

Abstract
In nondiabetic rat models of renal disease, angiotensin II (Ang II) perpetuates podocyte injury and promotes progression to end-stage kidney disease. Herein, we wanted to explore the role of Ang II in diabetic nephropathy by a translational approach spanning from in vitro to in vivo rat and human studies, and to dissect the intracellular pathways involved. In isolated perfused rat kidneys and in cultured human podocytes, Ang II down-regulated nephrin expression via Notch1 activation and nuclear translocation of Snail. Hairy enhancer of split-1 was a Notch1-downstream gene effector that activated Snail in cultured podocytes. In vitro changes of the Snail/nephrin axis were similar to those in renal biopsy specimens of Zucker diabetic fatty rats and patients with advanced diabetic nephropathy, and were normalized by pharmacological inhibition of the renin-angiotensin system. Collectively, the present studies provide evidence that Ang II plays a relevant role in perpetuating glomerular injury in experimental and human diabetic nephropathy via persistent activation of Notch1 and Snail signaling in podocytes, eventually resulting in down-regulation of nephrin expression, the integrity of which is crucial for the glomerular filtration barrier.
During the past 25 years, the burden of diabetes mellitus has almost doubled worldwide, and projection for the future is alarming.1 The increasing diabetes prevalence will also inevitably result in increasing proportions of deaths from cardiovascular disease2 and increased prevalence and associated consequences of other complications, such as nephropathy, a major cause of illness in diabetes.3 The nature and amount of glomerular capillary protein trafficking and resulting proteinuria are prognostic markers for cardiovascular and renal outcomes.
As documented in animal models of streptozotocin-induced diabetes,4 the enhanced intraglomerular capillary pressure is the initiating early event of glomerular damage and proteinuria in this disease. Ficoll clearance studies in diabetic rats found that enhanced intraglomerular capillary pressure stretched glomerular walls, which further led to direct glomerular cell injury and impaired the selective function of the glomerular capillary, an effect explained by the appearance of large pores that exceed the sizes observed in normal conditions and allowed increased filtration of plasma proteins.5,6 Mechanical strain also increases angiotensin II (Ang II) production and the expression of angiotensin type 1 receptors in podocytes7 that line the outer aspect of the glomerular capillary barrier in vivo,8,9 potentially contributing to further sustaining the glomerular hypertension-induced damage in diabetes. However, there is also evidence that, independent of its hemodynamic effect, Ang II may directly impair the glomerular barrier-sieving function, possibly through inhibition of nephrin expression,10,11 the essential protein component of the glomerular slit diaphragm, an elaborate multiprotein network bridging adjacent podocyte foot processes.10,12 This observation has been confirmed in previous studies in diabetic animals showing that blockade of Ang II synthesis/activity preserved in the glomeruli the expression of nephrin and prevented overt proteinuria.6,13 Thus, in diabetes, a pathogenetic relationship between Ang II and early proteinuria via functional podocyte alteration through modulation of nephrin protein level has been suggested. Nevertheless, whether Ang II would contribute to establish persistent podocyte dysfunction after the initial cell injury induced by glomerular hypertension, eventually allowing disease progression in diabetic kidneys, remains ill defined. This possibility is supported by recent studies in nondiabetic rat models of progressive renal disease showing that Ang II is essential to perpetuate podocyte injury and promote progression to end-stage kidney disease.14
Moreover, the molecular signaling pathways by which Ang II down-regulates the nephrin level in the podocytes in diabetes are unknown. Evidence is available that the expression of the intracellular domain of Notch1, a family of transmembrane receptors that mediate transcription of target genes,15 is increased in podocytes of diabetic mice.16,17 Because Notch1 operates by controlling the expression of the transcription factor, Snail,18 which has been shown to repress nephrin expression in rodent podocytes,19 we hypothesize that the Notch1/Snail pathway could be a key intracellular signal coupling Ang II to nephrin in diabetes.
In the studies reported herein, we addressed these questions and showed consistently in in vitro cell culture, in vivo rat model of diabetic nephropathy, and in patients with type 2 diabetes that Ang II induced persistent down-regulation of nephrin expression in podocytes via Notch1/Snail signals. Taken together, our data implicate an important role for the Ang II Notch1/Snail axis in the pathogenesis of diabetic nephropathy.
Materials and Methods
Cell Culture and Experimental Design
Conditionally immortalized human podocytes were grown and differentiated as previously described.20 Podocytes were maintained for 24 hours in serum-free conditions and then incubated with medium or with 100 nmol/L Ang II (Calbiochem, La Jolla, CA) at different time intervals. Nephrin protein expression and mRNA transcript levels were evaluated in Ang II–treated podocytes after 3 hours by immunofluorescence and real-time RT-PCR, respectively. γ-Secretase inhibitor (GSI) X (1 μmol; Calbiochem), was added to the cells 1 hour before and during the incubation with Ang II. The intracellular domain of Notch1 (ICN1) and Snail nuclear translocation were assessed by immunofluorescence analysis at different time intervals. In additional experiments, Snail mRNA and protein expression levels were evaluated in podocytes transfected with control nontarget small-interfering RNA (siNULL) or siRNA specific for hairy enhancer of split-1 (siHES1).
Quantitative Real-Time PCR
Total RNA was isolated from human podocytes by TRIzol Reagent (Invitrogen, Life Technologies Italia, Monza, Italy), according to the manufacturer's instruction. Contaminating genomic DNA was removed by RNase-free DNase (Promega, Ingelheim, Germany) for 1 hour at 37°C. Purified RNA (2 μg) was reverse transcribed using 50 ng of random hexamers and 50 U of SuperScript II Reverse Transcriptase (Invitrogen) for 1 hour at 42°C. No enzyme was added for reverse transcriptase negative controls.
