

Abstract
Elucidating biological and pathological functions of protein lysine acetyltransferases (KATs) greatly depends on the knowledge of the dynamic and spatial localization of their enzymatic targets in the cellular proteome. Here we report the design and application of chemical probes for facile labeling and detection of substrates of the three major human KAT enzymes. In this approach, we attempted to create engineered KATs in junction with synthetic Ac-CoA surrogates to effectively label KAT substrates even in the presence of competitive nascent cofactor acetyl-CoA. The functionalized and transferable acyl moiety of the Ac-CoA analogs further allowed the labeled substrates to be probed with alkynyl or azido-tagged fluorescent reporters by the copper-catalyzed azide–alkyne cycloaddition. The synthetic co-factors, in combination with either native or rationally engineered KAT enzymes, provide a versatile chemical biology strategy to label and profile cellular targets of KATs at the proteomic level.
Dynamic lysine acetylation of proteins is involved in a variety of fundamental biological processes including epigenetic programing, cell cycle, apoptosis, metabolism, and signal transduc-tion1. Acetylation is introduced by protein lysine acetyltransferases (KATs), also oft-referred to as histone acetyltransferases (HATs) and protein acetyltransferases (PATs), which transfer the acetyl group from the co-substrate acetyl-coenzyme A (acetyl-CoA, Ac-CoA) to the epsilon-amino group of specific lysine residues in proteins. Over the past decade, several KAT members have been identified and characterized both genetically and biochemically. Based on the sequence and structural similarities, KAT enzymes are classified into several major families, including the GCN5/PCAF family, the MYST family, and the p300/CBP2.
Recent biochemical and proteomic studies have revealed the existence of hundreds to thousands of acetylated proteins throughout the cell, which suggests that acetylation takes part in nearly every facet of cell physiology1b,1e,1f,3. Furthermore, significant amounts of evidence point toward that altered KAT expression and activity are characteristic to inflammation, diabetes, cancer, neurological disorders, and many other diseases4. While the importance of KATs in physiology and disease is well recognized, functional annotation of KAT enzymes in regulating key biological pathways is rather understudied. Especially, how the acetylome of individual KAT enzymes distinguishes from one another and how the substrate distribution of KATs is affected by different cellular contexts remain to be clarified. A clear biochemical and structural understanding of KAT substrate specificity and the impact of lysine acetylation in (patho)physiology is in great demand.
Elucidation of the molecular targets of KATs is a key step toward fully dissecting the epigenetic roles of KATs in gene regulation and their functions beyond the chromatin biology realm. Mass spectrometry (MS)-based technologies have provided a great deal of information about acetylated proteins1e,3a,3b, albeit with little on enzyme-substrate correlations. Protein microarray is another appealing approach in KAT substrate identification3c. Recently, synthetic Ac-CoA analogs have been explored to identify KAT substrates, which provides a great chemical biology strategy to interrogate the acetylome of particular KATs5. In line with this chemical biology paradigm, we attempted to create engineered KATs in junction with synthetic Ac-CoA surrogates to establish bioorthogonal probes to investigate cellular substrates of KAT enzymes. A panel of Ac-CoA analogs with alkynyl or azido functional groups were synthesized as cofactor substitutes for selective labeling of KAT substrates. Meanwhile, the active site of the key KATs was engineered in order to expand the cofactor binding capability of the enzymes to accommodate the bulkier synthetic cofactors (Fig. 1). By screening the activities of KAT enzymes to individual Ac-CoA analog, several enzyme-cofactor pairs were identified and applied to the investigation of cellular substrates of KATs.
Figure 1.

Acetyl-CoA analogs combined with a protein-engineering approach to label KAT substrates.
A set of Ac-CoA analogs was synthesized to provide potential active cofactors for the engineered KATs (Supplementary Table S1). These compounds include 4-pentynoyl CoA (4PY-CoA), 5-hexynoyl CoA (5HY-CoA), 6-heptynoyl CoA (6HY-CoA), 3-azidopropanoyl CoA (3AZ-CoA) and 4-azidobutanoyl CoA (4AZ-CoA). The chemical coupling of carboxylic acid with HS-CoA was achieved with N,N′-dicyclohexylcarbodiimide (DCC) with slight modification from the literature procedure6 (Supplementary Fig. S1). We noted that 4PY-CoA was previously utilized as a cofactor for p300 catalysis5b,6. The installed alkynyl and azido functional groups will facilitate subsequent detection and characterization of labeled KAT substrates.
