

Abstract
Despite advances in antiretroviral therapy that have revolutionized HIV disease management, effective control of the HIV infection pandemic remains elusive. Beyond the classic non-B endemic areas, HIV-1 non-B subtype infections are sharply increasing in previous subtype B homogeneous areas such as Europe and North America. As already known, several studies have shown that, among non-B subtypes, subtypes C and D were found to be more aggressive in terms of disease progression. Luckily, the response to antiretrovirals against HIV-1 seems to be similar among different subtypes, but these results are mainly based on small or poorly designed studies. On the other hand, differences in rates of acquisition of resistance among non-B subtypes are already being observed. This different propensity, beyond the type of treatment regimens used, as well as access to viral load testing in non-B endemic areas seems to be due to HIV-1 clade specific peculiarities. Indeed, some non-B subtypes are proved to be more prone to develop resistance compared to B subtype. This phenomenon can be related to the presence of subtype-specific polymorphisms, different codon usage, and/or subtype-specific RNA templates. This review aims to provide a complete picture of HIV-1 genetic diversity and its implications for HIV-1 disease spread, effectiveness of therapies, and drug resistance development.
1. Introduction
Thirty years have passed after discovering human immunodeficiency virus (HIV), the etiological agent of the acquired immunodeficiency syndrome (AIDS) [1–4].
Two types of HIV are known: the most common HIV-1, which is responsible to the worldwide AIDS epidemic, and the immunologically distinct HIV-2 [5], which is much less common and less virulent [6, 7] but produces clinical findings similar to HIV-1 [8]. The HIV-1 type itself includes four groups M (main), O (outlier), N (non-M, non-O), and P [9–12], which have different geographic distributions but all produce similar clinical symptoms. The M group further splits into 9 subtypes (A through J) [13–15], as well as at least 58 circulating recombinant forms (CRFs, http://www.hiv.lanl.gov/content/sequence/HIV/CRFs/CRFs.html, last accessed 06 May 2013) and multiple unique recombinant forms (URFs).
The vast majority of reports on drug resistance deal with HIV-1 subtype B infections in developed countries, and this is largely due to historical delays in access to antiretroviral therapy on a worldwide basis.
Advances in antiretroviral therapy have revolutionized HIV management and the control of the spread of regional epidemics [16–18]. Currently, a combination of several antiretroviral agents, termed Highly Active Anti-Retroviral Therapy (HAART), has been highly effective in reducing the number of HIV particles in the blood stream (as measured by a blood test called viral load) and delaying disease progression. Clinical trials and observational studies have shown profound reductions in morbidity and mortality in patients infected with HIV as a result of combination antiretroviral therapy [16, 19–27]. Of relevance, advances in HIV treatment have had a positive impact on all the affected demographic and behavioral risk groups, with an expected longevity for HIV-infected patients that is now 73 years [23]. Moreover, it should be considered that, thanks to the recent expansion in the number of antiretrovirals and antiretroviral classes, virological suppression has become achievable in most patients for whom numerous prior antiretroviral regimens had failed. In addition, antiretroviral therapy results in efficacious treatment of HIV-1, regardless of the viral subtype.
However, despite advances in antiretroviral therapy, some treatments still fail. A major cause of treatment failure is the development of drug resistance both in HIV-1 B and non-B subtypes [28–34]. The extreme variability and the high evolution rate of HIV-1 favour the development of antiretroviral resistance. Indeed, HIV-1 infection is characterized by a high degree of genetic variability within infected persons. This is explained by the fact that the virus population present at a certain time point within an infected person consists of a complex mixture of heterogeneous strains, termed “quasispecies” [35]. The heterogeneity of quasispecies is due to their different antigenic and phenotypic properties. They continuously compete among themselves for survival and propagation [36]. The subsequent overgrowth or dominance of a certain viral strain over another is largely determined by its relative adaptation to a given intrahost environment, a factor particularly relevant to the emergence of drug resistant variants. Indeed, the intrapatient virus population is a highly dynamic system, characterized by a high turnover rate and a high mutation rate [37, 38]. These evolutionary dynamics are the basis for a diversified population that can quickly generate drug-resistant variants in response to the therapy [39–42]. Escape mutants that have a selective advantage under therapy become dominant in the population and lead to an increasing virus production and eventually to therapy failure. The shifted population may be hit with a new drug combination, but finding such a potent regimen after treatment failure is challenging, since many accumulated mutations confer drug resistance not only to the administered drugs but also to structurally and functionally similar compounds [41, 43].
Identifying and understanding HIV-1 drug resistance therefore helps clinicians to avoid minimally active antiretrovirals in favor of newer drugs that are fully or nearly fully active [44–46]. For this reason, resistance testing has become an important diagnostic tool in the management of HIV infections [47–52]. With the aid of HIV resistance tests, antiretroviral treatment strategies can be improved. Pharmacoeconomic studies have shown that these tests are also cost effective [53, 54]. For several years, national and international HIV treatment guidelines have been recommending the use of resistance testing both in drug-naive and drug-treated patients [48–52].
Antiretroviral drug design, resistance research, and interpretation systems have been largely based on HIV-1 subtype B because of its predominance in the wealthy countries in which antiretroviral drugs were first introduced, as well as the availability of assays for drug resistance in such locations. However, it should be noted that HIV-1 B subtype represents only about 10% [55–57] of the overall subtypes in the world. Several studies showed that non-B subtype HIV-1 infections have been rapidly increasing over time in previously subtype B homogeneous areas such as Europe and North America [58–73]. For example, in France, Switzerland, and Italy, it is estimated that non-B infections constitute roughly 15%–35% of HIV-1 infections, with an increasing prevalence and complexity of intersubtype recombinants over the last years [59, 64, 66, 73–77]. Although non-B infections are infrequent in North America, a study in New York identified non-B infections in a few US citizens who never travelled abroad, suggesting that transmission of non-B subtype occurs in the United States independently of travel history [72]. Moreover, new HIV-1 strains are constantly emerging [78, 79].
Due to the spread of HIV-1 non-B subtypes and the introduction of antiretroviral drugs in resource-limited settings (known for the largest assortment of non-B subtypes), further knowledge concerning the responsiveness to antiretroviral therapy and HIV-1 drug resistance in non-B subtypes is required. In this regard, the development of specific mutations varies in different subtypes, and this can be explained mainly by the intrinsic properties of the virus and not only by a different pressure of antiretroviral drugs, as suggested in a recent study [76]. To date, the improvement of the genotypic resistance test in terms of sensitivity, cost effectiveness and detection of drug resistance in non-B subtypes is a major topic. In order to achieve this goal, the introduction of new affordable assays is crucial. Group M subtype independent genotyping assays for detection of HIV-1 drug resistance were recently developed. These assays could represent an alternative to commercial assays for HIV-1 drug resistance genotyping in routine diagnostics and for surveillance and monitoring of drug resistance in resource limited settings [80–82].
Several discrepancies are still present in the interpretation of resistance patterns present in HIV-1 non-B subtypes by using different interpretation algorithms. These discrepancies illustrate how hard it may be to reach an unambiguous conclusion, despite the efforts made over the last decade to interpret drug resistance in HIV-1 non-B subtypes [83–87]. Further and continuous refinement of interpretation algorithms is required to improve their prediction.
The increasing prevalence of HIV-1 non-B subtypes could have implications not only for the development of resistance but also for diagnosis [88], vaccine design [89], and the clinical management of HIV infection [65, 90, 91]. Moreover, patients infected by certain HIV-1 non-B subtypes present accelerated disease progression [92–97] and higher cognitive impairment [98].
In the light of the above mentioned, the aim of this review is to provide a complete picture about HIV-1 genetic diversity and its implications.
2. HIV-1 Genetic Diversity
HIV-1 is characterized by extensive genetic diversity. Mutational escape results in a remarkable degree of viral diversity within HIV-1 and in its adaptation to both immune activity and antiretroviral therapy. However, not all escape mutations are advantageous to the virus since some of them can severely affect viral fitness [99, 100]. The extensive genetic diversity of HIV-1 is due to its high replication rate, the error-prone reverse transcriptase, and recombination events that may occur during virus replication [101, 102].
2.1. Error-Prone Reverse Transcriptase Enzyme and High Replication Rate
The molecular basis of HIV-1 variability is a highly error-prone reverse transcriptase enzyme [103]. The activity of this enzyme, essential for viral replication, is specifically required for the conversion of single-stranded genomic RNA into double-stranded viral DNA, which is later integrated into the host genomic DNA [104]. For this reason, HIV-1 reverse transcriptase inhibitors are powerful inhibitors of HIV-1 replication and represent an important class of antiretroviral agents [105]. HIV-1 reverse transcriptase is a multifunctional enzyme that possesses RNA-dependent and DNA-dependent DNA polymerase activities as well as an RNase H activity that specifically degrades the RNA strand of RNA/DNA hybrids [104]. As an intrinsic property, and in contrast to other DNA polymerases, HIV-1 reverse transcriptase lacks a proofreading function. This error-prone nature of reverse transcriptase, together with the high rate of virus production sustained by HIV-1 infection in vivo, strongly contributes to the continuous generation of new viral variants [13, 106, 107].
The rate of nucleotide substitutions introduced by reverse transcriptase is approximately 10−4 per nucleotide per cycle of replication, which is equal to one nucleotide substitution per genome during a single replication cycle [108]. Insertions, deletions, and duplications also contribute to the genetic heterogeneity of HIV-1 [109].
HIV-1 has a rapid turnover, and it is estimated that approximately 109 virions per day are generated in an infected individual. The composite lifespan of plasma virus and virus-producing cells is very short with a half-life of approximately two days, and an almost complete replacement of wild-type strains by drug resistant virus occurs in plasma within 2–4 weeks [107]. During antiretroviral treatment, rapid viral turnover in combination with a high mutation rate is a primary factor behind the emergence of HIV variants with antiretroviral drug resistance.
2.2. Genetic Recombination
Genetic recombination is another important strategy by which HIV generates genetic diversity [109]. This process contributes strongly to high level multiple drug resistance [110–112]. Each retroviral particle contains two copies of single-stranded RNA, and template switches occur frequently during reverse transcription, thus generating mutations and recombination by intramolecular and intermolecular jumps. Recombination may link drug resistant mutations in HIV-1, leading to increased resistance to a particular drug [113], or the generation of multidrug resistant variants [110]. In addition, recombination may lead to the acquisition of mutations that compensate for a loss in viral fitness or replicative capacity due to previous acquisition of resistance mutations.
Since recombination can create a multiple drug resistant virus out of two single drug resistant strains, it is generally believed that the capacity of the virus to recombine facilitates the evolution of drug resistance [9, 110, 113–115]. This rapid evolution of drug resistance in HIV remains a major obstacle for HIV therapy.
Recombination is a strategy for viral rejuvenation, and it is likely that recombination between HIV strains may lead to the evolution of fitter forms and viral strains acquiring drug resistance to all major classes of HIV-1 inhibitors. A different scenario could be that a fitter virus can be generated by recombining parts of two parental genomes with lesser fitness, or alternatively a less fit virus can be generated by breaking up favourable combinations of mutations in the parental genomes. This interaction between recombination, mutations, and viral fitness is highly intricate, but nonetheless, recombination and its mechanisms, especially at the level of diverse subtypes, warrant further investigation. The potential for genetic differences among subtypes to yield different patterns of resistance-conferring mutations is supported by natural variation among HIV subtypes in genetic content (40% variation in the env gene, and 8–10% variation in the pol/gag genes). This issue acquires special relevance in view of the fact that the HIV pol gene is the major target for all major classes of anti-HIV drugs and most HIV strains show hotspots for recombination in gag-pol and env regions.
3. Origin of HIV-1 and Its Distribution
On the basis of genetic homology, HIV-1 has been classified into four groups: M (main), O (outlier), N (non-M, non-O), and the recently identified P [9–12]. The M group further splits into 9 subtypes (A through J) [13–15], as well as at least 58 circulating recombinant forms (CRFs, http://www.hiv.lanl.gov/content/sequence/HIV/CRFs/CRFs.html, last accessed 06 May 2013) and multiple unique recombinant forms (URFs). In general genetic variation is of 25–35% and 15–20% between and within subtypes, respectively. The genetic variation within some subtypes can determine distinct sequence clusters. For example, subtypes A and F have been subdivided into five (A1, A2, A3, A4, A5) and two subtypes (F1, F2), respectively.
CRFs are intersubtype recombinant HIV-1 genomes that have infected three or more subjects who are not epidemiologically related, so they can be assumed to make an epidemiologically relevant contribution to the HIV-1 M group epidemic. The CRFs are labelled with numbers rather than letters, and numbered in the order they were first adequately described as the first time. For example, CRF02_AG is the second CRF that has been described [116]. URF variants are widely distributed worldwide, with recombination breakpoints different from those found in CRFs.
Subtypes within the HIV-1 N and O groups are not yet clearly identified. Very few isolates of HIV-1 N group have been identified and sequenced from humans, while the diversity of sequences within the O group is nearly as great as the diversity of sequences in the M group.
Today, the HIV-1 M group, the cause of the worldwide pandemic, has a near global distribution, whereas the N and O groups are restricted to individuals of West African origin. The P group was recently identified in two individuals originating from Cameroon. The M and N HIV-1 groups have a common ancestor, one that is most closely related to the SIV strain found in chimpanzees (SIVcpz; Pan troglodytes troglodytes) [117, 118] that live mainly in Gabon, Cameroon, and the Republic of the Congo. This HIV-1 progenitor probably was passed from chimpanzees to human hunters through blood borne transmission [119, 120]. Recent evidence suggests that the HIV-1 O and P groups may have originated in wild gorillas, in which the closest relatives of these two groups have been identified [10, 121].
Phylogenetic analysis of HIV-1 and related viruses from nonhuman primates suggests that three independent transmission events, which happened in early 20th century, spawned the HIV-1 M, N, and O groups [119, 120]. In particular, the earliest direct evidence of HIV infection in humans was found retrospectively in a serum sample and in a lymph node biopsy specimen stored in 1959 and 1960, respectively, in Kinshasa in the Democratic Republic of the Congo [122, 123]. These samples provided direct evidence of the age of the HIV epidemic in humans. Moreover, they have been instrumental in the validation and extrapolations of molecular clock computer programs used to estimate the time to the most recent common ancestors (tMRCAs) and evolutionary rates of various HIV lineages [123, 124]. The HIV-1 M group appears to be the oldest HIV lineage in humans, with an estimated time to the tMRCA in the first two or three decades of the 1900s [123, 124]. In particular, the tMRCA shared between the M group and SIVcpz is estimated at 1853 (1799–1904) [125]. Cross-species transfer is therefore inferred to have taken place sometime between 1853 and the early 1900s, although there is some uncertainty, given the size of the confidence intervals.
HIV-1 M group subtypes can be considered as a result of the high error rate of reverse transcriptase enzyme during virus replication and selective pressure exerted by the immune system. Since their introduction into humans, HIV-1 M group subtypes have expanded rapidly in West and Central Africa and established multiple epidemics around the world. Regional expansion of HIV-1 has come in waves with the rapid emergence of specific subtypes due in part to specific modes and routes of transmission. For example, intravenous drug use in Southeast Asia in the mid-1980s and in Eastern Europe and Russia during the early 1990s led to the rapid spread of CRF01_AE and of subtype A, respectively [126, 127]. A similar expansion of subtype B HIV-1 transmission occurred among men who have sex with men in North America and Europe in the early 1980s. However, HIV-1 subtype C (the most dominant subtype in the world responsible for more than 50% of overall infections) appears to have slowly emerged throughout the world over the past 10 to 15 years as a consequence of multiple introductions [126, 128].
In recent years a substantial increase of recombinant forms has been observed as a consequence of the increased genetic complexity of the global epidemic [57, 59, 64, 66, 73–77, 129–131].
However, the prevalence of recombinant forms (estimated to be around the 20% [57]) is still underestimated. Indeed, genetic complexity is not always detected, and this is mainly due to the subtyping of only one genetic region and not of the full genome. Consequently, specimens previously considered “pure” variants may be classified as recombinants when additional viral genes are analyzed.