Amplification was performed on a 7300 Real Time PCR System with SYBR Green PCR Master Mix (Applied Biosystems, Life Technologies, Monza, Italia), according to the manufacturer's protocol. The following primers were used: 300 nmol/L human NEPHRIN forward 5′-CGTCTTGCTGAGGGCATCTC-3′ and reverse 5′-TGACTCGGTCCTCTTCCGAC-3′; 300 nmol/L human SNAIL forward 5′-TGCCCTGCGTCTGCGGAACC-3′ and reverse 5′-GGGCTTCTCGCCAGTGTGGG-3′; 300 nmol/L human HES1 forward 5′-AGCACAGAAAGTCATCAAAGCC-3′ and reverse 5′-ATGCCGCGAGCTATCTTTCT-3′; and 50 nmol/L 18S forward 5′-ACGGCTACCACATCCAAGGA-3′ and reverse 5′-CGGGAGTGGGGTAATTTGCG-3′. The ΔΔCT technique was used to calculate cDNA content in each sample using the cDNA expression in human podocyte control as calibrator.
Immunofluorescence Analysis on Cultured Cells
Podocytes were fixed in 2% paraformaldehyde (Electron Microscopy Science, Hatfield, PA) and 4% sucrose (Sigma-Aldrich, Milan, Italy) and then permeabilized with 0.3% Triton X-100 (Sigma-Aldrich). After blocking the non-specific binding sites, cells were incubated with rabbit polyclonal anti-nephrin antibody (1:200; Abcam, Cambridge, UK), rabbit polyclonal anti–Notch1-cleaved Val1744 antibody (1:200; Abcam) that detects endogenous levels of fragment of activated Notch1 (ICN1), or a rabbit polyclonal anti-Snail antibody (1:200; Abcam). After washing in PBS, cells were incubated with a goat anti-rabbit CY3-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) and then counterstained with DAPI (Sigma-Aldrich). Coverslips were mounted using Dako Fluorescence Mounting Medium (Dako Corporation, Carpentaria, CA). Samples were examined under confocal inverted laser microscopy (LSM 510 Meta; Zeiss, Jena, Germany). Quantification of ICN1 or Snail-positive nuclei per total number of cell nuclei, identified by DAPI staining, was assessed in 15 random fields per sample (n = 3 experiments).
Silencing of the HES1 Gene
Podocytes were transfected with 30 pmol Silencer Select predesigned siRNA specific for the HES1 sequence, NM_005524.3 (s6920, 5 nmol; Ambion, Life Technologies Italia, Monza, Italy), or with control nontarget siRNA (Silencer Select Negative Control 2 siRNA; Ambion) using the Amaxa Human NHDF Nucleofector Kit (VPE-1001; Lonza, Basel, Switzerland), according to the manufacturer's protocol. HES1 expression was evaluated 48 hours after transfection.
Isolated Perfused Rat Kidney Model
Isolated kidneys from adult male Sprague-Dawley rats (Charles River Laboratories Italia S.r.l., Calco, Italy) were perfused in a recirculating system at a constant pressure of 100 mmHg with an artificial cell-free medium, as previously described.21 Each kidney was allowed 15 to 20 minutes to equilibrate after beginning the perfusion. A 10-minute baseline urine collection and a perfusate sample were obtained at the end of the clearance period. Then, 16 ng/minute Ang II or vehicle (n = 4 for each group) was infused into the renal artery for 10 minutes, and both urine and perfusate samples were collected at the end of the clearance for creatinine assay. Urinary protein concentration and glomerular filtration rate were evaluated. The infusion time and Ang II concentration were chosen on the basis of a previous study showing that, after 10 minutes of infusion of 16 ng/minute Ang II, the urinary protein excretion rate was already significantly increased in respect to baseline values and preceded the decline of glomerular filtration rate.22 At the end of perfusion, kidney specimens were collected and processed for immunofluorescence evaluation.
In Vivo Study in Diabetic Rats
Two-month-old male Zucker diabetic fatty (ZDF) rats (ZDF/Gmi-fa/fa) and aged-matched nondiabetic lean rats (ZDF/Gmi-fa/+; Charles River Laboratories Italia S.r.l., Calco, Italy), were kept on a 12-hour light/dark cycle with free access to water. ZDF rats were maintained on Purina 5008 rat chow (protein, 26.8 kcal%; carbohydrate, 56.4 kcal%; and fat, 16.7 kcal%) to accelerate onset of diabetes. At 4 months, ZDF rats were randomized to receive the following daily, until month 8: vehicle (water, n = 3); and 1 mg/kg ramipril in the drinking water (n = 4). Four lean rats were used as controls. Systolic blood pressure was measured by tail plethysmography. Urinary protein excretion was measured by the Coomassie Blue method using a Cobas Mira plus autoanalyzer (Roche Diagnostic System, Basel, Switzerland). At the end of the study, the removed kidneys were fixed in Duboscq-Brazil and embedded in paraffin. Sections (3 μm thick; Ultrotome V, LKB, Bromma, Sweden) were stained with H&E and PAS reagents. At least 100 glomeruli were examined for each animal. The extent of glomerular damage was expressed as percentage of sclerotic glomeruli. Tubular changes (atrophy, casts, and dilation) were graded from 0 to 4 (0, no changes; 1, changes affecting <25% of the sample; 2, changes affecting 25% to ≤50% of the sample; 3, changes affecting >50% to ≤75% of the sample; and 4, changes affecting >75% to 100% of the sample). Renal biopsy specimens were simultaneously processed and analyzed by the same pathologist (E.G.), who was unaware of the nature of the experimental groups.
Animal Care
Animal care and treatment were conducted according to institutional guidelines in compliance with national (Decreto Legislativo number 116, Gazzetta Ufficiale supplement 40, 18 Febbraio 1992, Circolare number 8, Gazzetta Ufficiale 14 Luglio 1994) and international (European Economic Community Council Directive 86/609, OJL358-1, December 1987; Guide for the Care and Use of Laboratory Animals, US National Research Council, 1996) laws and policies. Animal studies were submitted to and approved by the Institutional Animal Care and Use Committee of Mario Negri Institute (Milan, Italy).
