

Summary
SCF (Skp1-Cul1-Fboxes) E3 ligases are activated by ligation to the ubiquitin-like protein Nedd8, which is reversed by the deneddylating Cop9 Signalosome (CSN). However, CSN also promotes SCF substrate turnover through unknown mechanisms. Through biochemical and electron microscopy analyses, we determined molecular models of CSN complexes with SCFSkp2/Cks1 and SCFFbw7 and found that CSN occludes both SCF functional sites – the catalytic Rbx1-Cul1 C-terminal domain and the substrate receptor. Indeed, CSN binding prevents SCF interactions with E2 enzymes and a ubiquitination substrate, and inhibits SCF-catalyzed ubiquitin chain formation independent of deneddylation. Importantly, CSN prevents neddylation of the bound cullin, unless binding of a ubiquitination substrate triggers SCF dissociation and neddylation. Taken together, the results provide a model for how reciprocal regulation sensitizes CSN to the SCF assembly state, and inhibits a catalytically-competent SCF until a ubiquitination substrate drives its own degradation by displacing CSN, thereby promoting cullin neddylation and substrate ubiquitination.
Introduction
Cullin-RING ligases (CRLs) constitute the largest family of E3 ubiquitin ligases (Deshaies and Joazeiro, 2009). The modular CRL architecture explains their pervasive but highly specific functions. As exemplified by the archetypical Skp1-Cullin1-F-box (SCF) complexes, CRLs are nucleated by one of seven structurally elongated cullin protein scaffolds. The conserved cullin C-terminal domain assembles the CRL catalytic core by binding a RING-finger protein, typically Rbx1, which in turn promotes ubiquitin transfer from an associated E2 enzyme. The distal N-terminal end of the cullin binds a substrate receptor (SR) module, which recruits the ubiquitination target. The modular and variable SRs allow recognition of numerous substrates by the same CRL catalytic core.
Human SCF complexes use one of more than 60 SRs, which bind Skp1 through their F-box domain. Skp1 bridges the interaction of Cul1 and the SR, which in turn recruits specific ubiquitination substrates via a distinctive degron motif (Duda et al., 2011). As examples, Fbw7’s WD-40 domain recognizes the phosphorylated form of the cell-cycle regulator CyclinE (Hao et al., 2007), while Skp2 uses a leucine-rich repeat domain together with the co-receptor Cks1 to recruit a phosphorylated form of the cyclin-dependent kinase (Cdk) inhibitor p27 (Hao et al., 2005).
CRL catalytic activity is also controlled by covalent attachment of the ubiquitin-like protein Nedd8 to the cullin’s winged-helix B (WHB) domain. Similar to ubiquitination, neddylation is mediated by an enzymatic cascade, including the Nedd8-conjugating E2 enzyme Ubc12, which is activated by the RING-domain of Rbx1 with stimulation by Dcn1 (Kurz et al., 2005; Scott et al., 2011). Non-neddylated cullin-Rbx1 complexes can bind an inhibitor CAND1, which prevents neddylation and competes with SR-association (Goldenberg et al., 2004). Neddylation favors a conformational rearrangement of the cullin C-terminal domain and Rbx1, which prevents CAND1 binding and enhances CRL-mediated ubiquitination activity (Duda et al., 2008; Saha and Deshaies, 2008; Yamoah et al., 2008).
Cullins are deneddylated by the Cop9 Signalosome (CSN). CSN comprises eight different subunits, Csn1 through Csn8, named by descending molecular weight. Together they unleash the zinc-metalloprotease activity of Csn5 by an unknown mechanism (Sharon et al., 2009). The active site of Csn5 is located within its N-terminal MPN domain (Cope et al., 2002; Lyapina et al., 2001) and most probably functions similarly to the thermolysin-like mechanism described for the homologous de-ubiquitinating enzyme AMSH-LP (Sato et al., 2008). Csn6 also comprises an MPN domain, which however lacks the conserved zinc-coordinating residues, and is thus thought to serve a scaffolding function. The remaining six subunits are characterized by PCI-domains, comprising a C-terminal winged helix preceded by a bundle of bi-helical repeats (Dessau et al., 2008; Scheel and Hofmann, 2005). The PCI domains of Csn1, Csn2, Csn3 and Csn4 are preceded by long N-terminal extensions, predicted to contain further helical repeats (Enchev et al., 2010; Pick et al., 2009). There is a striking similarity between the CSN subunit composition and that of the lid sub-complex of the 26S proteasome, which also comprises two MPN- and six PCI-domain containing subunits with one-to-one sequence correspondence (Enchev et al., 2010; Pick et al., 2009).
Upon CSN-mediated SCF deneddylation, CAND1 has been implicated in exchange of SRs (Schmidt et al., 2009). Thus, SCF regulation has been thought of as a cycle of assembly with SRs, neddylation, substrate ubiquitination, CSN-mediated deneddylation, followed by CAND1-stimulated disassembly/re-assembly and/or neddylation (Bosu and Kipreos, 2008; Cope and Deshaies, 2003). However, in cells only a small subset of un-neddylated cullins is found in complex with CAND1 (Bennett et al., 2010), and it remains unclear how the neddylation/de-neddylation cycle is coordinated with substrate availability. Moreover, cellular CRLs exist in a wide range of assembly states, and upon inhibition of deneddylation, both neddylated and non-neddylated CRLs stably associate with CSN, with or without SR modules (Bennett et al., 2010; Olma et al., 2009). How the different CRL assembly and activation states influence CSN and vice-versa remains poorly understood. Specific signals and SR binding have been suggested to regulate CSN-CRL4 complex formation (Fischer et al., 2011; Groisman et al., 2003), but neither the structural basis nor the functional significance of forming stable complexes between CSN and the products of its enzymatic reaction are understood. Intriguingly CRL4 enzymes, which may not require neddylation for activity, can be inhibited through CSN by an unknown mechanism (Fischer et al., 2011). Similarly, a recent study reported that CSN can inhibit SCF activity through a yet unknown, non-catalytic mechanism and that the SCF assembly state influences the CSN deneddylation activity (Emberley et al., 2012). Collectively the available data imply that CSN and CAND1 regulate CRL function on multiple levels, whose relationships and mechanisms remain incompletely characterized.
To understand the functional implications of the CSN interactions with CRLs, we determined structures of several CSN and CSN-SCF complexes by electron microscopy. We applied a hybrid structural approach to obtain pseudo-atomic molecular models, which were further validated biochemically. Surprisingly, we found that CSN can occlude both SCF functional sites – for E2 enzymes and for ubiquitination substrates - which are located at opposite ends of the SCF complex. Consequently, CSN-SCF complex formation results in inhibited activation of Ubc12 by unneddylated SCF and of Cdc34 by neddylated SCF, and competes with binding to CAND1 and ubiquitination substrate. Our findings thus provide a structural and biochemical basis underlying non-catalytic regulation of SCFs by CSN and imply that ubiquitination substrates can trigger SCF activation.
Results
Structural electron microscopy analysis of CSN complexes
To gain structural and functional insights into CSN binding to fully assembled SCFs, we reconstituted various CSN-SCF complexes in vitro. In addition to wild-type CSN, we also produced a recombinant CSN complex harboring a Csn5 subunit with a H138A point mutation in its active site, which interferes with zinc chelation, is deneddylation-defective (Cope et al., 2002), and thus stably associates with neddylated SCFs.
Purified complexes of CSNCsn5H138A with neddylated SCFSkp2/Cks1 (referred to as CSNCsn5H138A-SCF~N8Skp2/Cks1) or SCFFbw7 (referred to as CSNCsn5H138A-SCF~N8Fbw7) (Figure 1A) were subjected to negative stain electron microscopy and single particle analysis. We determined the structure of the CSNCsn5H138A-SCF~N8Skp2/Cks1 complex by, ab initio angular reconstitution and iterative rounds of refinement (Figure 1B and S1A). To differentiate between the CSN and SCF components within the map we analyzed electron microscope images of negatively stained apo CSN complexes. We calculated the structure of apo CSN using an initial reference derived from the CSNCsn5H138A-SCF~N8Skp2/Cks1 structure (Figure 1C, Figure S1B). Comparison between the two maps showed that the apo CSN structure matched well a large portion of the CSNCsn5H138A-SCF~N8Skp2/Cks1 map allowing the segmentation of the latter into its CSN and SCF components (Figure 1D) confirmed by difference analysis (Figure S3A). The CSN density is characterized by a well-resolved mesh of discrete patches of elongated densities, consistent with our previous data (Enchev et al., 2010). The remaining region, which therefore corresponds to SCF~N8Skp2/Cks1, forms a separate elongated curved density, connected to CSN through both of its ends.
