

Abstract
Synaptotagmin-12 (Syt12) is an abundant synaptic vesicle protein that—different from other synaptic vesicle-associated synaptotagmins—does not bind Ca2+. Syt12 is phosphorylated by cAMP-dependent protein kinase-A at serine-97 in an activity-dependent manner, suggesting a function for Syt12 in cAMP-dependent synaptic plasticity. To test this hypothesis, we here generated (1) Syt12 knock-out mice and (2) Syt12 knockin mice carrying a single amino-acid substitution [the serine-97-to-alanine- (S97A)-substitution]. Both Syt12 knock-out mice and Syt12 S97A-knockin mice were viable and fertile, and exhibited no measurable change in basal synaptic strength or short-term plasticity as analyzed in cultured cortical neurons or in acute hippocampal slices. However, both Syt12 knock-out and Syt12 S97A-knockin mice displayed a major impairment in cAMP-dependent mossy-fiber long-term potentiation (LTP) in the CA3 region of the hippocampus. This impairment was observed using different experimental configurations for inducing and monitoring mossy-fiber LTP. Moreover, although the Syt12 knock-out had no effect on the short-term potentiation of synaptic transmission induced by the adenylate–cyclase activator forskolin in cultured cortical neurons and in the CA1 region of the hippocampus, both the Syt12 knock-out and the Syt12 S97A-knockin impaired the long-term increase in mossy-fiber synaptic transmission induced by forskolin. Thus, Syt12 is essential for cAMP-dependent presynaptic LTP at mossy-fiber synapses, and a single amino-acid substitution that blocks the cAMP-dependent phosphorylation of Syt12 is sufficient to impair the function of Syt12 in mossy-fiber LTP, suggesting that cAMP-dependent phosphorylation of Syt12 on serine-97 contributes to the induction of mossy-fiber LTP.
Introduction
Synaptotagmins are membrane proteins composed of a short N-terminal noncytoplasmic (usually intravesicular) sequence, a single transmembrane region, a linker sequence of variable length, and two C2-domains. Synaptotagmins were discovered with the identification of synaptotagmin-1 (Syt1), the Ca2+-sensor for neurotransmitter release in presynaptic nerve terminals (Perin et al., 1990). Mammals contain 16 genes encoding synaptotagmins. Eight of these synaptotagmins bind Ca2+ via their C2-domains (Syt1-Syt3, Syt5–7, Syt9, and Syt10), whereas the other eight synaptotagmins do not [Syt4, Syt8, Syt11–16 (Pang and Südhof, 2010)]. Among the Ca2+-binding synaptotagmins, Syt1, Syt2, and Syt9 function as fast Ca2+-sensors for neurotransmitter release and neuroendocrine exocytosis, and Syt10 acts as a Ca2+-sensor for IGF-1 exocytosis (Geppert et al., 1994; Xu et al., 2008; Cao et al., 2011). The function of the Ca2+-independent synaptotagmins, however, remains largely unclear.
Among Ca2+-independent synaptotagmins, only Syt4 has been extensively studied. In different contexts, Syt4 was found to act as an activator or as an inhibitor of exocytosis (Littleton et al., 1999; Ferguson et al., 2000, 2004; Wang et al., 2001; Machado et al., 2004; Zhang et al., 2004; Yoshihara et al., 2005; Ting et al., 2006; Barber et al., 2009; Zhang et al., 2011). No clear function for Syt4 in mammals has emerged, and its localization in mammals continues to be uncertain. In contrast, in Drosophila conclusive evidence revealed a function of Syt4 in postsynaptic exocytosis (Yoshihara et al., 2005; Barber et al., 2009). Among the other Ca2+-independent synaptotagmins, only Syt12 has been studied in some detail (Maximov et al., 2007a). Syt12 was found to be colocalized with Syt1 and bound to Syt1 on synaptic vesicles. Syt12 is phosphorylated by cAMP-dependent protein kinase A (PKA) at a single residue (serine-97), suggesting that Syt12 may modulate Ca2+-triggering of release by Syt1 in a cAMP-dependent manner (Maximov et al., 2007a). However, these studies were performed exclusively in vitro, and no further tests of Syt12 function were presented.
Here, we have examined the function of Syt12 using a combined genetic and electrophysiological approach. We generated and analyzed Syt12 knock-out (KO) and Syt12 knockin (KI) mice. The latter carry a single alanine substitution of serine-97, the PKA-dependent phosphorylation site of Syt12 (referred to as the Syt12 S97A substitution). Our data reveal that Syt12 is not essential for basal synaptic transmission, and does not detectably contribute to short-term plasticity, but is required for cAMP-dependent mossy fiber LTP (mfLTP), a presynaptic form of long-term plasticity that is induced by PKA activation (Huang et al., 1994; Weisskopf et al., 1994). Our results demonstrate that both the Syt12 S97A substitution and the Syt12 KO severely impair mfLTP, and that Syt12 is required for the long-term increase in mossy-fiber synaptic transmission that is induced by forskolin and may mimic mfLTP. Thus, our results suggest that Syt12 is at least one of the elusive PKA-targets postulated to mediate LTP at the mossy fiber to CA3 pyramidal-cell synapse, and thereby address a long-standing problem that has hindered our understanding of this important form of synaptic plasticity.
Materials and Methods
Generation of Syt12 S97A-substituted KI and Syt12 KO mice.
Syt12 S97A KI and Syt12 KO mice were generated by standard procedures (Rosahl et al., 1993; Ho et al., 2006) using homologous recombination in embryonic stem cells targeting exon 4 of the Syt12 gene. In brief, a ∼15 kb genomic clone containing exon 4 up to the 3′ untranslated region (UTR) was isolated using a 32P-labeled probe for exon 4 of Syt12, which was amplified with primers YJ0601 (GCTGTCACCTAGCGATGGTGCC) and YJ0602 (CACGATCTGT TCGTCTGGCAGC). The targeting vector contained a Ser-97-Ala mutation in exon 4, and exon 4–6 were flanked with loxP sites for cre recombination; neomycin resistance gene flanked by frt sites was introduced at a nonconserved region between exon 5 and 6; and a diphtheria toxin (DT) expressing cassette was inserted at the end of 3′ UTR. Homologous recombination was achieved by electroporation of the linearized targeting construct in R1 embryonic stem cells (Nagy et al., 1993), and positive clones were isolated by drug selection and screened by Southern blotting and PCR genotyping. Clones with confirmed homologous recombination were then injected into C57BL/6 blastocysts. Southern blotting was performed with 32P labeled probes that anneal to the genomic sequences outside the 5′ and 3′ arms of the targeting vector: AseI restriction digest results in a 10.3 kb fragment for wild-type (WT) DNA, and 3.6 kb for mutant clones with additional AseI site that was introduced with loxP and S97A point-mutation (Fig. 1B); SpeI sites for 3′ downstream gives 12.3 kb for WT and 7.2 kb for mutant clone with additional SpeI introduced with neo cassette (data not shown). Flp recombinase transgenic mice (Dymecki, 1996) were used to remove the neomycin resistance cassette for Syt12 S97A KI, followed by breeding to protamine-cre transgenic mice (O'Gorman et al., 1997) to generate constitutive Syt12 KO. The floxed, flp recombined Syt12 S97A allele was genotyped by PCR with oligonucleotide primers YJ08109 (TAGGCTGTCAAGTCCAATAGGTCC) and YJ08110 (TACAGAAATAGTCTGTC ACCCTGG). This reaction results in a 218 bp WT band and a 317 bp floxed band. The Syt12 KO allele was genotyped with YJ0601 (GCTGTC ACCTAGCGATGGTGCC) and YJ0602 (CACGATCTGTTCGTCTGGCAGC) to identify a 447 bp WT band, and with YJ07068 (ATGTCCTTAAGTGCTGGTCCC) and YJ08109 to detect a 297 bp mutant band. All animal experiments were performed according to institutional guidelines. The Syt12 S97A KI mouse line was submitted to the Jackson Laboratories and is freely available to the community. Animal procedures adhered to National Institutes of Health guidelines and were approved by our respective Institutional Animal Care and Use Committees.
Figure 1.

Generation of Syt12 S97A KI and Syt12 KO mice. A, Gene targeting strategy for Syt12 S97A KI and Syt12 KO mice. In the targeting vector, we introduced a serine-97 to alanine (S97A) point mutation into exon 4 (asterisk), and inserted a neomycin resistance cassette (neo) flanked by frt recombination sites into an evolutionarily nonconserved region of the intron between exons 6 and 7. Mice produced with the targeted allele were crossed with mice expressing flp recombinase to delete the neomycin resistance cassette, resulting in Syt12 S97A KI mice. Cre recombination in the Syt12 S97A KI mice in the germline was used to derive constitutive Syt12 KO mice (DT, diphtheria toxin-expressing cassette; A, AseI restriction sites used for Southern blotting). B, Southern blot analysis of genomic DNA from heterozygous embryonic stem cells after homologous recombination of the Syt12 targeting vector (+/ki) and nonrecombined control cells (+/+). A 5′ outside probe was used after AseI digest (WT band, 12.3 kb, mutant band, 3.6 kb). C, Survival ratio of offspring from heterozygous matings of Syt12 S97A KI mice (left) and of Syt12 KO mice (right). The gray shaded area indicates the expected Mendelian ratio [n = 83 mice total for the Syt12 S97A KI analysis (+/+: 15, +/KI: 50, KI/KI: 18 mice) and n = 120 mice total for the Syt12 KO analysis (+/+: 33, +/KO: 58, KO/KO: 29)].