The GCN5/PCAF KATs are key players in biological acetylation. Their structures have been well characterized which provides atomic details of the Ac-CoA binding site 2a,7. Examination of the crystal structure of hGCN5—Ac-CoA complex (PDB 1Z4R, ref.8) revealed several conserved bulky residues in the Ac-CoA binding pocket of the enzyme, i.e. L531, M534, I576, F578, T612, F622, and Y645 (Fig. 2a). To expand the cofactor binding pocket so that it can tolerate larger size acyl groups, we replaced each of these residues with smaller amino acid residues, i.e. alanine or glycine. Furthermore, based on the initial enzymatic screening data, several double-point mutants were generated to further boost the acyl-transferring activity of the engineered GCN5.
Figure 2. Identification of enzyme-cofactor pairs for GCN5 substrate labeling.
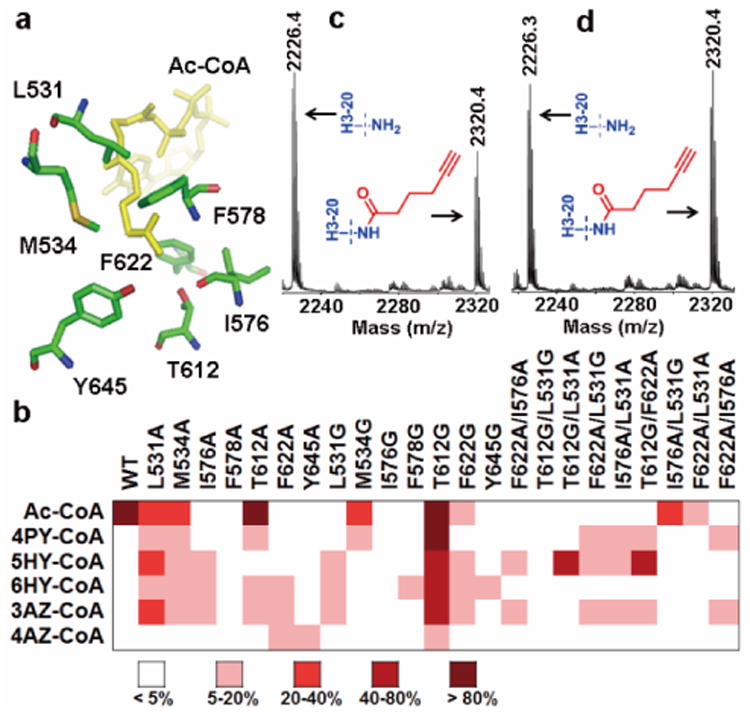
(a) The bulky catalytic residues in the active site of GCN5 (PDB 1Z4R)8. (b) Heat map of engineered GCN5 activities to different Ac-CoA analogs (see also Supplementary Fig. S3f). (c) MALDI-MS of H3-20 peptide modified by GCN5-T612G/F622A and 5HY-CoA (d) MALDI-MS of H3-20 peptide modified by the GCN5-T612G/L531A-5HY-CoA pair.