Figure 1.

Reconstitution and single particle Electron Microscope analysis of CSN-SCF complexes. (A) Coomassie-stained SDS-PAGE of recombinant CSNCsn5H138A and reconstituted CSN-SCF complexes after gel filtration. (B – F) Surface views of EM density maps of (B) CSNCsn5H138A-SCF~N8Skp2/Cks1, (C) CSNCsn5H138A, (D) CSNCsn5H138A-SCF~N8Skp2/Cks1 segmented into its SCF~N8Skp2/Cks1 (purple) and CSN (grey) subcomplexes. (E) CSNCsn5H138A-SCF~N8Fbw7, (F) CSNCsn5H138A-SCF~N8Fbw7 segmented into its CSNCsn5H138A-SCF~N8Fbw7 (purple) and CSN (grey) subcomplexes. See also Figures S1 and S3.
We next investigated whether this CSN-binding mode is structurally conserved among the SCF family by analysis of CSNCsn5H138A-SCF~N8Fbw7. The CSNCsn5H138A-SCF~N8Skp2/Cks1 structure was used as an initial reference for the analysis of CSNCsn5H138A-SCF~N8Fbw7 (Figure S1C). Indeed, the resulting structure (Figure 1E) is overall very similar to CSNCsn5H138A-SCF~N8Skp2/Cks1. Difference analysis with the apo CSN map indicated that SCF~N8Fbw7 also employs both its ends to bind CSN and adopts a comparable position (Figure 1F and Figure S3A).
Molecular models for CSN and CSN-SCF complexes
Given similarity of CSN and the 26S proteasome lid sub-complex (Pick et al., 2009), we considered recent EM studies defining molecular boundaries of the proteasome lid and proposing pseudo-atomic models with constituent subunits for the fission and budding yeast as well as for human lid sub-complexes (da Fonseca et al., 2012; Lander et al., 2012; Lasker et al., 2012). A comparison of the CSN region of the CSNCsn5H138A-SCF~N8Skp2/Cks1 map and the human lid (Figure S2A) reveals substantial similarity. We thus assigned individual subunits within the CSN density to locations corresponding to their homologues in the lid. Atomic models for all human CSN subunits from I-TASSER (Roy et al., 2010) (Figure S2B) were docked into the CSN density. The resulting model (Figure 2) is characterized by a close match between the protein density and the atomic coordinates. There is little density unoccupied by the docked models and there is no significant spatial overlap between the docked coordinates. Furthermore, the atomic models and the corresponding density segments of the CSNCsn5H138A-SCF~N8Skp2/Cks1 map demonstrate strong correlation (Figure S2D).
Figure 2.
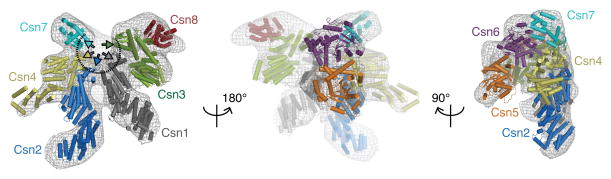
Molecular model for CSN. The CSN segment from the CSNCsn5H138A-SCF~N8Skp2/Cks1 map is shown as grey mesh. Left - PCI cluster side view. A dotted arc and color-coded arrows indicate the approximately coplanar positions of the winged-helix domains. MPN subunits are omitted for clarity. Center - opposite side, characterized by a protrusion formed by the two MPN domain subunits, Csn 5 and 6. Right - edge-on to the coplanar PCI cluster. The protrusion formed by the Ccn5 and 6 MPN subunits is left of the PCI cluster. Csn6 is more closely integrated with the PCI cluster while Csn5 is angled away. See also Figure S2.
In the model, the PCI subunits in CSN form an approximately coplanar surface (Figure 2, right hand panel). Their major interaction interfaces are formed from the C-terminal winged-helix domains that form an arc (Figure 2, left hand panel), from which the extended N-terminal domains radiate to form the characteristic ribbon-like densities in the map. In the model, the four longest subunits (Csn1, 2, 3 and 4) are docked into the central region of the arc, which is capped at each end by the shorter Csn7 and Csn8 subunits. The N-terminal peripheral density assigned to Csn4 in the model is less well recovered, possibly due to conformational heterogeneity in this region.
In contrast to the PCI subunits, the MPN subunits Csn5 and Csn6 are predicted to adopt globular conformations (Figure S2B). In the model, these two subunits form a protrusion on the side opposite to the PCI subunits (Figure 2, center and right hand panels), with Csn6 located over the center of the winged-helix arc and Csn5 offset in the direction of the Csn1 and Csn2 N-terminal domains and extending away from the plane of the PCI subunit cluster.
To test the assignments for Csn2 and Csn5, we produced CSN sub-complexes lacking the Csn5 subunit, CSNΔCsn5, or solely containing the PCI-domain of Csn2, CSNCsn2ΔN1–269 (Figure S2C, S1D, S4F, G). CSNΔCsn5 formed a complex with SCF~N8Skp2/Cks1 (Figure S3D, 4C). We subjected these complexes to electron microscopy and single particle analysis. We analyzed CSNΔCsn5-SCF~N8Skp2/Cks1 using the CSNCsn5H138A-SCF~N8Skp2/Cks1 structure as an initial reference (Figure S1E, S3B, C). The major difference between the refined CSNΔCsn5-SCF~N8Skp2/Cks1 and CSNCsn5H138A-SCF~N8Skp2/Cks1 maps is that the former lacks density in the region assigned to Csn5 (Figure S3B) that matches well with the Csn5 atomic model. Similarly, we analyzed CSNCsn2ΔN1–269 with the apo CSN model as a reference (Figure S1F). The resulting map closely resembles the apo CSN structure but lacks a peripheral, extended and curled density (Fig S3B), which matches well the location and dimensions of the segment in which we docked the predicted TPR-like fold of the PCI-associated module of Csn2.
Figure 4.

CSN-SCF interactions analyzed by analytical size exclusion chromatography. Indicated SCF complexes were tested with equimolar amounts of (A, D, E) CSN harboring the Csn5H138A active site mutation, (B) CSN harboring an N-terminally truncated Csn2 and Csn5H138A or (C) a CSN complex lacking Csn5. Input and peak fractions (numbered) were blotted with antibodies indicated to the right. Fractions in which particular complexes were eluted are indicated above each panel. Neddylated complexes are labeled with ~N8. See also Figure S4.
Having identified the CSN region in our maps of the CSN-SCF complexes and modeled individual CSN subunits, we interpreted the remaining density in terms of the known atomic structures of SCF components. We generated models for the neddylated SCF complexes based on the SCFSkp2(F-box domain) structure (Zheng et al., 2002) but using a model for the neddylated conformation of the C-terminal domain of Cul11–690 (Duda et al., 2008) and the structures of the respective substrate receptors Skp1-Skp2/Cks1 (Hao et al., 2005; Schulman et al., 2000) or Skp1-Fbw7 (Hao et al., 2007). We docked these as rigid bodies into the corresponding densities of the CSNCsn5H138A-SCF~N8Skp2/Cks1 and CSNCsn5H138A-SCF~N8Fbw7 maps (Figure 3) with close agreement in both cases. The extreme C-terminal domain of Cul1 (Cul1691–776), encompassing Helix29 and WHB, forms one structural entity of similar size to Rbx1 and Nedd8, and all three are known to be flexibly oriented to each other (Calabrese et al., 2011; Duda et al., 2008; Duda et al., 2012; Fischer et al., 2011), thus precluding their definitive docking in the map. Nevertheless, after assigning the CSN and Cul11–691/Skp1/Skp2/Cks1 segments, there is a portion of density left unaccounted for (Figure S3D), which matches well a neddylated WHB domain of Cul1 as well as Rbx1. Thus, a putative model is shown in Figure S3E–F, demonstrating that WHB~Nedd8 and Rbx1 can in principle be accommodated in the groove between Csn5 and Csn2 (see also Figure 6A).
