

Abstract
Silver N-heterocyclic carbene complexes have been shown to have great potential as antimicrobial agents, affecting a wide spectrum of both Gram-positive and Gram-negative bacteria. A new series of three silver carbene complexes (SCCs) based on 4,5,6,7-tetrachlorobenzimidazole has been synthesized, characterized, and tested against a panel of clinical strains of bacteria. The imidazolium salts and their precursors were characterized by elemental analysis, mass spectrometry, 1H and 13C NMR spectroscopy, and single crystal X-ray diffraction. The silver carbene complexes, SCC32, SCC33, and SCC34 were characterized by elemental analysis, 1H and 13C NMR spectroscopy, and single crystal X-ray diffraction. These complexes proved highly efficacious with minimum inhibitory concentrations (MICs) ranging from 0.25 to 6 μg mL−1. Overall, the complexes were effective against highly resistant bacteria strains, such as methicillin-resistant Staphylococcus aureus (MRSA), weaponizable bacteria, such as Yersinia pestis, and pathogens found within the lungs of cystic fibrosis patients, such as Pseudomonas aeruginosa, Alcaligenes xylosoxidans, and Burkholderia gladioli. SCC33 and SCC34 also showed clinically relevant activity against a silver-resistant strain of Escherichia coli based on MIC testing.
Introduction
Silver has been used throughout history for its antimicrobial properties. Silver coins were once placed into containers to prevent contamination of the water, and some surgical textbooks recommended silver being included with a surgeon’s tools to treat ulcers and abscesses.1–3 Silver nitrate was used in the 19th century to promote wound healing on burn victims.4 This practice continued with Fox’s advent of silver sulfadiazine in 1965, which is still used in burn wards worldwide.5,6 Recently, silver has been used in several different applications. Catheters, surgical instruments, and surgical masks have been embedded with silver to prevent nosocomial infections.7–9 Sutures and wound dressings have also been embedded with silver, allowing for a 50–60% reduction in infections.10,11 Silver itself has no known use within the body and is rapidly excreted, making it attractive as an internalized therapeutic agent.2
Widespread use of silver has led to bacterial resistance to silver.12 This has been observed in burn wards using silver salts and in hospitals that utilize silver-coated catheters.13,14 Expression of resistance is erratic in its occurrence; however several different clinical strains have been isolated.15 The plasmid pMG101 has been shown to bestow resistance to several heavy metals including the silver ion, and when introduced into E. coli allows for growth in >0.6 mM solution of silver ions.16,17 In addition, the plasmid confers resistance to several organic-based antibiotics, including ampicillin, chloramphenicol, tetracycline, streptomycin, and sulphonamides, making it the standard model for antibacterial resistance.18
N-Heterocyclic carbene (NHC) studies first began with the metallation of imidazol-2-ylidenes by Wanzlick and Öfele in 1968.19,20 Stemming from this research, Arduengo discovered and isolated the first stable free carbene in 1991, inspiring a plethora of research.21 Since then, NHCs have proven to be versatile ligands in both transition metal and main group coordination chemistry.22,23 The first silver carbene complex was synthesized by Arduengo in 1993 for use in catalysis.24 Arduengo’s method required harsh conditions and often led to decomposition of the metal complex. To remedy this problem, Wang and Lin developed a synthetic method that generated the carbene in situ by using a silver salt as a base.22 Silver carbene complexes (SCCs) are currently being explored due to their high efficacy against antibiotic-resistant bacteria, biosafety level 3 bacteria, and various types of cancer.25–29
Two main traits are prevalent in the previous studies of the SCCs. Modification of the substituents on the ligand can improve the efficacy of the drugs by aiding in drug transport across the cell membrane, increasing the viable dose, or enabling targeting of the drug.30 Stability of the SCCs has been improved by the use of electron withdrawing groups at the 4 and 5 positions of the imidazolin-2-ylidene ligand.27 Following these trends, we have synthesized three new silver NHC complexes expanding upon these two traits. Concurrent with our research, Özdemir and Tacke have used benzimidazole to form antimicrobial NHC complexes with silver.31,32 The benzene ring of benzimidazole allows for the addition of two more of the sigma-withdrawing pi-donating chlorine atoms than imidazole; increasing the stability of the NHCs. Herein, we report the synthesis of three SCCs based upon 4,5,6,7-tetrachlorobenzimidazole. These three SCCs have been successfully synthesized, characterized, and tested for their antimicrobial activity.
Results and discussion
Synthesis of new SCCs
4,5,6,7-Tetrachlorobenzimidazole (1) was synthesized based on a literature procedure.33 Benzimidazole was dissolved in a mixture of nitric and hydrochloric acids and allowed to reflux for 36 h to produce 1. Synthesis of (1,3-dimethyl-4,5,6,7-tetrachlorobenzimidazole-2-ylidene)silver(I) acetate (SCC32) was performed as shown in Scheme 1. The imidazolium salt was prepared via methylation of 1 with excess iodomethane to give 1,3-dimethyl-4,5,6,7-tetrachlorobenzimidazole iodide (2).28 The reaction of 2 with silver acetate gave the water-insoluble SCC32 in a 76% yield. (1-Hydroxyethyl-3-methyl-4,5,6,7-tetrachlorobenzimidazole-2-ylidene)silver(I) acetate (SCC33) was synthesized in a similar fashion (Scheme 2). Deprotonation of 1 with potassium hydroxide followed by the addition of 2-iodoethanol yielded 1-Hydroxyethyl-4,5,6,7-tetrachlorobenzimidazole (3). Methylation of 3 with excess iodomethane and subsequent reaction with two equivalents of silver acetate gave SCC33 in a 90% yield (Scheme 2). This addition of the hydroxyethyl group allowed for increased water solubility. Synthesis of the lipophilic compound (1-methylnapthyl-3-methyl-4,5,6,7-tetrachlorobenzimidazole-2-ylidene)silver(I) acetate (SCC34) is illustrated in Scheme 3. Compound 1 was deprotonated with potassium carbonate and reacted with one equivalent of 2-(bromomethyl) naphthalene to produce 1-methylnaphthyl-4,5,6,7-tetrachlorobenzimidazole (5). Compound 5 was methylated with methyl trifluoromethanesulfonate under an inert atmosphere and combined with tetra-n-butylammonium iodide to form 6. This was subsequently reacted with two equivalents of silver acetate to form SCC34 in a 37% yield.