4. Development of Resistance to Antiretroviral Therapy among Different HIV-1 Subtypes
The drugs currently approved for clinical use by the US Food and Drug Administration (FDA) and used to treat HIV infection belong to 6 distinct classes (http://www.fda.gov/ForConsumers/byAudience/ForPatientAdvocates/HIVandAIDSActivities/ucm118915.htm, last accessed 06 May 2013): (1) seven nucleoside and one nucleotide reverse transcriptase inhibitors (NRTIs); (2) five nonnucleoside reverse transcriptase inhibitors (NNRTIs); (3) nine protease inhibitors (PIs); (4) one fusion inhibitor (FI); (5) one CC chemokine receptor 5 (CCR5) antagonist; (6) two integrase inhibitors (INIs). Each of these drug classes act at different steps in the HIV replication cycle (Figure 1) [41]. The NNRTIs and NRTIs block the reverse transcription of the viral RNA genome in cDNA, which is catalysed by the reverse transcriptase. The PIs block protease mediated maturation of released virions. The FIs and CCR5 antagonists block HIV entry into cells. The INIs block the integration of viral genome into the DNA of the host cell.
Figure 1.
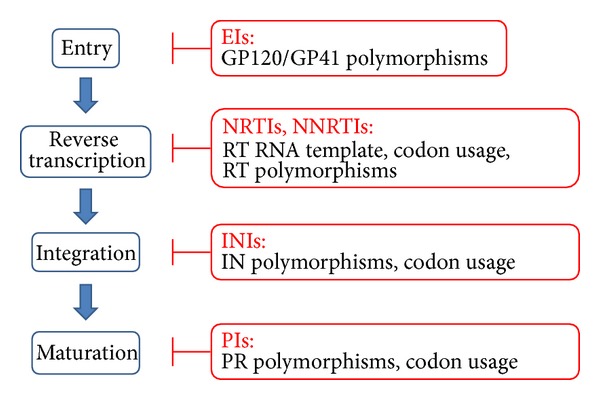
Steps of HIV-1 life cycle targeted by antiretroviral drugs and relative impact of HIV-1 subtype on resistance development. Blue boxes represent the crucial steps of HIV-1 life cycle targeted by antiretrovirals. Red boxes report the drug classes available in clinics and the HIV-1 genetic characteristics related to subtype potentially involved in resistance development. EIs: Entry inhibitors; IN: integrase; INI: integrase inhibitors; NRTIs: nucleotide reverse transcriptase inhibitor; NNRTI: non-NRTIs; RT: reverse transcriptase; PR: protease.
Although it seems that combination antiretroviral regimens are effective against all HIV-1 group M subtypes, there is emerging evidence of subtype differences in drug resistance relevant to antiretroviral strategies in different parts of the world. For this purpose, extensive sampling of HIV genetic diversity and ad hoc analyses are required to tailoring of antiretroviral therapies in different parts of the world. HIV-1 non-B subtypes present clade-specific substitutions in positions related to drug resistance [132] that could accelerate the emergence of drug-resistant viruses, change or induce alternative pathways of resistance, influence viral replicative capacity in vitro [133], impair the interpretation of genotypic resistance algorithms [70, 77, 134–137], and affect drug-binding affinity [138, 139].
4.1. Subtype Propensities to Develop Mutations
Specific mutation development varies with different subtypes because of the intrinsic properties of the virus as well as the different pressures of antiretroviral drugs. In particular, three factors are involved in this phenomenon.
Intersubtype differences in codon usage: Subtype differences in nucleotide and mutational motifs, which are defined as the number of transitions or transversions needed to develop resistance to different classes of antiretroviral drugs, may affect the genetic barrier for resistance, as shown in the development of some mutations in different HIV-1 proteins, such as (i) mutations associated with resistance to NNRTIs at the codon 106 of the reverse transcriptase in C and B subtypes (see Section 4.2.2) [140, 141]; (ii) mutations at the protease codons 74 in C subtypes, 82 in G subtypes, and 89 in several non-B subtypes such as C and CRF02_AG (see Section 4.2.3) [87, 142–145]; (iii) mutations in the integrase gene at codons 140, 148, 151, 157, and 160 (see Section 4.2.4) [146, 147].
Intersubtype amino acid differences involved in minor structural changes in the targets of therapy: In this situation, different mutations emerge under the same drug pressure (e.g., the protease mutation D30N; see Section 4.2.3).
Intersubtype differences in sequence motifs favoring nucleotide substitutions involved in drug resistance (e.g., the reverse transcriptase mutation K65R; see Section 4.2.1).
4.2. Impact of HIV-1 Subtypes on the Different Antiretrovirals
The following subsections describe the impact of HIV-1 subtypes on the different classes of antiretrovirals so far available. Table 1 provides an overview of the resistance associated mutations which are related to subtype diversity.
Table 1.
Main drug resistance mutations observed in different HIV-1 subtypes.
| Drug class | Subtype | Polymorphisms or mutations or positions associated with drug resistance | Drug(s) related | Comments | References |
|---|---|---|---|---|---|
| Reverse transcriptase | |||||
|
| |||||
| C | K65R | d4T, ddI, ABC, TDF | Preferential selection | [14, 15, 148–152, 215, 216] | |
| NRTI | C | K70E | d4T, ddI, AZT | High prevalence in subtype C endemic area | [153] |
| B | D67N | d4T, AZT | Preferential selection | [148, 149, 151] | |
|
| |||||
| G | A98S | NNRTIs | Common polymorphism | [15, 173] | |
| B, C, F, CRF02_AG | K103N | EFV, DLV, NPV | Lower frequency in subtype C compared to B, F, and AG subtype | [76, 168, 169, 172] | |
| B, C | V106M | EFV, NVP | Lower genetic barrier in subtype C in comparison with subtype B | [140, 141, 171] | |
| NNRTI | C | E138K | ETR | Preferential selection under drug pressure in subtype C | [177, 178] |
| C | G190A | NNRTIs | High frequency in subtype C | [140] | |
| A, B | Y181C | ETR | Preferential selection under drug pressure on A and B subtypes | [178] | |
| C | Y181C, Y188L | EFV, DLV, NPV | Higher frequency in subtype C | [76, 168, 169, 172] | |
| C | N348I | ETR | Higher frequency in subtype C at etravirine failure | [179, 180] | |
|
| |||||
| Protease | |||||
|
| |||||
| CRF02_AG | G17E, I64M | NFV, ATV, IDV | Hypersusceptibility in CRF02_AG | [193] | |
| B, C, F, G, CRF01_AE | D30N | NFV | Lower prevalence in C, F, G, and CRF01_AE subtypes under NFV pressure compared to subtype B | [186, 187] | |
| Non-B | M36I | PIs | Natural polymorphism | [133, 139, 191] | |
| PI | A, C, D, F, G, CRF02_AG | 10, 13, 14, 20, 53, 63, 67, 73, 74, 77, 82, 88, 89 | PIs | Natural polymorphisms | [87, 132, 133, 142–145, 194] |
| A, C, F, CRF01_AE | L89M | ATV, LPV, NFV | Natural polymorphism that may lead to the L89T mutational pathway | [139, 192, 224] | |
| CRF02_AG | L89V | FPV, DRV, LPV | Higher prevalence in CRFF02_AG compared to subtype B | [76, 143] | |
| B, C, F, G, CRF01_AE, CRF02_AG | N88S, L90M | ATV, NFV | Higher prevalence in C, F, G, CRF01_AE, and CRF02_AG subtypes compared to subtype B | [186, 187, 194] | |
| C | T74P | TPV | Higher prevalence in subtype C in comparison to subtype B | [76] | |
| Non-B | V82A/M/F/S | PIs | High prevalence in some non-B subtypes at failure | [194] | |
|
| |||||
| Integrase | |||||
|
| |||||
| B, C | N155H, E92Q | RAL, EVG | >10-fold resistance in subtype B in comparison to subtype C | [201] | |
| Non-B | T97A, V151I, G163R | RAL | High frequency in non-B subtypes endemic area | [203, 204] | |
| INI | Non-B | L101I, T124A | DTG | Higher frequency in non-B subtypes in comparison to subtype B | [198] |
| C, CRF02_AG | G118R | RAL | Emerging at RAL failure in subtype CRF02_AG | [202] | |
| B | R263K | DTG | Preferential selection under drug pressure | [206] | |
|
| |||||
| Env gene | |||||
|
| |||||
| FI | C | N42S, L54M, A67T | T20 | Higher frequency in subtype C in comparison to B | [211] |
| CCR5 inhibitors | C | R315Q, A316T | MVC, VCV | Higher frequency in subtype C in comparison to B | [211] |
ABC: abacavir; ATV: atazanavir; AZT: zidovudine; d4T: stavudine; ddI: didanosine; DRV: darunavir; DTG: dolutegravir; ETR: etravirine; EFV: efavirenz; EVG: elvitegravir; FI: fusion inhibitor; FPV: fosamprenavir; INI: integrase inhibitor; LPV: lopinavir; MVC: maraviroc; NFV: nelfinavir; NRTI: nucleotide reverse transcriptase inhibitor; NNRTI: non-NRTI; NVP: nevirapine; PI: protease inhibitor; RAL: raltegravir; TDF: tenofovir; T20: enfuvirtide; VCV: vicriviroc.
4.2.1. Nucleotide (Side) Reverse Transcriptase Inhibitors (NRTIs)
The impact of subtype in terms of emergence of resistance in the NRTI antiretroviral class is mainly due to the more rapid selection of primary resistance mutations in HIV-1 subtype C infected patients than in those infected by A subtype B virus.
Some of the differences in rates of acquisition of the mutation K65R or thymidine analogue mutations (TAMs) are doubtless due to treatment regimens and disease stage, as well as access to viral load testing in many developing countries. However, one study suggests that increased rates of K65R acquisition in subtype C may be due to the nature of the subtype C RNA template. The lysine to arginine mutation at position 65 (K65R) is a major mutation that confers broad high-level resistance to most NRTIs, except zidovudine [34]. The nucleotide sequence at codons 64-65-66 (containing a homopolymeric stretch of adenine bases) differs between subtypes B and subtype C viruses. This leads to reverse transcriptase pausing during the synthesis of double-stranded DNA from the single-stranded DNA intermediate template, a process that is template-specific but independent of the reverse transcriptase enzyme [14, 15, 148]. Subsequent misalignment of the subtype C template and primer leads to the AAG to AGG change responsible for the K65R mutation [149] (Figure 2). On the contrary, subtype B templates are prone to frequent pausing at codon 67, facilitating the generation of mutation D67N and TAMs instead of K65R (Figure 2, Table 1) [148, 150, 151].
Figure 2.

Different subtype associated RNA templates and development of NRTIs resistance in B and C subtypes. The figure represents the differences between subtype B (a) and subtype C (b), observed at nucleotide level in reverse transcriptase codons 64-65-66. AZT: zidovudine, d4T: stavudine, ddI: didanosine, ABC: abacavir, and TDF: tenofovir (modified by [15]).
A recent study showed a very high rate (>65%) of K65R for patients infected by subtype C who failed tenofovir-based first-line regimens in South Africa [152]. Furthermore, a rate of 7 to 15% of K65R and/or K70E mutations was observed in subtype C endemic area in patients failing regimens including stavudine, didanosine, or zidovudine [153]. Studies from Israel reported a high frequency of K65R with treatment failure in subtype C viruses from Ethiopian immigrants [154], while in Indian patients a rate of K65R around 10–12% at failure of first line combination therapy including stavudine, lamivudine, and nevirapine was observed [155]. By using ultrasensitive pyrosequencing, variants carrying K65R mutation showed a higher frequency in subtype C infected patients than those infected by subtype B (1.04% versus 0.25%). However, these results were not duplicated using the limiting dilution clonal sequencing approaches [156].
In addition to the recruitment for monitoring K65R prevalence in patients with subtype C HIV infection, also the TAM pathway (67N/70R/215Y) deserves further attention. Indeed, in patients infected by subtype C from Botswana, India, South Africa, and Malawi TAMs were also observed at treatment failure with zidovudine and didanosine [153, 155, 157, 158]. Larger numbers of patients and followups will be required to determine whether any consistent effect of the emergence of K65R in subtype C is clinically relevant.
4.2.2. Nonnucleotide Reverse Transcriptase Inhibitors (NNRTIs)
The emergence of mutations associated with resistance to NNRTIs occurs after a single dose of nevirapine (sdNVP) [159–161], a procedure recommended for the prevention of mother-to-child HIV transmission (PMTCT) by initial guidelines from the World Health Organization (WHO) [162, 163]. Replacement of sdNVP may be necessary to reduce infant HIV infection risk and drug resistance development. Indeed, high frequency of drug resistance was observed in 69% of pregnant women infected with subtype C, 36% of those with subtype D, 19% of those with subtype A, and 21% of those with the CRF02_AG, despite the absence of resistance before the administration of antiretroviral therapy [163–167]. Moreover, by using ultrasensitive sequencing techniques, several studies revealed that in patients infected by subtype C a higher prevalence of nevirapine-resistance mutations (K103N and Y181C) was found, reaching 70% to 87%, as compared with 42% of patients with subtype A with resistant viruses [168, 169]. These findings underscore the role of viral subtype in facilitating the development of drug resistance, which is exacerbated by the fact that resistance to NNRTIs can also be transmitted through breast feeding [170].
The propensity for subtype C viruses to develop resistance also affects the NNRTI class. Indeed, the V106M mutation is commonly selected in subtype C viruses (in approximately 30% of patients) after exposure to nevirapine or efavirenz, whereas a V106A substitution is only rarely selected (in approximately 5% of patients) by these two NNRTIs in other subtypes (Table 1).
This peculiarity of subtype C results from the fact that V106 is encoded by GTA in subtype B viruses and GTG in subtype C viruses [141, 171]. A single G-to-A transition at the first position of codon 106 in subtype C viruses results in V106M, which confers high-level resistance to efavirenz and nevirapine. In contrast, in subtype B viruses, V106M requires two nucleotide substitutions (GTA-ATG) and therefore occurs infrequently [140, 141].
K103N and Y181C mutations remain important in both subtypes B and C, with K103N occurring in 40% of subtype B and 29% of subtype C viruses and Y181C occurring in 23% of subtype B and 12% of subtype C viruses [172]. In a recent Italian study, the prevalence of mutation K103N in NNRTI-treated patients was lower in subtype C infected patients compared with patients infected by B, F, or CRF02_AG subtypes (Table 1). Conversely, the mutations Y181C and Y188L were found with higher frequency in patients infected with subtype C compared to patients infected with subtype B [76].
Another substitution that is seen more frequently among patients with a subtype C virus is G190A, which is also a result of naturally occurring G190A/S polymorphisms [140]. Reverse transcriptase polymorphisms at residue 98 are common in subtype G, but they are also associated with NNRTI resistance in subtype B (Table 1) [15, 173].
The second-generation NNRTI etravirine has demonstrated good efficacy across subtypes [174]. A recent study highlighted the fact that HIV-1 genetic variant does not significantly influence the genotypic prediction of etravirine susceptibility in infected individuals failing efavirenz containing regimens [175]. However, among the etravirine resistance associated mutations, some polymorphic substitutions resulted in drug-naive individuals infected with non-B subtypes, especially CRF02_AG [176]. Noteworthy, in a recent study it was found that the mutation E138K was the first mutation to emerge in either subtype B, C, or CRF02_AG clinical isolates under etravirine pressure (Table 1) [177]. Another study confirmed this finding for subtype C, while a preferential selection of Y181C for the subtype B virus was observed (Table 1) [178].
A novel mutation in the C-terminal domain of reverse transcriptase (N348I) has recently been reported to reduce susceptibility to etravirine in subtypes A, B, and C (Table 1) [179]. In particular, in a South-African clinical trial, the N348I mutation was observed in 24% of subtype C infected patients failing a first-line NNRTI regimen (most commonly with nevirapine) [180]. This mutation is not included in standard mutation lists or algorithms, but more data are urgently required to determine its clinical relevance.