Figure 3.

Molecular models for (A) CSNCsn5H138A-SCF~N8Skp2/Cks1 and (B) CSNCsn5H138A-SCF~N8Fbw7. Brown and purple (A) or purple (B) arrows in the central views indicate contact points between the SCF substrate receptors and CSN. Black arrows in the right hand views indicate the basic canyon region of Cul1. See also Figures S1 and S3.
Figure 6.

CSN-SCF binding regulates ubiquitination and neddylation. (A) Zoomed-in surface view of CSNCsn5H138A-SCF~N8Skp2/Cks1, color-coded as Figure 5A, showing overlay of all reported Rbx1 conformations without (left) or with docked Cdc34 model (right). Cul1411–690 (CTD without Helix29 and WHB) shown in green, Rbx1 and Cds34 orientations shown as hues of red and blue. (B) In vitro ubiquitination of p-p27 (left) CyclinEphospho-peptide (right),assayed in the absence (−) or presence (+) of catalytically-inactive CSNCsn5H138A. Unmodified and poly-ubiquitinated substrates (−(Ub)n) detected by immunoblotting with p-p27- and biotin-antibodies respectively. (C) Pulse-chase [32P]~ubiquitin (Ub) transfer from Cdc34 to lysine-less ubiquitin (UbK0) in the presence (+) or absence (−) of CSNCsn5H138A and neddylated Cul1/Rbx1 (upper panel) or neddylated SCFSkp2/Cks1 (lower panel). Formation of di-Ub was assayed by autoradiography (left hand panels), and quantified by plotting the percentage (%) of di-Ub formation as a function of time (right hand panels). (D) Deneddylation of Cul1CTD~N8/Rbx1 by CSN assayed as described in Figure S6B in the presence (+) or absence (−) of wild type (WT) or Rbx1-interction deffective R547A mutant Glmn. (E) Neddylation (~N8) of Cul1SCE/Rbx1 measured by immunoblotting with Cul1 antibodies, in the presence (+) or absence (−) of CSNCsn5H138A, Skp1/Skp2/Cks1 and CyclinA/Cdk2 and p-p27. See also Figures S6 and S7.
Both SCF complexes adopt a peripheral position, approaching CSN through their substrate recognition and E2-binding ends. At the E2-binding end, the Cul1 C-terminal domain (CTD) and the region of the map modeled as the N-terminal TPR-like domain of Csn2 are connected by a continuous density, which appears to provide the major interaction between SCF and CSN in both complexes (Figure 3). At the substrate recognition end of the CSNCsn5H138A-SCF~N8Skp2/Cks1 complex the C-terminal α-helices of Skp2 are located between the regions of the map modeled as the N-terminal domains of Csn3 and Csn1 (Figure 3A, middle panel brown and purple arrows). Analogously, the WD40 repeat domain of Fbw7 is located in the corresponding region in the CSNCsn5H138A-SCF~N8Fbw7 map (Figure 3B, purple arrows). On the other hand, for both CSNCsn5H138A-SCF~N8Skp2/Cks1 and CSNCsn5H138A-SCF~N8Fbw7 complexes the region into which the Cul1 N-terminal domain and the Skp1 subunit are docked is widely separated from CSN. Hence it appears unlikely that there is any significant interaction between CSN and either the Cul1 N-terminal domain or the Skp1 subunit.
Determinants and implications of CSN-SCF interactions
To test and further explore the observed CSN-SCF interactions, various CSN and SCF complexes were mixed at 1:1 molar ratios, and association was examined by co-elution over analytical size exclusion chromatography (Figure 4). For comparison, data of the isolated CSN, SCF complexes or different mutant complexes are shown in Figure S4A–E.
Our structural model predicts that CSN predominantly binds to the C-terminal portion of Cul1. Indeed, the co-elution profiles observed between CSNCsn5H138A and a complex containing only the CTD of Cul1 and Rbx1 (Cul1CTD/Rbx1) with and without neddylation were comparable to the respective full-length Cul1/Rbx1 variants (Figure 4A). On the other hand, modulating the Cul1 CTD influenced CSN-binding. In the presence of CSNCsn5H138A, Cul1/Rbx1 migrated in two peaks, one corresponding to a complex with CSN, and the other corresponding to free Cul1/Rbx1 (Figure 4A), while a greater proportion of the neddylated Cul1/Rbx1 (Cul1~N8/Rbx1) complex co-purified with CSNCsn5H138A (Figure 4A). Similarly, neddylated Cul1CTD/Rbx1 complexes showed increased comigration with CSNCsn5H138A compared to un-neddylated controls. Our structural model implies a potentially tenuous contact between CSN and the neddylated WHB domain of Cul1 (WHB~Nedd8, Figure S3C), and indeed, an intact WHB domain was required for binding of CSN and Cul1/Rbx1 complexes (Figure 4A). However, the WHB domain in isolation does not bind CSN sufficiently strongly to be detected by gel filtration (Figure S4B).
The structural analysis predicts that the Cul1 CTD approaches Csn2 to form a major interaction (Figure 3). Indeed, CSNCsn2ΔN1–269 lacking the N-terminal 269 residues of Csn2, and mutants with lesser truncations, failed to co-elute with Cul1~N8/Rbx1 (data not shown) and displayed decreased deneddylation activity, whereas complexes lacking Csn5 generally behaved similarly to CSNCsn5H138A (Figure 4, S2, S4). Introduction of the Csn5H138A active site mutation to CSNCsn2ΔN1–269 did not restore binding (Figure 4B).
In addition to the CSN-Cul1 CTD interface, we observe a more tenuous density connecting CSN and the SCF SRs (Figure 3A,B). The isolated SR modules Skp1/Skp2/Cks1 and Skp1/Fbw7 did not co-elute with CSN, although the SRs appeared to enhance Cul1/Rbx1 association with CSN, since stoichiometric co-elution of CSN and SCFSkp2/Cks1 or SCFFbw7 over gel filtration did not require neddylation (Figure 4D, E). Moreover, the observed interaction of CSN-SCFSkp2/Cks1 was not substantially affected by deletion of either WHB or the Rbx1 RING domain (Figure 4E). However, SCFSkp2/Cks1 did not co-elute with CSNCsn2ΔN1–104 (Figure S4I), confirming that the availability of SR is not sufficient to compensate for the requirement for the Csn2 N-terminal region. The CSN-SCF interaction was further explored by incubating pre-formed CSN-SCFSkp2/Cks1 complexes with CAND1 and analyzing the mix by gel filtration. Interestingly, we observed separation into CSN, CAND1-Cul1/Rbx1 and Skp1/Skp2/Cks1 complexes (Figure S4K).
CSN can compete with ubiquitination substrate for SCF binding
Since CSN approaches substrate receptors in SCF complexes, we examined the potential influence of a ubiquitination substrate. The SCFSkp2/Cks1 substrate p27 is phosphorylated prior to SCF-binding by cyclin-dependent kinases (Cdks). CyclinA/Cdk2 binds Skp2 and Cks1, and thus forms a complex with SCFSkp2/Cks1 and phospho-p27 (p-p27). p27 interacts both with Cdk2 and Skp2/Cks1 but only binding to the latter requires its phosphorylation (Hao et al., 2005; Spruck et al., 2001). The atomic coordinates of CyclinA/Cdk2 (Russo et al., 1996) can be docked onto the SCFSkp2/Cks1 structure without steric clashes with other protein densities of our map (Figure 5A). In contrast, modeling the phospho-p27 (p-p27) N- and C-terminal fragments (Hao et al., 2005; Russo et al., 1996) raised the possibility that p-p27 and CSN might bind SCFSkp2-CyclinA/Cdk2/Cks1 in a mutually exclusive manner (Figure 5A, arrow). Likewise, the proximity of CSN to Fbw7 may preclude a full-length substrate protein or protein complex from co-existing with CSN on SCFFbw7 (Figure S6F).