Generation of phospho-specific Syt12 antibody.
Fourteen amino acid peptides containing the phosphorylated Ser97 (CPPSRKGS97LSIEDT) were synthesized in the Protein Chemistry Technology Center at UT Southwestern Medical Center (Dallas, TX). Synthesized peptides were coupled with keyhole limpet hemocyanin (KLH) (Südhof et al., 1989) and injected into two rabbits for three consecutive times with interinjection intervals of 1 week. The blood was collected from the rabbits, and the red blood cells were separated from the antiserum by centrifugation. The antisera were tested by regular Western blotting procedures (see Protein quantification of brain homogenates, below) of tissue homogenates. The patterns of the bands were compared with band patterns of available Syt12 antibody (U5400), and antisera revealing positive signals were used for all experiments. Antibody specificity was unequivocally confirmed in the Syt12 KI and KO animals.
Protein quantification of brain homogenates.
Brain tissue from three adult (P30) male littermate mice per genotype was isolated and homogenized in PBS, 10 mm EDTA, 1 mm PMSF, and proteinase inhibitor mixture tablet (Roche). Brain lysates (40 μg) were separated by SDS-PAGE, and immunoblotting was performed with standard methods as described previously (Rosahl et al., 1995). Radioisotope 125I-labeled secondary antibodies were used for quantitative analyses followed by Typhoon PhosphorImager (GE Health Care Life Sciences) detection with GDP dissociation inhibitor (GDI) or valosin-containing protein (VCP) as internal controls. The following primary antibodies were used: liprin (4396, 1:5000), GDI (81.2, 1:2000, Synaptic Systems), VCP (443B, 1:1000), glial fibrillary acidic protein (GFAP; 172.002, 1:5000, Synaptic Systems), synapsin (46.1, 1:1000, Synaptic Systems), β-actin (1:1000, Sigma), 14-3-3 (E12, 1:1000, Santa Cruz Biotechnology), Cpx-1/2 (122 002, 1:1000, Synaptic Systems), RIM and Raphillin (U1565, 1:2000), Munc13-1 (CL266 B1, 1:1000, Synaptic Systems), ELKS (P224, 1:500); Syt 12 (U5400, 1:250), phospho-Syt 12 (342C, 1:250), Stx 1 (HPC-1, 1:2000, Synaptic Systems), synaptosomal-associated protein 25 (SNAP-25; 71.1, 1:2000, Synaptic Systems), cysteine-string protein (CSP; R809, 1:1000), Syb 2 (69.1, 1:2500, Synaptic Systems), Syt 1 (41.1, 1:2000, Synaptic Systems), and Rab3 (42.1, 1:2000, Synaptic Systems).
Neuronal cultures.
Neuronal cultures were obtained from mouse cortex as described previously (Yang et al., 2010). Briefly, mouse cortices were dissected from postnatal day 0 (P0) of Syt12 KO or littermate wild-type mice, dissociated by papain digestion (10 U/ml, with 1 μm Ca2+ and 0.5 μm EDTA) for 20 min at 37°C, plated on Matrigel-coated circular glass coverslips (12 mm diameter), and cultured in MEM (Invitrogen) supplemented with 2% B27 (Invitrogen), 0.5% w/v glucose, 100 mg/L transferrin, 5% fetal bovine serum, and 2 μm Ara-C (Sigma). Neurons were analyzed at DIV14–16. For overexpression experiments, cortical neurons were infected with lentiviruses expressing wild-type Syt12, Syt12 S97A mutation, or control virus at DIV5 and analyzed 9–10 d later (Maximov et al., 2007a).
Electrophysiological recordings in cultured neurons.
Recordings were performed in whole-cell patch-clamp mode (Yang et al., 2010). Evoked synaptic responses were triggered by a bipolar electrode placed 100–150 μm from the soma of neurons recorded. Patch pipettes were pulled from borosilicate glass capillary tubes (Warner Instruments) using a PC-10 pipette puller (Narishige). The resistance of pipettes filled with intracellular solution varied between 3 and 5 MOhm. After achieving the whole-cell configuration and equilibration of the intracellular pipette solution, the series resistance was compensated to 8–10 MOhm. Synaptic currents were monitored with a Multiclamp 700A amplifier (Molecular Devices). The frequency, duration, and magnitude of the extracellular stimulus were controlled with a Model 2100 Isolated Pulse Stimulator (A-M Systems) synchronized with Clampex 9.2 data acquisition software (Molecular Devices). The whole-cell pipette solution contained the following (in mm): 120 CsCl, 5 NaCl, 1 MgCl2, 10 HEPES, 10 EGTA, 0.3 Na-GTP, 3 Mg-ATP and 5 QX-314 (pH 7.2, adjusted with CsOH). The bath solution contained the following (in mm): 140 NaCl, 5 KCl, 2 MgCl2, 2 CaCl2, 10 HEPES-NaOH pH 7.4, 10 glucose. IPSCs and EPSCs were pharmacologically isolated by adding the AMPA and NMDA receptor blockers CNQX (10 μm) and AP-V (50 μm) or the GABAA-receptor blocker picrotoxin (100 μm) to the extracellular solution. Spontaneous mIPSCs and mEPSCs were monitored in the presence of tetrodotoxin (TTX; 1 μm) to block action potentials. Miniature events were analyzed in Clampfit 10 (Molecular Devices) using the template matching search and a minimal threshold of 5 pA, and each event was visually inspected for inclusion or rejection by an experimenter blind to the recording condition. Statistical analyses were performed with Student's t tests comparing test to control samples analyzed in the same experiments.
Electrophysiology in hippocampal slices.
Syt12 WT and S97A littermate mice aged 3–6 weeks of both sex, as well as Syt12 WT and KO littermates, were shipped from Stanford University to the Albert Einstein College of Medicine. The recipient investigator was blind to genotype. Acute, transverse hippocampal slices (400 μm thick) were prepared with a VT1200S microtome (Leica). Hippocampi were isolated in ice-cold cutting solution containing the following (in mm): 215 sucrose, 20 glucose, 26 NaHCO3, 4 MgCl2, 4 MgSO4, 1.6 NaH2PO4, 1 CaCl2, and 2.5 KCl. Slices were then placed, at room temperature, in a holding chamber containing 50% cutting solution and 50% artificial CSF (ACSF) recording solution containing the following (in mm): 124 NaCl, 26 NaHCO3, 10 glucose, 2.5 KCl, 1 NaH2PO4, 2.5 CaCl2, and 1.3 MgSO4. After 30 min, the 1:1 solution was switched to ACSF. For some mossy fiber LTP experiments, ASCF containing 4 CaCl2 and 4 MgCl2 was used to reduce CA3 excitability. All solutions were equilibrated for 30 min with 95% O2 and 5% CO2, pH 7.4. Slices recovered at room temperature for at least 1 h before transfer to a submersion-type recording chamber with temperature-controlling elements (Warner Instruments) and were perfused with ACSF at ∼2 ml/min using a peristaltic pump (Rainin). Experiments were performed at room temperature.