To date, the efficacy and the resistance patterns of the novel second-generation NNRTI rilpivirine seem to be similar across the different subtypes [181, 182].
A study focused on HIV-1 subtype CRF01_AE showed that the presence of mutations associated with resistance to rilpivirine are uncommon in patients infected by this subtype and failing a first-line NNRTI-containing regimen [183]. However, cross-resistance between rilpivirine and etravirine was high. Moreover, approximately 60% of patients had a high level of etravirine resistance, thus suggesting that the role of etravirine in HIV-1 subtype CRF01_AE infected patients in second-line therapy is limited in late NNRTI failure settings [183].
NNRTI resistance associated mutations such as Y181I, Y188L, G190A, K101A, V106I, and V179I are natural polymorphisms in HIV-2, thus HIV-2 proves to be naturally resistant to all NNRTIs [184].
4.2.3. Protease Inhibitors (PIs)
Nonpolymorphic substitutions in the protease gene have a greater impact on baseline susceptibility to PIs than polymorphic mutations [185]. However, intersubtype amino acid differences can create subtle structural differences in the targets of therapy. For example, subtype B infected patients receiving nelfinavir are more likely to develop D30N than those with viruses belonging to subtypes C, F, G and CRF01_AE, which are more likely to develop L90M or N88S (Table 1) [186, 187]. More specifically, by comparing B and G subtypes, even in cases where the first mutation is L90M in both these subtypes, subsequent mutations differ significantly. Indeed, in subtype B, L63P is the second mutation and occurs in almost all cases, suggesting that the progression of resistance is dependent on the emergence of this mutation, followed by the selection of V77I and other mutations. Differently, in subtype G, in almost 100% of cases, L89I follows the emergence of L90M, suggesting a role in subtype G similar to that of L63P in subtype B. In this regard, findings indicate that the association of L89I with L90M may further increase the PI resistance of subtype G viruses when compared with L90M alone [188]. The third emerging mutation can be either A71V or I54V.
Although subtype C isolates from Ethiopian immigrants to Israel favoured the L90M pathway, patients with subtype C viruses in Botswana did have D30N, suggesting that subtype C viruses from Ethiopia and Southern Africa might be different [186, 189]. The basis for the higher preponderance of D30N in subtype B than in other subtypes may be related to the flexibility of the protease flap region and destabilization of the PI complex in subtype B, whereas an accessory N83T mutation may be needed to rescue fitness and confer resistance in subtype C [142, 190].
Recent evidence has suggested that the polymorphism at codon 36 in the protease gene (M36 in subtype B and I36 in most other non-B subtypes) affects both the patterns of resistance that emerge under drug pressure and viral replication capacity (Table 1) [191]. Similarly, the M89 polymorphism in subtypes A, C, and CRF01_AE (L89 in subtype B) preferentially leads to the emergence under drug pressure of the M89T mutation, which confers high-level resistance to nelfinavir, atazanavir, and lopinavir [192]. There is also in vitro evidence that CRF2_AG viruses with the 17E/64M polymorphisms demonstrate hypersusceptibility to certain PIs (nelfinavir, atazanavir, and indinavir) [193].
Differences in polymorphisms in the protease gene have been reported among several non-B subtypes; these include the following protease residues: 10, 20, and 63 in subtype A; 20, 53, 63, 74, and 82 in subtype C; 13 and 20 in subtype D; 10, 14, 20, and 77 in subtype F; 20, 67, 73, 82, and 88 in subtype G; 20, 63, 82, and 89 in the CRF01_AE; 20 and 89 in the CRF02_AG (Table 1) [194]. In particular, in patients treated with PIs, L89V was predominantly found in CRF02_AG, while the tipranavir resistance mutation T74P was predominantly found in the C subtype in comparison to B subtype [76]. The mutation V82M was mainly found in subtype G while the mutations V82A/F/S were mainly present in other subtypes; the mutation N88S was found in subtypes C and CRF02_AG, while N88D was present in subtype B [194]. In southern Brazil, scientists reported a lower relative frequency of primary resistance to PIs in subtype C rather than in subtype B [195].
Despite clear evidence for preferential emergence, subtype diversity may not limit the initial benefits of antiretroviral therapy [196].
Diminished susceptibilities among wild-type isolates have been found for CRF02_AG recombinant viruses in three different studies of nelfinavir and atazanavir [138, 173, 194]. A study suggested that distortions in the K26 pocket of the A/G protease may be responsible for a lower binding energy of nelfinavir and lower susceptibility of A/G viruses [138].
The protease and gag genes coevolve as a functional unit when HIV is subjected to drug pressure from PIs. Mutations in gag can act as compensatory substitutions that can increase both rates and levels of resistance to PIs, as well as viral replication capacity [197]. No genotypic system for the determination of drug resistance to PIs currently monitors gag, despite the fact that relevant mutations in gag can increase resistance by a factor of 2 to 2.5, depending on the subtype. It is likely that different subtypes could develop compensatory gag mutations at different rates; this might ultimately justify the genotyping of gag in resistance testing.
4.2.4. Integrase Inhibitors (INIs)
The primary mutations in the integrase gene associated with INI resistance are E92Q, Y143R/C, Q148K/R/H, and N155H. The residues associated with primary resistance seem to be highly conserved across subtypes, but polymorphisms at the sites of secondary resistance mutations are more common in non-B subtypes [147, 198–200].
Signature subtype differences in integrase at codons 140, 148, 151, 157, and 160 among HIV subtypes may affect the genetic barrier for resistance [146]. These variations predict higher genetic barriers to the development of G140S and G140C mutations in subtypes C, CRF02_AG, and A/CRF01_AE, as well as higher genetic barriers to V151I in subtypes CRF02_AG and CRF01_AE [146, 147].
Regarding the impact of subtype on primary resistance development, it was recently observed that the subtype B integrase enzyme with the N155H mutation (±E92Q) exhibited increased resistance to raltegravir compared to the subtype C enzyme (Table 1) [201]. In addition a new potential resistance pathway for raltegravir might include the mutation G118R, a substitution associated with lower susceptibility to raltegravir in CRF_02AG subtypes [202].
Notably, polymorphisms such as T97A, V151I, and G163R, already known as raltegravir secondary resistance mutations, showed a considerable prevalence (≥9%) in several recent studies also analyzing non-B subtype infected patients [203, 204]. The additional INI-resistance associated mutations K156N and S230N mutations were more frequent in B subtype while V72I, L74I, T125A, V201I, and T206S were more frequent in certain non-B subtypes [200, 205]. No differences in phenotypic raltegravir resistance were found in several non-B isolates tested [196].
Dolutegravir is a second-generation INI which recently entered the battery of antiretrovirals against HIV-1 infection. It showed a higher genetic barrier to resistance and retains activity against viruses with resistance to raltegravir or elvitegravir. Interestingly, among the mutations associated with in vitro resistance to dolutegravir, the mutations L101I and T124A were polymorphic and significantly more prevalent in patients with non-B subtypes [200]. In addition, mutation R263K seems to be preferentially selected in subtype B under dolutegravir pressure (Table 1) [206].
4.2.5. Entry Inhibitors (EIs)
Among the EIs today available in clinics, both enfuvirtide (targeting gp41 HIV-1 protein) and maraviroc (targeting cellular CCR5 receptors) have shown antiviral activity against HIV-1 B and non-B subtypes [207, 208]. However, the high level of diversity (20 to 40%) in the env region predicts that this class of drugs will probably have higher potential for natural and emergent drug resistance. Clinical data have shown that the FI enfuvirtide is active against non-B subtypes owing to a highly conserved binding domain, although HIV-2 and HIV-1 class O viruses have natural resistance against this drug [209, 210]. So far, some studies suggest that there are significant differences in baseline susceptibility to HIV EIs among the predominant HIV-1 subtypes [211, 212]. For instance, it was found that in drug-naive patients many more mutations associated with resistance to EIs occur in subtype C compared with subtype B strains (Table 1) [211].
5. Clinical and Biological Relevance of HIV-1 Genetic Diversity
Due to the worldwide spread of non-B viruses and the introduction of antiretroviral drugs in developing countries (known for the largest assortment of non-B subtypes), further knowledge concerning responsiveness HIV-1 drug resistance in non-B subtypes is required. Moreover, it should be taken into account that the increasing prevalence of HIV-1 non-B subtypes could have several implications not only for the development of resistance but also for other topics such as the response to antiretroviral therapy, disease progression, and transmission.
5.1. Impact of HIV-1 Subtypes on the Drug Resistance
There is a solid body of evidence indicating that the type and degree of HIV-1 resistance to NRTIs, NNRTIs, and PIs vary between different subtypes [136, 186, 213, 214]. The development of nelfinavir resistance in subtypes B and G represents a classic example of this phenomenon. A different level of resistance has been observed among different subtypes. Indeed, the recombinant form CRF02_AG is more susceptible to nelfinavir and ritonavir than subtypes C and F; subtype G is more sensitive to tipranavir and lopinavir than other subtypes [135], and the subtype C has accelerated risk in developing resistance to tenofovir [189, 215, 216].
An explanation for the extreme variability of HIV-1 subtypes in the response to antiretrovirals can be given by the presence of some polymorphisms that can influence both the emergence of drug-resistance mutations and the response to drugs. For example, polymorphisms at residues 20 and 36 of HIV-1 protease decrease the genetic barrier to tipranavir resistance in subtypes A, C, F, and G [133], while nucleotide heterogeneity at 64 and 65 positions in the reverse transcriptase accelerates development of K65R in subtype C [150, 189]. In some cases, drug exposure may lead to amplification of such polymorphisms as A98G/S in reverse transcriptase and M36I, K20I, and L89M in protease, leading to a potential for resistance [139].
It is commonly accepted that, as a result of a high degree of polymorphism in the protease gene, drug-naive patients infected with non-B subtypes present a certain degree of resistance to the PIs. However, the data which support this assumption are largely controversial, as highlighted by two studies that showed conflicting results. Indeed, in a study [143] analysing the HIV-1 phenotype from 42 patients (19, subtype G; seven, subtype C; six, subtype F; 10, CRF02_AG) by the Antivirogram Assay (Virco BVBA), no differences in baseline susceptibility to any PI were found, with the exception of an unexpected hypersusceptibility to nelfinavir with CRF02_AG. On the contrary, a second study, investigating drug susceptibility of 39 isolates from treatment-naive Ghanaian patients [138], mostly infected with CRF02_AG, showed reduced susceptibility to several PIs, especially nelfinavir. This reduced in vitro susceptibility is not in agreement with the available clinical data, which show no difference in time to undetectable viral load among different subtypes [217–219].
Continuous research on the role of polymorphisms in the development of drug resistance is therefore necessary. Studies are needed to assess genotypes both before and after therapy in the context of possible associations between polymorphisms and drug resistance. This area of research could include polymorphism variability in variants of the same subtype in different geographic regions. This information might improve the efficacy of certain drug combinations over others in the context of second- or third-line therapeutic strategies. Polymorphisms should improve current algorithms for interpretation of genotyping results in a subtype-independent way.
As access to antiretroviral therapy in resource limited settings increases, where non-B subtypes are predominant, it remains imperative to establish appropriate treatment strategies for long-term clinical benefit that limits the emergence of drug resistance. The use of nontoxic, effective antiretroviral drugs should yield excellent clinical responsiveness, regardless of the viral subtype. Subtype differences, suboptimal therapies, and deficiencies in health care delivery systems can create conditions for accelerated development of resistance. Urgent recruitment for low-cost viral-load monitoring is needed to prevent and detect drug resistance, as well as to avoid unnecessary treatment switches [220, 221].
Unfortunately, pooling of resistance data often masks the role of regional differences in viral subtypes and antiretroviral therapies in the development of drug resistance. In resource-poor settings, such studies have used different NRTI backbones (e.g., combinations of zidovudine and didanosine, zidovudine and lamivudine, or stavudine and lamivudine). In parts of Africa, treatment failure has been reported in as many as 40% of patients after 2 years [222]. Of note, resistance rates in India to two drug classes were observed in more than 80% of patients who failed their first-line regimen using combinations of NRTIs and NNRTIs [223].
Larger longitudinal studies are necessary to determine the response to first-line combinations of antiretroviral drugs. The availability of genotypic resistance testing needs to be expanded to include all countries in which antiretroviral drugs are used.
Cross-resistance among drugs is important, especially in settings where treatment options may be limited. Relatively few in vitro comparative data are available for PIs in non-B subtypes, even if such data may be crucial for an understanding of cross-resistance to this class of antiretrovirals [191, 224]. Such data are important, since PIs are often the only available option for drug sequencing in resource-limited settings after the failure of first- or second-line treatment. Noteworthy, PIs are drugs with a high genetic barrier. This means that resistance to PIs usually requires the presence of large numbers of resistance mutations. For this reason, there is an advantage of using PIs to avoid early resistance development. Therefore, differences among subtypes with regard to drug resistance are likely to be more important for NRTIs, NNRTIs, and INIs than for PIs.
5.2. Impact of HIV-1 Subtypes on Response to Antiretroviral Therapy
Our knowledge of the impact of HIV-1 subtypes on responses to antiretroviral therapy is very limited. Most studies have been small or poorly designed, and only a few have had an evaluation period longer than 48 weeks. An important limitation of these studies was to compare subtype B with non-B subtypes grouped together, a necessary oversimplification when dealing with small numbers of patients infected with non-B subtypes [217–219, 225–227]. Moreover, no prospective, randomised trials specifically designed to address this question exist. A study by the Paediatric European Network for Treatment of AIDS (PENTA 5 trial) [219] investigated the response to therapy of 113 children infected with 7 different subtypes (A, 15%; B, 41%; C, 16%; D, 9%; F, 5%; G, 2%; H, 1%) and the two CRFs CRF01_AE (5%) and CRF02_AG (7%). No significant differences were observed at 48 weeks of treatment. However, these results should be used with caution because the study did not have the statistical power to rule out minor differences between subtypes. Moreover, only four drugs were used (zidovudine, lamivudine, abacavir, and nelfinavir). Although polymorphisms at resistance-associated positions were highly prevalent in non-B subtypes, no negative impact of the mutations in virological responses was found; therefore, the authors concluded that polymorphisms at resistance-associated positions in subtype B could not necessarily be interpreted as conferring resistance in non-B subtypes.
Frater et al. retrospectively analysed the virological response in 362 patients: 265 Europeans infected with subtype B and 97 Africans infected with non-B subtypes [218]. Subtype was presumed from ethnic and epidemiological data, with confirmation further extrapolated from genotyping the samples from 60% of the Africans and 30% of the European patients. There was a significant imbalance between the two groups in several important parameters, including gender, transmission-risk groups, CD4 cell counts, and antiretroviral regimens used. Results showed no difference in time to undetectable viral load or recovery of CD4 count; however, a significant difference was observed in viral load over time, with a continuous increase in the African group after 9 months, suggesting a higher rate of therapy failure. Based on indirect evidence (i.e., the number of resistance mutations at time of failure), the authors concluded that the difference in virological response was mainly due to poorer adherence in the African group rather than the infecting viral subtype. As the study was not controlled for adherence, its conclusions remain controversial.
A study which analysed retrospectively B and non-B subtypes (mainly focused on pure subtype A or recombinants with a subtype A protease) showed no differences in the time to undetectable viral load or in the viral load measured over time [217]. However, the gain in CD4 count was significantly lower in the non-B group, which showed an increase of 161 cells/mm3, compared with 236 cells/mm3 in the subtype B group. These results were consistent with those of Camacho, who compared retrospectively the pathways to nelfinavir resistance in 101 patients (46 subtype B, 55 subtype G) failing a first-line regimen including this PI [228]. At the time of failure, the only parameter significantly different between the two groups was the CD4 count (423 cells/mm3 in the subtype B group and 360 cells/mm3 in the subtype G group; P < 0.001). No explanation was given for this observation.