To identify the engineered enzyme forms that are active to the Ac-CoA substitutes, the entire panel of Ac-CoA analogs was screened in histone modification reactions catalyzed by both the wild-type (WT) and engineered GCN5 proteins. Typically, eachGCN5 was incubated with the N-terminal 20-aa H3 peptide (H3-20) and individual analogs. After the reaction, the mixtures were treated with 7-diethylamino-3-(4′-Maleimidylphenyl)-4-Methylcoumarin (CPM), a fluorogenic probe that becomes fluorescent upon reacting with the sulfhydryl group in HS-CoA9. The fluorescence screening data were quantitated (Supplementary Fig. S3) and presented in the heat map format in Fig. 2b. As expected, WT-GCN5 showed a strong activity toward Ac-CoA, the cognate cofactor of KATs, but was inert toward all the Ac-CoA substitutes. On the other hand, several engineered GCN5, e.g. GCN5-L531A/G, -I576A, -T612A/G, and -F622A, exhibited appreciable activities to the synthetic analogs at varied degrees. In particular, the single mutant GCN5-T612G was active toward all the analogs with 4AZ-CoA being the weakest. To further improve the activity and selectivity of the engineered GCN5 to the synthetic analogs rather than the nascent cofactor, several double-point mutants were generated by combining the most active single mutants (T612G, L531A, L531G, I576A, and F622A). As shown in Fig. 2b, most of these double mutants retained certain activities toward the analogs. Especially, GCN5-T612G/F622A and -T612G/L531A mutants exhibited excellent activity toward 5HY-CoA. MALDI-MS analysis of the reaction mixtures further confirmed that H3-20 peptide can be efficiently modified by GCN5-T612G/F622A and -T612G/L531A with 5HY-CoA (Fig. 2c, 2d, and additional MS data in Fig. S5). These data highlight that GCN5-T612G/F622A and -T612G/L531A in paired with 5HY-CoA are excellent enzyme-cofactor pairs for selective chemical labeling of GCN5 substrates.
The MYST proteins represent another major KAT family in the higher organisms10. To create chemical probes for identifying substrates of the MYST family, we engineered the active site of the MYST member MOF in a similar manner as described above for GCN5. In the crystal structure of MOF—Ac-CoA complex (PDB 2Y0M, ref.11), the acetyl moiety of the cofactor is encircled by bulky residues V314, I317, I333, P349, P352 and L353 (Fig. 3a). H273 is another potential residue that may affect the enzyme activity12. We performed mutation of each residue to Ala or Gly to expand the enzyme active site for acyl-CoA binding.
Figure 3. Identification of enzyme-cofactor pairs for MOF substrate labeling.
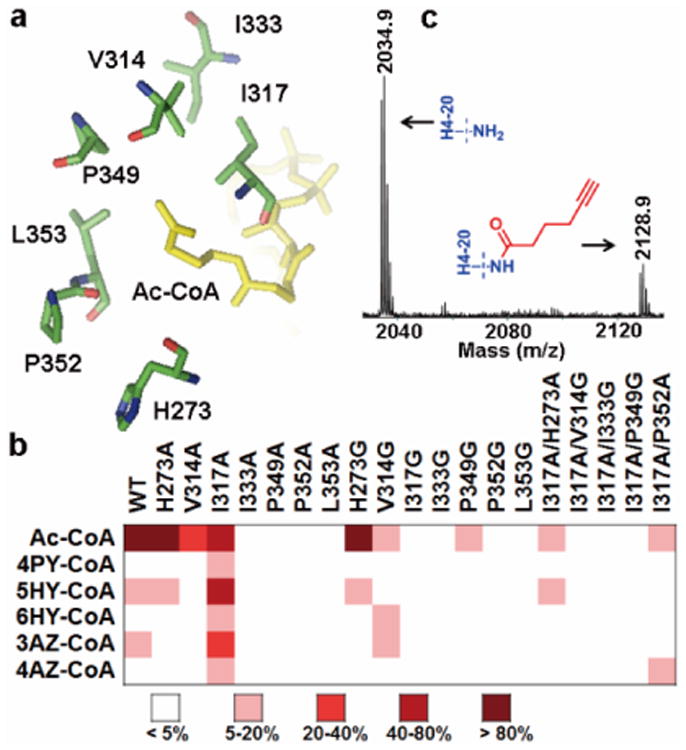
(a) Key residues surrounding the active site of MOF (PDB 2Y0M)11. (b) Heat map of engineered MOF activities to Ac-CoA analogs (see also Supplementary Fig. S4e). (c) MALDI-MS of H4-20 peptide modified by MOF-I317A/H273A-5HY-CoA pair.