To monitor synaptic transmission and plasticity, conventional field and whole-cell recordings were made with a Multi-Clamp 700A or 700B amplifier (Molecular Devices). For field recordings, patch-type stimulating and recording pipettes (∼2.6–4.0 mΩ) were filled with ACSF and 1 m NaCl, respectively. For whole-cell recordings, pipettes were filled with intracellular recording solution containing the following (in mm): 131 Cs-gluconate, 8 NaCl, 1 CaCl2, 10 EGTA, 10 HEPES, 10 glucose, 5 Mg-ATP, and 0.4 Na-GTP (pH 7.25, 291 mmol/L). Series resistance (∼6–25 MΩ) was monitored during experiments with a −5 mV, 80 ms voltage step. Neurons exhibiting > 20% change in series resistance were excluded from analysis. Excitatory extracellular field potentials (fEPSP) were recorded from the CA1 region of the hippocampus by stimulating Schaffer collaterals in stratum radiatum with monopolar square-wave pulses (100–200 μs) delivered through a stimulus isolator (Isoflex; AMPI). The peak amplitude of the fEPSP was analyzed during the experiments in which basal synaptic transmission was characterized, whereas fEPSP slope was analyzed for the input/output analysis. For extracellular mossy fiber-to-CA3 pyramidal cell recordings, the upper or lower blade of the dentate gyrus was stimulated (just at the hilar border) and field potentials were recorded in stratum lucidum in CA3. To monitor IPSCs from CA1 pyramidal cells, whole-cell recordings were obtained and neurons voltage-clamped at +10 mV. Inhibitory fibers located in stratum radiatum were stimulated while glutamatergic synaptic transmission was blocked using NMDA and AMPA/KA receptor antagonists (25 μm d-APV and 10 μm NBQX, respectively). To isolate mIPSCs, 1 μm TTX was additionally bath applied. To induce long-term depression of inhibition (iLTD), we used a theta burst stimulation (TBS) protocol consisting of 10 bursts of 5 stimuli (100 Hz within each burst, 200 ms interburst interval) repeated 4 times, 5 s apart. For whole-cell recordings from CA3 pyramidal neurons to monitor mossy fiber synaptic transmission and plasticity, NMDA receptor-mediated EPSCs were pharmacologically isolated with 10 μm NBQX and 100 μm picrotoxin. Cells were voltage clamped at +30–40 mV to reduce the voltage-dependent Mg2+ block of the NMDA receptor. Under these recording conditions, the stimulation intensity required to evoke an EPSC in the depolarized cell is reduced, thus limiting the likelihood of recruiting recurrent CA3 network activity. For experiments shown in Figure 6, 20 mm BAPTA was included in the internal solution to prevent NMDA receptor desensitization, thus allowing a more accurate measurement of short-term plasticity dynamics (Tong et al., 1995). For long-term plasticity, single test pulses were elicited every 20 s. Two stimuli spaced 40 ms apart were occasionally delivered during the baseline and post-LTP induction to monitor paired-pulse ratio (PPR) changes. mfLTP was elicited in normal ACSF or 4/4 ACSF with two trains of synaptic activity, each train containing 125 pulses at 25 Hz, spaced apart by 20 s. At the end of each individual mossy fiber experiment, 1 μm DCG-IV, a group II mGluR agonist, was bath applied to determine the relative contribution of mossy fiber synaptic inputs. DCG-IV reduced responses by >85%. Mossy fiber responses were measured after subtraction of the remaining responses after DCG-IV application. The magnitude of long-term plasticity was determined by comparing baseline-averaged responses before induction with the last 30 min (see Fig. 7) or the last 10 min (see Figs. 8, 9) of the experiment. Example traces are averages of at least 30 consecutive sweeps taken from a single representative experiment.
Figure 6.

Characterization of excitatory synaptic transmission at mossy fiber synapses in acute slices of the hippocampal CA3 region from WT and Syt12 S97A KI mice (all data represent whole-cell recordings of NMDA receptor-mediated EPSCs). A, High-frequency facilitation triggered with a presynaptic 25 Hz stimulus burst. Representative traces are shown on top, and summary graphs of the EPSC amplitudes on the bottom (normalized to first pulse in the burst after subtracting the baseline current that was reached before each new pulse). B, Low-frequency facilitation induced by changing the baseline stimulation frequency from 0.05 Hz to 0.5 Hz. Traces (above) were taken from a representative experiment whose time points match the numbers indicated in the summary graph (below). C, Measurements of the PPR. Representative traces of two sequential EPSCs evoked with increasing interstimulus intervals (top) and summary graphs of the resulting paired-pulse ratio (bottom) were obtained from slices from littermate WT and S97A KI mice. Inter-stimulus intervals of 20, 40, 200, 500, and 1000 ms were tested. Data are means ±SEM. Number of slices/mice tested are indicated in parenthesis. No significant difference in any of the comparisons was found using Student's t test (p > 0.05).
Figure 7.

The Syt12 S97A KI and the Syt12 KO impair mfLTP as monitored by extracellular field recordings. A, Representative traces of mossy fiber field-EPSPs (fEPSPs) before (trace 1) and after (trace 2) induction of mfLTP in acute slices from littermate WT and Syt12 S97A KI mice. mfLTP was induced by two 25 Hz/5 s stimulus trains separated by 20 s and applied in the presence of the NMDA-receptor antagonist d-APV (50 μm) that was washed out immediately after induction. After each individual mfLTP experiment, the slice was exposed to 1 μm DCG-IV, a group II mGluR agonist, to determine the relative contribution of mossy fiber synaptic inputs to the fEPSP (gray traces). B, Summary data of mossy fiber fEPSP amplitudes plotted as a function of time before and after mfLTP induction (arrow). Hatched box indicates the period when the slice was exposed to d-APV. For clarity, the DCG-IV block data were removed from the summary graph. Numbers indicate time-points for the traces shown in A. C, Distributions for the magnitude of LTP observed in each genotype, with circles indicating the values observed in individual experiments (WT, 187.6 ± 8.4% of baseline; S97A KI, 143.2 ± 8.5% of baseline; p < 0.003 by Student's t test). D–F, Same as A–C, except that mfLTP was examined in Syt12 WT and KO littermate mice. Circles in F indicate the values observed in individual experiments (WT 202.0 ± 10.7% of baseline; KO 145.6 ± 11.3% of baseline; p < 0.005 by Student's t test). Data shown are means ±SEM. Number of slices/mice tested are indicated in parenthesis. Horizontal brackets in B and E indicate the time window where the magnitude of LTP was measured. Given the large magnitude of PTP observed in extracellular recordings, the first three points after the stimulus trains were digitally removed to facilitate visualization of LTP. These points were not different between groups.
Figure 8.
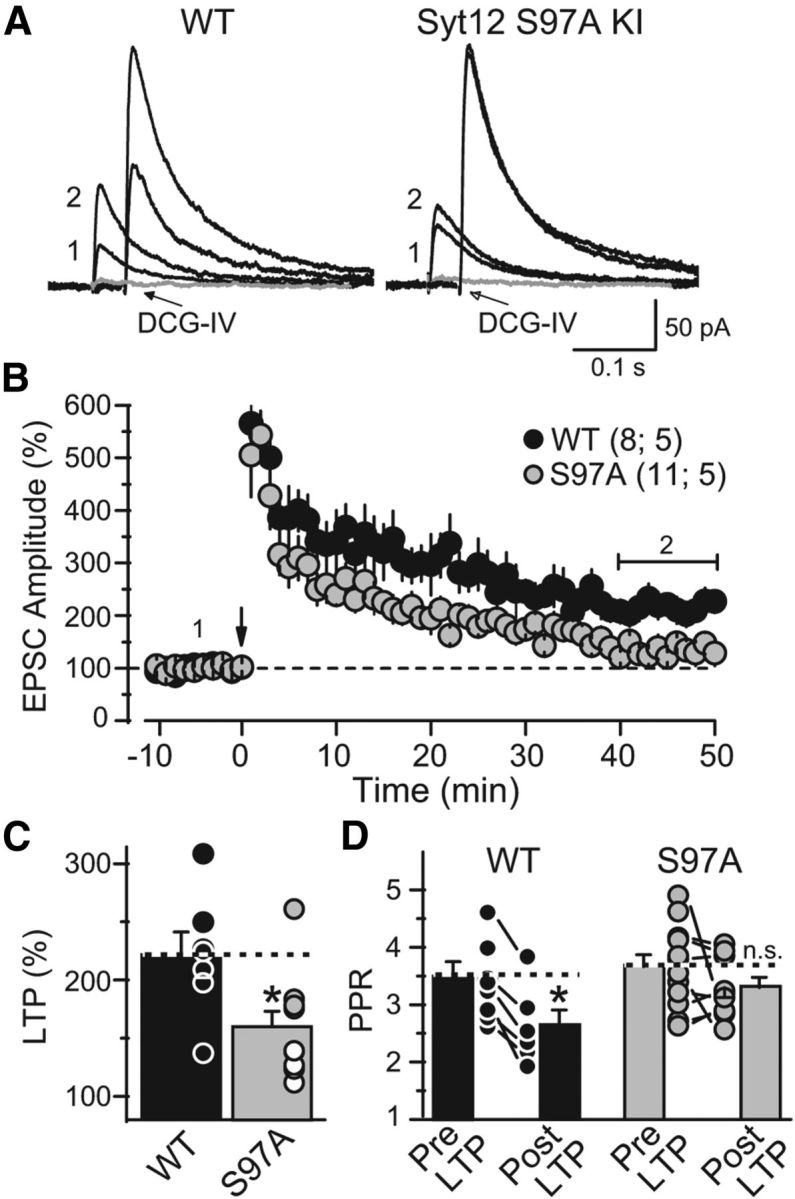
The Syt12 S97A KI impairs mfLTP as monitored by whole-cell recordings. A, Representative averaged traces of NMDA-receptor EPSCs before (trace 1) and after induction of mfLTP (trace 2). mfLTP was induced by two 25 Hz/5 s stimulus trains separated by 20 s. After each individual mfLTP experiment, the slice was exposed to 1 μm DCG-IV, a group II mGluR agonist, to determine the relative contribution of mossy fiber synaptic inputs to the recorded EPSC (gray traces). B, Summary data of mossy fiber NMDA-receptor EPSC amplitudes plotted as a function of time before and after mfLTP induction (arrow). For clarity, the DCG-IV block data were removed from the summary graph. Numbers indicate time-points for the traces shown in A. The horizontal bracket indicates the time window where the magnitude of LTP was measured. C, Distribution of mfLTP magnitude for each genotype (WT 221.7 ± 19.7% of baseline; S97A 159.9 ± 13.4% of baseline; p = 0.016). Circles indicate the values observed in individual experiments. D, Paired-pulse ratio measured with a 40 ms interstimulus interval in WT and S97A KI neurons before and after mfLTP induction; note the significant decrease in WT but not in KI neurons (WT: pre-LTP 3.5 ± 0.2; post-LTP 2.6 ± 0.2; p = 0.039; S97A: pre-LTP 3.6 ± 0.2; post-LTP 3.3 ± 0.2; p > 0.05). Data are means ±SEM. Number of slices/mice tested are indicated in parenthesis.