Scheme 1.

Synthesis of SCC32.
Scheme 2.

Synthesis of SCC33.
Scheme 3.

Synthesis of SCC34.
For all compounds, 1H and 13C NMR spectroscopy was used to confirm the formation of the imidazolium cation and the SCC. The loss of the resonance of the imidazolium cation proton from the 1H NMR spectra of 2, 4, and 6 (occurring at 9.89, 9.94, and 9.46 ppm, respectively) was the most informative change in the spectra of all three silver complexes. The most informative change in the 13C NMR spectra was a shift of the imidazolium cation carbon resonance from around 148 ppm to around 190 ppm, signifying the formation of the silver complexes. All silver complexes were also verified by single crystal X-ray diffraction. Single crystals of some of the SCC precursors were obtained and the X-ray crystallographic data is available in the ESI.†
Single crystals of SCC32, SCC33, and SCC34 suitable for X-ray diffraction were obtained from the slow evaporation of a concentrated sample in chloroform, dry methylene chloride, and methanol, respectively (Fig. 1–3). The asymmetric unit of these contain one SCC. Additionally, the asymmetric unit of SCC33 contains a disordered solvent molecule, which forms a network of weak hydrogen bonding with three molecules of SCC33. The asymmetric unit of SCC34 also contains a solvent molecule; however the solvent interacts with only two molecules of SCC34.
Fig. 1.

Thermal ellipsoid plot of SCC32 with thermal ellipsoids drawn at 50% probability. Hydrogen atoms were removed for clarity.
Fig. 3.

Thermal ellipsoid plot of SCC34 with thermal ellipsoids drawn at 50% probability. Hydrogen atoms and solvent methanol molecule were removed for clarity.
All bond lengths and angles between the metal center to the carbene carbon and oxygen atoms of the acetate are consistent with previously reported silver carbene complexes.25,26 Both SCC33 and SCC34 show short Ag–Ag contacts (3.25 Å and 3.26 Å, respectively), however this is not observed in SCC32. Additionally, while the metal center–acetate bond length of SCC32 was consistent with previously reported structures, it was found to be longer than that of SCC33 and SCC34 (2.155 Å compared to 2.104 Å and 2.122 Å). The lack of the Ag–Ag contact appears to be the reason for this increase in bond length.
In vitro efficacy determination of new SCCs
The minimum inhibitory concentra tion (MIC) and minimum bactericidal concentration (MBC) of SCC32, SCC33, and SCC34 against strains of Pseudomonas aeruginosa, Alcaligenes xylosoxidans, Stenotrophomonas maltophilia, Staphylococcus aureus, Yersinia pestis, Burkholderia gladioli, Burkholderia multivorans, and Escherichia coli species were determined by broth microdilution (Table 1). The MIC90 of SCC32, SCC33, and SCC34 were 1 μg mL−1, 2 μg mL−1, and 4 μg mL−1, respectively, for non-silver-resistant organisms tested. The MIC for the J53 strain of E. coli lacking the silver-resistant plasmid was 0.5 μg mL−1 for both SCC32 and SCC33 and 2 μg mL−1 for SCC34. In contrast, the MIC of SCC32, SCC33, and SCC34 for J53 containing the silver-resistant plasmid, pMG101, was >20, 8, and 10 μg mL−1, respectively. Therefore, the antimicrobial activity of the SCCs can be primarily attributed to the silver functionality. Following the determination of the MICs, the minimum bactericidal concentrations for each of the SCCs against the panel of bacteria was determined (Table 1). With the exception of the silver-resistant E. coli strain, all three SCCs appeared to be bactericidal for all of the strains tested. The bactericidal concentrations for all three SCCs were observed to be ≤6 μg mL−1, thus indicating that the SCCs are capable of killing numerous bacterial strains at clinically achievable concentrations. These compounds show comparable or enhanced activity when compared to previously reported silver complexes tested against the same clinical strains.26,28,34 Interestingly, the growth of the silver-resistant E. coli J53+pMG101 strain was found to be inhibited by SCC33 and SCC34, but not by SCC32, thus indicating that the substituents present on the 4,5,6,7-tetrachlorobenzimidazole carrier molecule may play an important synergistic role and may contribute to the antimicrobial activity of the therapeutic molecule. Furthermore, SCC34 was found to be bactericidal against the same strain. Subsequent testing showed the imidazolium cation precursors had no activity against the E. coli J53+pMG101 strain, though 6 showed some activity against the two MRSA strains. The primary structural difference between the two SCCs is the presence of the methylnaphthyl substituent on the carrier molecule for SCC34 as compared to the hydroxyethyl substituent in the case of SCC33. These results point towards structure-dependent antimicrobial efficacy of the SCCs. Furthermore, the results of SCC34 against the J53+pMG101 strain compare favorably to those of SCC23 (Fig. 4), currently the most effective SCC against the J53+pMG101 strain (with MIC values of 10 and 6 μg mL−1 respectively).34 This suggests that the efficacy against the silver-resistant J53+pMG101 strain is a result of the presence of methylnaphthyl substituents on the imidazolium carriers.
Table 1.