Activities of each MOF mutant toward the synthetic Ac-CoA analogs were screened through enzymatic modification of the histone H4 tail peptide containing the first 20 aa residues (H4-20) by using the same fluorescent assay as described for GCN5 (Supplementary Fig. S4). A heat map of MOF activity with respect to individual Ac-CoA analogs was generated and shown in Fig. 3b. Among the tested MOF mutants, H273A and H273G recognized 5HY-CoA, and V314G recognized 6HY-CoA and 3AZ-CoA. More strikingly, MOF-I317A was active toward all the Ac-CoA analogs. In order to further improve the selectivity and activity of MOF to the Ac-CoA analogs, several double-point mutants were produced based on MOF-I317A which had a marked activity to the analogs. The enzymatic assays revealed that, compared to the single mutant MOF-I317A, the activity of all the double mutants toward Ac-CoA and the analogs decreased by different degrees (Fig. 3b). This likely suggests that MOF could not tolerate more drastic mutations in its active site. We noted that MOF-I317A/H273A retained about 6% activity to 5HY-CoA (Fig. 3b). The activities of MOF-I317A and -I317A/H273A in taking the Ac-CoA analogs were further corroborated in the MS experiments (Fig. 3c, and Fig. S6).
The clickable feature of the alkyne- or azido-containing Ac-CoA analogs provides a unique advantage for selective labeling, visualization, and further characterization of KAT targets by using the copper-catalyzed azide–alkyne cycloaddition (CuAAC) chemistry (Fig. 1)13. First, we performed fluorescent detection of H3 protein modification mediated by the engineered GCN5. The recombinant H3 protein was incubated with each analog in the presence of GCN5-WT, -T612G, -T612G/F622A or -T612G/L531A. Next, the mixture was probed with azide- or alkyne-coupled tetramethylrhodamine (TAMRA) dyes with the CuAAC reaction. The reaction mixture was resolved on SDS-PAGE and the gel was scanned for fluorescence imaging. As shown in Fig. 4a, no labeling was observed in the reactions catalyzed by WT-GCN5, which further confirmed that WT-GCN5 was inert to the synthetic analogs. The three GCN5 mutants T612G, T612G/F622A, and T612G/L531A, showed particularly strong labeling activity when paired with 5HY-CoA. GCN5-T612G also showed activity toward several other analogs such as 4PY-CoA, 6HY-CoA, and 3AZ-CoA, which is consistent with the screening results in the heat map (Fig. 2b). In the labeling reactions, strong fluorescence was only observed when H3 was subjected to the enzyme-catalyzed modification, but barely observed in the enzyme-negative controls, which excludes the possibility of nonspecific reactions. These results again support that both GCN5-T612G/F622A—5HY-CoA and T612G/L531A—5HY-CoA are excellent enzyme-cofactor pairs for labeling of GCN5/PCAF substrates.
Figure 4. In-gel fluorescent detection of KAT substrates with the bioorthogonal chemical probes.
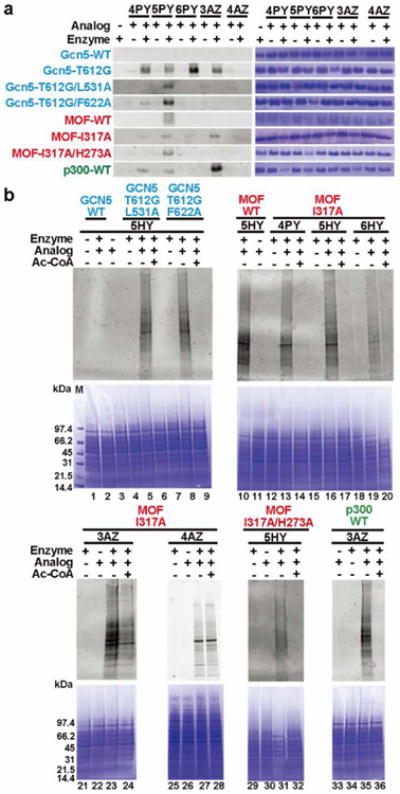
(a) Histone labeling by KATs and Ac-CoA analogs. Commassie blue staining was shown in right panel. The whole gel images were shown in Supplementary Fig. S8. (b) Cell lysate labeling by KATs and selected Ac-CoA analogs. The lower panel shows the commassie blue staining.