Figure 9.

Forskolin-induced potentiation of mossy fiber synaptic transmission in Syt12 WT, S97A, and KO mice examined using whole-cell recordings. A, Representative traces (top) and summary graphs (bottom) of NMDA-receptor-mediated EPSCs recorded from pyramidal neurons in the CA3 region of the hippocampus in slices from littermate WT and Syt12 S97A KI mice. The summary graph depicts the normalized EPSC amplitude as a function of time before and after addition of 10 μm forskolin that was applied as indicated by the gray bar. Numbers indicate time-points for the traces shown above. DCG-IV (1 μm) was bath applied at the end of each individual experiment to determine the contribution of mossy fibers to synaptic transmission. For clarity, the DCG-IV block data were removed from the summary graph. The magnitude of forskolin-induced, long-term enhancement was significantly different between groups (WT 251.8 ± 37.5% of baseline; S97A 152.9 ± 20.8% of baseline; p = 0.040), whereas the short-term enhancement (min 5 to min 15) was not (WT 325 ± 45% of baseline; S97A 270 ± 37% of baseline; p = 0.356). B, Same as A except that slices were from littermate Syt12 KO and WT mice (forskolin-induced long-term enhancement: WT 207.8 ± 33.8% of baseline; KO 104.2 ± 12.7% of baseline; p = 0.027; short-term enhancement 353 ± 48% of baseline; KO 260 ± 53% of baseline; p = 0.153). Data are plotted as means ±SEM. Number of slices/mice tested are indicated in parenthesis. Horizontal brackets in the bottom panels indicate the time window where the magnitude of forskolin-induced potentiation was measured.
Data acquisition and statistical analysis.
Stimulation and acquisition were controlled by custom software. Output signals from field and whole-cell recordings were filtered at 2.4 kHz, acquired at 5 kHz, and stored online in IgorPro (Wavemetrics). All data are shown as means ±SEM. Statistics were performed using OriginPro (Origin Lab), and statistical significance was determined by the Student's t test (two tailed distribution, paired) unless otherwise stated. All analyses compared mutant (the Syt12 KO or Syt12 S97A substitution KI) to WT neurons examined in littermate mice in the same experiments by an experimenter who was blind to the genotype; only statistically significant differences are noted, and a lack of statistically significant difference is not specifically mentioned.
Reagents.
Stock solutions were prepared in water or DMSO, depending on the manufacturers' recommendation, and stored at −20°C. Upon experimentation, reagents were bath applied following dilution into ACSF. Final DMSO concentration was <0.01% total volume. NBQX, d- APV, and TTX were purchased from Ascent Scientific/Abcam. BAPTA and DCG-IV were purchased from Tocris Bioscience. Picrotoxin and salts for making ACSF and internal solution were purchased from Sigma.
Results
Generation of Syt12 S97A KI and Syt12 KO mice
We constructed a targeting vector in which we mutated serine-97 in exon 4 to alanine, and additionally inserted loxP sites into the introns 5′ of exon 4 and 3′ of exon 6 (Fig. 1A). The design of the targeting vector was guided by the goal of introducing a point mutation substituting alanine for serine-97, and of additionally allowing an out-of-frame deletion of exons (i.e., producing a KO).
We used the targeting vector for homologous recombination in embryonic stem cells, and identified correctly recombined clones with Southern blotting (Fig. 1B). We then generated mutant mice from three clones, crossed the mutant mice to transgenic mice expressing flp recombinase in the germ line to delete the neomycin resistance cassette (Fig. 1A), and bred the resulting S97A-KI mice to homozygosity. In addition, we deleted Syt12 exons 4–6 in the mutant mice by crossing them to transgenic mice expressing cre recombinase in the germ line, and also bred the resulting Syt12 KO mice to homozygosity. We systematically analyzed the survival of homozygous Syt12 S97A-KI and KO mice resulting from heterozygous matings in these crosses, but detected no impairment in survival caused by either the Syt12 S97A substitution or the Syt12 KO (Fig. 1C).
Immunoblotting analyses of homozygous Syt12 S97A-KI mice with generic and phospho-specific Syt12 antibodies revealed that the Syt12 S97A substitution abolished serine-97 phosphorylation as expected, but did not detectably alter the overall expression of Syt12 (Fig. 2A). Again as expected, we could not detect any Syt12 protein in Syt12 KO mice, demonstrating that the genetic manipulations of the Syt12 gene worked as intended (Fig. 2A).
Figure 2.
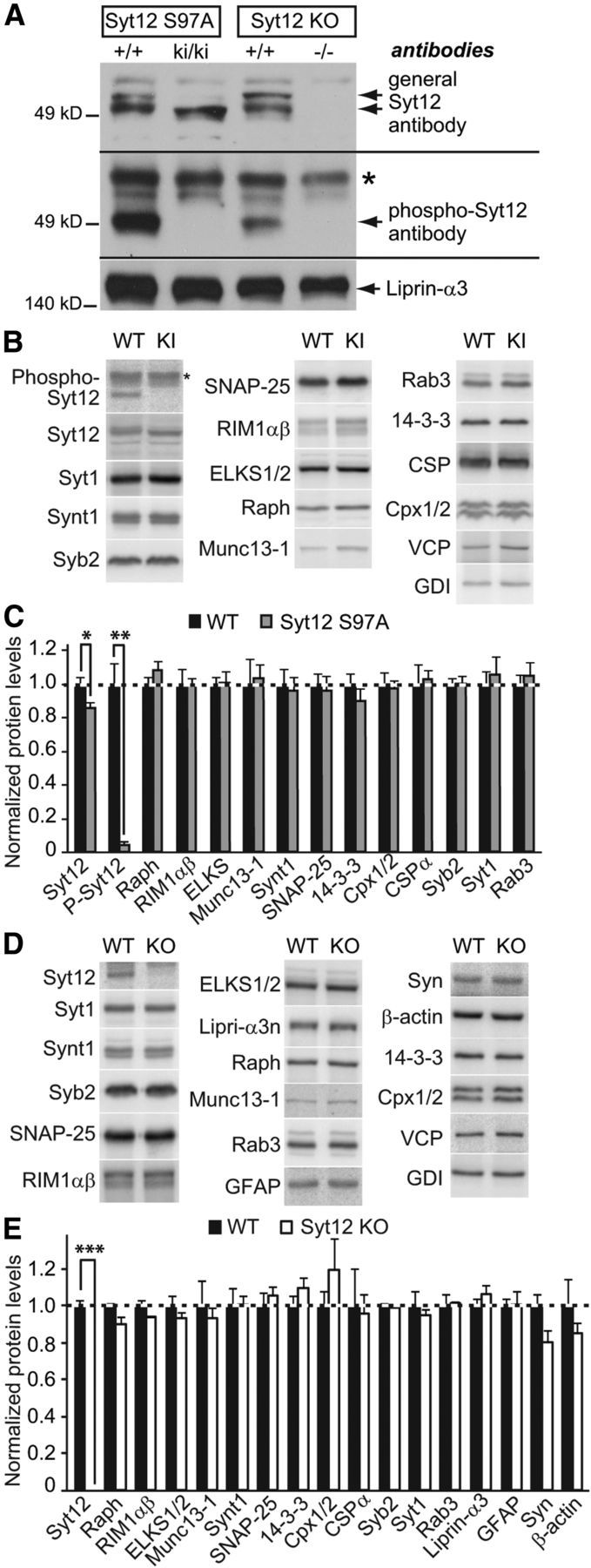
Characterization of protein expression in Syt12 KI and KO mice. A, Immunoblotting analysis of whole brain homogenates from Syt12 S97A KI and Syt12 KO mice using antibodies to Syt12 (top blot; upper band in +/+ samples corresponds to phosphorylated Syt12 and lower band to nonphosphorylated Syt12) and to phospho-Syt12 that is phosphorylated on serine-97 (* denotes nonspecific band). Note the loss of Syt12 immunoreactivity with phospho-specific antibodies against Syt12 in the S97A KI sample. Liprin-α3 antibodies were used as a loading control. B–E, Quantitative immunoblotting analysis of brain protein levels in littermate WT control mice and Syt12 S97A KI (B and C) or Syt12 KO (D and E). B and D depict representative blots visualized by PhosphorImager; C and E, quantification of protein levels using 125I-labeled secondary antibodies (n = 3 independent experiments). Cpx1/2, Complexin 1/2; ELKS1/2, Rab6-interacting protein; Raph, Raphilin; Syn, Synapsin; Syb2, Synaptobrevin-2; Synt1, Syntaxin-1). Statistical differences were assessed by Student's t test (*p < 0.05; **p < 0.01; ***p < 0.001).