Geretti et al. assessed virologic and immunologic responses to starting HAART in a large cohort of 2116 patients, with the specific objective of comparing outcomes in patients with subtype B infection and those with subtype C, subtype A, CRF02_AG, and subtype D infection, the predominant non-B subtypes circulating in the United Kingdom [229]. Overall, 1906 (90%) patients achieved viral load undetectability within 12 months after they started HAART, of whom 335 (18%) subsequently experienced virologic rebound. In adjusted analyses, viral load suppression occurred more rapidly in patients infected with subtype C (hazard ratio (95% confidence interval, CI) 1.16 [1.01–1.33], P = 0.04) and subtype A (1.35 [1.04–1.74], P = 0.02) relative to subtype B infection. The virologic rebound occurred marginally faster in patients with subtype C infection (1.40 [1.00–1.95], P = 0.05), but the hazard of virologic rebound was similar with other subtypes. Although patients infected by subtype B viruses showed higher baseline CD4 cell counts and maintained the advantage throughout therapy, CD4 cell count recovery occurred at similar rates with all subtypes. These findings suggest that HAART achieves good outcomes regardless of the infecting subtype.
The effect of pretreatment HIV-1 drug resistance on the response to first-line combination antiretroviral therapy in Sub-Saharan Africa has recently been evaluated in a large prospective cohort [230]. Pretreatment drug resistance results were available for 2579 (94%) of 2733 participants. Among them, 123 (5%) had pretreatment drug resistance to at least one prescribed drug, and 52 (2%) had pretreatment drug resistance and received fully active antiretroviral therapy. More than 50% of participants for whom it was possible to determine the subtype were infected by HIV-1 subtype C (1405 patients, 54%), while 638 (25%) harboured subtype A, 296 (11%) D, 117 (5%) A/G recombinant, 68 (3%) G, 48 (2%) other recombinants, seven (<1%) other subtypes, and five (<1%) B. Compared with participants without pretreatment drug resistance, the odds ratio (OR) for virological failure (OR (95% CI): 2.13 [1.44–3.14], P < 0.0001) and acquired drug resistance (2.30 [1.55–3.40]; P < 0.0001) was increased in participants with pretreatment drug resistance to at least one prescribed drug but not in those with pretreatment drug resistance and fully active antiretroviral therapy. CD4 cell count increased less in participants with pretreatment drug resistance than in those without (35 cells per μL difference after 12 months; 95% CI 13–58; P = 0.002). HIV-1 subtype was not associated either with acquired drug resistance or virological failure.
Further ad hoc studies are needed to better evaluate the therapeutic implications of specific non-B subtype differences in terms of long-term efficacy of different antiviral regimens.
5.3. Impact of HIV-1 Subtypes on Disease Progression and Viral Transmission
Several studies on disease progression showed that, among non-B subtypes, subtypes C and D were found to be more aggressive, followed by G, AE, AG, and A, the least aggressive of all HIV-1 subtypes [92–97].
In this regard, it should be noted that because the different HIV-1 subtypes are not uniformly dispersed, comparisons of virulence and transmissibility are hampered by potential confounders, such as ethnic, socioeconomic, and other epidemiological factors. The report by Kiwanuka et al. [96] concerning this topic provided a good opportunity to compare rates of disease progression associated with these different subtypes within a similar population, thanks to the cocirculation of HIV-1 subtypes A and D, and several intersubtype recombinants in the Rakai district of Uganda. In particular, Kiwanuka et al. compared rates of progression among HIV-1 seroconverters who were followed as part of the Rakai Health Sciences Program [96]. Subjects identified during 1997–2002 were followed through 2004, when antiretroviral therapy became available in Rakai district. The primary end point was the time to achievement of a CD4 cell count of ⩽250 cells/mm3 or death due to AIDS. Progression to a CD4 cell count of ⩽250 cells/mm3 was significantly less common among subjects infected with HIV-1 subtype A (20%), compared with subjects infected with subtype D (40%), recombinant forms (40%), or multiple subtypes (53%) (P = 0.03). Death from AIDS was also less common among subjects infected with HIV-1 subtype A. These differences were reflected in the longer time to AIDS onset for subjects infected with HIV-1 subtype A (8.05 years) compared with those infected with non-A subtypes (D = 6.49 years; recombinant forms = 5.57 years; multiple subtypes = 5.80 years). By multivariable models (adjusting for viral load, age, and sex), subjects infected with non-A subtypes more likely progressed to AIDS compared with those infected with HIV-1 subtype A. Similarly, subjects infected with non-A subtypes had an approximately 6–8-fold greater risk of death from AIDS. Viral factors that could explain some of the HIV-1 pathogenicity are higher ex vivo replicative capacity, higher genetic diversity, and CXCR4 coreceptor usage [231–233]. In this regard, a less common emergence of CXCR4-using (X4) variants was found in HIV-1 subtype A infection compared with HIV-1 subtype D infection, thus explaining the apparent lower virulence of HIV-1 A [234, 235].
A European study showed how subtype D has a fourfold higher rate of CD4 count decline, in the absence of antiretroviral therapy, compared with other subtypes in the study (A, B, C, and CRF02_AG, which had similar rates of CD4 loss), even when adjusted for baseline CD4 count [236].
Subtype D was also associated with higher rates of dementia in individuals with advanced immunosuppression if compared with subtype A [98]. A study by Kiwanuka et al. showed how in Uganda subtype A viruses have a significantly higher rate of heterosexual transmission than subtype D viruses [237]. The faster disease progression and the lower rate of heterosexual transmission of subtype D compared with subtype A can explain the changes in the proportions of subtypes A and D observed over time in Uganda and Kenya [238, 239]. In particular, a significant decrease in the prevalence of subtype D with a concurrent increase of subtype A was observed.
5.4. Immune Response according to Different HIV-1 Subtypes
Despite the importance of viral characteristics in determining the rate of HIV-1 disease progression, recent findings from genomic studies show that host genetic factors also play a crucial role. The genetic determinants that influence susceptibility to HIV-1 and limit AIDS vary in different populations and among individuals. Meta-analyses of large cohort studies have identified several genetic variants that regulate HIV cell entry (particularly chemokine coreceptors and their ligands with copy number variations), acquired and innate immunity (major histocompatibility complex (MHC), Killer immunoglobulin-like receptors (KIRs), and cytokines), and others (TRIM5-α and APOBEC3G) that influence the outcome of HIV infection [240–242]. Of the various genes that contribute toward host genetic propensity, MHC turns out to be the major contributor because it is responsible both for restriction of cytotoxic T lymphocyte (CTL) epitopes and for the emergence of CTL escape mutants. Leukocyte antigen (HLA) alleles have been shown to be associated with the rate of disease progression in Africans and Caucasians [243–245].
The interplay of viral, immune, and host genetic factors in the control of HIV-1 replication has been recently evaluated in HIV controllers [246, 247]. Similar efforts should be undertaken in diverse human populations infected with a variety of HIV-1 subtypes in order to understand fully the complex interplay of virus and host in AIDS pathogenesis.
6. Summary and Conclusions
HIV diversity plays a central role in the HIV pandemic; for this reason, it is today imperative that global molecular epidemiology surveillance is continued and improved using rigorous sampling strategies. The improved knowledge of the significance of non-B subtypes for resistance evolution and interpretation, in response to antiretroviral therapy, disease progression and vaccine design are becoming mandatory today not only because antiretroviral therapy is being introduced in countries where non-B subtypes are driving the epidemic but also because the number of infections by these variants is increasing sharply in previously subtype B homogeneous areas such as Europe and North America [57–69, 71–73, 248]. It is reassuring that current antiretroviral strategies appear to be effective against a broad spectrum of HIV subtypes. However, further ad hoc studies in larger and homogenous populations infected by different specific non-B subtypes are required to better evaluate their therapeutic implications in terms of long-term efficacy of different antiviral regimens. Further elucidation of viral polymorphisms and properties associated with transmission, viral load set point, and disease progression may lead to new approaches to disease prevention and treatment.
Efforts should be undertaken in diverse human populations infected with a variety of HIV-1 subtypes in order to fully understand the complex interplay of HIV and host in AIDS pathogenesis.
References
- 1.Barre Sinoussi F, Chermann JC, Rey F. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS) Science. 1983;220(4599):868–871. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- 2.Coffin J, Haase A, Levy JA, et al. Human immunodeficiency viruses. Science. 1986;232(4751):p. 697. doi: 10.1126/science.3008335. [DOI] [PubMed] [Google Scholar]
- 3.Gallo RC, Sarin PS, Gelmann EP. Isolation of human T-cell leukemia virus in acquired immune deficiency syndrome (AIDS) Science. 1983;220(4599):865–867. doi: 10.1126/science.6601823. [DOI] [PubMed] [Google Scholar]
- 4.Levy JA, Hoffman AD, Kramer SM. Isolation of lymphocytopathic retroviruses from San Francisco patients with AIDS. Science. 1984;225(4664):840–842. doi: 10.1126/science.6206563. [DOI] [PubMed] [Google Scholar]
- 5.Clavel F, Guétard D, Brun-Vézinet F, et al. Isolation of a new human retrovirus from West African patients with AIDS. Science. 1986;233(4761):343–346. doi: 10.1126/science.2425430. [DOI] [PubMed] [Google Scholar]
- 6.Ariyoshi K, Schim van der Loeff M, Berry N, Jaffar S, Whittle H. Plasma HIV viral load in relation to season and to Plasmodium falciparum parasitaemia. AIDS. 1999;13(9):1145–1146. doi: 10.1097/00002030-199906180-00023. [DOI] [PubMed] [Google Scholar]
- 7.Ariyoshi K, Jaffar S, Alabi AS, et al. Plasma RNA viral load predicts the rate of CD4 T cell decline and death in HIV-2-infected patients in West Africa. AIDS. 2000;14(4):339–344. doi: 10.1097/00002030-200003100-00006. [DOI] [PubMed] [Google Scholar]
- 8.Wilkins A, Ricard D, Todd J, Whittle H, Dias F, Da Silva AP. The epidemiology of HIV infection in a rural area of Guinea-Bissau. AIDS. 1993;7(8):1119–1122. doi: 10.1097/00002030-199308000-00015. [DOI] [PubMed] [Google Scholar]
- 9.Burke DS. Recombination in HIV: an important viral evolutionary strategy. Emerging Infectious Diseases. 1997;3(3):253–259. doi: 10.3201/eid0303.970301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plantier JC, Leoz M, Dickerson JE, et al. A new human immunodeficiency virus derived from gorillas. Nature Medicine. 2009;15(8):871–872. doi: 10.1038/nm.2016. [DOI] [PubMed] [Google Scholar]
- 11.Robertson DL, Anderson JP, Bradac JA, et al. HIV-1 nomenclature proposal. Science. 2000;288(5463):55–57. doi: 10.1126/science.288.5463.55d. [DOI] [PubMed] [Google Scholar]
- 12.Vallari A, Holzmayer V, Harris B, et al. Confirmation of putative HIV-1 group P in Cameroon. Journal of Virology. 2011;85(3):1403–1407. doi: 10.1128/JVI.02005-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perrin L, Kaiser L, Yerly S. Travel and the spread of HIV-1 genetic variants. The Lancet Infectious Diseases. 2003;3(1):22–27. doi: 10.1016/s1473-3099(03)00484-5. [DOI] [PubMed] [Google Scholar]
- 14.Lessells R, Katzenstein D, De OT. Are subtype differences important in HIV drug resistance? Current Opinion in Virology. 2012;2(5):636–643. doi: 10.1016/j.coviro.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wainberg MA, Brenner BG. The impact of HIV genetic polymorphisms and subtype differences on the occurrence of resistance to antiretroviral drugs. Molecular Biology International. 2012;2012:10 pages. doi: 10.1155/2012/256982.256982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palella FJ, Jr., Delaney KM, Moorman AC, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. The New England Journal of Medicine. 1998;338(13):853–860. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- 17.Montaner JS, Lima VD, Barrios R, et al. Association of highly active antiretroviral therapy coverage, population viral load, and yearly new HIV diagnoses in British Columbia, Canada: a population-based study. The Lancet. 2010;376(9740):532–539. doi: 10.1016/S0140-6736(10)60936-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnston KM, Levy AR, Lima VD, et al. Expanding access to HAART: A cost-effective approach for treating and preventing HIV. AIDS. 2010;24(12):1929–1935. doi: 10.1097/QAD.0b013e32833af85d. [DOI] [PubMed] [Google Scholar]
- 19.Chiasson MA, Berenson L, Li W, et al. Declining HIV/AIDS mortality in New York City. Journal of Acquired Immune Deficiency Syndromes and Human Retrovirology. 1999;21(1):59–64. doi: 10.1097/00126334-199905010-00008. [DOI] [PubMed] [Google Scholar]
- 20.Hogg RS, O’Shaughnessy MV, Gataric N, et al. Decline in deaths from AIDS due to new antiretrovirals. The Lancet. 1997;349(9061):p. 1294. doi: 10.1016/S0140-6736(05)62505-6. [DOI] [PubMed] [Google Scholar]
- 21.Detels R, Muñoz A, McFarlane G, et al. Effectiveness of potent antiretroviral therapy on time to AIDS and death in men with known HIV infection duration. Journal of the American Medical Association. 1998;280(17):1497–1503. doi: 10.1001/jama.280.17.1497. [DOI] [PubMed] [Google Scholar]
- 22.Mocroft A, Vella S, Benfield TL, et al. Changing patterns of mortality across Europe in patients infected with HIV-1. The Lancet. 1998;352(9142):1725–1730. doi: 10.1016/s0140-6736(98)03201-2. [DOI] [PubMed] [Google Scholar]
- 23.Moore RD, Keruly JC, Bartlett JG. Improvement in the health of HIV-infected persons in care: reducing disparities. Clinical Infectious Disease. 2012;55(9):1242–1251. doi: 10.1093/cid/cis654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Panos G, Samonis G, Alexiou VG, Kavarnou GA, Charatsis G, Falagas ME. Mortality and morbidity of HIV infected patients receiving HAART: a cohort study. Current HIV Research. 2008;6(3):257–260. doi: 10.2174/157016208784324976. [DOI] [PubMed] [Google Scholar]
- 25.Sterne JAC, Hernán MA, Ledergerber B, et al. Long-term effectiveness of potent antiretroviral therapy in preventing AIDS and death: a prospective cohort study. The Lancet. 2005;366(9483):378–384. doi: 10.1016/S0140-6736(05)67022-5. [DOI] [PubMed] [Google Scholar]
- 26.Vittinghoff E, Scheer S, O’Malley P, Colfax G, Holmberg SD, Buchbinder SP. Combination antiretroviral therapy and recent declines in AIDS incidence and mortality. Journal of Infectious Diseases. 1999;179(3):717–720. doi: 10.1086/314623. [DOI] [PubMed] [Google Scholar]
- 27.Wong KH, Chan KC, Lee SS. Delayed progression to death and to AIDS in a Hong Kong cohort of patients with advanced HIV type 1 disease during the era of highly active antiretroviral therapy. Clinical Infectious Diseases. 2004;39(6):853–860. doi: 10.1086/423183. [DOI] [PubMed] [Google Scholar]
- 28.Clavel F, Hance AJ. HIV drug resistance. The New England Journal of Medicine. 2004;350(10):1023–1035. doi: 10.1056/NEJMra025195. [DOI] [PubMed] [Google Scholar]
- 29.Deeks SG. Determinants of virological response to antiretroviral therapy: implications for long-term strategies. Clinical Infectious Diseases. 2000;30(supplement 2):S177–S184. doi: 10.1086/313855. [DOI] [PubMed] [Google Scholar]
- 30.Karmochkine M, Si A, Piketty C, et al. The cumulative occurrence of resistance mutations in the HIV-1 protease gene is associated with failure of salvage therapy with ritonavir and saquinavir in protease inhibitor-experienced patients. Antiviral Research. 2000;47(3):179–188. doi: 10.1016/s0166-3542(00)00110-8. [DOI] [PubMed] [Google Scholar]
- 31.Miller V, Phillips A, Rottmann C, et al. Dual resistance to zidovudine and lamivudine in patients treated with zidovudine-lamivudine combination therapy: association with therapy failure. Journal of Infectious Diseases. 1998;177(6):1521–1532. doi: 10.1086/515304. [DOI] [PubMed] [Google Scholar]