MIC/MBC of tetrachlorobenzimidazole-based SCCs against clinical isolate bacterial strains
| Bacterial species isolate | SCC32 (μg mL−1)
|
SCC33 (μg mL−1)
|
SCC34 (μg mL−1)
|
|||
|---|---|---|---|---|---|---|
| MIC | MBC | MIC | MBC | MIC | MBC | |
| Pseudomonas aeruginosa | ||||||
| PA 01-V | 0.5 | 6 | 2 | 2 | 2 | 4 |
| PA M57-15 | 1 | 2 | 1 | 4 | 0.5 | 2 |
| PA RR05 | 0.5 | 1 | 0.5 | 1 | 0.5 | 2 |
| PA HP3 | 0.25 | 1 | 2 | 6 | 0.5 | 6 |
| PA LF05 | 0.5 | 2 | 0.5 | 1 | 1 | 2 |
| Alcaligenes xylosoxidans | ||||||
| AX 22 | 0.5 | 2 | 0.5 | 2 | 1 | 4 |
| AX RE05 | 0.25 | 0.5 | 0.5 | 1 | 1 | 1 |
| Stenotrophomonas maltophilia | ||||||
| SM AH08 | 0.5 | 1 | 0.5 | 6 | 4 | 6 |
| Methicillin-resistant Staphylococcus aureus (MRSA) | ||||||
| SA LL06 | 1 | 6 | 1 | 2 | 4 | 6 |
| SA EH06 | 1 | 6 | 1 | 4 | 2 | 6 |
| Yersinia pestis | ||||||
| YP1-CO92-LCR- | 1 | 1 | 0.5 | 1 | 1 | 1 |
| Burkholderia gladioli | ||||||
| BG 80 | 1 | 4 | 2 | 4 | 1 | 2 |
| Burkholderia multivorans | ||||||
| BM 54 | 0.25 | 0.5 | 2 | 4 | 4 | 6 |
| Escherichia coli | ||||||
| J53 | 0.5 | 1 | 0.5 | 1 | 2 | 2 |
| J53+pMG101 | >20 | >20 | 8 | >20 | 10 | 15 |
Fig. 4.

Schematic representation of SCC23.
Experimental
General considerations
All reactions were carried out under aerobic conditions unless otherwise specified. Benzimidazole, 2-iodoethanol, iodomethane, methyl trifluoromethanesulfonate, tetra-n-butylammonium iodide, and silver acetate were purchased from Alfa Aesar. 2-(Bromomethyl)naphthalene was purchased from Waterstone Technologies. All solvents were purchased from Fisher Scientific. All reagents were used as received without further purification. All solvents were used as received with the exception of the dry methylene chloride used in the synthesis of 6, which was collected using a PurSolv solvent purification system by Innovative Technologies. Melting points were obtained on a MelTemp apparatus. 1H and 13C NMR spectra were collected on a Varian 500 MHz instrument with all spectra referenced to deuterated solvents. Mass spectrometry was performed by the University of Akron mass spectrometry lab. Elemental Analysis was performed by the University of Illinois at Urbana-Champaign. FTIR data was collected on a Thermo Scientific Nicolet iS5 Infrared Spectrometer. Crystals of the compounds were coated in paratone oil, mounted on a CryoLoop and placed on a goniometer under a stream of nitrogen. Crystal structure data sets were collected on a Bruker APEX CCD diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) or a Bruker APEX II Duo CCD system equipped with a Cu ImuS micro-focus source (λ = 1.54178 Å). The unit cell was determined by using reflections from three different orientations. The data was integrated using SAINT.35,36 An empirical absorption correction and other corrections were applied to the data using multi-scan SADABS.35 Structure solution, refinement, and modelling were accomplished by using the Bruker SHELXTL package.36,37 The structure was determined by full-matrix least-squares refinement of F2 and the selection of the appropriate atoms from the generated difference map. Hydrogen atom positions were calculated and Uiso(H) values were fixed according to a riding model.
Synthesis of 4,5,6,7-tetrachlorobenzimidazole (1)
Benzimidazole (5.0 g, 42 mmol) was dissolved into 1.2 L of concentrated hydrochloric acid. Concentrated nitric acid (600 mL) was added and the solution was refluxed for 36 h. After cooling to room temperature, a light yellow precipitate formed. This precipitate was collected and recrystallized from cold ethanol twice for purification, leaving 9.6 g of a white powder (90% yield). Mp: 321–324 °C. Found: C, 32.9; H, 0.5; N, 10.6 Calc. for C7H2Cl4N2: C, 32.85; H, 0.8; N, 10.95%. FTIR (KBr, cm−1) ν(CN) 1249, ν(CCl) 620. 1H NMR (500 MHz, DMSO-d6) 7.79 (1H, s, NH) 13C{1H} NMR (125 MHz, DMSO-d6) 156.2 (NCHN), 143.5 (Ar–Cl), 118.6 (Ar–Cl), 117.6 (Ar). MS: m/z = 256.8 (theor. for C7H3Cl4N2+ = 256.905).
Crystals suitable for single crystal X-ray diffraction were grown from a concentrated ethanol solution. Crystal data for 1: C7H2Cl4N2, Mr = 255.91, triclinic, a = 7.4984(19) Å, b = 15.060(4) Å, c = 16.612(4) Å, α = 108.611(4)°, β = 94.721(4)°, γ = 90.265(4)°, V = 1770.9(8) Å3, T = 100(2) K, space group P1̄, Z = 8, μ(Mo Kα) = 1.280 mm−1, 13 921 reflections measured, 7025 independent reflections (Rint = 0.0275). The final R1 values were 0.0377 (I > 2σ(I)). The final wR(F2) values were 0.1055 (I > 2σ(I)). The final R1 values were 0.0449 (all data). The final wR(F2) values were 0.1227 (all data). The goodness of fit on F2 was 1.084.