We next tested whether the Syt12 S97A substitution or the Syt12 KO caused a major effect on brain structure or composition. Overall size and morphology of mutant brains did not reveal obvious changes. Analyses of the levels of a series of synaptic proteins by quantitative immunoblotting using 125I-tagged secondary antibodies confirmed that Syt12 is deleted in Syt12 KO mice, but failed to identify major changes in any protein tested (Figs. 2B–2E). These experiments thus suggest that the Syt12 S97A substitution and the Syt12 KO did not cause a major restructuring of the brain.
Cortical neurons cultured from Syt12 KO mice do not exhibit major changes in synaptic transmission
To analyze the functional consequences of the Syt12 mutations, we first tested in cultured cortical neurons the effect of the Syt12 KO on basal synaptic transmission and on the potentiation of synaptic transmission by forskolin (Fig. 3). We performed these measurements in cultured cortical neurons because this preparation allows quantitative measurements of basal synaptic parameters (Maximov et al., 2007b). We analyzed the Syt12 KO rather than the S97A substitution because we expected that any phenotype of S97A-KI mice should also be present in KO mice, whereas the reverse may not necessarily be true. Furthermore, we examined the effect of forskolin because our previous studies had shown that Syt12 overexpression altered a neuron's response to forskolin (Maximov et al., 2007a), suggesting that the Syt12 KO may also do so.
Figure 3.

Syt12 is not required for spontaneous or evoked neurotransmitter release in cortical neurons cultured from littermate WT and Syt12 KO mice. A, B, Representative traces (top) and summary graphs of the frequency and amplitudes (bottom) of mEPSCs (A) and or mIPSCs (B) with or without forskolin treatment (50 μm for 1 h), respectively. C, D, Representative traces (C) and summary graphs of the amplitudes (D) of EPSCs and IPSCs that were evoked by a single test pulse. E, Representative traces of IPSCs evoked by a 10 Hz stimulus train applied for 1 s. F, Plot of IPSC amplitudes during the 10 Hz stimulus train normalized to the first peak amplitude and plotted against the stimulus number. Data are means ±SEM; numbers of cells/independent cultures analyzed are shown in the bars. Statistical assessments were performed by Student's t test (*p < 0.05; **p < 0.01).
Measurements of the spontaneous miniature EPSC (mEPSC) frequency or amplitude failed to reveal a difference between neurons obtained from littermate WT and Syt12 KO mice, and application of forskolin produced the same increase in mEPSC frequency in WT and KO neurons (Fig. 3A). Similarly, the frequency and amplitude of spontaneous miniature IPSCs (mIPSCs) was not changed in Syt12 KO neurons, and the increase of mIPSC frequency induced by forskolin was also unaffected by the Syt12 KO (Fig. 3B). Furthermore, no difference in amplitude of evoked EPSCs or of evoked IPSCs was noted between WT and Syt12 KO neurons (Fig. 3C, 3D). Finally, IPSCs elicited by stimulus trains (10 Hz applied for 1 s) exhibited the same peak amplitude, delayed release [the continuing release after the end of the stimulus train, defined as all release after 100 ms after the last stimulus (Maximov and Südhof, 2005)], and short-term plasticity in WT and Syt12 KO neurons [Fig. 3E,F; total charge transfer, Control = 2.21 ± 0.21 nC (12 cells/3 cultures); Syt12 KO = 2.44 ± 0.17 nC (12 cells/3 cultures); delayed release, Control = 0.63 ± 0.05 nC (12 cells/3 cultures); Syt12 KO = 0.57 ± 0.11 nC (12 cells/3 cultures)]. Together, these results suggest that the Syt12 KO does not impair fundamental parameters of excitatory or inhibitory synaptic transmission in cultured cortical neurons.
These results were surprising considering the large enhancement of the forskolin-induced increase in mIPSC frequency that we previously observed upon overexpression of WT but not S97A-mutant Syt12, which suggested that the level of Syt12 regulates neurotransmitter release by a mechanism involving PKA-dependent phosphorylation of serine-97 (Maximov et al., 2007a). Thus, we asked whether a change in the precise experimental conditions between the earlier work and the current experiments may have contributed to the apparent discrepancy in results. In addition, we inquired whether forskolin-dependent changes in mEPSC frequency may also be potentially sensitive to Syt12 levels. To address these questions, we analyzed mEPSC and mIPSC frequencies and amplitudes in WT neurons that were infected with control lentivirus or lentiviruses overexpressing either WT or S97A-mutant Syt12.
We found that Syt12 overexpression had no detectable effect on mEPSC frequency or amplitude without or with addition of forskolin (Fig. 4A). Syt12 overexpression did, however, increase the mIPSC frequency but not amplitude by nearly 50% when mIPSCs were stimulated by forskolin; this effect was blocked by the S97A-substitution in Syt12 (Fig. 4B). These results confirm that increasing the Syt12 concentration enhances the mIPSC frequency at least upon activation of adenylate cyclase by forskolin, and that this effect depends on serine-97 in Syt12. Moreover, these results show that this effect is specific for mIPSCs and not observed with mEPSCs. Since the Syt12 KO and the Syt12 S97A KI do not alter the forskolin-induced increase in mIPSC or mEPSC frequency (Fig. 3) and since Syt12 overexpression does not affect the forskolin-induced increase in mEPSC frequency (Fig. 4), the overexpression data cannot be interpreted to reflect a direct and essential role for Syt12 in the PKA-dependent activation of spontaneous release. Instead, these data suggest that the Syt12 overexpression effect may be an indirect consequence of a physiological role of Syt12 in a different PKA-dependent regulation of neurotransmitter release that is not directly related to spontaneous mini release.
Figure 4.

Reassessment of the effect of overexpression of wild-type and S97A-mutant Syt12 on spontaneous mini release in cultured cortical neurons. A, B, Representative traces (left) and summary graphs of the frequency and amplitudes (right) of mEPSCs (A) and mIPSCs (B) measured as a function of the addition of forskolin (50 μm). Neurons infected with a control lentivirus and lentiviruses overexpressing wild-type Syt12 (Syt12WT) or S97A-mutant Syt12 (Syt12S97A) were analyzed. Data shown are means ±SEM; numbers of cells/independent cultures analyzed are shown in the bars. Statistical assessments were performed by Student's t test (*p < 0.05; **p < 0.01; ***p < 0.001).
Normal synaptic strength in the CA1 region of the hippocampus of Syt12 S97A KI and Syt12 KO mice
Although monitoring synaptic transmission in cultured neurons allows determination of fundamental synaptic parameters, important properties of normal synaptic transmission may be lost in neurons cultured from newborn animals. Thus, we next asked whether the Syt12 S97A substitution or the Syt12 KO may affect synaptic transmission in situ in juvenile mice. For these experiments, we chose the CA1 region of the hippocampus, arguably one of the best understood brain areas in terms of synaptic transmission.
Input–output curves of excitatory synaptic transmission at Schaffer collateral synapses, measured by extracellular field recordings, detected no difference between slices obtained from littermate WT and either Syt12 S97A KI or KO mice (Fig. 5A,C). Similarly, measurements of the paired-pulse ratio—a form of short-term synaptic plasticity measured as the relative strength of the second of two consecutive synaptic events, and monitored as a function of the interstimulus interval—revealed no change, suggesting that there is no alteration in release probability (Fig. 5B,D).
Figure 5.

Characterization of excitatory and inhibitory synaptic transmission in acute slices of the hippocampal CA1 region from WT, Syt12 S97A KI, and Syt12 KO mice. A, Input/output relationship of excitatory synapses examined using extracellular field potential recordings in slices from littermate WT and Syt12 S97A KI mice. Representative traces, averaged over multiple sweeps in one slice, are shown on top for the stimulus intensities tested (5, 10, 20, 40, and 60 V). The fast component of the voltage response is the fiber volley (FV) and the remainder of the response is the synaptic potential (fEPSP). The positivity observed with strong stimulation intensities is the population spike. A summary plot of the field EPSP slope versus the fiber volley amplitude is shown below. B, Short-term plasticity at excitatory synapses assessed by field recording measurements of the paired-pulse ratio (PPR) in slices from WT and Syt12 S97A KI mice. Representative traces for paired-pulses at 20, 50, 200, 400, and 800 ms interstimulus intervals (ISI) are shown on top, with the fast and slow components of the responses again corresponding to the fiber volley and fEPSP, respectively, and a summary plot of the data is shown below. C, D, Same as A and B, except that littermate WT and Syt12 KO mice were analyzed. E, Input/output relationship at inhibitory synapses in slices from littermate WT and Syt12 S97A KI mice, assessed by whole-cell recordings. Representative traces, averaged over multiple sweeps in one slice, are shown on top, and a summary plot of the IPSC amplitude versus the stimulation strength is shown below. F, Same as B, except that PPR was measured by whole-cell recordings at inhibitory synapses. G, H, Same as E and F, except that slices from WT and Syt12 KO mice were analyzed. Data shown are means ±SEM. Number of slices/mice tested are indicated in parenthesis. No significant difference in any of the comparisons was found using Student's t test (p > 0.05).