To probe protein acetylation catalyzed by the MYST KATs, both single-point mutant MOF-I317A and double-point mutant MOF-I317A/H273A were applied to label recombinant H4 in the presence of the synthetic Ac-CoA analogs. Again, following the enzymatic reaction, CuAAC chemistry was applied for fluorescent detection. As shown in Fig. 4a, MOF-I317A and -I317A/H273A showed a clear histone labeling activity when 5HY-CoA was used as the acyl donor. These encouraging results demonstrate that both MOF-I317A and -I317A/H273A in combination with 5HY-CoA are suitable enzyme-cofactor matches to label substrates of the MYST KATs. 3AZ-CoA was also a very effective cofactor for labeling by MOF-I317A.
Having validated the activities of the engineered GCN5 and MOF in conjugation with the synthetic cofactors on their cognate histone substrates, we attempted to profile the proteome-wide targets of KATs from the cellular contexts. As a principle of concept, we performed fluorescent labeling of KAT substrates from human embryonic kidney 293T cells. The cells were lysed in the M-PER® buffer, treated with the KAT-cofactor pairs, then labeled with fluorescent reporters via the CuAAC and visualized on SDS-PAGE. Multiple modified protein bands were readily visualized on in-gel fluorescence of the cell lysate treated with GCN5-T612G/L531A-5HY-CoA and T612G/F622A-5HY-CoA pairs (lane 5 and 8 in Fig. 4b). The addition of Ac-CoA suppressed the fluorescent labeling, which was in good agreement with the competitive nature of 5HY-CoA with respect to Ac-CoA (lane 6 and 9 in Fig. 4b). The lack of fluorescence in WT-GCN5-5HY-CoA pairing (lane 2 in Fig. 4b) as well as in the enzyme-negative control (lane 3 in Fig. 4b) further supports the matching characteristic of the engineered KAT-cofactor pairs and thereby endogenous GCN5 in the 293T cells did not interfere with the assay.
We also performed fluorescent labeling of protein substrates of the MYST KATs with the engineered MOF and cofactor substitutes. Both MOF-I317A and -I317A/H273A, which showed appreciable activity in histone modification assays, were used in conjugation with selected Ac-CoA analogs for target labeling. On the fluorescent image of the SDS-PAGE gel, multiple labeled lanes were observed when the cell lysate was treated with MOF-I317A in the presence of the synthetic analogs (lane 13, 16, 19, 23, 27 in Fig. 4b). Consistent with the results of the histone modification assays, MOF-I317A showed labeling activity toward the tested analogs, with 5HY-CoA and 3AZ-CoA being particularly strong. As expected, the double-point mutant MOF-I317A/H273A effectively labeled the cell lysate with 5HY-CoA (lane 31 in Fig. 4b)
The other important KAT family in the mammalian system is p300/CBP. So far a clear p300/Ac-CoA complex structure is not yet available, which makes it difficult to determine what residues should be selected for active site engineering. Given this limitation, we tested the histone H4 peptide labeling activity of p300 HAT domain protein with each synthetic Ac-CoA analog. Strikingly, in addition to the previously reported active cofactor 4PY-CoA5b,6, the azido-containing analog 3AZ-CoA was found to be strongly recognized by p300 HAT domain (Supplementary Fig. S7). This data suggests that 3AZ-CoA is another excellent Ac-CoA surrogate to identify protein targets of the p300/CPB family KATs. To validate this result, we examined p300 by pairing it with the synthetic analogs for labeling the H4 protein. The recombinant H4 protein was mixed with different analogs and p300 HAT domain, followed by CuAAC with an alkyne/azide-conjugated TAMRA dyes. As seen in Fig. 4a, most efficient labeling was observed in the p300—3AZ-CoA pair. As a control, there was no modification for the enzyme-negative sample. Also, p300 paired with the other analogs gave much weaker histone labeling activity than the p300—3AZ-CoA pair. Replacing the p300 HAT domain with full-length p300 yielded similar results (Supplementary Fig. S8c). We further tested fluorescent labeling of p300 substrates from the 293T cell lysate with the p300—3AZ-CoA pair (lane 35 in Fig. 4b). Multiple protein bands were shown as p300 substrates. Addition of Ac-CoA abolished the labeling, supporting that 3AZ-CoA is a highly competitive substitute for the native cofactor.