Synthesis of 1,3-dimethyl-4,5,6,7-tetrachlorobenzimidazolium iodide (2)
Compound 1 (1 g, 4 mmol) was dissolved into 15 mL of DMF and potassium carbonate (1.1 g, 7.7 mmol) was added. The mixture was allowed to stir for 30 min to ensure deprotonation, and the excess base was filtered off. Iodomethane (4.0 mL, 43 mmol) was added and the reaction was heated at 80 °C overnight. The volatile components were removed via rotary evaporation and the resulting solid was washed with water to yield a yellow solid (0.70 g, 41% yield). Mp: 242–244 °C. Found: C, 26.3; H, 1.5; N, 6.5 Calc. for C9H7Cl4N2I1: C, 26.2; H, 1.7; N, 6.8%. FTIR (KBr, cm−1) ν(CN) 1270, ν(CCl) 666. 1H NMR (500 MHz, DMSO-d6) 9.89 (1H, s, NCHN), 4.28 (6H, s, CH3). 13C{1H} NMR (125 MHz, DMSO-d6) 148.6 (NCN), 130.9 (Ar–Cl), 129.6 (Ar–Cl), 119.1 (Ar), 37.7 (CH3). MS: m/z = 284.8 (theor. for C9H7Cl4N2+ = 284.9).
Synthesis of (1,3-dimethyl-4,5,6,7-tetrachlorobenzimidazole-2-ylidene)silver(I) acetate (SCC32)
Compound 2 (0.61 g, 1.5 mmol) was dissolved into 40 mL of methylene chloride. Silver acetate (0.49 g, 3.0 mmol) was added, and the reaction was stirred for 30 min. A yellow precipitate, presumed to be silver iodide, was filtered off, and the solvent was removed via rotary evaporation to give an off-white product (0.51 g, 76% yield). Mp: 253–256 °C. Found: C, 29.2; H, 1.8; N, 6.1 Calc. for C9H7Cl4N2I1: C, 26.2; H, 1.7; N, 6.8%. FTIR (KBr, cm−1) ν(C=O) 17.19, ν(CN) 1272, ν(CCl) 616. 1H NMR (500 MHz, DMSO-d6) 4.31 (6H, s, NCH3), 1.81 (3H, s, CH3). 13C{1H} NMR (125 MHz-DMSO-d6) 179.2 (NCN), 172.6 (COO), 135.2 (Ar–Cl), 131.5 (Ar–Cl), 116.8 (Ar–Cl), 21.5 (CH3). Crystals suitable for single crystal X-ray diffraction were grown from a concentrated chloroform solution.
Crystal data for SCC32: C11H9Cl4N2O2Ag1, Mr = 450.87, monoclinic, a = 8.404(2) Å, b = 5.5910(16) Å, c = 15.250(5) Å, β = 101.844(4)°, V = 701.3(4) Å3, T = 100(2) K, space group P2 (1), Z = 2, μ(Mo Kα) = 2.198 mm−1, 5411 reflections measured, 2767 independent reflections (Rint = 0.0427). The final R1 values were 0.0367 (I > 2σ(I)). The final wR(F2) values were 0.0809 (I > 2σ(I)). The final R1 values were 0.0415 (all data). The final wR(F2) values were 0.0833 (all data). The flack parameter was 0.02(4). The goodness of fit on F2 was 0.999.
Synthesis of 1-hydroxyethyl-4,5,6,7-tetrachlorobenzimidazole (3)
Compound 1 (1.7 g, 6.5 mmol) was placed in a Chemglass screw cap pressure tube and dissolved in 10 mL of THF. Potassium hydroxide (0.44 g, 7.8 mmol) was added and the mixture was heated to 90 °C for 30 min. 2-Iodoethanol (2 mL, 25.6 mmol) was added, the pressure tube was closed, and the reaction was heated at 90 °C for 72 h. The reaction was cooled and a white precipitate, presumed to be potassium iodide, was filtered off and the remaining solvent was removed via rotary evaporation. The resulting solid was washed with cold ethanol and then benzene to produce a white powder (1.46 g, 75% yield). Mp: 254–255 °C. Found: C, 35.9; H, 1.7; N, 8.9 Calc. for C9H6Cl4N2O1: C, 36.0; H, 2.0; N, 9.3%. FTIR (KBr, cm−1) ν(OH) 3294, ν(CN) 1307 and 1064, ν(CCl) 671. 1H NMR (500 MHz, DMSO-d6) 8.40 (1H, s, NCHN), 4.98 (1H, t, OH), 4.55 (2H, t, CH2), 3.75 (2H, td, CH2). 13C{1H} NMR (125 MHz, DMSO-d6) 149.4 (NCHN), 141.3 (Ar–Cl), 129.7 (Ar–Cl), 125.4 (Ar–Cl), 124.1 (Ar–Cl), 122.4 (Ar), 114.9 (Ar), 60.3 (CH2), 48.8 (CH2). MS: m/z = 300.9 (theor. for C9H6Cl4N2O1+ = 299.97).
Synthesis of 1-hydroxyethyl-3-methyl-4,5,6,7-tetrachlorobenzimidazolium iodide (4)
Compound 3 (1.0 g, 3.6 mmol) was dissolved in 17 mL of DMF. Iodomethane (1.00 mL, 10.8 mmol) was added and the solution was heated at 90 °C for 20 h. The solution was then cooled and a light yellow precipitate was collected via filtration and then immediately washed with cold ethanol to give a light yellow powder (1.59 g, 62% yield). Mp: 218–220 °C. Found: C, 27.1; H, 1.9; N, 6.1 Calc. for C10H9Cl4N2O1I1: C, 27.2; H, 2.05; N, 6.3%. FTIR (KBr, cm−1) ν(OH) 3304, ν(CN) 1305 and 1075, ν(CCl) 673. 1H NMR (500 MHz, DMSO-d6) 9.94 (1H, s, NCHN), 5.17 (1H, t, OH), 4.79 (2H, t, CH2), 4.33 (3H, s, CH3), 3.85 (2H, t, CH2). 13C {1H} NMR (125 MHz, DMSO-d6) 148.1 (NCN), 130.7 (Ar–Cl), 130.5 (Ar–Cl), 129.5 (Ar–Cl), 128.5 (Ar–Cl), 118.7 (Ar), 118.2 (Ar), 58.9 (CH2), 52.1 (CH2), 37.3 (CH3). MS: m/z = 312.9 (theor. for C7H3Cl4N2+ = 312.9).