We also examined inhibitory synaptic events in CA1 pyramidal neurons as assessed by whole-cell recordings. During systematic probing of inhibitory synapses in the CA1 region of Syt12 S97A-KI or KO mice, we detected no phenotype either in evoked IPSCs (Fig. 5E,G), or paired-pulse ratio (Fig. 5F,H). Thus, similar to what we observed in cultured cortical neurons, we found no significant phenotype induced by Syt12 mutations in acute slices of the CA1 region of the hippocampus.
Normal short-term plasticity at hippocampal mossy-fiber synapses of Syt12 S97A KI mice
We next examined transmission at mossy fiber to CA3 pyramidal neuron synapses (mf-CA3) in Syt12 S97A KI mice. These synapses exhibit uniquely robust short-term plasticity, such as frequency facilitation and paired-pulse facilitation, and a characteristic form of presynaptic LTP (Nicoll and Schmitz, 2005). To better assess mf-CA3 short-term plasticity, we monitored the NMDA-receptor mediated component of transmission under conditions of reduced network excitability (i.e., by blocking AMPA/kainate receptor-mediated transmission and using medium containing 4 mm Ca2+/4 mm Mg2+). These recording conditions reduce the likelihood of eliciting polysynaptic contamination generated by auto-associational/commissural fiber inputs that also synapse onto CA3 pyramidal cells, thus facilitating a more accurate measurement of mossy fiber synaptic transmission (Nicoll and Schmitz, 2005). We observed no differences in frequency facilitation—elicited by 5 stimuli, 25 Hz bursts, or by stepping the stimulation rate from 0.05 to 0.5 Hz—or paired-pulse facilitation between Syt12 S97A KI and WT mice (Fig. 6). Thus, lack of Syt12 phosphorylation (at S97) does not seem to play a role in short-term plasticity at mossy-fiber synapses.
Decreased LTP in mossy-fiber synapses of the CA3 region of the hippocampus of Syt12 S97A KI and Syt12 KO mice
We next examined LTP at mossy fiber synapses (mfLTP), a presynaptic form of long-term plasticity (Nicoll and Schmitz, 2005) whose induction requires activation of the cAMP/PKA cascade (Huang et al., 1994; Weisskopf et al., 1994). To this end, we first performed extracellular field recordings (in medium containing 4 mm Ca2+/4 mm Mg2+) in acute slices from Syt12 S97A-KI mice and Syt12 KO mice. We found a significant reduction (∼50%) in the magnitude of mfLTP in both mutant mice (Fig. 7), as analyzed in a time window of 50–80 min post-induction. While the magnitude of post-tetanic potentiation (PTP; e.g., the first 5 min post-tetanus) also seemed to be reduced, such a change is difficult to interpret given the large contamination of extracellular responses with population action potentials that transiently occur in response to strong presynaptic activity (Nicoll and Schmitz, 2005).
In a further series of experiments, we probed the effect of the Syt12 S97A KI on mfLTP by intracellular recordings under experimental conditions designed to limit network excitability as in Figure 6. Again, we observed a ∼50% decrease in the extent of mfLTP in S97A KI mice (Fig. 8A–8C), as analyzed at 40–50 min following LTP induction. We also measured the paired pulse ratio of synaptic responses before and after mfLTP induction (Fig. 8D). Consistent with its presynaptic expression, mfLTP in WT mice was associated with a decrease in PPR, whereas mfLTP was not accompanied by a significant change in PPR in S97A KI mice (Fig. 8D). Of note, under these recording conditions of reduced excitability, no changes in PTP were observed. Together, these results show that Syt12 S97A KI and the Syt12 KO mice exhibit the same synaptic impairment in LTP, indicating that the KO phenotype reflects a lack of activation of Syt12 by phosphorylation of serine-97.
Decreased forskolin-induced long-term increase in synaptic transmission in mossy-fiber synapses of Syt12 S97A-KI and KO mice
mfLTP is known to depend on activation of PKA by cAMP. However, our previous studies in mutant mice such as those lacking Rab3A (Castillo et al., 1997) and RIM1α (Castillo et al., 2002) failed to detect deficits in the potentiation of transmitter release at mf-CA3 synapses by the adenylate-cyclase activator forskolin. Thus, we tested whether Syt12 mutant mice were normally responsive to forskolin at mossy fiber synapses. The experimental conditions were as in Figure 6, but with normal ACSF in the bath solution. While forskolin initially increased synaptic responses in WT and mutant synapses to a similar extent, both the Syt12 S97A KI and the Syt12 KO nearly abolished the long-term increase in synaptic responses induced by an application of forskolin (Fig. 9), as analyzed at 50–60 min following the start of forskolin application. Together, these results suggest that forskolin activates multiple separate processes to enhance synaptic transmission, of which only the long-term process is affected by the Syt12 mutations, which is similarly impaired by the Syt12 KO and S97A KI.
Syt12 is not required for cAMP-dependent synaptic plasticity at inhibitory synapses
Given the impairment of both mfLTP and of forskolin-induced potentiation of mf-CA3 synaptic transmission, as well as the fact that Syt12 is a PKA substrate (Maximov et al., 2007a), we tested whether Syt12 might be involved in other forms of synaptic plasticity that also depend on cAMP/PKA signaling. We first studied the forskolin-induced increase in mIPSC frequency in acute slices from the CA1 area of the hippocampus. Similar to the results obtained in cultured neurons (Fig. 3B), we detected no difference between WT and Syt12 KO neurons (Fig. 10A,B). We next examined iLTD, a cAMP-dependent long-term presynaptic form of plasticity at inhibitory synapses (Chevaleyre and Castillo, 2003) that requires retrograde endocannabinoid signaling, RIM1α, and phosphorylation of an as-yet-unidentified PKA target (Chevaleyre et al., 2007; Kaeser et al., 2008). We found no difference in the magnitude of iLTD in WT and Syt12 KO slices (Fig. 10C). In addition, the adenylate cyclase activator forskolin induced similar potentiation of IPSCs in both groups (Fig. 10D). It is therefore unlikely that Syt12 plays a role in cAMP-dependent changes of inhibitory synaptic transmission such as iLTD.
Figure 10.
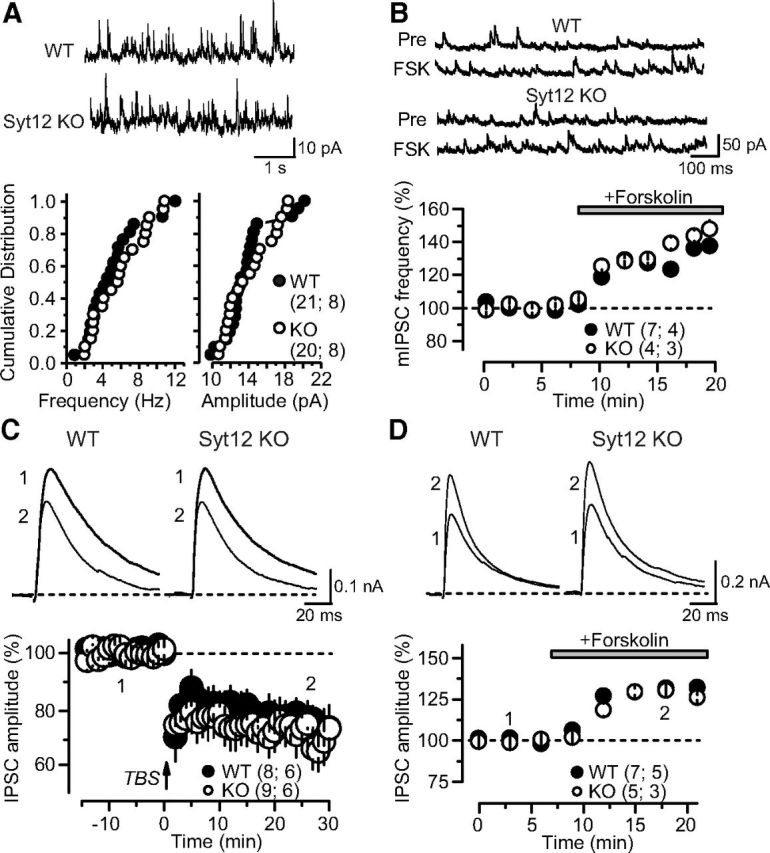
No PKA-dependent changes in inhibitory synaptic strength in the hippocampal CA1 region of WT and Syt12 KO mice. A, Measurements of inhibitory spontaneous mIPSCs reveal that there is no difference in the mIPSC frequency or amplitude between WT and Syt12 KO neurons obtained from littermate mice. Representative traces are depicted on top, and cumulative probability plots of the mIPSC frequency (left) and amplitude (right) at the bottom. B, The adenylate cyclase activator forskolin (20 μm) produces a similar enhancement of mIPSC frequency in WT and Syt12 KO hippocampus. Representative traces are shown on top, and a plot of the normalized mIPSC frequency as a function of time after forskolin application is shown on the bottom. C, iLTD is similarly induced in WT and Syt12 KO neurons. Representative traces are shown on top, and summary graphs of the normalized IPSC amplitude on the bottom. Numbers in the representative traces correspond to the indicated times in the summary. iLTD was triggered using TBS at time 0, as indicated by the arrow. D, Forskolin (20 μm) produces a similar acute enhancement of evoked IPSCs in WT and Syt12 KO hippocampus. Representative traces are shown on top, and summary graphs of the normalized IPSC amplitude as a function of time after forskolin addition is shown on the bottom. Data are plotted as means ±SEM. Number of slices/mice tested are indicated in parenthesis. No statistically significant differences between WT and Syt12 KO mice were detected in any of the parameters analyzed here.