Figure 5.

CSN-SCF interactions in the presence of ubiquitination substrate. (A) Views of the CSNCsn5H138A-SCF~N8Skp2/Cks1 map are shown with CSN as an orange surface, SCFSkp2/Cks1~N8 as a gray mesh and the atomic coordinates as in Figure 3A. Docked Cdk2 (red), CyclinA (blue) and N- and C-terminal segments of p-p27 (yellow) are indicated by circles. A potential steric clash of p-p27 with the CSN density is indicated by a dashed yellow curve and an arrow on the left. (B) Neddylated (~N8) or non-neddylated SCFSkp2 complexes were incubated with CyclinA/Cdk2/Cks1 and p-p27, in the presence or absence of equimolar amounts of deneddylation-defective CSNCsn5H138A and analyzed by size exclusion chromatography. See also Figure S5.
To test whether CSN and p-p27 compete for SCF-binding, we first performed analytical size exclusion chromatography with equimolar CSNCsn5H138A and SCFSkp2-p-p27/CyclinA/Cdk2/Cks1 in both neddylated and non-neddylated states (Figure 5B). Indeed, CSNCsn5H138A formed a stable complex with SCF~N8Skp2-CyclinA/Cdk2/Cks1, and p-p27 was excluded from the CSN complexes containing Cul1 (Figure 5B and Figure S5). In the absence of neddylation, SCFSkp2-CyclinA/Cdk2/Cks1 segregated as mutually exclusive complexes with both CSN and p-p27 in a dynamic equilibrium, most likely due to the less stable complex of CSN with non-neddylated SCFSkp2-CyclinA/Cdk2/Cks1 (Figure 5B).
CSN-mediated deneddylation depends on the SCF assembly state
To explore the functional effects of different SCF assembly states, we examined CSN deneddylation activity toward different neddylated Cul1/Rbx1 complexes. As endogenous and recombinant CSN displayed similar deneddylation activities (Figure S6A), all assays were performed with recombinant CSN. Consistent with the finding that the N-terminal domain of Cul1 is not necessary for CSN binding, deneddylation was comparable for neddylated full-length Cul1/Rbx1 (Cul1fl/Rbx1), Cul1CTD/Rbx1, and split-and-coexpressed Cul1/Rbx1 (referred to here as Cul1SCE/Rbx1) as substrates in our assays (Figure S6A). The latter is obtained by co-expression in bacteria of the Cul1 N-terminal domain (referred to as Cul1NTD), the Cul1 C-terminal domain (referred to as Cul1CTD), and Rbx1 as a total of three separate polypeptides, which assemble into a Cul1SCE/Rbx1 complex whose structural and biochemical properties resemble those of full-length Cul1/Rbx1 (Duda et al., 2008; Goldenberg et al., 2004; Saha and Deshaies, 2008; Zheng et al., 2002).
Interestingly, the deneddylation activity toward Cul1SCE~N8/Rbx1 was decreased upon addition of stoichiometric amounts of Skp1/Skp2/Cks1 or Skp1/Fbw7 (Figure S6B). Adding CyclinA/Cdk2 strengthened the inhibitory effect of Skp1/Skp2/Cks1, an effect maintained upon further addition of in vitro phosphorylated or unmodified p27 (Figure S6B). These results, which are consistent with a recent biochemical study (Emberley et al., 2012), suggest that Cul1/Rbx1 binding to Skp1/F-box modules, or their complexes with partner proteins, attenuates the deneddylation activity of CSN, possibly through product inhibition. We interpret the lack of deneddylation activity in the presence of substrate as due to the lack of association between substrate bound SCFs and CSN (Figure 4B).
CSN-SCF binding interferes with Cdc34 activity
CSN has been shown to impede CRL4 auto-ubiquitination independently of deneddylation (Fischer et al., 2011). To gain structural insights into whether and how CSN might impact SCF-mediated ubiquitination, we docked the E2 Cdc34 onto the Rbx1 RING domain based on available structures of other E2-RING E3 complexes (Zheng et al., 2000). Although the orientation of Rbx1’s RING domain relative to the cullin subunit is known to be flexible (Calabrese et al., 2011; Duda et al., 2008; Fischer et al., 2011), Rbx1 can be positioned as a rigid body together with neddylated Cul1CTD (Duda et al., 2008) with a reasonable fit in the EM map (Figure S3F). Strikingly, with Rbx1’s RING domain in this orientation, or in any other previously reported conformation, steric hindrance would prevent the simultaneous interaction of Cul1/Rbx1 with CSN and Cdc34 (Figure 6A). Moreover, part of the CSN density appears in close proximity to the region most likely corresponding to the basic canyon of Cul1, which recruits the specialized acidic tail of Cdc34 with high affinity (Kleiger et al., 2009) (Figure 3, black arrows in right panels). Structural modeling thus predicts that CSN binding prevents a catalytic interaction of Cul1/Rbx1 and Cdc34. As evident from analytical size exclusion (Figure 5B and S5), CSN and p-p27 binding to neddylated SCFSkp2/Cks1 is mutually exclusive. Thus, under the equillibrium conditions of the ubiquitination assays, CSNCsn5H138A or CSNΔCsn5 are expected to stongly reduce the occupancy time of both p-p27 and Cdc34. Combined with the low efficiency of Cdc34 as a priming E2, addition of these CSN constructs resulted in a decreased length of the poly-ubiquitin chains assembled by SCFSkp2/Cdc34 on in vitro phosphorylated full-length p27 (p-p27; Figure 6B, left and Figure S6C, D). CSNCsn2ΔN1–269 only partially prevented p-p27 ubiquitination in this lower molecular weight region, likely due to deneddylation by the relatively high and stoichiometric CSN:SCF concentrations used in this assay (Figure S6C).
In cells CSN binding to neddylated SCFs would most likely result in very rapid deneddylation (cf. Figure S6B). To test whether CSN binding can exert an inhibitory effect on non-neddylated SCF, we used a non-neddylatable Cul1 construct with an Arg-to-Lys mutation of its neddylation site (Cul1K720R/Rbx1), which shows residual ubiquitination activity at long time-points (Duda et al., 2008; Saha and Deshaies, 2008). This effect was strongly attenuated by stoichiometric addition of CSNCsn5H138A (Figure S6D). The decreased poly-ubiquitin chain length observed upon addition of CSNCsn5H138A was not due to an obvious contamination with a de-ubiquitinating enzyme (Figure S6E). Moreover, CSNCsn5H138A counteracts SCF~N8Fbw7-activation of processive Cdc34-mediated ubiquitination of a short Cyclin E phosphopeptide (Figure 6B, right). Notably, due to its small size, this Cyclin E peptide may circumvent the steric clash within the SCFFbw7-CSN complex as shown by analytical size exclusion chromatography (Figure S6F, G).
Because the data suggest that CSN interferes with ubiquitin-transfer activity of Cdc34 independent of substrate-SR interactions, we performed pulse-chase assays to monitor SCF-mediated activation of Cdc34’s intrinsic ubiquitin transfer activity. Briefly, in the pulse reaction, we generated a thioester-linked Cdc34 conjugate with a 32P-labeled lysineless (K0) version of ubiquitin. Use of lysine-less ubiquitin prevents polyubiquitin chains from forming during the pulse. In the chase, we added unlabeled wild-type ubiquitin, and monitored ligation of radiolabeled K0 ubiquitin by appearance of di-ubiquitin chains. Consistent with previous studies, Cdc34-mediated di-ubiquitin synthesis was stimulated by all complexes containing neddylated Cul1/Rbx1 (Figure 6C and Figure S7A). In addition to neddylated Cul1/Rbx1 itself, this includes neddylated Cul1CTD/Rbx1, SCFSkp2/Cks1, and SCFFbw7 (Saha and Deshaies, 2008). Paralleling the effects on substrate ubiquitination, addition of CSNCsn5H138A and CSNΔCsn5, but not CSNCsn2ΔN1–269, markedly decreased di-ubiquitin formation (Figure 6C and Figure S7A).