Crystals suitable for X-ray diffraction were grown from a concentrated chloroform solution. Crystal data for 4: C10H9Cl4N2O1I1, Mr = 441.89, monoclinic, a = 9.2405(9) Å, b = 6.9153(7) Å, c = 22.244(2) Å, β = 99.331(2)°, V = 1402.6(2) Å3, T = 100(2) K, space group P2(1)/c, Z = 4, μ(Mo Kα) = 3.033 mm−1, 11 737 reflections measured, 3346 independent reflections (Rint = 0.0311). The final R1 values were 0.0257 (I > 2σ(I)). The final wR(F2) values were 0.0630 (I > 2σ(I)). The final R1 values were 0.0276 (all data). The final wR(F2) values were 0.0639 (all data). The goodness of fit on F2 was 1.087.
Synthesis of (1-hydroxyethyl-3-methyl-4,5,6,7-tetrachlorobenzimidazole-2-ylidene) silver(I) acetate (SCC33)
Compound 4 (0.16 g, 0.37 mmol) was dissolved in hot ethanol. The solution was cooled to room temperature, silver acetate (0.13 g, 0.75 mmol) was added and the reaction was stirred for 1 h. A yellow precipitate, presumed to be silver iodide, was filtered off and the volatiles were removed via rotary evaporation to give a white solid (0.16 g, 90% yield). Mp: 210–212 °C. Found: C, 30.3; H, 2.1; N, 5.7 Calc. for C12H11Cl4N2O3Ag1: C, 30.0; H, 2.3; N, 5.8%. FTIR (KBr, cm−1) ν(C=O) 1707, ν(CN) 1324 and 1081, ν(CCl) 679. 1H NMR (500 MHz, DMSO-d6) 8.40 (1H, s, NCHN), 4.98 (1H, t, OH), 4.55 (2H, t, CH2), 3.75 (2H, dt, CH2). 13C{1H} NMR (125 MHz, DMSO-d6) 197.0 (NCN), 131.6 (Ar–Cl), 130.9 (Ar–Cl), 128.1 (Ar–Cl), 127.8 (Ar–Cl), 116.8 (Ar), 116.2 (Ar), 60.9 (CH2), 53.3 (CH2), 23.3 (CH3).
Crystals suitable for single crystal X-ray diffraction were grown from a concentrated dry methylene chloride solution. Crystal data for SCC33: C13H13Cl6N2O3Ag1, Mr = 565.82, monoclinic, a = 8.616(3) Å, b = 13.028(5) Å, c = 17.176(6) Å, β = 92.684(5)°, V = 1925.9(12) Å3, T = 100(2) K, space group P2(1)/c, Z = 4, μ(Mo Kα) = 1.951 mm−1, 14 692 reflections measured, 3909 independent reflections (Rint = 0.0908). The final R1 values were 0.0528 (I > 2σ(I)). The final wR(F2) values were 0.1296 (I > 2σ(I)). The final R1 values were 0.0655 (all data). The final wR(F2) values were 0.1389 (all data). The goodness of fit on F2 was 1.015.
Synthesis of 1-methylnaphthyl-4,5,6,7-tetrachlorobenzimidazole (5)
Compound 1 (0.5 g, 2 mmol) was dissolved into 3 mL of DMF. Potassium carbonate (0.30 g, 2.2 mmol) was added and the mixture was stirred for 30 min to ensure deprotonation. 2-(Bromomethyl)naphthalene (0.44 g, 2.0 mmol) was then added and the reaction was heated at 80 °C for 1 h. The precipitate was then filtered off and the volatiles were removed via rotary evaporation. The resulting yellow solid was washed with methanol to yield a white powder (0.43 g, 55% yield). Mp: 175–177 °C. Found: C, 54.4; H, 2.3; N, 7.0 Calc. for C18H10Cl4N2: C, 54.6; H, 2.5; N, 7.1%. FTIR (KBr, cm−1) ν(CN) 1307 and 1104, ν(CCl) 665. 1H NMR (500 MHz, DMSO-d6) 8.78 (1H, s, NCHN), 7.90 (3H, m, Ar), 7.50 (2H, m, Ar), 7.47 (1H, d, Ar), 7.33 (1H, d, Ar), 5.97 (2H, s, CH2). 13C{1H} NMR (125 MHz, DMSO-d6) 149.8 (NCN), 141.9 (Ar–Cl), 135.5 (Ar–Cl), 133.3 (Ar–Cl), 132.8 (Ar–Cl), 130.2 (Ar), 128.9 (Ar), 128.2 (Ar), 127.9 (Ar), 126.8 (Ar), 126.6 (Ar), 126.3 (Ar), 125.2 (Ar), 125.1 (Ar), 124.7 (Ar), 123.2 (NC), 115.5 (NC), 49.9 (CH2).
Crystals suitable for single crystal X-ray diffraction were grown from a concentrated chloroform solution. Crystal data for 5: C18H10Cl4N2, Mr = 396.08, triclinic, a = 4.979(5) Å, b = 12.523(13) Å, c = 13.184(13) Å, α = 84.139(16)°, β = 87.926 (16)°, γ = 83.410(16)°, V = 812.1(14) Å3, T = 100(2) K, space group P1̄, Z = 2, μ(Mo Kα) = 0.730 mm−1, 6080 reflections measured, 3170 independent reflections (Rint = 0.0531). The final R1 values were 0.0596 (I > 2σ(I)). The final wR(F2) values were 0.1484 (I > 2σ(I)). The final R1 values were 0.0830 (all data). The final wR(F2) values were 0.1622 (all data). The goodness of fit on F2 was 0.937.