Patient Enrollment
Nineteen subjects with a known history of type 2 diabetes mellitus, diagnosed according to the criteria of the World Health Organization, were enrolled in the study. Eight patients not receiving any renin-angiotensin system blocking drugs as part of their anti-hypertensive therapy and 11 patients treated with the angiotensin-converting enzyme (ACE) inhibitor, ramipril (treatment started at 2.5 mg/day and then up titrated to 5, 10, and 15 mg/day according to renal function and tolerability), from at least 1 month, were studied. The predefined inclusion criteria were as follows: renal tissues obtained from biopsy specimens of the patients who were admitted for diagnostic reasons to the Nephrology Unit (Azienda Ospedaliera Papa Giovanni XXIII, Bergamo, Italy) (serum creatinine, <4 mg/dL; proteinuria, >0.5 g/24 hours), for at least 6 months; histological diagnosis of diabetic glomerulopathy without other concomitant glomerular disease; and no evidence of renovascular disease, obstructive uropathy, systemic disease, and other possibly confounding conditions. Five age- and sex-matched non-proteinuric nondiabetic controls undergoing nephrectomy for kidney adenocarcinoma, with proteinuria <0.5 g/24 hours and without histological evidence of glomerular disease, were also included. Written informed consent was obtained from all of the patients enrolled in the study.
Morphometric Analysis at Transmission Electron Microscopy
Renal specimens were fixed with 2.5% glutaraldehyde in 0.1 mol/L cacodylate buffer (pH 7.4) for 4 hours at 4°C, washed in cacodylate buffer, and then post-fixed with a solution of tannic acid–glutaraldehyde (1% tannic acid and 1% glutaraldehyde in 0.1 mol/L phosphate buffer, pH 7.4) for 2 hours to increase the contrast and definition of extracellular structures in the glomerulus and reveal the isoporous substructure of the slit diaphragm.23 Kidney fragments were then processed, and ultrastructural evaluations were performed by transmission electron microscopy (Morgagni 268D; Philips, Brno, Czech Republic). An average of 50 images of three glomeruli per animal was digitized (final magnification, ×56,000) and processed with image-processing software (NIH Image, version 1.6; NIH, Bethesda, MD). For each image, the length of the glomerular basement membrane was calculated using a computer-assisted morphometric unit, as previously described.23 Epithelial filtration slit frequency was evaluated as number of slits observed/micrometer of glomerular basement membrane length. The presence of the filamentous structure in the slits observed in each animal was evaluated and expressed as a percentage.
Podocyte Count
Podocytes were identified using the antibody directed against WT1, the podocyte-specific marker.13 All glomeruli included in each biopsy specimen (from 3 to 15) were acquired by confocal laser-scanning microscope (LSM 510 Meta) and examined by an observer (P.R.) unaware of the identity of samples. The estimation of the average number of podocytes per glomerulus was determined by using morphometric analysis proposed by Weibel,24 as previously reported.25
Immunohistochemical Analysis on Renal Tissues
Acetone-fixed cryosections were subjected to antigen retrieval and incubated with goat anti-rat/human nephrin (1:100; Santa Cruz Biotechnology Inc., Santa Cruz, CA), rabbit anti-rat/human Snail (1:200; Abcam), rabbit or mouse anti-rat/human WT1 (1:50; Santa Cruz Biotechnology Inc.), and rabbit polyclonal to Notch1-cleaved Val1744 antibody (1:50; Abcam), followed by the specific fluorescein isothiocyanate– or Cy3-conjugated secondary antibodies. Cell nuclei were labeled by DAPI, and the glomerular tuft was labeled by wheat germ agglutinin. Fluorescence was examined by an inverted confocal laser-scanning microscope (LSM 510 Meta). For nephrin and Snail immunofluorescence, the signal intensity was graded on a scale of 0 to 3 (nephrin expression: 0, no staining; 1, highly fragmented; 2, fragmented; and 3, linear signal; Snail expression: 0, no staining; 1, spotted; 2, weak; and 3, strong diffusion).
Duboscq-Brazil fixed and paraffin-embedded human kidney sections (3 μm thick) were used. After antigen retrieval, the sections were incubated with the primary antibodies to Notch1-cleaved Val1744, human zonula occludens protein (ZO)-1 (1:25; Zymed Laboratories, San Francisco, CA), and human CD2AP (1:100; Santa Cruz Biotechnology Inc.). Then, specimens were incubated with the biotinylated species-specific secondary antibodies, avidin-biotin peroxidase complex solution, and developed with diaminobenzidine. Slides were counterstained with hematoxylin and observed using light microscopy. Seven glomeruli, on average, in each sample were analyzed by an investigator (P.R.) unaware of biopsy groups. For each glomerulus, Notch1 activation was expressed as percentage of ICN1-positive cell nuclei per total number of cell nuclei identified in a hematoxylin-stained kidney section. Negative controls were obtained by omitting the primary antibody on adjacent sections. All renal tissue sections were analyzed by the same pathologist (P.R.), in a single-blinded manner.
Statistical Analysis
Results are reported as means ± SEM. Statistical comparisons were analyzed by the nonparametric Kruskal-Wallis test for multiple comparisons or analysis of variance with Bonferroni correction or Student's t-test for unpaired data. Linear regression analysis was performed between immunohistochemistry scores for nephrin and degree of proteinuria. P < 0.05 was considered significant. In the correlation analysis, a Spearman ρ correlation coefficient was used.
Results
Ang II Affects Glomerular Filtration Function and Structure in Isolated Perfused Rat Kidneys by Lowering Nephrin
The role of Ang II in progressive disruption of glomerular perm-selectivity to macromolecules is well established in ex vivo models, in which Ang II enhances filtration of molecules with radii ≥34 Å and increases protein excretion.21,22 To investigate whether Ang II induces proteinuria by direct modulation of glomerular nephrin expression and eventually podocyte foot process injury, we used an ex vivo model of isolated perfused rat kidney.21 The reasoning behind this approach is that, with this tool, hemodynamic modifications can be minimized by artificially maintaining a constant renal perfusion pressure of 100 mmHg throughout the experiment; thus, changes in glomerular capillary permeability are more likely to correspond to a direct effect of Ang II on the capillary wall function. Ang II was infused during 10 minutes into the renal artery of the isolated kidney preparation. A group of kidneys infused with vehicle was taken for comparison. Consistent with our previous reports,21,22 Ang II significantly increased urinary protein excretion compared with baseline values (32.4 ± 4.1 versus 11.7 ± 1.6 μg/minute; P < 0.01) (Figure 1A). At variance, vehicle infusion did not change the protein excretion rate (13.4 ± 1.9 μg/minute) that was comparable to that found at baseline (Figure 1A). No significant change in glomerular filtration rate was observed after Ang II (baseline, 0.93 ± 0.10 mL/minute; post-Ang II, 0.90 ± 0.10 mL/minute) or vehicle (baseline, 0.89 ± 0.10 mL/minute; post-vehicle, 0.84 ± 0.10 mL/minute).