- 32.Praparattanapan J, Kotarathitithum W, Chaiwarith R, et al. Resistance-associated mutations after initial antiretroviral treatment failure in a large cohort of patients infected with HIV-1 subtype CRF01_AE. Current HIV Research. 2012;10(8):647–652. doi: 10.2174/157016212803901356. [DOI] [PubMed] [Google Scholar]
- 33.Wainberg MA, Rooke R, Tremblay M, et al. Clinical significance and characterization of AZT-resistant strains of HIV-1. Canadian Journal of Infectious Diseases and Medical Microbiology. 1991;2(1):5–11. doi: 10.1155/1991/124860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parikh UM, Mellors JW. Pretreatment HIV-1 drug resistance is strongly associated with virologic failure in HIV-infected patients receiving partly active antiretroviral regimens. Future Microbiology. 2012;7(8):929–932. doi: 10.2217/fmb.12.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meyerhans A, Cheynier R, Albert J, et al. Temporal fluctuations in HIV quasispecies in vivo are not reflected by sequential HIV isolations. Cell. 1989;58(5):901–910. doi: 10.1016/0092-8674(89)90942-2. [DOI] [PubMed] [Google Scholar]
- 36.Fisher AG, Ensoli B, Looney D, et al. Biologically diverse molecular variants within a single HIV-1 isolate. Nature. 1988;334(6181):444–447. doi: 10.1038/334444a0. [DOI] [PubMed] [Google Scholar]
- 37.Domingo E, Holland JJ. RNA virus mutations and fitness for survival. Annual Review of Microbiology. 1997;51:151–178. doi: 10.1146/annurev.micro.51.1.151. [DOI] [PubMed] [Google Scholar]
- 38.Nowak M, May R. Virus Dynamics. Oxford University Press; 2000. [Google Scholar]
- 39.Frost SDW, McLean AR. Quasispecies dynamics and the emergence of drug resistance during zidovudine therapy of HIV infection. AIDS. 1994;8(3):323–332. doi: 10.1097/00002030-199403000-00005. [DOI] [PubMed] [Google Scholar]
- 40.Nowak MA, Bonhoeffer S, Shaw GM, May RM. Anti-viral drug treatment: dynamics of resistance in free virus and infected cell populations. Journal of Theoretical Biology. 1997;184(2):203–217. doi: 10.1006/jtbi.1996.0307. [DOI] [PubMed] [Google Scholar]
- 41.Tang MW, Shafer RW. HIV-1 antiretroviral resistance: scientific principles and clinical applications. Drugs. 2012;72(9):1–25. doi: 10.2165/11633630-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Charpentier C, Dwyer DE, Mammano F, Lecossier D, Clavel F, Hance AJ. Role of minority populations of human immunodeficiency virus type 1 in the evolution of viral resistance to protease inhibitors. Journal of Virology. 2004;78(8):4234–4247. doi: 10.1128/JVI.78.8.4234-4247.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hertogs K, Bloor S, Kemp SD, et al. Phenotypic and genotypic analysis of clinical HIV-1 isolates reveals extensive protease inhibitor cross-resistance: a survey of over 6000 samples. AIDS. 2000;14(9):1203–1210. doi: 10.1097/00002030-200006160-00018. [DOI] [PubMed] [Google Scholar]
- 44.Ciancio BC, Trotta MP, Lorenzini P, et al. The effect of number of mutations and of drug-class sparing on virological response to salvage genotype-guided antiretroviral therapy. Antiviral Therapy. 2003;8(6):611–616. [PubMed] [Google Scholar]
- 45.Ross L, Liao Q, Gao H, et al. Impact of HIV type 1 drug resistance mutations and phenotypic resistance profile on virologic response to salvage therapy. AIDS Research and Human Retroviruses. 2001;17(15):1379–1385. doi: 10.1089/088922201753197042. [DOI] [PubMed] [Google Scholar]
- 46.Haupts S, Ledergerber B, Böni J, et al. Impact of genotypic resistance testing on selection of salvage regimen in clinical practice. Antiviral Therapy. 2003;8(5):443–454. [PubMed] [Google Scholar]
- 47.Ceccherini-Silberstein F, Cento V, Calvez V, Perno CF. The use of human immunodeficiency virus resistance tests in clinical practice. Clinical Microbiology and Infection. 2010;16(10):1511–1517. doi: 10.1111/j.1469-0691.2010.03353.x. [DOI] [PubMed] [Google Scholar]
- 48.Hirsch MS, Günthard HF, Schapiro JM, et al. Antiretroviral drug resistance testing in adult HIV-1 infection: 2008 recommendations of an International AIDS Society-USA panel. Clinical Infectious Diseases. 2008;47(2):266–285. doi: 10.1086/589297. [DOI] [PubMed] [Google Scholar]
- 49.Thompson MA, Aberg JA, Hoy JF, et al. Antiretroviral treatment of adult HIV infection: 2012 recommendations of the International Antiviral Society-USA panel. Journal of the American Medical Association. 2012;308(4):387–402. doi: 10.1001/jama.2012.7961. [DOI] [PubMed] [Google Scholar]
- 50.Vandamme AM, Camacho RJ, Ceccherini-Silberstein F, et al. European recommendations for the clinical use of HIV drug resistance testing: 2011 update. AIDS Review. 2011;13(2):77–108. [PubMed] [Google Scholar]
- 51.Guidelines for the clinical management and treatment of HIV infected adults in Europe, Version 6. 2011, http://www.europeanaidsclinicalsociety.org. [PubMed]
- 52.Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. 2012, http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf.
- 53.Corzillius M, Mühlberger N, Sroczynski G, Jaeger H, Wasem J, Siebert U. Cost effectiveness analysis of routine use of genotypic antiretroviral resistance testing after failure of antiretroviral treatment for HIV. Antiviral Therapy. 2004;9(1):27–36. [PubMed] [Google Scholar]
- 54.Sendi P, Günthard HF, Simcock M, Ledergerber B, Schüpbach J, Battegay M. Cost-effectiveness of genotypic antiretroviral resistance testing in HIV-infected patients with treatment failure. PLoS ONE. 2007;2(1, article e173) doi: 10.1371/journal.pone.0000173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taylor BS, Hammer SM. The challenge of HIV-1 subtype diversity. The New England Journal of Medicine. 2008;359(18):1965–1966. doi: 10.1056/NEJMc086373. [DOI] [PubMed] [Google Scholar]
- 56.UNAIDS. Global report: UNAIDS report on the global AIDS epidemic 2010. 2010, http://www.unaids.org/documents/20101123_GlobalReport_em.pdf.
- 57.Hemelaar J. The origin and diversity of the HIV-1 pandemic. Trends in Molecular Medicine. 2012;18(3):182–192. doi: 10.1016/j.molmed.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 58.Achkar JM, Burda ST, Konings FAJ, et al. Infection with HIV type 1 group M non-B subtypes in individuals living in New York City. Journal of Acquired Immune Deficiency Syndromes. 2004;36(3):835–844. doi: 10.1097/00126334-200407010-00011. [DOI] [PubMed] [Google Scholar]
- 59.Barin F, Courouce AM, Pillonel J, Buzelay L. Increasing diversity of HIV-1(M) serotypes in French blood donors over a 10-year period (1985–1995) AIDS. 1997;11(12):1503–1508. doi: 10.1097/00002030-199712000-00015. [DOI] [PubMed] [Google Scholar]
- 60.Brennan CA, Yamaguchi J, Devare SG, Foster GA, Stramer SL. Expanded evaluation of blood donors in the United States for human immunodeficiency virus type 1 non-B subtypes and antiretroviral drug-resistant strains: 2005 through 2007. Transfusion. 2010;50(12):2707–2712. doi: 10.1111/j.1537-2995.2010.02767.x. [DOI] [PubMed] [Google Scholar]
- 61.Carr JK, Osinusi A, Flynn CP, Gilliam BL, Maheshwari V, Zhao RY. Two independent epidemics of HIV in Maryland. Journal of Acquired Immune Deficiency Syndromes. 2010;54(3):297–303. doi: 10.1097/QAI.0b013e3181e0c3b3. [DOI] [PubMed] [Google Scholar]
- 62.Cuevas M, Fernandez-Garcia A, Sanchez-Garcia A, et al. Incidence of non-B subtypes of HIV-1 in Galicia, Spain: high frequency and diversity of HIV-1 among men who have sex with men. Euro Surveillance. 2009;14(47) doi: 10.2807/ese.14.47.19413-en. [DOI] [PubMed] [Google Scholar]
- 63.de Mendoza C, Garrido C, Poveda E, et al. Changes in drug resistance patterns following the introduction of HIV type 1 non-B subtypes in Spain. AIDS Research and Human Retroviruses. 2009;25(10):967–972. doi: 10.1089/aid.2008.0166. [DOI] [PubMed] [Google Scholar]
- 64.Frange P, Galimand J, Vidal N, et al. New and old complex recombinant HIV-1 strains among patients with primary infection in 1996–2006 in France: the French ANRS CO06 primo cohort study. Retrovirology. 2008;5, article 69 doi: 10.1186/1742-4690-5-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Holguín A, de Mulder M, Yebra G, López M, Soriano V. Increase of non-B subtypes and recombinants among newly diagnosed HIV-1 native spaniards and immigrants in Spain. Current HIV Research. 2008;6(4):327–334. doi: 10.2174/157016208785132455. [DOI] [PubMed] [Google Scholar]
- 66.Lai A, Riva C, Marconi A, et al. Changing patterns in HIV-1 non-B clade prevalence and diversity in Italy over three decades. HIV Medicine. 2010;11(9):593–602. doi: 10.1111/j.1468-1293.2010.00832.x. [DOI] [PubMed] [Google Scholar]
- 67.Lai A, Simonetti FR, Zehender G, et al. HIV-1 subtype F1 epidemiological networks among Italian heterosexual males are associated with introduction events from South America. PLoS One. 2012;7(8) doi: 10.1371/journal.pone.0042223.e42223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Locateli D, Stoco PH, De Queiroz ATL, et al. Molecular epidemiology of HIV-1 in Santa Catarina State confirms increases of subtype c in southern Brazil. Journal of Medical Virology. 2007;79(10):1455–1463. doi: 10.1002/jmv.20955. [DOI] [PubMed] [Google Scholar]
- 69.Salemi M, de Oliveira T, Ciccozzi M, Rezza G, Goodenow MM. High-resolution molecular epidemiology and evolutionary history of HIV-1 subtypes in Albania. PLoS ONE. 2008;3(1) doi: 10.1371/journal.pone.0001390.e1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Snoeck J, Kantor R, Shafer RW, et al. Discordances between interpretation algorithms for genotypic resistance to protease and reverse transcriptase inhibitors of human immunodeficiency virus are subtype dependent. Antimicrobial Agents and Chemotherapy. 2006;50(2):694–701. doi: 10.1128/AAC.50.2.694-701.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vermund SH, Leigh-Brown AJ. The HIV epidemic: high-income countries. Cold Spring Harbor Perspectives in Medicine. 2012;2(5) doi: 10.1101/cshperspect.a007195.a007195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weidle PJ, Ganea CE, Irwin KL, et al. Presence of human immunodeficiency virus (HIV) type 1, group M, non-B subtypes, Bronx, New York: a sentinel site for monitoring HIV genetic diversity in the United States. Journal of Infectious Diseases. 2000;181(2):470–475. doi: 10.1086/315253. [DOI] [PubMed] [Google Scholar]
- 73.Yerly S, Vora S, Rizzardi P, et al. Acute HIV infection: impact on the spread of HIV and transmission of drug resistance. AIDS. 2001;15(17):2287–2292. doi: 10.1097/00002030-200111230-00010. [DOI] [PubMed] [Google Scholar]
- 74.Ciccozzi M, Santoro MM, Giovanetti M, et al. HIV-1 non-B subtypes in Italy: a growing trend. New Microbiol. 2012;35(4):377–386. [PubMed] [Google Scholar]
- 75.Fernández-García A, Pérez-Álvarez L, Cuevas MT, et al. Identification of a new HIV type 1 circulating BF intersubtype recombinant form (CRF47_BF) in Spain. AIDS Research and Human Retroviruses. 2010;26(7):827–832. doi: 10.1089/aid.2009.0311. [DOI] [PubMed] [Google Scholar]
- 76.Santoro MM, Alteri C, Ronga L, et al. Comparative analysis of drug resistance among B and the most prevalent non-B HIV Type 1 subtypes (C, F, and CRF02_AG) in Italy. AIDS Research and Human Retroviruses. 2012;28(10):1285–1293. doi: 10.1089/aid.2011.0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yebra G, de MM, Martin L, et al. Most HIV type 1 non-B infections in the Spanish cohort of antiretroviral treatment-naive HIV-infected patients (CoRIS) are due to recombinant viruses. Journal of Clinical Microbiology. 2012;50(2):407–413. doi: 10.1128/JCM.05798-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Galimand J, Frange P, Rouzioux C, et al. Short communication: evidence of HIV type 1 complex and second generation recombinant strains among patients infected in 1997–2007 in France: ANRS CO06 PRIMO cohort. AIDS Research and Human Retroviruses. 2010;26(6):645–651. doi: 10.1089/aid.2009.0201. [DOI] [PubMed] [Google Scholar]
- 79.Lihana RW, Ssemwanga D, Abimiku A, Ndembi N. Update on HIV-1 diversity in Africa: a decade in review. AIDS Review. 2012;14(2):83–100. [PubMed] [Google Scholar]
- 80.Aitken SC, Kliphuis A, Wallis CL, et al. Development and evaluation of an assay for HIV-1 protease and reverse transcriptase drug resistance genotyping of all major group-M subtypes. Journal of Clinical Virology. 2012;54(1):21–25. doi: 10.1016/j.jcv.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 81.Fokam J, Salpini R, Santoro MM, et al. Performance evaluation of an in-house human immunodeficiency virus type-1 protease-reverse transcriptase genotyping assay in Cameroon. Archives of Virology. 2011;156(7):1235–1243. doi: 10.1007/s00705-011-0982-3. [DOI] [PubMed] [Google Scholar]
- 82.Saravanan S, Vidya M, Balakrishanan P, et al. Evaluation of two human immunodeficiency virus-1 genotyping systems: ViroSeq™ 2.0 and an in-house method. Journal of Virological Methods. 2009;159(2):211–216. doi: 10.1016/j.jviromet.2009.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Costagliola D, Descamps D, Assoumou L, et al. Prevalence of HIV-1 drug resistance in treated patients: a French nationwide study. Journal of Acquired Immune Deficiency Syndromes. 2007;46(1):12–18. doi: 10.1097/QAI.0b013e318074eb73. [DOI] [PubMed] [Google Scholar]
- 84.Eberle J, Gurtler L. The evolution of drug resistance interpretation algorithms: ANRS, REGA and extension of resistance analysis to HIV-1 group O and HIV-2. Intervirology. 2012;55(2):128–133. doi: 10.1159/000332009. [DOI] [PubMed] [Google Scholar]
- 85.Frentz D, Boucher CAB, Assel M, et al. Comparison of HIV-1 genotypic resistance test interpretation systems in predicting virological outcomes over time. PLoS ONE. 2010;5(7) doi: 10.1371/journal.pone.0011505.e11505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rhee SY, Jeffrey Fessel W, Liu TF, et al. Predictive value of HIV-1 genotypic resistance test interpretation algorithms. Journal of Infectious Diseases. 2009;200(3):453–463. doi: 10.1086/600073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.van de Vijver DAMC, Wensing AMJ, Åsjö B, et al. HIV-1 drug-resistance patterns among patients on failing treatment in a large number of European countries. Acta Dermatovenerologica Alpina, Pannonica et Adriatica. 2010;19(4):3–9. [PubMed] [Google Scholar]
- 88.Candotti D, Adu-Sarkodie Y, Davies F, et al. AIDS in an HIV-seronegative Ghanaian woman with intersubtype A/G recombinant HIV-1 infection. Journal of Medical Virology. 2000;62(1):1–8. doi: 10.1002/1096-9071(200009)62:1<1::aid-jmv1>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 89.Zhang M, Foley B, Schultz AK, et al. The role of recombination in the emergence of a complex and dynamic HIV epidemic. Retrovirology. 2010;7, article 25 doi: 10.1186/1742-4690-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rouet F, Chaix ML, Nerrienet E, et al. Impact of HIV-1 genetic diversity on plasma HIV-1 RNA quantification: usefulness of the Agence Nationale de Recherches sur le SIDA second-generation long terminal repeat-based real-time reverse transcriptase polymerase chain reaction test. Journal of Acquired Immune Deficiency Syndromes. 2007;45(4):380–388. doi: 10.1097/QAI.0b013e3180640cf5. [DOI] [PubMed] [Google Scholar]
- 91.Senechal B, James VL. Ten years of external quality assessment of human immunodeficiency virus type 1 RNA quantification. Journal of Clinical Microbiology. 2012;50(11):3614–3619. doi: 10.1128/JCM.01221-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Baeten JM, Chohan B, Lavreys L, et al. HIV-1 subtype D infection is associated with faster disease progression than subtype A in spite of similar plasma HIV-1 loads. Journal of Infectious Diseases. 2007;195(8):1177–1180. doi: 10.1086/512682. [DOI] [PubMed] [Google Scholar]
- 93.Kaleebu P, Ross A, Morgan D, et al. Relationship between HIV-1 Env subtypes A and D and disease progression in a rural Ugandan cohort. AIDS. 2001;15(3):293–299. doi: 10.1097/00002030-200102160-00001. [DOI] [PubMed] [Google Scholar]
- 94.Kaleebu P, French N, Mahe C, et al. Effect of human immunodeficiency virus (HIV) type 1 envelope subtypes A and D on disease progression in a large cohort of HIV-1-positive persons in Uganda. Journal of Infectious Diseases. 2002;185(9):1244–1250. doi: 10.1086/340130. [DOI] [PubMed] [Google Scholar]
- 95.Kanki PJ, Hamel DJ, Sankalé JL, et al. Human immunodeficiency virus type 1 subtypes differ in disease progression. Journal of Infectious Diseases. 1999;179(1):68–73. doi: 10.1086/314557. [DOI] [PubMed] [Google Scholar]