Synthesis of 1-methylnapthyl-3-methyl-4,5,6,7-tetrachlorobenzimidazolium iodide (6)
Synthesis of 6 was conducted under anaerobic conditions. Compound 5 (0.77 g, 2.0 mmol) was dissolved into 30 mL of dry methylene chloride combined with methyl trifluoromethanesulfonate (0.25 mL, 2.3 mmol). The resulting mixture was refluxed for 20 h, during which time a precipitate formed. The precipitate was collected, washed with benzene, and redissolved in methanol that contained tetra-n-butylammonium iodide (0.59 g, 1.5 mmol). A white solid that precipitated out was then collected and washed with cold ethanol to yield a white powder (0.54 g, 50% yield). Mp: 187–189 °C. Found: C, 42.4; H, 2.2; N, 5.0 Calc. for C19H13Cl4N2I1: C, 42.4; H, 2.4; N, 5.2%. FTIR (KBr, cm−1) ν(CN) 1305 and 1104, ν(CCl) 669. 1H NMR (500 MHz, DMSO-d6) 9.46 (1H, s, NCHN), 7.98 (4H, m, Ar), 7.57 (3H, m, Ar), 5.66 (2H, s, NCH2), 3.84 (3H, s, NCH3). 13C{1H} NMR (125 MHz, DMSO-d6) 148.3 (NCN), 132.7 (Ar–Cl), 132.6 (Ar–Cl), 131.8 (Ar–Cl), 130.8 (Ar–Cl), 129.8 (Ar), 128.6 (Ar), 128.5 (Ar), 127.7 (Ar), 127.6 (Ar), 126.6 (Ar), 125.9 (Ar), 124.7 (Ar), 52.2 (CH2), 37.7 (CH3). MS: m/z = 408.8 (theor. for C19H13Cl4N2+ = 408.98).
Crystals suitable for single crystal X-ray diffraction were grown from a concentrated solution of chloroform. Crystal data for 6: C19H13Cl4N2I1, Mr = 538.01, monoclinic, a = 24.500(3) Å, b = 12.9443(12) Å, c = 14.1288(13) Å, β = 117.003(2)°, V = 3992.3(7) Å3, T = 100(2) K, space group C2/c, Z = 8, μ(Mo Kα) = 2.146 mm−1, 17 009 reflections measured, 4569 independent reflections (Rint = 0.0396). The final R1 values were 0.0330 (I > 2σ(I)). The final wR(F2) values were 0.0798 (I > 2σ(I)). The final R1 values were 0.0404 (all data). The final wR(F2) values were 0.0850 (all data). The goodness of fit on F2 was 1.048.
Synthesis of (1-methylnapthyl-3-methyl-4,5,6,7-tetrachlorobenzimidazole-2-ylidene) silver(I) acetate (SCC34)
Compound 6 (0.54 g, 1.0 mmol) was dissolved into 40 mL of hot methanol and silver acetate (0.33 g, 2.0 mmol) was added. The reaction was stirred for 1 h at room temperature. A yellow solid, presumed to be silver iodide, was filtered off and the volatiles were removed via rotary evaporation to obtain a white powder (0.21 g, 37% yield). Mp: 197–198 °C. Found: C, 43.75; H, 3.0; N, 4.7 Calc. for C21H15Cl4N2O2Ag1: C, 43.7; H, 2.6; N, 4.85%. FTIR (KBr, cm−1) ν(C=O) 1714, ν(CN) 1272 and 1033, ν(CCl) 671. 1H NMR (500 MHz, DMSO-d6) 7.79 (4H, m, Ar), 7.34 (3H, m, Ar), 6.191 (2H, s, CH2), 4.38 (3H, s, CH3), 1.78 (3H, s, CH3). 13C{1H} (125 MHz, DMSO-d6) 197.5 (NCN), 175.6 (COO), 134.8 (Ar–Cl), 132.8 (Ar–Cl), 132.3 (Ar–Cl), 131.9 (Ar–Cl), 130.8 (Ar), 128.4 (Ar), 128.3 (Ar), 128.3 (Ar), 128.2 (Ar), 128.1 (Ar), 127.7 (Ar), 127.5 (Ar), 54.0 (CH2), 23.3 (CH3), 15.1 (CH3).
Crystals suitable for single crystal X-ray diffraction were grown from a concentrated methanol solution. Crystal data for SCC34: C22H19Cl4N2O3Ag1, Mr = 609.06, triclinic, a = 10.1480 (4) Å, b = 11.5407(4) Å, c = 12.1076(5) Å, α = 106.806(1)°, β = 105.315(1)°, γ = 111.769(1)°, V = 1145.37(8) Å3, T = 100(2) K, space group P1̄, Z = 2, μ(Cu Kα) = 11.596 mm−1, 7216 reflections measured, 3486 independent reflections (Rint = 0.0227). The final R1 values were 0.0239 (I > 2σ(I)). The final wR(F2) values were 0.0662 (I > 2σ(I)). The final R1 values were 0.0243 (all data). The final wR(F2) values were 0.0667 (all data). The goodness of fit on F2 was 1.323.
Bacteria
The laboratory strain PAO1-V was provided by Dr Maynard Olson (University of Washington, Seattle). The mucoid clinical isolate of P. aeruginosa PA M57-15 was provided by Dr Thomas Ferkol (Washington University, St. Louis, MO). The PA HP3, PA RR05, PA LF05, AX RE05, AX 22, SM AH08, SA EH06, SA LL06, YP1-1CO92-LCR-, BG 80, and BM 54 strains were cultured from the sputum of cystic fibrosis patients at St. Louis Children’s Hospital. The silver-sensitive and silver-resistant E. coli strains, J53 and J53+pMG101 respectively, were provided by Dr Simon Silver (University of Chicago, Chicago, IL). The J53 strain is known to be sensitive to killing by silver cations and serves as a positive control. In contrast, the J53+pMG101 strain is a J53 derivative that harbors the pMG101 plasmid originally conferring silver resistance to a burn ward isolate of Salmonella and serves as a negative control. All bacterial strains were maintained as glycerol stocks at −80 °C.