Finally, we used Glmn to probe the accessibility of the RING-domain of Rbx1 in the presence or absence of CSN. Glmn can stably interact with the Rbx1 RING domain (Duda et al., 2012), and as with Cdc34, docking its structure onto Rbx1 suggested a steric clash with CSN (Figure S7B). Indeed, addition of Glmn resulted in inhibition of deneddylation, probably due to CSN’s inability to bind the Cul1~N8/Rbx1-Glmn complex. Consistently, an Arg547 to Ala mutant Glmn impaired for Rbx1 binding did not inhibit CSN deneddylation activity (Figure 6D).
CSN- and ubiquitination substrate-binding differentially regulate SCF neddylation
The close structural integration of CSN with the Cul1 CTD-Rbx1 region raises the possibility that CSN-binding might also affect neddylation. Indeed, addition of CSNCsn5H138A moderately decreased neddylation of Cul1SCE/Rbx1, and substantially reduced neddylation of Cul1SCE/Rbx1 complexed with Skp1/Skp2/Cks1 (Figure 6E). Addition of Skp1/Skp2/Cks1 as well as CyclinA/Cdk2 in the absence of CSNCsn5H138A, on the other hand, had no major inhibitory effect on Cul1 neddylation (data not shown). Importantly however, addition of CyclinA/Cdk2 in complex with in vitro phosphorylated p27 relieves the CSNCsn5H138A-mediated inhibition of neddylation (Figure 6E). These experiments support the notion that CSN inhibits neddylation of bound SCF complexes by blocking access to Ubc12, implying that CSN couples SCF neddylation with substrate availability.
Discussion
Recent studies showing that over 30% of cellular CRLs exist in stable complexes with CSN demonstrates that CRLs and CSN do not only interact as short-lived catalytic intermediates (Olma et al., 2009; Bennett et al., 2010). Here we report molecular models of CSN in complex with two fully assembled SCFs, which provide a structural framework for understanding stable CSN-SCF species, and combined with biochemical assays reveal unexpected non-catalytic modes of SCF regulation by CSN.
Molecular architecture of CSN
The proposed molecular model for CSN provides a first glimpse into the overall three-dimensional arrangement of the eight CSN subunits. We have experimentally validated the global locations of Csn2 and Csn5 and docked homology models into the corresponding segments (Figure S2D). Although the docking statistics for the remaining subunits appears of similarly high quality, we note that the models are only predictions and their exact conformation and/or orientation cannot be determined at the resolution of the present study. Nevertheless, our model is supported by the strong similarity between the density distribution of the CSN complex described here and that of its homologue, the lid of the 26S proteasome, whose subunit arrangement has been established at higher resolution (Figure 2A) (da Fonseca et al., 2012; Lander et al., 2012; Lasker et al., 2012). Furthermore, our model is supported by most available subunit interaction data. For example the subunit proximities match well with the tandem mass spectrometry analysis of recombinantly produced and reconstituted CSN (Sharon et al., 2009) as well as additional subunit contacts, including Csn1-Csn4 and Csn2-Csn4 (Serino et al., 2003; Tsuge et al., 2001). In our model, interaction between Csn1 and Csn2 involves their C-terminal winged helix domains in agreement with the experimental observation that their PCI domains are sufficient for incorporation into the complex, whereas their N-terminal portions do not co-purify with CSN (Tsuge et al., 2001; Yang et al., 2002). In fact a splice variant of Csn2, known as Alien, is missing the winged-helix domain and functions as a CSN-independent co-repressor of the thyroid hormone receptor (Tenbaum et al., 2003). Indeed, our model is consistent with the major interactions between the elongated PCI-domain containing subunits Csn1, 2, 3, and 4 being through their winged-helix domains (Figure 2). Interestingly, although the overall molecular architecture of CSN and the proteasome lid are strikingly similar, these two complexes appear to recognize their substrates by different structural elements. While CSN requires the N-terminus of Csn2 to position neddylated cullin CTD in proximity to Csn5 (Figure 3), the proteasome paralogue of Csn2, Rpn6, is a scaffold, linking the lid to the 20S core. Recognition of poly-ubiquitinated substrates relies on non-lid subunits such as Rpn10 and Rpn13.
Molecular determinants of the CSN-SCF interaction
No structural information for a CSN-CRL interaction was previously available. Extrapolating from X-ray crystallographic data for other CRL binding partners, one might have predicted either a relatively narrow interface, as observed between Cul1/Rbx1 and E2 enzymes and/or Dcn1, or a very broad interface engulfing most of the cullin as for Cand1 (Duda et al., 2011). However, our structural and biochemical analysis revealed that CSN localizes to both SCF functional sites. In our model, this two-pronged interaction between CSN and SCF involves primarily the Csn2/Csn5 region and the cullin CTD, as well as the Csn1/Csn3 region and the SCF substrate receptor (Figure 3).
Interestingly, our data revealed that Csn5 is dispensable for assembly of the remaining seven subunits, consistent with its peripheral location in the complex (Figure 2) and the fact that such Csn5-free CSN sub-complex can still bind SCF but not deneddylate it (Figure 4C and S4G). Furthermore, cullin neddylation stabilized the CSN-SCF interaction, even in the absence of Csn5 (Figure 4), suggesting that a Csn5-Nedd8 contact is not essential for the interaction. It seems likely that Csn2 and perhaps other CSN subunits preferentially bind to the neddylated conformation of the cullin CTD and/or Rbx1. Notably, although Csn5 harbors a catalytic JAMM motif, the subunit has to be assembled in the CSN complex to observe deneddylation (Sharon et al., 2009). Our molecular model thus suggests an unexpected explanation: Csn5 itself cannot recognize neddylated SCF substrates but apparently requires Csn2 and the rest of the complex to position the Nedd8 moiety attached to the CRL correctly in its active site. Moreover, this result implies that in contrast to Csn2, depletion of Csn5 or addition of chemical Csn5 inhibitors may not affect the formation rate or stability of CSN-CRL complexes in cells. Importantly, in our in vitro assays loss of Csn5 had no effect on the non-catalytic modes of CSN-mediated SCF-inhibition. We therefore caution against using Csn5 depletion or chemical inhibition as the sole means to inactivate CSN function.
We also identified CSN density segments modeled as Csn1 and Csn3 as approaching the SCF substrate receptors (SRs), consistent with binding of a beta-barrel SR and Csn1 (Tsuge et al., 2001). Indeed, we observed small structural differences in the densities assigned to the N-termini of Csn1 and Csn3 between the apo CSN and the CSN-SCF complexes (Figure S3A), which may indicate conformational re-arrangements on SCF binding and are reminiscent of the conformational changes reported for binding of the lid to the proteasome (Lander et al., 2012). However, this interaction appears less substantial than the Csn2-dependent Cul1 CTD interaction (Figure 3) and when examined by analytical size exclusion chromatography (Figure 4B) and pull-down assays (not shown), CSN does not appreciably associate with SRs in the absence of Cul1/Rbx1. Nevertheless, the presence of SRs increases Cul1/Rbx1 association with CSN (Figure 4). Intriguingly, CSN can preferentially form complexes with CRL subsets, determined partly by the identity of the SRs (Olma et al., 2009). Further studies are needed to shed light on the structural basis for such preferences and their functional implications.