Figure 1.

Infusion of Ang II in rat isolated perfused kidneys affects glomerular barrier function and integrity. A: Proteinuria levels at baseline (white bars) and after a 10-minute infusion with 16 ng/minute Ang II or vehicle (grey bars). ∗P < 0.01 versus baseline. B: Representative images of nephrin immunofluorescence (red) in glomeruli of rats after Ang II (left panel) or vehicle (right panel) infusion. C: Transmission electron micrographs of the capillary wall ultrastructure of rats infused with Ang II (left panel) or vehicle (right panel). Arrowhead, podocyte effacement; arrows, podocyte slits with or without the linear diaphragm between neighboring podocytes.
The increase of proteinuria after the short-term Ang II infusion was associated with modifications in the components of the glomerular capillary barrier and in the podocyte structure. After Ang II, the immunofluorescence signal for the slit diaphragm protein, nephrin, was markedly altered in 50% of glomeruli examined, exhibiting a heterogeneous expression with a punctuate pattern or loss of protein at focal areas within glomeruli (Figure 1B). In glomeruli of vehicle-infused kidneys, the immunofluorescence signal for nephrin was unaffected, with a typical predominant linear pattern along the peripheral capillary loops (Figure 1B). The Ang II–induced changes in nephrin localization were associated with ultrastructural evidence of podocyte damage. Transmission electron microscopy analysis showed mild Ang II–induced foot process effacement (frequency of filtration slit pores, 2.36 ± 0.1 slits/μm glomerular basement membrane versus 2.74 ± 0.2 slits/μm after vehicle infusion) (Figure 1C). In noneffaced regions, the percentage of intact pores spanned by the linear image of slit diaphragm was significantly lower in Ang II– than in vehicle-infused kidneys (57.3% ± 6.4% versus 79.3% ± 3.5%; P < 0.05) (Figure 1C).
Ang II Down-Regulates Nephrin Expression via Activation of the Notch1-Snail Signaling Pathway in Human Cultured Podocytes
To elucidate the intracellular molecular mechanism linking Ang II to nephrin down-regulation, we focused on Notch1 and its downstream effector, Snail, a negative regulator of nephrin expression in podocytes. In untreated cells, nephrin immunostaining showed a punctuated pattern, with peripheral distribution that was markedly reduced after 3 hours' exposure to Ang II (Figure 2A). Similarly, nephrin mRNA expression in Ang II–treated podocytes was significantly lower than in control untreated podocytes (P < 0.01) (Figure 2B). The role of Notch1 in Ang II–induced down-regulation of nephrin was assessed in podocytes treated with the γ-secretase inhibitor, GSI X, which blocks the proteolytic activation of Notch1.17 As shown in Figure 2B, the addition of GSI X to the cell culture medium blunted the lowering effect of Ang II on nephrin mRNA expression, although it was unable to completely restore the mRNA expression to normal levels (Figure 2B), suggesting the involvement of Notch1 signaling in Ang II–promoted down-regulation of nephrin mRNA in podocytes.
Figure 2.

Ang II reduces nephrin expression through the involvement of Notch1 in cultured human podocytes. A: Representative images of nephrin staining in human podocytes exposed for 3 hours to 100 nmol/L Ang II. B: Nephrin mRNA expression in human podocytes exposed to Ang II alone or in combination with 1 μmol/L γ-secretase inhibitor, GSI X. ∗P < 0.01 versus control podocytes; †P < 0.01 versus Ang II alone. C, top panel: Representative images of ICN1 staining (red) and cell nuclei (DAPI, blue). Bottom panel: Quantification of the percentage of nuclei positive for ICN1 at different time intervals. Transforming growth factor (TGF)-β (5 ng/mL for 120 minutes) was used as a positive control. ∗∗P < 0.001 versus control podocytes.
Because activation of Notch1 results from the cleavage of the transmembrane domain at Val 1744, with the release of ICN1 that translocates into the nucleus, we analyzed the presence of ICN1 nuclear staining in Ang II–treated podocytes at different time intervals. In control cells, ICN1 weakly localized in the cytoplasm (Figure 2C). Ang II increased ICN1 expression and promoted protein translocation from the cytoplasm to the nucleus, with a peak at 30 minutes (Figure 2C). Nuclear ICN1 staining in response to Ang II persisted over time until 120 minutes, an effect that was confirmed by quantification of the percentage of ICN1-positive nuclei (Figure 2C). Transforming growth factor-β, herein used as a positive control, markedly increased the activation and nuclear translocation of ICN1, similar to Ang II (Figure 2C).
We next examined the involvement of activated Notch1 in the Snail signaling pathway in cultured podocytes. Immunofluorescence experiments showed that Snail protein localized in the cytoplasm of untreated podocytes (Figure 3A). Ang II induced Snail protein expression and translocation into the nucleus, starting from 30 minutes, reaching a peak at 60 minutes (Figure 3A). The γ-secretase inhibitor, GSI X, markedly reduced the enhanced Snail protein nuclear translocation in response to Ang II (Figure 3A). Consistently, GSI X normalized Snail gene expression in Ang II–treated podocytes (Figure 3B).
Figure 3.