In vitro antimicrobial activity
Minimum inhibitory concentrations were determined by broth microdilution method as previously described by a standard Clinical and Laboratory Standards Institute (CLSI) protocol.38 Briefly, bacteria were streaked from frozen glycerol stocks onto TSA or blood agar plates and incubated overnight at 37 °C. Colonies from the fresh plates were suspended in the CLSI standard M–H broth to an optical density at 650 nm (OD650) of 0.2 and grown at 37 °C in a shaking incubator at 200 rpm to an OD650 of 0.4, which corresponds to ~5 × 108 colony forming units (CFU) mL−1. The bacteria were diluted in the broth to a concentration of 105 in 100 μL, which was added to triplicate wells of a 96-well plate, containing 100 μL of SCC32, SCC33 or SCC34 solution at various concentrations. The dilutions for SCCs were prepared from 1 mg mL−1 stocks in DMSO with the final solution composition being 95% sterile water and 5% DMSO by volume. Solutions containing 95% sterile water and 5% DMSO but no SCCs were used as the appropriate control. The final concentrations tested were 0.125, 0.25, 0.5, 1, 2, 4, 6, 8, 10, 15, and 20 μg mL−1. The plates were incubated overnight at 37 °C. The MIC was the lowest of these concentrations, at which each of the triplicate wells in each 96-well plate was clear after 16–24 h incubation. Each triplicate measurement was performed at least in duplicate for a minimum of six separate measurements. The MBC of various SCCs was determined by plating the wells with growth inhibition on TSA or blood agar plates and noting the lowest concentration that resulted in no growth after an overnight incubation at 37 °C.
Conclusions
The data presented show a large increase in activity against bacteria in comparison to previously reported benzimidazole-based silver carbene complexes. Like previously reported SCCs that contain electron withdrawing groups, all new SCCs showed single-digit μg mL−1 concentration MICs in vitro. The addition of a hydroxyethyl substituent allowed for relative water solubility, providing a method of treatment that does not require any carrier for the drug. SCC33 inhibited growth of the silver-resistant J53+pMG101 E. coli strain, while SCC34 showed bactericidal activity against silver-resistant bacteria. This demonstrates the ability of the imidazolium cation carrier to enhance the antimicrobial activity of the silver cation.
Supplementary Material
Fig. 2.
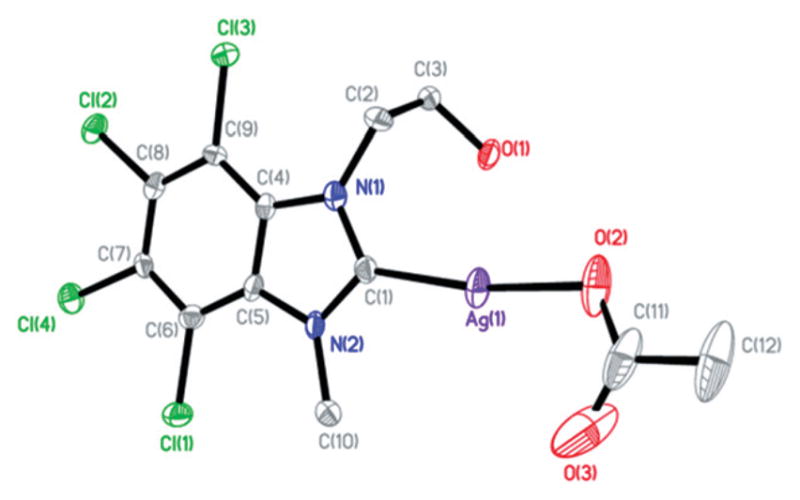
Thermal ellipsoid plot of SCC33 with thermal ellipsoids drawn at 50% probability. Hydrogen atoms and solvent methylene chloride molecule were removed for clarity.
Acknowledgments
We wish to thank The University of Akron, the National Institute of Health-National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK0825), National Institute of General Medical Sciences (R01 GM86895-01), National Heart Lung and Blood Institute (NHLBI-PEN HHSN268201000046C), and the National Institutes of Allergies and Infectious Diseases for financial support. We also thank The Ohio Board of Regents and The National Science Foundation (CHE-9977144, CHE-0341701 and DMR-0414599) for funds used to purchase the NMR instruments used in this work. We additionally thank the NSF (CHE-0840446, CHE-0116041) for the funds used to purchase the Bruker Apex II Duo Imus Mo/Cu Kappa and the Apex CCD X-ray diffractometers. We would also like to thank labmates and the staff of the University of Akron Magnetic Resonance Center (especially V. Dudipala and S. Stakleff), without whose help this work would not have been possible.
Footnotes
Electronic supplementary information (ESI) available. CCDC 845904–845910. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2dt00055e
Contributor Information
Carolyn L. Cannon, Email: carolyn.cannon@utsouthwestern.edu.
Wiley J. Youngs, Email: youngs@uakron.edu.