CSN regulates SCF activity by multiple mechanisms
The functional significance of the prevalent CSN-CRL complexes found in cells has remained poorly understood. The structural models presented here suggest a surprisingly complex, multi-layered mechanism of CSN-mediated inhibition of SCF activity. Apart from deneddylation, our data suggest that CSN-binding exerts a double-pronged attack on both SCF functional sites by sterically hindering productive interactions of SCF with other factors. Indeed, we demonstrate that CSN competes with the Rbx1 RING-interactors Glmn (Duda et al., 2012; Tron et al., 2012), the ubiquitin E2 enzyme Cdc34 and the Nedd8-E2 Ubc12, as well as a ubiquitination substrate. Other RING-interactors have also been shown to interfere with CSN-mediated deneddylation (Emberley et al., 2012). Consistently, we showed that CSN-binding is sufficient to inhibit SCF-mediated ubiquitination, even when using catalytically-inactive CSNCsn5H138A complexes. Importantly, wild type CSN has been reported to inhibit CRL4 ubiquitination activity by an unknown, non-catalytic mechanism (Fischer et al., 2011), suggesting that CSN inhibition mediated by steric hindrance is conserved across the CRL family.
CSN and CAND1 differentially regulate CRL assembly and activity
Very little is known about the regulation of CSN activity. Unexpectedly, we observe that the assembly of Cul1/Rbx1 with substrate receptors also down-regulates the deneddylation activity of CSN, implying that SR-free cullins are better deneddylation substrates. This is consistent with findings that binding to SRs correlates with increased Cul1/Rbx1 neddylation in cells (Chew and Hagen, 2007; Kawakami et al., 2001). Taken together, our data provide the structural and mechanistic basis for a two-branched model to regulate SCF assembly and ubiquitination activity by CSN and CAND1, which may have evolved to regulate distinct Cul1/Rbx1 assemblies. CAND1 recognizes non-neddylated Cul1/Rbx1 complexes and its interaction with Cul1/Rbx1 is in a dynamic equilibrium with the Cul1/Rbx1-Skp1/F-box interaction, thereby facilitating re-assembly of non-neddylated cullins with substrate receptor modules (Bornstein et al., 2006; Schmidt et al., 2009; Siergiejuk et al., 2009) (Figure S4K; DCS and BAS, unpublished results). We presume that there are no protein factors favoring directionality in the equilibrium. On the other hand, CSN regulates the ubiquitination activity of assembled SCFs through the multiple mechanisms detailed above. Importantly, in addition to deneddylation, the CSN-SCF interaction inhibits Rbx1-Ubc12 mediated Cul1 neddylation, thereby sequestering and protecting assembled SCF complexes in an inactive state.
CRL activation requires dissociation of CSN-CRL complexes
Our work implies that the prevalent stably bound SCF-CSN complexes in cells (Bennett et al., 2010) are in a reciprocally inactivated state, which raises the question as to how SCF-CSN complexes dissociate. Formation of CSN-CRL4s plays regulatory roles in the cellular response to DNA damage (Groisman et al., 2003) – a complex cellular process, meticulously regulated by post-translational modifications. It is thus conceivable that for example phosphorylation of the CSN-CRL binding interface could influence complex formation. Indeed, several kinases associate with CSN, and have been shown to phosphorylate Csn2 and Csn7 (Sun et al., 2002; Uhle et al., 2003; Wilson et al., 2001). Moreover, Csn1, Csn3 and Csn8 have also been shown to be phosphorylation targets (Fang et al., 2008; Matsuoka et al., 2007). It will thus be important to investigate whether post-translational modifications could disrupt interaction interfaces between CSN and CRLs and thus play broad roles in negatively regulating their association.
Interestingly, our data suggest that CSN and the ubiquitination substrate p27 compete for SCFSkp2/Cks1-binding (Figure 5). Similarly, addition the SCFFbw7-substrate phospho-CyclinE/Cdk2 downregulates deneddylation (Emberley et al., 2012). Moreover, CRL4 binding to chromatin-located substrates has been proposed to trigger to CSN-CRL4 dissociation (Fischer et al., 2011). Thus, the levels of ubiquitination substrates might also regulate CSN-CRL dissociation in vivo. Consistent with this notion, substrate-bound CRLs are fully neddylated in cells (Read et al., 2000). Here we showed that substrate promotes Cul1 neddylation even in the presence of catalytically-inactive CSN. In cells, substrate-favored dissociation of CSN-SCF complexes could both allow neddylation, and further promote poly-ubiquitination and proteasomal degradation of the substrate. CSN binding may prevent a futile neddylation/de-neddylation cycle by coupling cullin neddylation to substrate availability. Unidirectionality may be further programmed by CSN’s failure to deneddylate substrate-bound SCFs. Our data suggest that the molecular architecture of CSN is better suited to stably bind fully assembled SCFs rather than SR-free cullins. We therefore propose that CSN acts as a sensor for catalytically assembled SCFs, and protects these complexes from disassembly, neddylation and ubiquitin-mediated degradation until critical amounts of the cognate ubiquitination substrate have accumulated. In this model, CSN would not only work as an inhibitor of SCF activity, but also promote SCF function by maintaining their assembly in a state that ensures rapid and efficient substrate turnover.
Experimental Procedures
Cloning, Expression, Purification and Immunoblotting
Full description is provided in the Supplemental Experimental Procedures.
Analytical Size Exclusion Chromatography
Protein concentrations were measured with a NanoDrop (Thermo Scientific) in 6 M guanidine. Equimolar (1 μM) CSN and Cul1/Rbx1 variants were mixed on ice for 10 min in 15 mM HEPES, pH 7.8, 150 mM NaCl, 1% glycerol, 1 mM DTT. Skp1/Skp2/Cks1, p27/CyclinA/Cdk2 and/or CAND1 were added in slight excess (1.2 μM). Nedd8, the WHB domain of Cul1 were added at 5 μM. 200 μl protein mixture was injected onto a Superose 6 10/300 size exclusion column (GE Healthcare); the run was performed at 4 °C, collecting 48 500 μl fractions. Aliquots were analyzed by SDS PAGE on 4–12% or 12% gels and immunoblotting.
Neddylation and deneddylation assays
Cul1/Rbx1 purified from insect cells (Enchev et al., 2010) and all other Cul1/Rbx1 constructs were neddylated by incubating 8 μM Cul1fl/Rbx1; 500 nM APPBP1-Uba3; 1 μM Ubc12; 10 μM Nedd8 at room temperature for 10 min in 50 mM Tris/HCl, pH 7.6, 100 mM NaCl, 2.5 mM MgCl2, 150 μM ATP. After addition of 10 mM DTT, products were purified over a Superdex 200 column (Duda et al., 2008). Neddylation assays involving CSNCsn5H138A addition contained 25 nM APPBP1/Uba3, 125 nM Ubc12, 125 nM Cul1/Rbx1, +/− 125 nM Skp1/Skp2/Cks1, +/− 1 μM or 3 μM CyclinA/Cdk2 or CyclinA/Cdk2/p-p27 in 25mM Tris pH 7.0, 50mM NaCl, 10mM MgCl2, 1.5mM ATP, incubated on ice for 20 min. CSNCsn5H138A was then added at 500 nM and incubated on ice for 5 min. Reactions were initiated by addition of 20 μM Nedd8. Aliquots were analyzed by SDS PAGE on 4–12% gels and immunoblotting. Deneddylation assays were performed in 50mM Tris, 50mM NaCl pH=7.6 at room temperature using 2 nM CSN variants and 150 nM neddylated Cul1/Rbx1 variants and Skp1/F-box. 800nM CyclinA/Cdk2, CyclinA/Cdk2/p27, or CyclinA/Cdk2/p-p27 was added to ensure complex formation with Skp1/Skp2.
Ubiquitination assays
Ubiquitination assays were performed at room temperature with 200 nM SCF variants and 100 nM UbE1, 500 nM Cdc34 and 50 μM ubiquitin. p27 was phosphorylated in vitro for 30 min at 30 °C by mixing 4 μM CyclinA/Cdk2 and p27 in 40 mM Tris/HCl, pH 7.6, 10 mM MgCl2, 1 mM ATP and 1 mM DTT. 200 nM phospho-p27/CyclinA/Cdk2 or 5 μM biotinylated CyclinE phospho-peptide were used as substrates. 500 nM (Figure 6B and S6C) or 200 nM (Figure S6D) CSN variants were added as indicated. Aliquots were analyzed by SDS PAGE on 4–12% gels immunoblotting.