- 96.Kiwanuka N, Laeyendecker O, Robb M, et al. Effect of human immunodeficiency virus type 1 (HIV-1) subtype on disease progression in persons from Rakai, Uganda, with incident HIV-1 infection. Journal of Infectious Diseases. 2008;197(5):707–713. doi: 10.1086/527416. [DOI] [PubMed] [Google Scholar]
- 97.Vasan A, Renjifo B, Hertzmark E, et al. Different rates of disease progression of HIV type 1 infection in Tanzania based on infecting subtype. Clinical Infectious Diseases. 2006;42(6):843–852. doi: 10.1086/499952. [DOI] [PubMed] [Google Scholar]
- 98.Sacktor N, Nakasujja N, Skolasky RL, et al. HIV subtype D is associated with dementia, compared with subtype A, in immunosuppressed individuals at risk of cognitive impairment in Kampala, Uganda. Clinical Infectious Diseases. 2009;49(5):780–786. doi: 10.1086/605284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ariën KK, Abraha A, Quiñones-Mateu ME, Kestens L, Vanham G, Arts EJ. The replicative fitness of primary human immunodeficiency virus type 1 (HIV-1) group M, HIV-1 group O, and HIV-2 isolates. Journal of Virology. 2005;79(14):8979–8990. doi: 10.1128/JVI.79.14.8979-8990.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Troyer RM, McNevin J, Liu Y, et al. Variable fitness impact of HIV-1 escape mutations to cytotoxic T lymphocyte (CTL) response. PLoS Pathogens. 2009;5(4) doi: 10.1371/journal.ppat.1000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhuang J, Jetzt AE, Sun G, et al. Human immunodeficiency virus type 1 recombination: rate, fidelity, and putative hot spots. Journal of Virology. 2002;76(22):11273–11282. doi: 10.1128/JVI.76.22.11273-11282.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ramirez BC, Simon-Loriere E, Galetto R, Negroni M. Implications of recombination for HIV diversity. Virus Research. 2008;134(1-2):64–73. doi: 10.1016/j.virusres.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 103.Roberts JD, Bebenek K, Kunkel TA. The accuracy of reverse transcriptase from HIV-1. Science. 1988;242(4882):1171–1173. doi: 10.1126/science.2460925. [DOI] [PubMed] [Google Scholar]
- 104.Götte M, Li X, Wainberg MA. HIV-1 reverse transcription: a brief overview focused on structure-function relationships among molecules involved in initiation of the reaction. Archives of Biochemistry and Biophysics. 1999;365(2):199–210. doi: 10.1006/abbi.1999.1209. [DOI] [PubMed] [Google Scholar]
- 105.de Clercq E. Antivirals and antiviral strategies. Nature Reviews Microbiology. 2004;2(9):704–720. doi: 10.1038/nrmicro975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Coffin JM. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science. 1995;267(5197):483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- 107.Wei X, Ghosh SK, Taylor ME, et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373(6510):117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 108.Nowak M. HIV mutation rate. Nature. 1990;347(6293):p. 522. doi: 10.1038/347522a0. [DOI] [PubMed] [Google Scholar]
- 109.Hu WS, Temin HM. Retroviral recombination and reverse transcription. Science. 1990;250(4985):1227–1233. doi: 10.1126/science.1700865. [DOI] [PubMed] [Google Scholar]
- 110.Moutouh L, Corbeil J, Richman DD. Recombination leads to the rapid emergence of HIV-1 dually resistant mutants under selective drug pressure. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(12):6106–6111. doi: 10.1073/pnas.93.12.6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wain-Hobson S, Renoux-Elbé C, Vartanian JP, Meyerhans A. Network analysis of human and simian immunodeficiency virus sequence sets reveals massive recombination resulting in shorter pathways. Journal of General Virology. 2003;84(4):885–895. doi: 10.1099/vir.0.18894-0. [DOI] [PubMed] [Google Scholar]
- 112.Yusa K, Kavlick MF, Kosalaraksa P, Mitsuya H. HIV-1 acquires resistance to two classes of antiviral drugs through homologous recombination. Antiviral Research. 1997;36(3):179–189. doi: 10.1016/s0166-3542(97)00053-3. [DOI] [PubMed] [Google Scholar]
- 113.Kellam P, Larder BA. Retroviral recombination can lead to linkage of reverse transcriptase mutations that confer increased zidovudine resistance. Journal of Virology. 1995;69(2):669–674. doi: 10.1128/jvi.69.2.669-674.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gu Z, Gao Q, Faust EA, Wainberg MA. Possible involvement of cell fusion and viral recombination in generation of human immunodeficiency virus variants that display dual resistance to AZT and 3TC. Journal of General Virology. 1995;76(10):2601–2605. doi: 10.1099/0022-1317-76-10-2601. [DOI] [PubMed] [Google Scholar]
- 115.Monno L, Appice A, Cavaliere R, et al. Highly active antiretroviral therapy failure and protease and reverse transcriptase human immunodeficiency virus type I gene mutations. Journal of Infectious Diseases. 1999;180(2):568–571. doi: 10.1086/314909. [DOI] [PubMed] [Google Scholar]
- 116.Carr JK, Salminen MO, Albert J, et al. Full genome sequences of human immunodeficiency virus type 1 subtypes G and A/G intersubtype recombinants. Virology. 1998;247(1):22–31. doi: 10.1006/viro.1998.9211. [DOI] [PubMed] [Google Scholar]
- 117.Gao F, Balles E, Robertson DL, et al. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature. 1999;397(6718):436–441. doi: 10.1038/17130. [DOI] [PubMed] [Google Scholar]
- 118.Nerrienet E, Santiago ML, Foupouapouognigni Y, et al. Simian immunodeficiency virus infection in wild-caught chimpanzees from Cameroon. Journal of Virology. 2005;79(2):1312–1319. doi: 10.1128/JVI.79.2.1312-1319.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Keele BF, Van Heuverswyn F, Li Y, et al. Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science. 2006;313(5786):523–526. doi: 10.1126/science.1126531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Santiago ML, Rodenburg CM, Kamenya S, et al. SIVcpz in wild chimpanzees. Science. 2002;295(5554, article 465) doi: 10.1126/science.295.5554.465. [DOI] [PubMed] [Google Scholar]
- 121.Van Heuverswyn F, Li Y, Neel C, et al. Human immunodeficiency viruses: SIV infection in wild gorillas. Nature. 2006;444(7116):p. 164. doi: 10.1038/444164a. [DOI] [PubMed] [Google Scholar]
- 122.Zhu T, Korber BT, Nahmias AJ, Hooper E, Sharp PM, Ho DD. An African HIV-1 sequence from 1959 and implications for the of the epidemic. Nature. 1998;391(6667):594–597. doi: 10.1038/35400. [DOI] [PubMed] [Google Scholar]
- 123.Worobey M, Gemmel M, Teuwen DE, et al. Direct evidence of extensive diversity of HIV-1 in Kinshasa by 1960. Nature. 2008;455(7213):661–664. doi: 10.1038/nature07390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Korber B, Muldoon M, Theiler J, et al. Timing the ancestor of the HIV-1 pandemic strains. Science. 2000;288(5472):1789–1796. doi: 10.1126/science.288.5472.1789. [DOI] [PubMed] [Google Scholar]
- 125.Wertheim JO, Worobey M. Dating the age of the SIV lineages that gave rise to HIV-1 and HIV-2. PLoS Computational Biology. 2009;5(5) doi: 10.1371/journal.pcbi.1000377.e1000377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ariën KK, Vanham G, Arts EJ. Is HIV-1 evolving to a less virulent form in humans? Nature Reviews Microbiology. 2007;5(2):141–151. doi: 10.1038/nrmicro1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lau KA, Wang B, Saksena NK. Emerging trends of HIV epidemiology in Asia. AIDS Reviews. 2007;9(4):218–229. [PubMed] [Google Scholar]
- 128.Essex M. Human immunodeficiency viruses in the developing world. Advances in Virus Research. 1999;53:71–88. doi: 10.1016/s0065-3527(08)60343-7. [DOI] [PubMed] [Google Scholar]
- 129.Chen JHK, Wong KH, Chen Z, et al. Increased genetic diversity of HIV-1 circulating in Hong Kong. PLoS ONE. 2010;5(8) doi: 10.1371/journal.pone.0012198.e12198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liu Y, Li L, Yang S, et al. Identification and characterization of two new HIV type 1 unique (B/C) recombinant forms in China. AIDS Research and Human Retroviruses. 2011;27(4):445–451. doi: 10.1089/aid.2010.0212. [DOI] [PubMed] [Google Scholar]
- 131.Kuiken C, Foley B, Leitner T, et al. HIV Sequence Compendium 2010. Theoretical Biology and Biophysics Group, Los Alamos National Laboratory; 2010. [Google Scholar]
- 132.Kantor R, Katzenstein D. Polymorphism in HIV-1 non-subtype b protease and reverse transcriptase and its potential impact on drug susceptibility and drug resistance evolution. AIDS Reviews. 2003;5(1):25–35. [PubMed] [Google Scholar]
- 133.Holguín A, Suñe C, Hamy F, Soriano V, Klimkait T. Natural polymorphisms in the protease gene modulate the replicative capacity of non-B HIV-1 variants in the absence of drug pressure. Journal of Clinical Virology. 2006;36(4):264–271. doi: 10.1016/j.jcv.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 134.Champenois K, Bocket L, Deuffic-Burban S, et al. Expected response to protease inhibitors of HIV-1 non-B subtype viruses according to resistance algorithms. AIDS. 2008;22(9):1087–1089. doi: 10.1097/QAD.0b013e3282ff629b. [DOI] [PubMed] [Google Scholar]
- 135.Marcelin AG, Masquelier B, Descamps D, et al. Tipranavir-ritonavir genotypic resistance score in protease inhibitor-experienced patients. Antimicrobial Agents and Chemotherapy. 2008;52(9):3237–3243. doi: 10.1128/AAC.00133-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Martínez-Cajas JL, Pant-Pai N, Klein MB, Wainberg MA. Role of genetic diversity amongst HIV-1 non-B subtypes in drug resistance: a systematic review of virologic and biochemical evidence. AIDS Reviews. 2008;10(4):212–223. [PubMed] [Google Scholar]
- 137.Poonpiriya V, Sungkanuparph S, Leechanachai P, et al. A study of seven rule-based algorithms for the interpretation of HIV-1 genotypic resistance data in Thailand. Journal of Virological Methods. 2008;151(1):79–86. doi: 10.1016/j.jviromet.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 138.Kinomoto M, Appiah-Opong R, Brandful JAM, et al. HIV-1 proteases from drug-naive West African patients are differentially less susceptible to protease inhibitors. Clinical Infectious Diseases. 2005;41(2):243–251. doi: 10.1086/431197. [DOI] [PubMed] [Google Scholar]
- 139.Velazquez-Campoy A, Todd MJ, Vega S, Freire E. Catalytic efficiency and vitality of HIV-1 proteases from African viral subtypes. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(11):6062–6067. doi: 10.1073/pnas.111152698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Grossman Z, Istomin V, Averbuch D, et al. Genetic variation at NNRTI resistance-associated positions in patients infected with HIV-1 subtype C. AIDS. 2004;18(6):909–915. doi: 10.1097/00002030-200404090-00008. [DOI] [PubMed] [Google Scholar]
- 141.Brenner B, Turner D, Oliveira M, et al. A V106M mutation in HIV-1 clade C viruses exposed to efavirenz confers cross-resistance to non-nucleoside reverse transcriptase inhibitors. AIDS. 2003;17(1):F1–F5. doi: 10.1097/00002030-200301030-00001. [DOI] [PubMed] [Google Scholar]
- 142.Gonzalez LMF, Brindeiro RM, Aguiar RS, et al. Impact of nelfinavir resistance mutations on in vitro phenotype, fitness, and replication capacity of human immunodeficiency virus type 1 with subtype B and C proteases. Antimicrobial Agents and Chemotherapy. 2004;48(9):3552–3555. doi: 10.1128/AAC.48.9.3552-3555.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Abecasis AB, Deforche K, Snoeck J, et al. Protease mutation M89I/V is linked to therapy failure in patients infected with the HIV-1 non-B subtypes C, F or G. AIDS. 2005;19(16):1799–1806. doi: 10.1097/01.aids.0000188422.95162.b7. [DOI] [PubMed] [Google Scholar]
- 144.van de Vijver DA, Wensing AMJ, Angarano G, et al. The calculated genetic barrier for antiretroviral drug resistance substitutions is largely similar for different HIV-1 subtypes. Journal of Acquired Immune Deficiency Syndromes. 2006;41(3):352–360. doi: 10.1097/01.qai.0000209899.05126.e4. [DOI] [PubMed] [Google Scholar]
- 145.Palma AC, Araújo F, Duque V, Borges F, Paixão MT, Camacho R. Molecular epidemiology and prevalence of drug resistance-associated mutations in newly diagnosed HIV-1 patients in Portugal. Infection, Genetics and Evolution. 2007;7(3):391–398. doi: 10.1016/j.meegid.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 146.Maïga AI, Malet I, Soulie C, et al. Genetic barriers for integrase inhibitor drug resistance in HIV type-1 B and CRF02_AG subtypes. Antiviral Therapy. 2009;14(1):123–129. [PubMed] [Google Scholar]
- 147.Brenner BG, Lowe M, Moisi D, et al. Subtype diversity associated with the development of HIV-1 resistance to integrase inhibitors. Journal of Medical Virology. 2011;83(5):751–759. doi: 10.1002/jmv.22047. [DOI] [PubMed] [Google Scholar]
- 148.Coutsinos D, Invernizzi CF, Xu H, Brenner BG, Wainberg MA. Factors affecting template usage in the development of K65R resistance in subtype C variants of HIV type-1. Antiviral Chemistry and Chemotherapy. 2010;20(3):117–131. doi: 10.3851/IMP1443. [DOI] [PubMed] [Google Scholar]
- 149.Coutsinos D, Invernizzi CF, Moisi D, et al. A template-dependent dislocation mechanism potentiates K65R reverse transcriptase mutation development in subtype C variants of HIV-1. PLoS ONE. 2011;6(5) doi: 10.1371/journal.pone.0020208.e20208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Coutsinos D, Invernizzi CF, Xu H, et al. Template usage is responsible for the preferential acquisition of the K65R reverse transcriptase mutation in subtype C variants of human immunodeficiency virus type 1. Journal of Virology. 2009;83(4):2029–2033. doi: 10.1128/JVI.01349-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Harrigan PR, Sheen CW, Gill VS, et al. Silent mutations are selected in HIV-1 reverse transcriptase and affect enzymatic efficiency. AIDS. 2008;22(18):2501–2508. doi: 10.1097/QAD.0b013e328318f16c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sunpath H, Wu B, Gordon M, et al. High rate of K65R for antiretroviral therapy-naive patients with subtype C HIV infection failing a tenofovir-containing first-line regimen. AIDS. 2012;26(13):1679–1684. doi: 10.1097/QAD.0b013e328356886d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hosseinipour MC, van Oosterhout JJG, Weigel R, et al. The public health approach to identify antiretroviral therapy failure: high-level nucleoside reverse transcriptase inhibitor resistance among Malawians failing first-line antiretroviral therapy. AIDS. 2009;23(9):1127–1134. doi: 10.1097/QAD.0b013e32832ac34e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Turner D, Shahar E, Katchman E, et al. Prevalence of the K65R resistance reverse transcriptase mutation in different HIV-1 subtypes in Israel. Journal of Medical Virology. 2009;81(9):1509–1512. doi: 10.1002/jmv.21567. [DOI] [PubMed] [Google Scholar]
- 155.Deshpande A, Jauvin V, Magnin N, et al. Resistance mutations in subtype C HIV type 1 isolates from Indian patients of Mumbai receiving NRTIs plus NNRTIs and experiencing a treatment failure: resistance to AR. AIDS Research and Human Retroviruses. 2007;23(2):335–340. doi: 10.1089/aid.2006.0183. [DOI] [PubMed] [Google Scholar]
- 156.Varghese V, Wang E, Babrzadeh F, et al. Nucleic acid template and the risk of a PCR-induced HIV-1 drug resistance mutation. PLoS ONE. 2010;5(6) doi: 10.1371/journal.pone.0010992.e10992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Barth RE, Wensing AM, Tempelman HA, Moraba R, Schuurman R, Hoepelman AI. Rapid accumulation of nonnucleoside reverse transcriptase inhibitor-associated resistance: evidence of transmitted resistance in rural South Africa. AIDS. 2008;22(16):2210–2212. doi: 10.1097/QAD.0b013e328313bf87. [DOI] [PubMed] [Google Scholar]
- 158.Novitsky V, Wester CW, DeGruttola V, et al. The reverse transcriptase 67N 70R 215Y genotype is the predominant TAM pathway associated with virologic failure among HIV type 1C-infected adults treated with ZDV/ddI-containing HAART in Southern Africa. AIDS Research and Human Retroviruses. 2007;23(7):868–878. doi: 10.1089/aid.2006.0298. [DOI] [PubMed] [Google Scholar]
- 159.McConnell MS, Stringer JSA, Kourtis AP, Weidle PJ, Eshleman SH. Use of single-dose nevirapine for the prevention of mother-to-child transmission of HIV-1: does development of resistance matter? The American Journal of Obstetrics and Gynecology. 2007;197(3) supplement:S56–S63. doi: 10.1016/j.ajog.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 160.Eshleman SH, Mracna M, Guay LA, et al. Selection and fading of resistance mutations in women and infants receiving nevirapine to prevent HIV-1 vertical transmission (HIVNET 012) AIDS. 2001;15(15):1951–1957. doi: 10.1097/00002030-200110190-00006. [DOI] [PubMed] [Google Scholar]
- 161.WHO HIV Drug resistance report 2012. 2012, http://apps.who.int/iris/bitstream/10665/75183/1/9789241503938_eng.pdf.