Functions of individual KATs are closely associated with their acetylomes in the biological milieus. Thus, identifying and profiling cellular substrates of KATs is of vital significance for understanding their roles in physiology and disease, which can be accelerated by developing innovative chemical biology probes14. In this study, we demonstrated the success of using rationally engineered proteins in conjunction with synthetic Ac-CoA analogs, as new chemical tools, to efficiently label KAT substrates. For the GCN5/PCAF family, we found that both GCN5-T612G/F622A—5HY-CoA and GCN5-T612G/L531A—5HY-CoA are superior matching pairs for selective labeling of substrates of GCN5/PCAF KATs. For the MYST family, we identified MOF-I317A and MOF-I317A/H273A that showed marked activity with 5HY-CoA. MOF-I317A was also very active toward 3AZ-CoA. For the KAT p300, in addition to 4PY-CoA, a previously identified cofactor for the enzyme, we identified 3AZ-CoA as another efficient cofactor for chemical labeling of p300 substrates. Advancing from these new findings, several further studies and improvement would be warranted. For instance, thus far a clear biochemical and structural understanding of how different KAT families showed varied abilities in recognizing the Ac-CoA analogs is not available. A systematic kinetic study will quantitatively evaluate the efficiencies of the KATs (both wt- and mutant forms) for the synthetic cofactors to assure competent significance of the bioorthogonal probes relative to the endogenous counterpart. Also, in the current version, the KAT chemical probes are limited to labeling protein substrates in cell lysates. Creation of cell-permeable Ac-CoA analogs could be valuable for direct interrogation of intracellular acetylation. The use of functionalized acetate may also be a practical approach5b. In conclusion, we identified for each of the three major families of human KAT proteins the optimal enzyme-cofactor pairs that can be applied to investigate new acetylation substrates. The synthetic Ac-CoA analog cofactors in conjunction with their matched KATs are expected to be versatile probes to identify KAT targets from homogenized cellular and tissue specimens, thereby expanding our chemical tool repertoires for functional annotation of the acetylome in higher organisms.
Supplementary Material
Acknowledgments
This work was supported by AHA Grant 12GRNT12070056 and NIH Grant R01GM086717.
Footnotes
Supporting Information Available: Experimental details for chemical synthesis, DNA mutation and protein expression, enzymatic assays, and additional data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Selvi RB, Kundu TK. Biotechnol J. 2009;4:375. doi: 10.1002/biot.200900032. [DOI] [PubMed] [Google Scholar]; (b) Smith KT, Workman JL. Nat Biotechnol. 2009;27:917. doi: 10.1038/nbt1009-917. [DOI] [PubMed] [Google Scholar]; (c) Strahl BD, Allis CD. Nature. 2000;403:41. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]; (d) Yang XJ, Seto E. Mol Cell. 2008;31:449. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Glozak MA, Sengupta N, Zhang XH, Seto E. Gene. 2005;363:15. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]; (f) Spange S, Wagner T, Heinzel T, Kramer OH. Int J Biochem Cell Biol. 2009;41:185. doi: 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 2.(a) Marmorstein R, Trievel RC. Biochim Biophys Acta. 2009;1789:58. doi: 10.1016/j.bbagrm.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vetting MW, LP SdC, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. Arch Biochem Biophys. 2005;433:212. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]; (c) Lee KK, Workman JL. Nat Rev Mol Cell Biol. 2007;8:284. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]; (d) Yang XJ. Nucleic Acids Res. 2004;32:959. doi: 10.1093/nar/gkh252. