thrombin inhibitors. Pathophysiol Haemost Thromb. 2003;33:173–183. doi: 10.1159/000081505. [DOI] [PubMed] [Google Scholar]
- 8.Liesenfeld K-H, Schäfer HG, Trocóniz IF, Tillmann C, Eriksson BI, Stangier J. Effects of the direct thrombin inhibitor dabigatran on ex vivo coagulation time in orthopaedic surgery patients: a population model analysis. Br J Clin Pharmacol. 2006;62:527–537. doi: 10.1111/j.1365-2125.2006.02667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuhn E, Lavielle M. Maximum likelihood estimation in nonlinear mixed effects models. Comput Stat Data Anal. 2005;49:1020–1038. [Google Scholar]
- 10.Weiss M. A novel extravascular input function for the assessment of drug absorption in bioavailability studies. Pharm Res. 1996;13:1547–1553. doi: 10.1023/a:1016039931663. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, Weiss M, D'Argenio DZ. A note on population analysis of dissolution-absorption models using the inverse Gaussian function. J Clin Pharmacol. 2008;48:719–725. doi: 10.1177/0091270008315956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weiss M, Sermsappasuk P, Siegmund W. Modeling the kinetics of digoxin absorption: enhancement by P-glycoprotein inhibition. J Clin Pharmacol. 2012;52:381–387. doi: 10.1177/0091270010396711. [DOI] [PubMed] [Google Scholar]
- 13.Mandema JW, Verotta D, Sheiner LB. Building population pharmacokinetic – pharmacodynamic models. I. Models for covariate effects. J Pharmacokinet Biopharm. 1992;20:511–528. doi: 10.1007/BF01061469. [DOI] [PubMed] [Google Scholar]
- 14.Brendel K, Comets E, Laffont C, Mentre F. Evaluation of different tests based on observations for external model evaluation of population analyses. J Pharmacokinet Pharmacodyn. 2010;37:49–65. doi: 10.1007/s10928-009-9143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liesenfeld KH, Lehr T, Dansirikul C, Reilly PA, Connolly SJ, Ezekowitz MD, Yusuf S, Wallentin L, Haertter S, Staab A. Population pharmacokinetic analysis of the oral thrombin inhibitor dabigatran etexilate in patients with non-valvular atrial fibrillation from the RE-LY trial. J Thromb Haemost. 2011;9:2168–2175. doi: 10.1111/j.1538-7836.2011.04498.x. [DOI] [PubMed] [Google Scholar]
- 16.Trocóniz IF, Tillmann C, Liesenfeld K-H, Schäfer HG, Stangier J. Population pharmacokinetic analysis of the new oral thrombin inhibitor dabigatran etexilate (BIBR 1048) in patients undergoing primary elective total hip replacement surgery. J Clin Pharmacol. 2007;47:371–382. doi: 10.1177/0091270006297228. [DOI] [PubMed] [Google Scholar]
- 17.Panhard X, Mentré F. Evaluation by simulation of tests based on non-linear mixed-effects models in pharmacokinetic interaction and bioequivalence cross-over trials. Stat Med. 2005;24:1509–1524. doi: 10.1002/sim.2047. [DOI] [PubMed] [Google Scholar]
- 18.Delavenne X, Laporte S, Demasles S, Mallouk N, Basset T, Tod M, Girard P, Mismetti P. Investigation of PK-PD drug-drug interaction between acenocoumarol and amoxicillin plus clavulanic acid. Fundam Clin Pharmacol. 2009;23:127–135. doi: 10.1111/j.1472-8206.2008.00642.x. [DOI] [PubMed] [Google Scholar]
- 19.Pare G. RELY- genetics: genetic determinants of dabigatran plasma levels and their relation to clinical response. Eur Heart J. 2012;33:abstr 5170. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.