Role of the Notch1 pathway on Snail activation in Ang II–treated podocytes. A: Snail protein expression in podocytes exposed to Ang II in the presence or absence of 1 μmol/L γ-secretase inhibitor, GSI X, at different time intervals. Representative images of Snail staining (red) and nuclei (DAPI, blue) and quantification of Snail-positive nuclei (%). ∗P < 0.001 versus control; †P < 0.001 versus Ang II. B: Snail mRNA expression in podocytes exposed to Ang II for 1 hour in the presence or absence of GSI X and in podocytes knocked down for HES1 with siHES1 or control nontarget siRNA (siNULL). ∗P < 0.001 versus control; †P < 0.001 versus Ang II or Ang II + siNULL. C: Representative images of Snail staining (red with DAPI in blue) in siNULL- or siHES1-transfected podocytes exposed to Ang II at different time intervals and relative quantification of Snail-positive nuclei (%). ∗P < 0.001 versus control; †P < 0.001 versus Ang II + siNULL.
To explore the intracellular pathway by which Notch1 activates Snail, we focused on HES1, a downstream target gene of Notch1 described to up-regulate Snail transcription in other cell systems.18 Ang II significantly increased HES1 mRNA expression after a 30-minute incubation in respect to unstimulated cells (1.5 ± 0.03 versus 1 ± 0.02; n = 3; P < 0.001). The functional role of HES1 was evaluated by knocking down its expression with specific siRNA in podocytes. The percentage of HES1 silencing was 80% in respect to cells transfected with siNULL. Silencing of HES1 prevented the up-regulation of Snail mRNA in Ang II–treated podocytes (Figure 3B). Consistently, nuclear staining of Snail in siHES1 podocytes exposed to Ang II was markedly reduced at 30 and 60 minutes compared with the strong Snail activation in siNULL podocytes (Figure 3C). This difference was also documented by the quantification of the percentage of Snail-positive nuclei in both siHES1- or siNULL-transfected podocytes (Figure 3C).
Ang II Inhibition Reduces Proteinuria and Increases Glomerular Nephrin Expression via Snail Modulation in Rats with Diabetic Nephropathy
Based on our in vitro findings, we hypothesized that the regulation of Notch1/Snail axis by Ang II occurred in vivo, contributing to perpetuate podocyte injury and nephropathy in diabetic rats. To test this hypothesis, we characterized the renal effect of Ang II synthesis inhibition with the ACE inhibitor, ramipril, in ZDF rats, a model that recapitulates human type 2 diabetes.26,27 Moreover, to closely mimic the human conditions, the diabetic animals were randomized to treatment with ramipril or vehicle at 4 months of age when overt nephropathy is already established. At baseline, in diabetic rats, systolic blood pressure averaged 132.7 ± 2.6 mmHg, and increased to 143.7 ± 4.3 mmHg at the end of follow-up. At 8 months, a significant (P < 0.05) reduction of systolic blood pressure was found in ramipril (121 ± 3.1 mmHg) compared with vehicle-treated rats.
As shown in Figure 4A, the urinary protein excretion rate at baseline was significantly (P < 0.01) higher in vehicle-treated diabetic rats than in controls, and further increased thereafter. On the contrary, in diabetic rats, ramipril stabilized proteinuria at baseline values; thus, at the age of 8 months, urinary protein excretion was similar to values measured before treatment, with a significant reduction with respect to the vehicle-treated group (P < 0.01) (Figure 4A). The increase of proteinuria in vehicle-treated diabetic rats paralleled the worsening of renal histological features, as for the presence of sclerotic glomeruli (score, 20.7% ± 2.0%) and tubular damage (score, 1.0 ± 0.1), lesions not detectable in controls (sclerotic glomeruli score, 0%; tubular damage score, 0; P < 0.01) (Figure 4B). Ramipril reduced glomerulosclerosis and tubular lesions to a significant extent (sclerotic glomeruli score, 8.3% ± 2.8%; tubular damage score, 0.5 ± 0.1; P < 0.05 versus vehicle-treated animals) (Figure 4B).
Figure 4.

Ramipril reduced proteinuria, improved renal histological features, and restored nephrin and Snail expression in ZDF rats. A: Urinary protein excretion rate in ZDF rats receiving vehicle or ACE inhibitor and controls measured at 4 (pretreatment) and 8 months. ∗P < 0.01 versus controls; †P < 0.01 versus untreated. B: Representative images of PAS reagent–stained kidney sections of control and diabetic animals that received ramipril or did not receive ramipril and were sacrificed at 8 months. C: Photomicrographs of nephrin (red) and Snail (green) immunofluorescence on frozen kidney sections. Nuclei were stained with DAPI.
Consistent with changes in the glomerular perm-selectivity, diabetic rats given vehicle showed a significant (P < 0.05) reduction of nephrin protein expression by immunofluorescence on frozen kidney sections (score, 1.30 ± 0.2) (Figure 4C) compared with controls (score, 2.3 ± 0.1) (Figure 4C). In contrast, ramipril treatment increased the expression of nephrin in the glomeruli of diabetic animals (score, 1.8 ± 0.1) (Figure 4C), but the levels of protein were lower than in nondiabetic controls.
Based on in vitro findings, the contribution of Snail in Ang II–induced changes in nephrin expression was investigated in diabetic rats. Snail expression was negligible (score, 0.7 ± 0.1) (Figure 4C) in glomeruli of nondiabetic controls, whereas it was significantly increased (P < 0.01) in the glomerular visceral epithelial layer of vehicle-treated diabetic rats (score, 1.8 ± 0.1) (Figure 4C). Concomitant with the increased nephrin expression induced by ramipril, in diabetic animals, the glomerular Snail expression was reduced (score, 1.1 ± 0.1) (Figure 4C). Overall, an inverse correlation was found between Snail and nephrin protein expression in the glomeruli that, however, did not reach statistical significance (r = −0.762, P = 0.06).