Di-ubiquitin formation assays
10 μM Cdc34 was loaded with 20 μM lysine-less [32P]-ubiquitin ([32P]-UbR7) with 100 nM ubiquitin E1 in 50 mM Hepes, pH 7.5, 200 mM NaCl, 10 mM MgCl2, 1.5 mM ATP at room temperature for 25 min. The loading reaction was quenched by the addition of EDTA to 50 mM. Discharge reactions were performed in 25 mM Tris pH7.6, 100 mM NaCl, 50 mM EDTA. 250 nM ubiquitin, 500 nM Cul1~N8/Rbx1 +/− a 1:1 mix of Skp1/F-box were incubated on ice for 30 min to equilibrate SCF complex formation. The mixtures were placed at room temperature, and after a 5-minute incubation, the indicated CSN variants were added to a final concentration of 1.25 μM followed by an additional 5-min incubation at room temperature. Discharge was initiated by the addition of Cdc34~[32P]UbR7 to a final concentration of 400 nM. Aliquots were analyzed on 4–12% gels, dried, and exposed to a phosphoimager screen, scanned on a StormImager and quantified using ImageQuant TL v2003.02.
Electron Microscopy and Single Particle Analysis
Electron microscopy data collection and analysis were similar to that described in (Enchev et al., 2010). Detailed description is given in the Supplemental Experimental Procedures.
Supplementary Material
Highlights.
Electron microscopy structures of CSN with neddylated SCFSkp2/Cks1 and SCFFbw7
Molecular models for CSN and CSN-SCF complexes
SCF assembly state influences CSN binding and deneddylation activity
Mechanistic insights into non-catalytic CSN inhibition of SCF ubiquitination activity
Acknowledgments
We thank D. Barford for providing access to equipment and laboratory space, F. Beuron for EM support and R. Deshaies and A. Smith for critical reading. RIE is funded by an ETH/Marie Curie fellowship. EPM is supported by the ICR, MP by the Swiss National Science Foundation, an ERC senior award and the ETHZ, and BAS by ALSAC, HHMI, and NIH R01GM069530. The authors declare no competing financial interests.
Footnotes
Accession Numbers
CSNCsn5H138A-SCF~N8Skp2/Cks1 is deposited in the EM Data Bank under accession code EMD-2173; CSNCsn5H138A-SCF~N8Fbw7 under EMD-2174; CSNΔCsn5-SCF~N8Skp2/Cks1 under EMD-2175; CSN under EMD-2176 and CSNCsn2ΔN1–269 under EMD-2177.
Supplemental Information includes seven Figures and Supplemental Experimental Procedures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bennett EJ, Rush J, Gygi SP, Harper JW. Dynamics of cullin-RING ubiquitin ligase network revealed by systematic quantitative proteomics. Cell. 2010;143:951–965. doi: 10.1016/j.cell.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein G, Ganoth D, Hershko A. Regulation of neddylation and deneddylation of cullin1 in SCFSkp2 ubiquitin ligase by F-box protein and substrate. Proc Natl Acad Sci U S A. 2006;103:11515–11520. doi: 10.1073/pnas.0603921103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosu DR, Kipreos ET. Cullin-RING ubiquitin ligases: global regulation and activation cycles. Cell division. 2008;3:7. doi: 10.1186/1747-1028-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese MF, Scott DC, Duda DM, Grace CR, Kurinov I, Kriwacki RW, Schulman BA. A RING E3-substrate complex poised for ubiquitin-like protein transfer: structural insights into cullin-RING ligases. Nature structural & molecular biology. 2011;18:947–949. doi: 10.1038/nsmb.2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew EH, Hagen T. Substrate-mediated regulation of cullin neddylation. J Biol Chem. 2007;282:17032–17040. doi: 10.1074/jbc.M701153200. [DOI] [PubMed] [Google Scholar]
- Cope GA, Deshaies RJ. COP9 signalosome: a multifunctional regulator of SCF and other cullin-based ubiquitin ligases. Cell. 2003;114:663–671. doi: 10.1016/s0092-8674(03)00722-0. [DOI] [PubMed] [Google Scholar]
- Cope GA, Suh GS, Aravind L, Schwarz SE, Zipursky SL, Koonin EV, Deshaies RJ. Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science. 2002;298:608–611. doi: 10.1126/science.1075901. [DOI] [PubMed] [Google Scholar]
- da Fonseca PC, He J, Morris EP. Molecular model of the human 26S proteasome. Molecular cell. 2012;46:54–66. doi: 10.1016/j.molcel.2012.03.026. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annual review of biochemistry. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- Dessau M, Halimi Y, Erez T, Chomsky-Hecht O, Chamovitz DA, Hirsch JA. The Arabidopsis COP9 signalosome subunit 7 is a model PCI domain protein with subdomains involved in COP9 signalosome assembly. Plant Cell. 2008;20:2815–2834. doi: 10.1105/tpc.107.053801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell. 2008;134:995–1006. doi: 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda DM, Olszewski JL, Tron AE, Hammel M, Lambert LJ, Waddell MB, Mittag T, Decaprio JA, Schulman BA. Structure of a Glomulin-RBX1-CUL1 Complex: Inhibition of a RING E3 Ligase through Masking of Its E2-Binding Surface. Molecular cell. 2012;47:371–382. doi: 10.1016/j.molcel.2012.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda DM, Scott DC, Calabrese MF, Zimmerman ES, Zheng N, Schulman BA. Structural regulation of cullin-RING ubiquitin ligase complexes. Current opinion in structural biology. 2011;21:257–264. doi: 10.1016/j.sbi.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emberley ED, Mosadeghi R, Deshaies RJ. Deconjugation of Nedd8 from Cul1 is directly regulated by Skp1-Fbox and substrate, and CSN inhibits deneddylated SCF by a non-catalytic mechanism. The Journal of biological chemistry. 2012 doi: 10.1074/jbc.M112.352484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enchev RI, Schreiber A, Beuron F, Morris EP. Structural insights into the COP9 signalosome and its common architecture with the 26S proteasome lid and eIF3. Structure. 2010;18:518–527. doi: 10.1016/j.str.2010.02.008. [DOI] [PubMed] [Google Scholar]
- Fang L, Wang X, Yamoah K, Chen PL, Pan ZQ, Huang L. Characterization of the human COP9 signalosome complex using affinity purification and mass spectrometry. J Proteome Res. 2008;7:4914–4925. doi: 10.1021/pr800574c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer ES, Scrima A, Bohm K, Matsumoto S, Lingaraju GM, Faty M, Yasuda T, Cavadini S, Wakasugi M, Hanaoka F, et al. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell. 2011;147:1024–1039. doi: 10.1016/j.cell.2011.10.035. [DOI] [PubMed] [Google Scholar]
- Goldenberg SJ, Cascio TC, Shumway SD, Garbutt KC, Liu J, Xiong Y, Zheng N. Structure of the Cand1-Cul1-Roc1 complex reveals regulatory mechanisms for the assembly of the multisubunit cullin-dependent ubiquitin ligases. Cell. 2004;119:517–528. doi: 10.1016/j.cell.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113:357–367. doi: 10.1016/s0092-8674(03)00316-7. [DOI] [PubMed] [Google Scholar]
- Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell. 2007;26:131–143. doi: 10.1016/j.molcel.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Hao B, Zheng N, Schulman BA, Wu G, Miller JJ, Pagano M, Pavletich NP. Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Mol Cell. 2005;20:9–19. doi: 10.1016/j.molcel.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Kawakami T, Chiba T, Suzuki T, Iwai K, Yamanaka K, Minato N, Suzuki H, Shimbara N, Hidaka Y, Osaka F, et al. NEDD8 recruits E2-ubiquitin to SCF E3 ligase. Embo J. 2001;20:4003–4012. doi: 10.1093/emboj/20.15.4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiger G, Saha A, Lewis S, Kuhlman B, Deshaies RJ. Rapid E2–E3 assembly and disassembly enable processive ubiquitylation of cullin-RING ubiquitin ligase substrates. Cell. 2009;139:957–968. doi: 10.1016/j.cell.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz T, Ozlu N, Rudolf F, O’Rourke SM, Luke B, Hofmann K, Hyman AA, Bowerman B, Peter M. The conserved protein DCN-1/Dcn1p is required for cullin neddylation in C. elegans and S. cerevisiae. Nature. 2005;435:1257–1261. doi: 10.1038/nature03662. [DOI] [PubMed] [Google Scholar]