- 162.Ciaranello AL, Perez F, Maruva M, et al. WHO 2010 guidelines for prevention of mother-to-child HIV transmission in Zimbabwe: Modeling clinical outcomes in infants and mothers. PLoS ONE. 2011;6(6) doi: 10.1371/journal.pone.0020224.e20224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Antiretroviral drugs for treating pregnant women and preventing HIV infection in infants. 2010, http://whqlibdoc.who.int/publications/2010/9789241599818_eng.pdf.
- 164.Chaix ML, Ekouevi DK, Rouet F, et al. Low risk of nevirapine resistance mutations in the prevention of mother-to-child transmission of HIV-1: agence Nationale de Recherches sur le SIDA Ditrame Plus, Abidjan, Côte d’Ivoire. Journal of Infectious Diseases. 2006;193(4):482–487. doi: 10.1086/499966. [DOI] [PubMed] [Google Scholar]
- 165.Eshleman SH, Hoover DR, Chen S, et al. Nevirapine (NVP) resistance in women with HIV-1 subtype C, compared with subtypes A and D, after the administration of single-dose NVP. Journal of Infectious Diseases. 2005;192(1):30–36. doi: 10.1086/430764. [DOI] [PubMed] [Google Scholar]
- 166.Eshleman SH, Church JD, Chen S, et al. Comparison of HIV-1 mother-to-child transmission after single-dose nevirapine prophylaxis among African women with subtypes A, C, and D. Journal of Acquired Immune Deficiency Syndromes. 2006;42(4):518–521. doi: 10.1097/01.qai.0000221676.22069.b8. [DOI] [PubMed] [Google Scholar]
- 167.Toni TD, Masquelier B, Lazaro E, et al. Characterization of nevirapine (NVP) resistance mutations and HIV type 1 subtype in women from Abidjan (Côte d’Ivoire) after NVP single-dose prophylaxis of HIV type 1 mother-to-child transmission. AIDS Research and Human Retroviruses. 2005;21(12):1031–1034. doi: 10.1089/aid.2005.21.1031. [DOI] [PubMed] [Google Scholar]
- 168.Flys TS, Chen S, Jones DC, et al. Quantitative analysis of HIV-1 variants with the K103N resistance mutation after single-dose nevirapine in women with HIV-1 subtypes A, C, and D. Journal of Acquired Immune Deficiency Syndromes. 2006;42(5):610–613. doi: 10.1097/01.qai.0000221686.67810.20. [DOI] [PubMed] [Google Scholar]
- 169.Johnson JA, Li JF, Morris L, et al. Emergence of drug-resistant HIV-1 after intrapartum administration of single-dose nevirapine is substantially underestimated. Journal of Infectious Diseases. 2005;192(1):16–23. doi: 10.1086/430741. [DOI] [PubMed] [Google Scholar]
- 170.Zeh C, Weidle PJ, Nafisa L, et al. HIV-1 drug resistance emergence among breastfeeding infants born to HIV-infected mothers during a single-arm trial of triple-antiretroviral prophylaxis for prevention of mother-to-child transmission: a secondary analysis. PLoS Medicine. 2011;8(3) doi: 10.1371/journal.pmed.1000430.e1000430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Loemba H, Brenner B, Parniak MA, et al. Genetic divergence of human immunodeficiency virus type 1 Ethiopian clade C reverse transcriptase (RT) and rapid development of resistance against nonnucleoside inhibitors of RT. Antimicrobial Agents and Chemotherapy. 2002;46(7):2087–2094. doi: 10.1128/AAC.46.7.2087-2094.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Shafer RW. Rationale and uses of a public HIV drug-resistance database. Journal of Infectious Diseases. 2006;194(supplement 1):S51–S58. doi: 10.1086/505356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sylla M, Chamberland A, Boileau C, et al. Characterization of drug resistance in antiretroviral-treated patients infected with HIV-1 CRF02_AG and AGK subtypes in Mali and Burkina Faso. Antiviral Therapy. 2008;13(1):141–148. [PubMed] [Google Scholar]
- 174.Vingerhoets J, Azijn H, Tambuyzer L, et al. Short communication: activity of etravirine on different HIV type 1 subtypes: in vitro susceptibility in treatment-naive patients and week 48 pooled DUET study data. AIDS Research and Human Retroviruses. 2010;26(6):621–624. doi: 10.1089/aid.2009.0239. [DOI] [PubMed] [Google Scholar]
- 175.Pereira-Vaz J, Duque V, Pereira B, et al. Prevalence of etravirine resistance associated mutations in HIV-1 strains isolated from infected individuals failing efavirenz: comparison between subtype B and non-B genetic variants. Journal of Medical Virology. 2012;84(4):551–554. doi: 10.1002/jmv.23232. [DOI] [PubMed] [Google Scholar]
- 176.Maïga AI, Descamps D, Morand-Joubert L, et al. Resistance-associated mutations to etravirine (TMC-125) in antiretroviral-naïve patients infected with non-B HIV-1 subtypes. Antimicrobial Agents and Chemotherapy. 2010;54(2):728–733. doi: 10.1128/AAC.01335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Asahchop EL, Oliveira M, Wainberg MA, Brenner BG, Toni TD, Tremblay CL. Characterization of the E138K resistance mutation in HIV-1 reverse transcriptase conferring susceptibility to etravirine in B and non-B HIV-1 subtypes. Antimicrobial Agents and Chemotherapy. 2011;55(2):600–607. doi: 10.1128/AAC.01192-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lai MT, Lu M, Felock PJ, et al. Distinct mutation pathways of non-subtype B HIV-1 during in vitro resistance selection with nonnucleoside reverse transcriptase inhibitors. Antimicrobial Agents and Chemotherapy. 2010;54(11):4812–4824. doi: 10.1128/AAC.00829-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.McCormick AL, Parry CM, Crombe A, et al. Impact of the N348I mutation in HIV-1 reverse transcriptase on nonnucleoside reverse transcriptase inhibitor resistance in non-subtype B HIV-1. Antimicrobial Agents and Chemotherapy. 2011;55(4):1806–1809. doi: 10.1128/AAC.01197-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Brehm JH, Koontz DL, Wallis CL, et al. Frequent emergence of N348I in HIV-1 subtype C reverse transcriptase with failure of initial therapy reduces susceptibility to reverse-transcriptase inhibitors. Clinical Infectious Disease. 2012;55(5):737–745. doi: 10.1093/cid/cis501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Cohen CJ, Molina JM, Cahn P, et al. Efficacy and safety of rilpivirine (TMC278) versus efavirenz at 48 weeks in treatment-naive HIV-1-infected patients: pooled results from the phase 3 double-blind randomized ECHO and THRIVE Trials. Journal of Acquired Immune Deficiency Syndromes. 2012;60(1):33–42. doi: 10.1097/QAI.0b013e31824d006e. [DOI] [PubMed] [Google Scholar]
- 182.Rimsky L, Vingerhoets J, Van Eygen V, et al. Genotypic and phenotypic characterization of HIV-1 isolates obtained from patients on rilpivirine therapy experiencing virologic failure in the phase 3 ECHO and THRIVE studies: 48-week analysis. Journal of Acquired Immune Deficiency Syndromes. 2012;59(1):39–46. doi: 10.1097/QAI.0b013e31823df4da. [DOI] [PubMed] [Google Scholar]
- 183.Bunupuradah T, Ananworanich J, Chetchotisakd P, et al. Etravirine and rilpivirine resistance in HIV-1 subtype CRF01_AE-infected adults failing non-nucleoside reverse transcriptase inhibitor-based regimens. Antiviral Therapy. 2011;16(7):1113–1121. doi: 10.3851/IMP1906. [DOI] [PubMed] [Google Scholar]
- 184.Ntemgwa ML, Toni TD, Brenner BG, Camacho RJ, Wainberg MA. Antiretroviral drug resistance in human immunodeficiency virus type 2. Antimicrobial Agents and Chemotherapy. 2009;53(9):3611–3619. doi: 10.1128/AAC.00154-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Rhee SY, Taylor J, Fessel WJ, et al. HIV-1 protease mutations and protease inhibitor cross-resistance. Antimicrobial Agents and Chemotherapy. 2010;54(10):4253–4261. doi: 10.1128/AAC.00574-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Grossman Z, Paxinos EE, Averbuch D, et al. Mutation D30N is not preferentially selected by human immunodeficiency virus type 1 subtype C in the development of resistance to nelfinavir. Antimicrobial Agents and Chemotherapy. 2004;48(6):2159–2165. doi: 10.1128/AAC.48.6.2159-2165.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Sukasem C, Churdboonchart V, Sukeepaisarncharoen W, et al. Genotypic resistance profiles in antiretroviral-naive HIV-1 infections before and after initiation of first-line HAART: impact of polymorphism on resistance to therapy. International Journal of Antimicrobial Agents. 2008;31(3):277–281. doi: 10.1016/j.ijantimicag.2007.10.029. [DOI] [PubMed] [Google Scholar]
- 188.Gonzalez LMF, Santos AF, Abecasis AB, et al. Impact of HIV-1 protease mutations A71V/T and T74S on M89I/V-mediated protease inhibitor resistance in subtype G isolates. Journal of Antimicrobial Chemotherapy. 2008;61(6):1201–1204. doi: 10.1093/jac/dkn099. [DOI] [PubMed] [Google Scholar]
- 189.Doualla-Bell F, Avalos A, Gaolathe T, et al. Impact of human immunodeficiency virus type 1 subtype C on drug resistance mutations in patients from Botswana failing a nelfinavir-containing regimen. Antimicrobial Agents and Chemotherapy. 2006;50(6):2210–2213. doi: 10.1128/AAC.01447-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Soares RO, Batista PR, Costa MGS, Dardenne LE, Pascutti PG, Soares MA. Understanding the HIV-1 protease nelfinavir resistance mutation D30N in subtypes B and C through molecular dynamics simulations. Journal of Molecular Graphics and Modelling. 2010;29(2):137–147. doi: 10.1016/j.jmgm.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 191.Lisovsky I, Schader SM, Martinez-Cajas JL, Oliveira M, Moisi D, Wainberg MA. HIV-1 protease codon 36 polymorphisms and differential development of resistance to nelfinavir, lopinavir, and atazanavir in different HIV-1 subtypes. Antimicrobial Agents and Chemotherapy. 2010;54(7):2878–2885. doi: 10.1128/AAC.01828-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Martinez-Cajas JL, Wainberg MA, Oliveira M, et al. The role of polymorphisms at position 89 in the HIV-1 protease gene in the development of drug resistance to HIV-1 protease inhibitors. Journal of Antimicrobial Chemotherapy. 2012;67(4):988–994. doi: 10.1093/jac/dkr582. [DOI] [PubMed] [Google Scholar]
- 193.Santos AF, Tebit DM, Lalonde MS, et al. Effect of natural polymorphisms in the HIV-1 CRF02_AG protease on protease inhibitor hypersusceptibility. Antimicrobial Agents Chemotherapy. 2012;56(5):2719–2725. doi: 10.1128/AAC.06079-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kantor R, Shafer RW, Katzenstein D. The HIV-1 Non-subtype B Workgroup: an international collaboration for the collection and analysis of HIV-1 non-subtype B data. Journal of the International AIDS Society. 2005;7(1):p. 71. doi: 10.1186/1758-2652-7-1-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Sprinz E, Netto EM, Patelli M, et al. Primary antiretroviral drug resistance among HIV type 1-infected individuals in Brazil. AIDS Research and Human Retroviruses. 2009;25(9):861–867. doi: 10.1089/aid.2009.0012. [DOI] [PubMed] [Google Scholar]
- 196.Kantor R, Katzenstein DA, Efron B, et al. Impact of HIV-1 subtype and antiretroviral therapy on protease and reverse transcriptase genotype: results of a global collaboration. PLoS Medicine. 2005;2:0325–0337. doi: 10.1371/journal.pmed.0020112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.van Maarseveen N, Boucher C. Resistance to protease inhibitors. In: Geretti AM, editor. Antiretroviral Resistance in Clinical Practice. 2006. [PubMed] [Google Scholar]
- 198.Garrido C, Soriano V, Geretti AM, et al. Resistance associated mutations to dolutegravir (S/GSK1349572) in HIV-infected patients: impact of HIV subtypes and prior raltegravir experience. Antiviral Research. 2011;90(3):164–167. doi: 10.1016/j.antiviral.2011.03.178. [DOI] [PubMed] [Google Scholar]
- 199.Fish MQ, Hewer R, Wallis CL, Venter WDF, Stevens WS, Papathanasopoulos MA. Natural polymorphisms of integrase among HIV type 1-infected South African patients. AIDS Research and Human Retroviruses. 2010;26(4):489–493. doi: 10.1089/aid.2009.0249. [DOI] [PubMed] [Google Scholar]
- 200.Sierra S, Lübke N, Walter H, et al. The SnoB study: frequency of baseline raltegravir resistance mutations prevalence in different non-B subtypes. Medical Microbiology and Immunology. 2011;200(4):225–232. doi: 10.1007/s00430-011-0194-1. [DOI] [PubMed] [Google Scholar]
- 201.Bar-Magen T, Donahue DA, McDonough EI, et al. HIV-1 subtype B and C integrase enzymes exhibit differential patterns of resistance to integrase inhibitors in biochemical assays. AIDS. 2010;24(14):2171–2179. doi: 10.1097/QAD.0b013e32833cf265. [DOI] [PubMed] [Google Scholar]
- 202.Malet I, Fourati S, Charpentier C, et al. The HIV-1 integrase G118R mutation confers raltegravir resistance to the CRF02_AG HIV-1 subtype. Journal of Antimicrobial Chemotherapy. 2011;66(12):2827–2830. doi: 10.1093/jac/dkr389. [DOI] [PubMed] [Google Scholar]
- 203.Monleau M, Aghokeng AF, Nkano BA, Chaix ML, ANRS Working Group Drug resistance mutations of HIV-1 non-B viruses to integrase inhibitors in treatment-naïve patients from sub-Saharan countries and discordant interpretations. AIDS Research and Human Retroviruses. 2012;28(9):1157–1160. doi: 10.1089/AID.2011.0326. [DOI] [PubMed] [Google Scholar]