Ang II Inhibition Improves Proteinuria and Restores Nephrin via Notch1-Snail Modulation in Patients with Diabetic Nephropathy
To examine the clinical relevance of the observation made in diabetic ZDF rats, we next assessed whether the glomerular nephrin/Snail axis was altered in type 2 diabetic patients with overt nephropathy, and inhibition of Ang II synthesis with ramipril enabled restoration of these pathways while reducing proteinuria. To this end, proteinuria and the glomerular expression of nephrin and Snail proteins were assessed in diabetic patients (not given renin-angiotensin system inhibitors/blockers, untreated diabetics), in nondiabetic control subjects, and in a group of age- and sex-matched diabetics given the ACE inhibitor, ramipril, for at least 1 month as part of their antihypertensive therapy. The duration of diabetes, control of glycemia as glycosylated hemoglobin and of blood pressure, and severity of chronic renal dysfunction were comparable in diabetic groups (Table 1). Untreated diabetic patients exhibited nephrotic range proteinuria (Table 1). Ramipril significantly reduced, but did not normalize, urinary protein excretion compared with untreated diabetic patients (Table 1).
Table 1.
Clinical Findings of Control Subjects and Diabetic Patients at Biopsy
| Variable | Controls (N = 5) | Untreated (N = 8) | Ramipril (N = 11) |
|---|---|---|---|
| Age (years) | 63.4 ± 3.5 | 64.3 ± 3.7 | 63 ± 2.3 |
| Male sex [no. (%)] | 4 (80) | 5 (62.5) | 9 (81.8) |
| Known duration of diabetes (months) | Not applicable | 141.8 ± 48.3 | 124.2 ± 31.4 |
| Glycosylated hemoglobin (%) | 3.2 ± 0.3 | 6.1 ± 0.6 | 6.6 ± 0.5 |
| Blood pressure (mmHg) | |||
| Systolic | 123.7 ± 3.8 | 151.3 ± 10.8 | 143.9 ± 6.4 |
| Diastolic | 78.4 ± 1.6 | 83.8 ± 5.3 | 83.1 ± 2.6 |
| Urinary protein excretion (g/day) | 0.1 ± 0.0 | 5.5 ± 1.1∗ | 2.2 ± 0.5† |
| Serum creatinine (mg/dL) | 1.1 ± 0.0 | 1.82 ± 0.4 | 1.7 ± 0.2 |
| Creatinine clearance rate (mL/minute per 1.73 m2) | 91.8 ± 1.8 | 48.9 ± 8.0 | 48.3 ± 5.6 |
| Triglycerides (mg/dL) | 127.3 ± 9.9 | 199.2 ± 50.3 | 179.1 ± 18.9 |
| Cholesterol (mg/dL) | 156.7 ± 13.8 | 227.7 ± 22.8 | 229.1 ± 15.9 |
P < 0.01 versus controls.
P < 0.05 versus untreated.
To test whether ramipril-induced reduction of proteinuria was associated with change in podocyte number, morphometric analysis of renal biopsy specimens was performed. The total number of podocytes per glomerulus was similar in untreated and ramipril-treated diabetic patients (533.67 ± 59.8 versus 557.50 ± 68.6), and comparable to controls (516 ± 31.1). However, untreated diabetics showed glomeruli with a mean volume significantly higher than controls (6.49 ± 0.5 × 106 μm3 versus 2.72 ± 0.2 × 106 μm3; P < 0.05), which translated in significant reduction of the volume density of podocytes (82.13 ± 6.3 versus 190.5 ± 5.3 × 106 μm3). Moreover, to cover the increased glomerular capillary surface, in untreated diabetic patients, the podocytes became hypertrophic (diabetics versus controls, 1.23 ± 0.1 versus 0.53 ± 0.1 × 104 μm3; P < 0.05). ACE inhibition did not reduce mean glomerular volume (7.5 ± 1.2 × 106 μm3) nor podocyte hypertrophy (1.45 ± 0.4 × 104 μm3); thus, the volume density of podocytes (85.23 ± 22.5 × 106 μm3) remained comparable to that of untreated diabetics. Similarly, ramipril failed to ameliorate renal tissue injury induced by diabetes, because the glomerulosclerosis score was comparable in treated and untreated patients at the histological examination (2.5 ± 0.1 versus 2.4 ± 0.2) (Figure 5).
Figure 5.
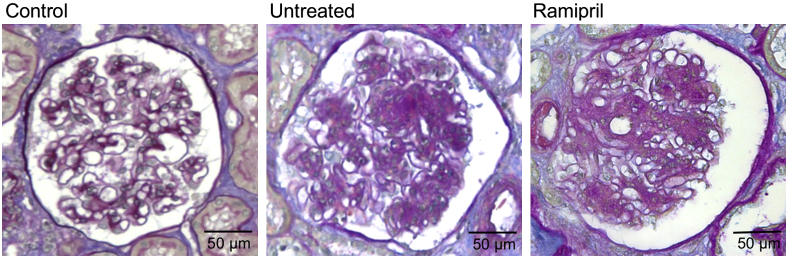
Ramipril did not reduce the area of sclerosis in glomeruli of patients with type 2 diabetes. Representative images of PAS reagent–stained kidney biopsy specimens from control and diabetic patients untreated or treated with ramipril. Scale bars are indicated.
Despite the absence of evident morphometric and histological changes in the glomeruli of ramipril-treated diabetic patients, the inhibition of Ang II synthesis improved the integrity of the glomerular capillary wall. Indeed, glomeruli of patients given ramipril showed higher nephrin protein expression than those of untreated diabetic patients (score, 1.4 ± 0.1 versus 0.7 ± 0.2; P < 0.05) (Figure 6A). Nevertheless, the nephrin signal score was still lower than in nondiabetic controls (2.8 ± 0.1) (Figure 6A). In a diabetic patient studied before and after ramipril treatment, the ACE inhibition doubled the nephrin protein expression score in glomeruli (0.5 to 1.0), in parallel with halving the urinary protein excretion rate (8.53 to 4.42 g/24 hours). More important, considering the combined cohorts of diabetics and controls, an inverse correlation was found between nephrin protein expression and degree of proteinuria (r = −0.495, P = 0.03), further suggesting that aberrant regulation of nephrin in the slit diaphragm of podocyte foot processes contributes to changes in the filtration barrier and proteinuria.
Figure 6.