- Lander GC, Estrin E, Matyskiela ME, Bashore C, Nogales E, Martin A. Complete subunit architecture of the proteasome regulatory particle. Nature. 2012;482:186–191. doi: 10.1038/nature10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasker K, Forster F, Bohn S, Walzthoeni T, Villa E, Unverdorben P, Beck F, Aebersold R, Sali A, Baumeister W. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:1380–1387. doi: 10.1073/pnas.1120559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyapina S, Cope G, Shevchenko A, Serino G, Tsuge T, Zhou C, Wolf DA, Wei N, Deshaies RJ. Promotion of NEDD-CUL1 conjugate cleavage by COP9 signalosome. Science. 2001;292:1382–1385. doi: 10.1126/science.1059780. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- Olma MH, Roy M, Le Bihan T, Sumara I, Maerki S, Larsen B, Quadroni M, Peter M, Tyers M, Pintard L. An interaction network of the mammalian COP9 signalosome identifies Dda1 as a core subunit of multiple Cul4-based E3 ligases. J Cell Sci. 2009;122:1035–1044. doi: 10.1242/jcs.043539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pick E, Hofmann K, Glickman MH. PCI complexes: Beyond the proteasome, CSN, and eIF3 Troika. Mol Cell. 2009;35:260–264. doi: 10.1016/j.molcel.2009.07.009. [DOI] [PubMed] [Google Scholar]
- Read MA, Brownell JE, Gladysheva TB, Hottelet M, Parent LA, Coggins MB, Pierce JW, Podust VN, Luo RS, Chau V, et al. Nedd8 modification of cul-1 activates SCF(beta(TrCP))-dependent ubiquitination of IkappaBalpha. Mol Cell Biol. 2000;20:2326–2333. doi: 10.1128/mcb.20.7.2326-2333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nature protocols. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo AA, Jeffrey PD, Patten AK, Massague J, Pavletich NP. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382:325–331. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
- Saha A, Deshaies RJ. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol Cell. 2008;32:21–31. doi: 10.1016/j.molcel.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Yoshikawa A, Yamagata A, Mimura H, Yamashita M, Ookata K, Nureki O, Iwai K, Komada M, Fukai S. Structural basis for specific cleavage of Lys 63-linked polyubiquitin chains. Nature. 2008;455:358–362. doi: 10.1038/nature07254. [DOI] [PubMed] [Google Scholar]
- Scheel H, Hofmann K. Prediction of a common structural scaffold for proteasome lid, COP9-signalosome and eIF3 complexes. BMC Bioinformatics. 2005;6:71. doi: 10.1186/1471-2105-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MW, McQuary PR, Wee S, Hofmann K, Wolf DA. F-box-directed CRL complex assembly and regulation by the CSN and CAND1. Mol Cell. 2009;35:586–597. doi: 10.1016/j.molcel.2009.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman BA, Carrano AC, Jeffrey PD, Bowen Z, Kinnucan ER, Finnin MS, Elledge SJ, Harper JW, Pagano M, Pavletich NP. Insights into SCF ubiquitin ligases from the structure of the Skp1–Skp2 complex. Nature. 2000;408:381–386. doi: 10.1038/35042620. [DOI] [PubMed] [Google Scholar]
- Scott DC, Monda JK, Bennett EJ, Harper JW, Schulman BA. N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science. 2011;334:674–678. doi: 10.1126/science.1209307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serino G, Su H, Peng Z, Tsuge T, Wei N, Gu H, Deng XW. Characterization of the last subunit of the Arabidopsis COP9 signalosome: implications for the overall structure and origin of the complex. Plant Cell. 2003;15:719–731. doi: 10.1105/tpc.009092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon M, Mao H, Boeri Erba E, Stephens E, Zheng N, Robinson CV. Symmetrical modularity of the COP9 signalosome complex suggests its multifunctionality. Structure. 2009;17:31–40. doi: 10.1016/j.str.2008.10.012. [DOI] [PubMed] [Google Scholar]
- Siergiejuk E, Scott DC, Schulman BA, Hofmann K, Kurz T, Peter M. Cullin neddylation and substrate-adaptors counteract SCF inhibition by the CAND1-like protein Lag2 in Saccharomyces cerevisiae. EMBO J. 2009;28:3845–3856. doi: 10.1038/emboj.2009.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruck C, Strohmaier H, Watson M, Smith AP, Ryan A, Krek TW, Reed SI. A CDK-independent function of mammalian Cks1: targeting of SCF(Skp2) to the CDK inhibitor p27Kip1. Molecular cell. 2001;7:639–650. doi: 10.1016/s1097-2765(01)00210-6. [DOI] [PubMed] [Google Scholar]
- Sun Y, Wilson MP, Majerus PW. Inositol 1,3,4-trisphosphate 5/6-kinase associates with the COP9 signalosome by binding to CSN1. J Biol Chem. 2002;277:45759–45764. doi: 10.1074/jbc.M208709200. [DOI] [PubMed] [Google Scholar]
- Tenbaum SP, Juenemann S, Schlitt T, Bernal J, Renkawitz R, Munoz A, Baniahmad A. Alien/CSN2 gene expression is regulated by thyroid hormone in rat brain. Dev Biol. 2003;254:149–160. doi: 10.1016/s0012-1606(02)00023-4. [DOI] [PubMed] [Google Scholar]
- Tron AE, Arai T, Duda DM, Kuwabara H, Olszewski JL, Fujiwara Y, Bahamon BN, Signoretti S, Schulman BA, DeCaprio JA. The glomuvenous malformation protein Glomulin binds Rbx1 and regulates cullin RING ligase-mediated turnover of Fbw7. Molecular cell. 2012;46:67–78. doi: 10.1016/j.molcel.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuge T, Matsui M, Wei N. The subunit 1 of the COP9 signalosome suppresses gene expression through its N-terminal domain and incorporates into the complex through the PCI domain. J Mol Biol. 2001;305:1–9. doi: 10.1006/jmbi.2000.4288. [DOI] [PubMed] [Google Scholar]
- Uhle S, Medalia O, Waldron R, Dumdey R, Henklein P, Bech-Otschir D, Huang X, Berse M, Sperling J, Schade R, et al. Protein kinase CK2 and protein kinase D are associated with the COP9 signalosome. Embo J. 2003;22:1302–1312. doi: 10.1093/emboj/cdg127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MP, Sun Y, Cao L, Majerus PW. Inositol 1,3,4-trisphosphate 5/6-kinase is a protein kinase that phosphorylates the transcription factors c-Jun and ATF-2. J Biol Chem. 2001;276:40998–41004. doi: 10.1074/jbc.M106605200. [DOI] [PubMed] [Google Scholar]
- Yamoah K, Oashi T, Sarikas A, Gazdoiu S, Osman R, Pan ZQ. Autoinhibitory regulation of SCF-mediated ubiquitination by human cullin 1’s C-terminal tail. Proc Natl Acad Sci U S A. 2008;105:12230–12235. doi: 10.1073/pnas.0806155105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Menon S, Lykke-Andersen K, Tsuge T, Di X, Wang X, Rodriguez-Suarez RJ, Zhang H, Wei N. The COP9 signalosome inhibits p27(kip1) degradation and impedes G1-S phase progression via deneddylation of SCF Cul1. Curr Biol. 2002;12:667–672. doi: 10.1016/s0960-9822(02)00791-1. [DOI] [PubMed] [Google Scholar]
- Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416:703–709. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]
- Zheng N, Wang P, Jeffrey PD, Pavletich NP. Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell. 2000;102:533–539. doi: 10.1016/s0092-8674(00)00057-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.