Notes and references
- 1.Davies RL, Etris SF. Catal Today. 1997;36:107. [Google Scholar]
- 2.Melaiye A, Youngs WJ. Expert Opin Ther Pat. 2005;15(2):125. [Google Scholar]
- 3.Chen X, Schluesener HJ. Toxicol Lett. 2008;176:1. doi: 10.1016/j.toxlet.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Klasen HJ. Burns. 2000;26:117. doi: 10.1016/s0305-4179(99)00108-4. [DOI] [PubMed] [Google Scholar]
- 5.Fox CL. Arch Surg. 1968;96:184. doi: 10.1001/archsurg.1968.01330200022004. [DOI] [PubMed] [Google Scholar]
- 6.Fox CL., Jr 3,761,590. US Pat. 1970 May 18;
- 7.Foss SW. 6,946,196. US Pat. 2005 Sep 20;
- 8.Son WK, Youk JH, Lee TS, Park WH. Macromol Rapid Commun. 2004;25:1632. [Google Scholar]
- 9.Xing Y, Xiojun Y, Dai J. J Sol-Gel Sci Technol. 2007;43:187. [Google Scholar]
- 10.Schottman TC, Hennessey PM, Gruening R. 7,008,979. US Pat. 2006 Mar 7;
- 11.Maneerung T, Tokura S, Rujiravanit R. Carbohydr Polym. 2008;72:43. [Google Scholar]
- 12.Hendrey AT, Stewart IO. Can J Microbiol. 1979;25:915. doi: 10.1139/m79-136. [DOI] [PubMed] [Google Scholar]
- 13.Klasen HJ. Burns. 2000;26:131–138. doi: 10.1016/s0305-4179(99)00116-3. [DOI] [PubMed] [Google Scholar]
- 14.Sampath LA, Chowdhury N, Caraos L, Modak SM. J Hosp Infect. 1995;30:201–210. doi: 10.1016/s0195-6701(95)90315-1. [DOI] [PubMed] [Google Scholar]
- 15.Percival SL, Bowler PG, Russell D. J Hosp Infect. 2005;60:1–7. doi: 10.1016/j.jhin.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 16.Gupta A, Matsui K, Lo JF, Silver S. Nat Med (N Y) 1999;5:183–188. doi: 10.1038/5545. [DOI] [PubMed] [Google Scholar]
- 17.Baquero F. Drug Resist Updates. 2001;4:93–105. doi: 10.1054/drup.2001.0196. [DOI] [PubMed] [Google Scholar]
- 18.McHugh SL, Moellering RC, Hopkins CC, Swartz MN. Lancet. 1975;305:235–240. doi: 10.1016/s0140-6736(75)91138-1. [DOI] [PubMed] [Google Scholar]
- 19.Wanzlick HW, Schonherr HJ. Angew Chem, Int Ed Engl. 1968;7:141. [Google Scholar]
- 20.Öfele K. J Organomet Chem. 1968;12:P42. [Google Scholar]
- 21.Arduengo AJ, Harlow RL, Kline M. J Am Chem Soc. 1991;113:361. [Google Scholar]
- 22.Garrison JC, Youngs WJ. Coord Chem Rev. 2005;105:3978. doi: 10.1021/cr050004s. [DOI] [PubMed] [Google Scholar]
- 23.Kascatan-Nebioglu A, Panzner MJ, Tessier CA, Cannon CL, Youngs WJ. Coord Chem Rev. 2007;251:884. [Google Scholar]
- 24.Arduengo AJ, Dias HVR, Calabrese JC, Davidson F. Organometallics. 1993;12:3405. [Google Scholar]
- 25.Kascatan-Nebioglu A, Melaiye A, Hindi K, Durmus S, Panzner MJ, Hogue LA, Mallett RJ, Hovis CE, Coughenour M, Crosby SD, Milsted A, Ely DL, Tessier CA, Cannon CL, Youngs WJ. J Med Chem. 2006;49:6811. doi: 10.1021/jm060711t. [DOI] [PubMed] [Google Scholar]
- 26.Panzner MJ, Hindi KM, Wright BD, Taylor JB, Han DS, Youngs WJ, Cannon CL. Dalton Trans. 2009:7308. doi: 10.1039/b907726j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hindi K, Siciliano T, Durmus S, Panzner M, Medvetz D, Reddy V, Hogue L, Hovis C, Hilliard J, Mallet R, Tessier C, Cannon C, Youngs WJ. J Med Chem. 2008;51:1577. doi: 10.1021/jm0708679. [DOI] [PubMed] [Google Scholar]
- 28.Panzner MJ, Deeraksa A, Smith A, Wright BD, Hindi KM, Kascatan-Nebioglu A, Torres AG, Judy BM, Hovis CE, Hilliard JK, Mallett RJ, Cope E, Estes DM, Cannon CL, Leid JG, Youngs WJ. Eur J Inorg Chem. 2009:1739. doi: 10.1002/ejic.200801159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Medvetz DA, Hindi KM, Panzner MJ, Ditto AJ, Yun YH, Youngs WJ. Met-Based Drugs. 2008:1. doi: 10.1155/2008/384010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knapp AR, Panzner MJ, Medvetz DA, Wright BD, Tessier CA, Youngs WJ. Inorg Chim Acta. 2010;364:125. doi: 10.1016/j.ica.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Özdemir İ, Gürbüz N, Doğan Ö, Günal S, Özdemir İ. Appl Organomet Chem. 2010;24:758. [Google Scholar]
- 32.Patil S, Deally A, Gleeson B, Müller-Bunz H, Paradisi F, Tacke M. Appl Organomet Chem. 2010;24:781. [Google Scholar]
- 33.Zień P, Bretner M, Zastąpiło K, Szyszka R, Shugar D. Biochem Biophys Res Commun. 2003;306:129. doi: 10.1016/s0006-291x(03)00928-8. [DOI] [PubMed] [Google Scholar]
- 34.Leid JG, Ditto AJ, Knapp A, Shah PN, Wright BD, Blust R, Cope EK, Christensen L, Clemons CB, Wilber JB, Young GW, Kang A, Panzner MJ, Cannon CL, Yun YH, Youngs WJ, Seckinger NM. J Antimicrob Chemother. 2012;67(1):138–148. doi: 10.1093/jac/dkr408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bruker . SMART, version 5.625, SAINT, version 6.22 and SHELXTL, (version 6.10) 1997. [Google Scholar]
- 36.Bruker . APEX II. Bruker AXS Inc; Madison, Wisconsin, USA: 2007. [Google Scholar]
- 37.Sheldrick GM. Acta Crystallogr. 2008;A64:112. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 38.Hindi KM, Ditto AJ, Panzner MJ, Medvetz DA, Tessier CA, Hovis CE, Han DS, Hilliard JK, Taylor JB, Yun YH, Cannon CL, Youngs WJ. Biomaterials. 2009;30:3771. doi: 10.1016/j.biomaterials.2009.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.