Ramipril restores nephrin and Snail expression in patients with type 2 diabetes to control levels. A: Representative images of immunofluorescence of nephrin (red) in glomeruli of diabetic patients, untreated or treated with ACE inhibitor. Control subjects were included for comparisons. B: Snail expression evaluated by immunofluorescence (green). C: Double immunofluorescence of nephrin and Snail. Insets, colocalization of nephrin with Snail (yellow, merged signal; twofold enlargement of boxed area).
In addition, the reduction of nephrin expression in the glomeruli of diabetic patients was not the mere consequence of reduced volume density of podocytes. Indeed, the expression of other podocyte proteins, such as ZO-1 and CD2AP, that are located at the cytoplasmic side of the slit diaphragm10 was similar in untreated diabetic patients and nondiabetic controls (ZO-1, 2.2 ± 0.4 versus 2.8 ± 0.1; CD2AP, 2.2 ± 0.3 versus 2.5 ± 0.1) (Supplemental Figure S1, A and B), with strong protein signals revealed along the capillary loops, with the exception of sclerotic areas.
Consistent with our findings in ZDF diabetic rats, in diabetic patients, the down-regulation of nephrin expression was associated with changes in the glomerular expression of the transcriptional regulator, Snail. Thus, the discontinuous and spotted signal of Snail documented in control subjects was significantly increased along the glomerular visceral epithelial layer of untreated diabetic patients (score, 1.0 ± 0.0 versus 2.4 ± 0.3; P < 0.01) (Figure 6B). Double immunofluorescence revealed an almost complete localization of Snail protein with the podocyte marker, WT1,13 indicating that the transcriptional regulator localized specifically in the podocytes (Supplemental Figure S2). Moreover, double-immunofluorescence analysis (Figure 6C) documented that Snail expression increased in those glomeruli in which nephrin expression was reduced (r = −0.6, P = 0.067), confirming the previously described role of Snail as a nephrin repressor.19 Inhibition of Ang II synthesis with ramipril significantly restored Snail expression signal to control levels (score, 1.4 ± 0.1; P < 0.01) (Figure 6, B and C).
We finally examined the podocyte expression and localization of active Notch1 in renal biopsy specimens from the untreated and ramipril-treated diabetic patients and from nondiabetic controls. In control subjects, a mild staining for ICN1 was observed in few glomerular cells, and only occasionally in the nuclei (Figure 7). At variance, in glomeruli of untreated diabetic patients, ICN1 expression increased and localized into the nuclei (Figure 7). ICN1 staining was confined to podocytes, as revealed by double immunostaining with WT1, a specific marker for these glomerular epithelial cells13 (Figure 7). Quantitative analysis of ICN1 in untreated diabetic patients showed a significantly higher percentage of positive nuclei than in controls (48.3% ± 6.4% versus 10.3% ± 3.9%; P < 0.05) (Figure 7). Ramipril reduced nuclear expression of ICN1 into the podocytes of diabetic patients in respect to untreated ones (Figure 7). Together, these data highlighted a key role of Notch1/Snail signaling in the Ang II–induced changes in nephrin expression, even in diabetic patients with overt nephropathy.
Figure 7.

Ramipril restores Notch1 expression in diabetic patients. Representative images of Notch1 glomerular expression assessed by ICN1 immunoperoxidase. Insets, twofold enlargement of boxed area. Arrows, mild and cytoplasmic signal in controls that increases and becomes nuclear in diabetic glomeruli. The staining is negative in the absence of primary antibody (minus). Double immunofluorescence of ICN1 (green) and WT1 (red) reveals Notch1 activation in podocytes of untreated diabetic patients. Arrows, colocalization of the two signals (yellow). The histogram shows the percentage of ICN1-positive nuclei. ∗P < 0.05 versus control group; †P < 0.01 versus diabetic patients.
Discussion
Stemming from previous evidence in nondiabetic rat models of renal disease that Ang II perpetuates podocyte injury and promotes progression to end-stage kidney disease,14 we explored whether this possibility could apply to the progressive renal dysfunction occurring in diabetic animals and humans. In particular, we studied the following: i) the Ang II contribution to persistent podocyte dysfunction, ii) the possible involvement of nephrin based on previous data and its relevance to perm-selective properties of the glomerular filtration barrier, and iii) the intracellular molecular signaling coupling Ang II to nephrin. The results of these studies form the basis of this report.
Building on our previous work,22 herein we showed that the infusion of Ang II into the renal artery of an ex vivo model of isolated perfused rat kidney resulted in foot process effacement with profound rearrangement of pore number and structure. This effect was achieved in a perfusion system in which the possible confounding factor of Ang II–induced increase in arterial blood pressure was artificially minimized by maintaining constant the renal perfusion pressure. Such structural changes were associated with proteinuria and reduction of the glomerular expression of nephrin, one of the major components of the slit diaphragm,28 as others have previously documented in the setting of in vitro cultured podocytes.11 These data indicate that Ang II directly promotes and is critically important for the perturbation of the podocyte barrier. Although we cannot exclude that other slit diaphragm proteins have a certain role, this ex vivo model suggests that the down-regulation of nephrin expression induced by Ang II is essential for podocyte dysfunction.
To our knowledge, the intracellular molecular mechanisms coupling Ang II to nephrin down-regulation have not been fully unraveled. Given the relationship between Notch1 and Snail axis18 and the pivotal role of Snail signal in modulating nephrin gene expression,19 we elected to investigate in cultured human podocytes these pathways and found that Notch1/Snail signaling is actually not dispensable to repress nephrin gene and protein induced by Ang II.