Discussion
In the present study, we generated two lines of mutant mice, KI mice containing the S97A-substitution in Syt12 that abolishes PKA-dependent phosphorylation of Syt12, and KO mice that lack Syt12 (Figs. 1, 2). We found that Syt12 in general, and serine-97 of Syt12 in particular, is essential for normal mfLTP in the CA3 region of the hippocampus (Figs. 7–9). This requirement was documented in both Syt12 S97A KI mice and in Syt12 KO mice, using both extracellular and intracellular recordings and different recording conditions. Moreover, we found that Syt12 is essential for the forskolin-induced long-term enhancement of mossy fiber synaptic transmission, but is dispensable for the forskolin-induced short-term potentiation of mf-CA3 transmission (Fig. 9). Thus, we conclude that the synaptic vesicle protein Syt12 is an intrinsic component of the machinery that mediates presynaptic mfLTP in the CA3 region of the hippocampus.
In total, we analyzed the phenotype of Syt12 S97A KI and Syt12 KO mice in five different synapses: excitatory and inhibitory synapses formed by cortical neurons cultured from newborn mice (Fig. 3), excitatory synapses formed by Schaffer collaterals and inhibitory synapses formed by interneurons in the CA1 region of the hippocampus (Figs. 5, 10), and excitatory synapses formed by mossy fibers in the CA3 region of the hippocampus (Figs. 6–9). Our results demonstrate that Syt12 is not essential for basal synaptic transmission or short-term plasticity in cultured cortical neurons or in acute slices from the CA1 region of the hippocampus, and is also not required for basal synaptic transmission or short-term plasticity in mossy-fiber synapses monitored in acute slice from the CA3 region of the hippocampus (Figs. 6, 8). Unexpectedly in view of our previous more indirect studies (Maximov et al., 2007a), the Syt12 KO had no effect on the forskolin-induced increase in mEPSC or mIPSC frequency in cultured neurons (Fig. 3), as well as mIPSC frequency or evoked IPSCs in acute slices (Fig. 10). However, we confirmed in independent experiments our previous results (Maximov et al., 2007a) that Syt12 overexpression can lead to an increase in mIPSC frequency when the frequency is enhanced by forskolin treatment, and that this effect depends on the presence of serine-97 as a PKA target (Fig. 4). Finally, the Syt12 KO did not alter iLTD [which is known to be PKA-dependent (Chevaleyre et al., 2007)] at inhibitory synapses of the CA1 region of the hippocampus (Fig. 10).
Viewed together, our results rule out an essential role for Syt12 in synaptic transmission as such, consistent with the lack of a survival phenotype or major changes in brain protein composition in the Syt12 KO and Syt12 S97A KI mice (Figs. 1, 2). The discrete and selective phenotype of the Syt12 mutant mice that is restricted to one particular synapse and one particular form of long-term synaptic plasticity is surprising given its abundance and widespread expression (Thompson, 1996; Maximov et al., 2007a). Although our results identify an essential role for Syt12 in the PKA-dependent regulation of synaptic strength during mfLTP, our results also show that Syt12 is not required for the PKA-dependent regulation of spontaneous mini release. This conclusion differs from our earlier hypothesis—based on overexpression experiments—that Syt12 may be involved in the PKA-dependent basal regulation of synaptic transmission (Maximov et al., 2007a). In hindsight, a plausible interpretation of the overexpression data (Fig. 4) is that they correctly indicated that PKA-dependent phosphorylation at serine-97 of Syt12 is an important regulatory step in neurotransmitter release, but that they did not reveal the nature of this regulatory step—namely the PKA-dependent regulation of presynaptic LTP—which could only be identified with the genetic approach used in the present study.
Other proteins were previously shown to be required for mfLTP, in particular Rab3A (Castillo et al., 1997) and RIM1α (Castillo et al., 2002), but the mechanisms of their involvement remains unclear (Fig. 11). mfLTP is induced by activation of PKA (Huang et al., 1994; Weisskopf et al., 1994) and involves phosphorylation of an unknown target protein that then increases presynaptic release probability (Xiang et al., 1994). Based on studies in cultured cerebellar neurons from RIM1α-deficient mice, we initially suggested that RIM1α might be the target for PKA in mfLTP (Lonart et al., 2003). Using KI mice in which the PKA-phosphorylated serine-413 of RIM1α was substituted by an alanine, however, we subsequently found that the phosphorylation of RIM1α by PKA is not essential for mfLTP (Kaeser et al., 2008), as confirmed by others (Yang and Calakos, 2010).
Figure 11.

Illustration of molecular pathways involved in presynaptic mfLTP induction. During repetitive activation of mossy fibers, Ca2+ accumulates in presynaptic nerve terminals outside of the immediate area of the active zone, thereby activating adenylate cyclase (AC) via calmodulin. Adenylate cyclase activation increases the presynaptic levels of cAMP which in turn activates protein kinase-A which phosphorylates Syt12 and possibly other targets. These targets then cooperate with RIM1α and Rab3A to increase the efficacy of the neurotransmitter release machinery, thereby producing a long-lasting enhancement of neurotransmitter release.
The fact that Syt12 S97A KI mice exhibit the same phenotype in mfLTP as Syt12 KO mice shows that PKA-dependent phosphorylation of serine-97 of Syt12 is required for mfLTP. The data presented here thus suggest that Syt12 represents at least one of the PKA-targets whose phosphorylation induces mfLTP (Fig. 11). Although our results do not necessarily mean that Syt12 is the only PKA target whose phosphorylation is required for mfLTP, these findings identify Syt12 as the currently only known PKA target for mfLTP. This conclusion is supported by the observation that Syt12 is essential for the long-lasting potentiation of synaptic transmission that is induced by a cAMP/PKA activator at mossy fiber-CA3 synapses (Fig. 9). Syt12 is in an excellent position to regulate synaptic exocytosis based on its localization to synaptic vesicles and its interaction with Syt1 (Maximov et al., 2007a). Although the mechanisms by which phosphorylation of Syt12 regulates mossy fiber synaptic transmission remain unexplored, it seems plausible that Syt12 phosphorylation may increase the presynaptic release probability of a synapse by altering the interaction of Syt12 with a target protein. No candidate Syt12 interacting protein is known in addition to Syt1 [whose binding to Syt12 is not affected by PKA-dependent phosphorylation; see the study by Maximov et al. (2007a)], making it difficult at present to test this hypothesis.
Our study raises important new questions, the most important of which probably regards the function of Syt12 in synapses not expressing PKA-dependent presynaptic forms of plasticity, such as mfLTP. The widespread expression of Syt12 throughout the brain (Thompson, 1996; Maximov et al., 2007a) raises the possibility that Syt12 may have additional regulatory functions in addition to controlling the PKA-dependent plasticity, but that these other functions are not apparent during basal release reactions. Our findings do not rule out the possibility that Syt12 performs other functions in synaptic transmission in addition to its essential role in mfLTP. For example, Syt12 could play a fundamental role in regulating Syt1 efficacy, but this role may be shared with other Ca2+-independent synaptotagmins. A systematic analysis of potentially redundant Ca2+-independent synaptotagmins, especially Syt13-Syt16, will be required to address this possibility in future studies. Another, possibly equally important question is what molecule mediates the PKA-dependent suppression of synaptic transmission during iLTD, which is another PKA-dependent form of presynaptic long-term plasticity but does not require Syt12 (Fig. 10). Clearly, Syt12 phosphorylation by PKA alone does not account for all presynaptic long-term plasticity mediated by PKA, and other PKA substrates are likely involved.
In summary, our data identify the PKA-dependent phosphorylation of Syt12 as an essential step in the expression of mfLTP, and thereby demonstrate that Syt12 regulates neurotransmitter release during mfLTP consistent with its localization to synaptic vesicles and its phosphorylation by PKA.