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Berndsen CE, Denu JM. Curr Opin Struct Biol. 2008;18:682. doi: 10.1016/j.sbi.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL. Science. 2010;327:1000. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning ZB, Zeng R, Xiong Y, Guan KL, Zhao S, Zhao GP. Science. 2010;327:1004. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lin YY, Lu JY, Zhang J, Walter W, Dang W, Wan J, Tao SC, Qian J, Zhao Y, Boeke JD, Berger SL, Zhu H. Cell. 2009;136:1073. doi: 10.1016/j.cell.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Science. 2009;325:834. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]; (e) Basu A, Rose KL, Zhang J, Beavis RC, Ueberheide B, Garcia BA, Chait B, Zhao Y, Hunt DF, Segal E, Allis CD, Hake SB. Proc Natl Acad Sci U S A. 2009;106:13785. doi: 10.1073/pnas.0906801106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Kim GW, Yang XJ. Trends Biochem Sci. 2011;36:211. doi: 10.1016/j.tibs.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 4.(a) Saha RN, Pahan K. Cell Death Differ. 2006;13:539. doi: 10.1038/sj.cdd.4401769. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Avvakumov N, Cote J. Oncogene. 2007;26:5395. doi: 10.1038/sj.onc.1210608. [DOI] [PubMed] [Google Scholar]; (c) Iyer A, Fairlie DP, Brown L. Immunol Cell Biol. 2012;90:39. doi: 10.1038/icb.2011.99. [DOI] [PubMed] [Google Scholar]
- 5.(a) Yu M, de Carvalho LP, Sun G, Blanchard JS. J Am Chem Soc. 2006;128:15356. doi: 10.1021/ja066298w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang YY, Ascano JM, Hang HC. J Am Chem Soc. 2010;132:3640. doi: 10.1021/ja908871t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang YY, Grammel M, Hang HC. Bioorg Med Chem Lett. 2011;21:4976. doi: 10.1016/j.bmcl.2011.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hodawadekar SC, Marmorstein R. Oncogene. 2007;26:5528. doi: 10.1038/sj.onc.1210619. [DOI] [PubMed] [Google Scholar]
- 8.Schuetz A, Bernstein G, Dong A, Antoshenko T, Wu H, Loppnau P, Bochkarev A, Plotnikov AN. Proteins. 2007;68:403. doi: 10.1002/prot.21407. [DOI] [PubMed] [Google Scholar]
- 9.Trievel RC, Li FY, Marmorstein R. Anal Biochem. 2000;287:319. doi: 10.1006/abio.2000.4855. [DOI] [PubMed] [Google Scholar]
- 10.Utley RT, Cote J. Curr Top Microbiol Immunol. 2003;274:203. doi: 10.1007/978-3-642-55747-7_8. [DOI] [PubMed] [Google Scholar]
- 11.Kadlec J, Hallacli E, Lipp M, Holz H, Sanchez-Weatherby J, Cusack S, Akhtar A. Nat Struct Mol Biol. 2011;18:142. doi: 10.1038/nsmb.1960. [DOI] [PubMed] [Google Scholar]
- 12.(a) Yuan H, Rossetto D, Mellert H, Dang W, Srinivasan M, Johnson J, Hodawadekar S, Ding EC, Speicher K, Abshiru N, Perry R, Wu J, Yang C, Zheng YG, Speicher DW, Thibault P, Verreault A, Johnson FB, Berger SL, Sternglanz R, McMahon SB, Cote J, Marmorstein R. Embo J. 2012;31:58. doi: 10.1038/emboj.2011.382. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sun B, Guo S, Tang Q, Li C, Zeng R, Xiong Z, Zhong C, Ding J. Cell Res. 2011;21:1262. doi: 10.1038/cr.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yang C, Wu J, Sinha SH, Neveu JM, Zheng YG. J Biol Chem. 2012;287:34917. doi: 10.1074/jbc.M112.359356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meldal M, Tornoe CW. Chem Rev. 2008;108:2952. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- 14.(a) Albaugh BN, Arnold KM, Denu JM. Chembiochem. 2011;12:290. doi: 10.1002/cbic.201000438. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Allis CD, Muir TW. Chembiochem. 2011;12:264. doi: 10.1002/cbic.201000761. [DOI] [PubMed] [Google Scholar]; (c) Yang YY, Hang HC. Chembiochem. 2011;12:314. doi: 10.1002/cbic.201000558. [DOI] [PubMed] [Google Scholar]; (d) Cole PA. Nat Chem Biol. 2008;4:590. doi: 10.1038/nchembio.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.