- 204.Iamarino A, de Melo FL, Braconi CT, de Andrade Zanotto PM. BF integrase genes of HIV-1 circulating in São Paulo, Brazil, with a recurrent recombination region. PLoS One. 2012;7(4) doi: 10.1371/journal.pone.0034324.e34324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Phuphuakrat A, Pasomsub E, Kiertiburanakul S, Chantratita W, Sungkanuparph S. HIV type 1 integrase polymorphisms in treatment-naive and treatment-experienced HIV type 1-infected patients in Thailand where HIV type 1 subtype A/E predominates. AIDS Research and Human Retroviruses. 2011;28(8):937–943. doi: 10.1089/AID.2011.0139. [DOI] [PubMed] [Google Scholar]
- 206.Quashie PK, Mesplede T, Han YS, et al. Characterization of the R263K mutation in HIV-1 integrase that confers low-level resistance to the second-generation integrase strand transfer inhibitor dolutegravir. Journal of Virology. 2012;86(5):2696–2705. doi: 10.1128/JVI.06591-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Melby T, Sista P, DeMasi R, et al. Characterization of envelope glycoprotein gp41 genotype and phenotypic susceptibility to enfuvirtide at baseline and on treatment in the phase III clinical trials TORO-1 and TORO-2. AIDS Research and Human Retroviruses. 2006;22(5):375–385. doi: 10.1089/aid.2006.22.375. [DOI] [PubMed] [Google Scholar]
- 208.Seclén E, del Mar González M, Lapaz M, et al. Primary resistance to maraviroc in a large set of R5-V3 viral sequences from HIV-1-infected patients. Journal of Antimicrobial Chemotherapy. 2010;65(12):2502–2504. doi: 10.1093/jac/dkq381. [DOI] [PubMed] [Google Scholar]
- 209.Aghokeng AF, Ewane L, Awazi B, et al. Enfuvirtide binding domain is highly conserved in non-B HIV type 1 strains from Cameroon, West Central Africa. AIDS Research and Human Retroviruses. 2005;21(5):430–433. doi: 10.1089/aid.2005.21.430. [DOI] [PubMed] [Google Scholar]
- 210.Poveda E, Rodes B, Toro C, Soriano V. Are fusion inhibitors active against all HIV variants? AIDS Research and Human Retroviruses. 2004;20(3):347–348. doi: 10.1089/088922204322996590. [DOI] [PubMed] [Google Scholar]
- 211.Araujo LA, Junqueira DM, de Medeiros RM, Matte MC, Almeida SE. Naturally occurring resistance mutations to HIV-1 entry inhibitors in subtypes B, C, and CRF31_BC. Journal of Clinical Virology. 2012;54(1):6–10. doi: 10.1016/j.jcv.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 212.Yu X, Yuan L, Huang Y, et al. Susceptibility of HIV-1 subtypes B′, CRF07_BC and CRF01_AE that are predominantly circulating in China to HIV-1 entry inhibitors. PLoS ONE. 2011;6(3) doi: 10.1371/journal.pone.0017605.e17605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ariyoshi K, Matsuda M, Miura H, Tateishi S, Yamada K, Sugiura W. Patterns of point mutations associated with antiretroviral drug treatment failure in CRF01_AE (subtype E) infection differ from subtype B infection. Journal of Acquired Immune Deficiency Syndromes. 2003;33(3):336–342. doi: 10.1097/00126334-200307010-00007. [DOI] [PubMed] [Google Scholar]
- 214.Wainberg MA, Zaharatos GJ, Brenner BG. Development of antiretroviral drug resistance. The New England Journal of Medicine. 2011;365(7):637–646. doi: 10.1056/NEJMra1004180. [DOI] [PubMed] [Google Scholar]
- 215.Brenner BG, Oliveira M, Doualla-Bell F, et al. HIV-1 subtype C viruses rapidly develop K65R resistance to tenofovir in cell culture. AIDS. 2006;20(9):F9–F13. doi: 10.1097/01.aids.0000232228.88511.0b. [DOI] [PubMed] [Google Scholar]
- 216.Miller MD, Margot N, McColl D, Cheng AK. K65R development among subtype C HIV-1-infected patients in tenofovir DF clinical trials. AIDS. 2007;21(2):265–266. doi: 10.1097/QAD.0b013e32801199ee. [DOI] [PubMed] [Google Scholar]
- 217.de Wit S, Boulmé R, Poll B, Schmit JC, Clumeck N. Viral load and CD4 cell response to protease inhibitor-containing regimens in subtype B versus non-B treatment-naive HIV-1 patients. AIDS. 2004;18(17):2330–2331. doi: 10.1097/00002030-200411190-00016. [DOI] [PubMed] [Google Scholar]
- 218.Frater AJ, Dunn DT, Beardall AJ, et al. Comparative response of African HIV-1-infected individuals to highly active antiretroviral therapy. AIDS. 2002;16(8):1139–1146. doi: 10.1097/00002030-200205240-00007. [DOI] [PubMed] [Google Scholar]
- 219.Pillay D, Walker AS, Gibb DM, et al. Impact of human immunodeficiency virus type 1 subtypes on virologic response and emergence of drug resistance among children in the Paediatric European Network for Treatment of AIDS (PENTA) 5 trial. Journal of Infectious Diseases. 2002;186(5):617–625. doi: 10.1086/342680. [DOI] [PubMed] [Google Scholar]
- 220.Rewari BB, Bachani D, Rajasekaran S, Deshpande A, Chan PL, Srikantiah P. Evaluating patients for second-line antiretroviral therapy in India: the role of targeted viral load testing. Journal of Acquired Immune Deficiency Syndromes. 2010;55(5):610–614. doi: 10.1097/QAI.0b013e3181f43a31. [DOI] [PubMed] [Google Scholar]
- 221.Phillips AN, Pillay D, Garnett G, et al. Effect on transmission of HIV-1 resistance of timing of implementation of viral load monitoring to determine switches from first to second-line antiretroviral regimens in resource-limited settings. AIDS. 2011;25(6):843–850. doi: 10.1097/QAD.0b013e328344037a. [DOI] [PubMed] [Google Scholar]
- 222.Nachega JB, Hislop M, Dowdy DW, Chaisson RE, Regensberg L, Maartens G. Adherence to nonnucleoside reverse transcriptase inhibitor-based HIV therapy and virologic outcomes. Annals of Internal Medicine. 2007;146(8):564–573. doi: 10.7326/0003-4819-146-8-200704170-00007. [DOI] [PubMed] [Google Scholar]
- 223.Richard N, Juntilla M, Abraha A, et al. High prevalence of antiretroviral resistance in treated Ugandans infected with non-subtype B human immunodeficiency virus type 1. AIDS Research and Human Retroviruses. 2004;20(4):355–364. doi: 10.1089/088922204323048104. [DOI] [PubMed] [Google Scholar]
- 224.Calazans A, Brindeiro R, Brindeiro P, et al. Low accumulation of L90M in protease from subtype F HIV-1 with resistance to protease inhibitors is caused by the L89M polymorphism. Journal of Infectious Diseases. 2005;191(11):1961–1970. doi: 10.1086/430002. [DOI] [PubMed] [Google Scholar]
- 225.Atlas A, Granath F, Lindström A, Lidman K, Lindbäck S, Alaeus A. Impact of HIV type 1 genetic subtype on the outcome of antiretroviral therapy. AIDS Research and Human Retroviruses. 2005;21(3):221–227. doi: 10.1089/aid.2005.21.221. [DOI] [PubMed] [Google Scholar]
- 226.Bannister WP, Ruiz L, Loveday C, et al. HIV-1 subtypes and response to combination antiretroviral therapy in Europe. Antiviral Therapy. 2006;11(6):707–715. [PubMed] [Google Scholar]
- 227.Alexander CS, Montessori V, Wynhoven B, et al. Prevalence and response to antiretroviral therapy of non-B subtypes of HIV in antiretroviral-naive individuals in British Columbia. Antiviral Therapy. 2002;7(1):31–35. [PubMed] [Google Scholar]
- 228.Camacho R. The significance of subtype-related genetic variability: controversies and unanswered questions. In: Geretti AM, editor. Antiretroviral Resistance in Clinical Practice. 2006. [PubMed] [Google Scholar]
- 229.Geretti AM, Harrison L, Green H, et al. Effect of HIV-1 subtype on virologic and immunologic response to starting highly active antiretroviral therapy. Clinical Infectious Diseases. 2009;48(9):1296–1305. doi: 10.1086/598502. [DOI] [PubMed] [Google Scholar]
- 230.Hamers RL, Schuurman R, Sigaloff KC, et al. Effect of pretreatment HIV-1 drug resistance on immunological,virological, and drug-resistance outcomes of first-line antiretroviral treatment in sub-Saharan Africa: a multicentre cohort study. The Lancet Infectious Disease. 2012;12(4):307–317. doi: 10.1016/S1473-3099(11)70255-9. [DOI] [PubMed] [Google Scholar]
- 231.Prado JG, Prendergast A, Thobakgale C, et al. Replicative capacity of human immunodeficiency virus type 1 transmitted from mother to child is associated with pediatric disease progression rate. Journal of Virology. 2010;84(1):492–502. doi: 10.1128/JVI.01743-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Solomon A, Lane N, Wightman F, Gorry PR, Lewin SR. Enhanced replicative capacity and pathogenicity of HIV-1 isolated from individuals infected with drug-resistant virus and declining CD4+ T-cell counts. Journal of Acquired Immune Deficiency Syndromes. 2005;40(2):140–148. doi: 10.1097/01.qai.0000173460.75322.93. [DOI] [PubMed] [Google Scholar]
- 233.Troyer RM, Collins KR, Abraha A, et al. Changes in human immunodeficiency virus type 1 fitness and genetic diversity during disease progression. Journal of Virology. 2005;79(14):9006–9018. doi: 10.1128/JVI.79.14.9006-9018.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Huang W, Eshleman SH, Toma J, et al. Coreceptor tropism in human immunodeficiency virus type 1 subtype D: high prevalence of CXCR4 tropism and heterogeneous composition of viral populations. Journal of Virology. 2007;81(15):7885–7893. doi: 10.1128/JVI.00218-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Kaleebu P, Nankya IL, Yirrell DL, et al. Relation between chemokine receptor use, disease stage, and HIV-1 subtypes A and D: results from a rural Ugandan cohort. Journal of Acquired Immune Deficiency Syndromes. 2007;45(1):28–33. doi: 10.1097/QAI.0b013e3180385aa0. [DOI] [PubMed] [Google Scholar]
- 236.Easterbrook PJ, Smith M, Mullen J, et al. Impact of HIV-1 viral subtype on disease progression and response to antiretroviral therapy. Journal of the International AIDS Society. 2010;13(1, article 4) doi: 10.1186/1758-2652-13-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kiwanuka N, Laeyendecker O, Quinn TC, et al. HIV-1 subtypes and differences in heterosexual HIV transmission among HIV-discordant couples in Rakai, Uganda. AIDS. 2009;23(18):2479–2484. doi: 10.1097/QAD.0b013e328330cc08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Rainwater S, DeVange S, Sagar M, et al. No evidence for rapid subtype C spread within an epidemic in which multiple subtypes and intersubtype recombinants circulate. AIDS Research and Human Retroviruses. 2005;21(12):1060–1065. doi: 10.1089/aid.2005.21.1060. [DOI] [PubMed] [Google Scholar]
- 239.Conroy SA, Laeyendecker O, Redd AD, et al. Changes in the distribution of HIV type 1 subtypes D and A in Rakai District, Uganda between 1994 and 2002. AIDS Research and Human Retroviruses. 2010;26(10):1087–1091. doi: 10.1089/aid.2010.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Ahuja SK, Kulkarni H, Catano G, et al. CCL3L1-CCR5 genotype influences durability of immune recovery during antiretroviral therapy of HIV-1-infected individuals. Nature Medicine. 2008;14(4):413–420. doi: 10.1038/nm1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Dolan MJ, Kulkarni H, Camargo JF, et al. CCL3L1 and CCR5 influence cell-mediated immunity and affect HIV-AIDS pathogenesis via viral entry-independent mechanisms. Nature Immunology. 2007;8(12):1324–1336. doi: 10.1038/ni1521. [DOI] [PubMed] [Google Scholar]
- 242.Kaur G, Mehra N. Genetic determinants of HIV-1 infection and progression to AIDS: susceptibility to HIV infection. Tissue Antigens. 2009;73(4):289–301. doi: 10.1111/j.1399-0039.2009.01220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Carrington M, O’Brien SJ. The influence of HLA genotype on AIDS. Annual Review of Medicine. 2003;54:535–551. doi: 10.1146/annurev.med.54.101601.152346. [DOI] [PubMed] [Google Scholar]
- 244.Grifoni A, Montesano C, Palma P, et al. Role of HLA-B alpha-3 domain amino acid position 194 in HIV disease progression. Molecular Immunology. 2012;53(4):410–413. doi: 10.1016/j.molimm.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 245.Naruto T, Gatanaga H, Nelson G, et al. HLA class I-mediated control of HIV-1 in the Japanese population, in which the protective HLA-B*57 and HLA-B*27 alleles are absent. Journal of Virology. 2012;86(19):10870–10872. doi: 10.1128/JVI.00689-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Deeks SG, Walker BD. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity. 2007;27(3):406–416. doi: 10.1016/j.immuni.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 247.Pereyra F, Addo MM, Kaufmann DE, et al. Genetic and immunologic heterogeneity among persons who control HIV infection in the absence of therapy. Journal of Infectious Diseases. 2008;197(4):563–571. doi: 10.1086/526786. [DOI] [PubMed] [Google Scholar]
- 248.Snoeck J, Van Laethem K, Hermans P, et al. Rising prevalence of HIV-1 non-B subtypes in Belgium: 1983–2001. Journal of Acquired Immune Deficiency Syndromes. 2004;35(3):279–285. doi: 10.1097/00126334-200403010-00009. [DOI] [PubMed] [Google Scholar]