Footnotes
This work was supported by a grant from NINDS (NS077906 to T.C.S.) and NIDA (DA17392 to P.E.C.). We thank I. Kornblum, E. Borowicz, J. Mitchell, L. Fan, and N. Huang for excellent technical assistance, Dr. Robert Hammer for electroporation and blastocyst injection of embryonic stem cells, and Dr. Pascal Kaeser for advice. We are grateful to the members of the Südhof, Castillo, and Malenka laboratories for comments and discussions.
The authors declare no competing financial interests.
References
- Barber CF, Jorquera RA, Melom JE, Littleton JT. Postsynaptic regulation of synaptic plasticity by synaptotagmin 4 requires both C2 domains. J Cell Biol. 2009;187:295–310. doi: 10.1083/jcb.200903098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P, Maximov A, Südhof TC. Activity-dependent IGF-1 exocytosis is controlled by the Ca(2+)-sensor synaptotagmin-10. Cell. 2011;145:300–311. doi: 10.1016/j.cell.2011.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Janz R, Südhof TC, Tzounopoulos T, Malenka RC, Nicoll RA. Rab3A is essential for mossy fibre long-term potentiation in the hippocampus. Nature. 1997;388:590–593. doi: 10.1038/41574. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Südhof TC, Malenka RC. RIM1α is required for presynaptic long-term potentiation. Nature. 2002;415:327–330. doi: 10.1038/415327a. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/S0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Heifets BD, Kaeser PS, Südhof TC, Castillo PE. Endocannabinoid mediated long-term plasticity requires cAMP/PKA signaling and RIM1α. Neuron. 2007;54:801–812. doi: 10.1016/j.neuron.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dymecki SM. Flp recombinase promotes site-specific DNA recombination in embryonic stem cells and transgenic mice. Proc Natl Acad Sci U S A. 1996;93:6191–6196. doi: 10.1073/pnas.93.12.6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson GD, Anagnostaras SG, Silva AJ, Herschman HR. Deficits in memory and motor performance in synaptotagmin IV mutant mice. Proc Natl Acad Sci U S A. 2000;97:5598–5603. doi: 10.1073/pnas.100104597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson GD, Wang H, Herschman HR, Storm DR. Altered hippocampal short-term plasticity and associative memory in synaptotagmin IV(−/−) mice. Hippocampus. 2004;14:964–974. doi: 10.1002/hipo.20013. [DOI] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Südhof TC. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- Ho A, Morishita W, Atasoy D, Liu X, Tabuchi K, Hammer RE, Malenka RC, Südhof TC. Genetic analysis of Mint/X11 proteins: essential presynaptic functions of a neuronal adaptor protein family. J Neurosci. 2006;26:13089–13101. doi: 10.1523/JNEUROSCI.2855-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Li XC, Kandel ER. cAMP contributes to mossy fiber LTP by initiating both a covalently mediated early phase and macromolecular synthesis-dependent late phase. Cell. 1994;79:69–79. doi: 10.1016/0092-8674(94)90401-4. [DOI] [PubMed] [Google Scholar]
- Kaeser PS, Kwon HB, Blundell J, Chevaleyre V, Morishita W, Malenka RC, Powell CM, Castillo PE, Südhof TC. RIM1α phosphorylation at serine-413 by protein kinase A is not required for presynaptic long-term plasticity or learning. Proc Natl Acad Sci U S A. 2008;105:14680–14685. doi: 10.1073/pnas.0806679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littleton JT, Serano TL, Rubin GM, Ganetzky B, Chapman ER. Synaptic function modulated by changes in the ratio of synaptotagmin I and IV. Nature. 1999;400:757–760. doi: 10.1038/23462. [DOI] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Südhof TC, Linden DJ. Phosphorylation of RIM1α by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell. 2003;115:49–60. doi: 10.1016/S0092-8674(03)00727-X. [DOI] [PubMed] [Google Scholar]
- Machado HB, Liu W, Vician LJ, Herschman HR. Synaptotagmin IV overexpression inhibits depolarization-induced exocytosis in PC12 cells. J Neurosci Res. 2004;76:334–341. doi: 10.1002/jnr.20072. [DOI] [PubMed] [Google Scholar]
- Maximov A, Südhof TC. Autonomous function of synaptotagmin 1 in triggering synchronous release independent of asynchronous release. Neuron. 2005;48:547–554. doi: 10.1016/j.neuron.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Maximov A, Shin OH, Liu X, Südhof TC. Synaptotagmin-12, a synaptic vesicle phosphoprotein that modulates spontaneous neurotransmitter release. J Cell Biol. 2007a;176:113–124. doi: 10.1083/jcb.200607021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Pang ZP, Tervo DG, Südhof TC. Monitoring synaptic transmission in primary neuronal cultures using local extracellular stimulation. J Neurosci Methods. 2007b;161:75–87. doi: 10.1016/j.jneumeth.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci U S A. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci. 2005;6:863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- O'Gorman S, Dagenais NA, Qian M, Marchuk Y. Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germ line of mice, but not in embryonic stem cells. Proc Natl Acad Sci U S A. 1997;94:14602–14607. doi: 10.1073/pnas.94.26.14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Südhof TC. Cell biology of Ca2+-triggered exocytosis. Curr Opin Cell Biol. 2010;22:496–505. doi: 10.1016/j.ceb.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perin MS, Fried VA, Mignery GA, Jahn R, Südhof TC. Phospholipid binding by a synaptic vesicle protein homologous to the regulatory region of protein kinase C. Nature. 1990;345:260–263. doi: 10.1038/345260a0. [DOI] [PubMed] [Google Scholar]
- Rosahl TW, Geppert M, Spillane D, Herz J, Hammer RE, Malenka RC, Südhof TC. Short-term synaptic plasticity is altered in mice lacking synapsin I. Cell. 1993;75:661–670. doi: 10.1016/0092-8674(93)90487-B. [DOI] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Südhof TC. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature. 1995;375:488–493. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- Südhof TC, Fried VA, Stone DK, Johnston PA, Xie XS. Human endomembrane H+ pump strongly resembles the ATP-synthetase of Archaebacteria. Proc Natl Acad Sci U S A. 1989;86:6067–6071. doi: 10.1073/pnas.86.16.6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CC. Thyroid hormone-responsive genes in developing cerebellum include a novel synaptotagmin and a hairless homolog. J Neurosci. 1996;16:7832–7840. doi: 10.1523/JNEUROSCI.16-24-07832.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JT, Kelley BG, Sullivan JM. Synaptotagmin IV does not alter excitatory fast synaptic transmission or fusion pore kinetics in mammalian CNS neurons. J Neurosci. 2006;26:372–380. doi: 10.1523/JNEUROSCI.3997-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong G, Shepherd D, Jahr CE. Synaptic desensitization of NMDA receptors by calcineurin. Science. 1995;267:1510–1512. doi: 10.1126/science.7878472. [DOI] [PubMed] [Google Scholar]
- Wang CT, Grishanin R, Earles CA, Chang PY, Martin TF, Chapman ER, Jackson MB. Synaptotagmin modulation of fusion pore kinetics in regulated exocytosis of dense-core vesicles. Science. 2001;294:1111–1115. doi: 10.1126/science.1064002. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Greenwood AC, Kairiss EW, Brown TH. Quantal mechanism of long-term potentiation in hippocampal mossy-fiber synapses. J Neurophysiol. 1994;71:2552–2556. doi: 10.1152/jn.1994.71.6.2552. [DOI] [PubMed] [Google Scholar]
- Xu J, Mashimo T, Südhof TC. Synaptotagmin-1, -2, and -9: Ca(2+) sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron. 2007;54:567–581. doi: 10.1016/j.neuron.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Yang X, Kaeser-Woo YJ, Pang ZP, Xu W, Südhof TC. Complexin clamps asynchronous release by blocking a secondary Ca2+-sensor via its accessory α-helix. Neuron. 2010;68:907–920. doi: 10.1016/j.neuron.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Calakos N. Acute in vivo genetic rescue demonstrates that phosphorylation of RIM1α serine 413 is not required for mossy fiber long-term potentiation. J Neurosci. 2010;30:2542–2546. doi: 10.1523/JNEUROSCI.4285-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara M, Adolfsen B, Galle KT, Littleton JT. Retrograde signaling by Syt 4 induces presynaptic release and synapse-specific growth. Science. 2005;310:858–863. doi: 10.1126/science.1117541. [DOI] [PubMed] [Google Scholar]
- Zhang G, Bai H, Zhang H, Dean C, Wu Q, Li J, Guariglia S, Meng Q, Cai D. Neuropeptide exocytosis involving synaptotagmin-4 and oxytocin in hypothalamic programming of body weight and energy balance. Neuron. 2011;69:523–535. doi: 10.1016/j.neuron.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Fukuda M, Van Bockstaele E, Pascual O, Haydon PG. Synaptotagmin IV regulates glial glutamate release. Proc Natl Acad Sci U S A. 2004;101:9441–9446. doi: 10.1073/pnas.0401960101. [DOI] [PMC free article] [PubMed] [Google Scholar]