The Notch1 signaling pathway is a short-range communication transducer that is involved in regulating many cellular processes during development and renewal of adult tissues.15 A key step in Notch1 activation is a series of proteolytic cleavages of Notch1 receptor by metalloprotease and γ-secretase that finally release the active Notch1 intracellular domain, which travels to the nucleus.15 The γ-secretase inhibitor, GSI X, which blocks the proteolytic activation of Notch1,17 actually blunted the Ang II effect of lowering podocyte nephrin mRNA expression. Although in mature kidney, little Notch1 can be detected,29 our present findings indicate that, at least in vitro, the Notch1 pathway can be activated in these cells under a particular setting of stimulation. This is consistent with previous reports in cultured mouse podocytes transduced with retroviral constructs expressing the active Notch1 intracellular domain, showing cell apoptosis induced by Notch1 activation.17 Among the targets of Notch1 signaling, there is the transcription factor, Snail, which is up-regulated after ICN1 recruitment into the nucleus and subsequent activation of downstream target genes, such as HES1.18 In keeping with these observations, our in vitro findings demonstrate that, in human podocytes, Ang II up-regulated the expression of the Snail gene via Notch1 activation of the transcriptional regulator, HES1. Collectively, these data and the finding that Ang II also promoted Snail protein translocation to the nucleus strongly suggest that Ang II–induced nephrin down-regulation in human podocytes depends on activation of Notch1/Snail signaling.
To assess the biological relevance of these findings, we used an in vivo model of diabetic nephropathy in ZDF/Gmi-fa/fa rats that closely mimics human type 2 diabetes.26,27 Data showed that ACE inhibition with ramipril, starting at 4 months of age when animals already had overt nephropathy, reduced proteinuria, ameliorated glomerular lesions, and increased the expression of nephrin protein along the glomerular capillary wall. Relevant to the involvement of the Notch1/Snail pathway in Ang II–induced down-regulation of nephrin gene expression, in our study, Snail protein signal was enhanced in the glomerular visceral epithelial layer in untreated diabetic rats, in parallel to reduction of nephrin protein. Moreover, glomerular capillary Snail expression was reduced by ramipril, which also normalized the nephrin level.
The translational relevance of the present study rests on the strong similarity of Notch1/Snail signaling activation in vitro, in diabetic rats, and in diabetic patients. Changes in the Snail/nephrin axis induced by Ang II, comparable to those found in rats, have also been documented herein in patients with advanced diabetic nephropathy and overt proteinuria. Mechanistic insight into these findings rests on a substantial increase of Notch1 expression in podocytes of diabetic patients, in parallel with reduction of nephrin protein level, consistent with the results of the previously described in vitro studies and on the demonstration that ramipril restored the Notch1 signal to normal.
Overall, our findings offer the rationale for using therapy that selectively targets Notch1 in diabetic nephropathy. However, safety concerns should be accounted for long-term treatment with γ-secretase inhibitors, given the essential physiological role of Notch1 in the hematopoietic system, the gastrointestinal tract, and the skin.30
How Ang II activated the Notch1 pathway in diabetic podocytes remains ill defined. Ang II binds angiotensin type 1 receptors that, coupled to heterotrimetric guanine nucleotide–binding protein G (G proteins), activate phospholipase C-β isoforms that hydrolyze phosphatidylinositol 4,5-biophosphate.31 Studies, however, have also established complementary pathways of Ang II–mediated intracellular signal transduction systems, such as activation of phosphatidylinositol 3-kinase,32,33 which stimulates the tyrosine phosphorylation of a distinct set of cellular proteins34,35 and leads to activation of the glycogen synthase kinase-3β; this, in turn, regulates the Notch1 level, as shown in the 293 HEK cell line.36 Taken together, our present study led us to propose cohesive intracellular molecular pathways of podocyte activation in diabetes, related to the contribution of Ang II to nephrin down-regulation (Figure 8).
Figure 8.

Ang II activates the Notch1/Snail signaling, which is responsible for the pathogenesis of proteinuria in diabetic nephropathy. Schematic drawing summarizes the molecular pathway by which Ang II, after binding with angiotensin type 1 receptor (AT1R), induces increased activation of Notch1. The active Notch1 (ICN1) via the downstream target gene, HES1, increases the expression of Snail and its translocation into the nucleus, leading to nephrin down-regulation. This series of events is responsible for the molecular podocyte lesions and loss of the glomerular permeability in diabetes.
In conclusion, we present evidence that, in diabetes, Ang II plays a relevant role to perpetuate glomerular injury after the initial insult of hyperglycemia and intraglomerular hypertension, via persistent activation of Notch1 and Snail signaling in the podocyte, eventually resulting in down-regulation of nephrin expression, the integrity of which is crucial for the glomerular filtration barrier. The consistency of findings in the rat model and in type 2 diabetic patients with nephropathy provides robust insights into novel molecular mechanisms underlying progression of kidney disease in diabetes. It also highlights the rationale for future investigations focused on strategies that modulate transduction pathways and foot process function to most effectively prevent renal disease progression in diabetic patients resistant to available treatments. This is particularly relevant considering that, in experimental diabetes rat models37 and in patients with type 2 diabetes,38–42 blockade of the renin-angiotensin system affords significant renoprotection only when treatment is established early in the course of kidney disease.
Acknowledgments
We thank Drs. Peter Mathieson and Moin Saleem (University of Bristol, Bristol, UK) for providing immortalized human podocytes, Dr. Sara Conti for transmission electron microscopy assessments, Dr. Daniela Corna for the excellent coordination of the in vivo studies, Prof. Antonino Lembo and Dr. Marlisa Capitanio for invaluable collaboration in obtaining kidney specimens of human controls, Luciana Prometti and Romana Stacchetti for helpful assistance in animal care, and Manuela Passera for help in preparing the manuscript.
Footnotes
Supported by the European Community's Seventh Framework Programme grant 241544 (SysKid) and a Fondazione Aiuti per la Ricerca sulle Malattie Rare fellowship (Bergamo, Italy) (P.R.).
E.G. and N.P. contributed equally to this work.
Supplemental Data
The expression of ZO-1 and CD2AP in diabetic patients, either treated with ramipril or untreated, was comparable to controls. A: Immunoperoxidase staining for ZO-1. No specific signal was obtained, omitting the primary antibodies (minus). B: Immunoperoxidase staining for CD2AP. No signal was observed in the negative control (minus). Scale bars are indicated.
Snail shows specific podocyte localization in human diabetic glomeruli. Representative image of the double immunofluorescence with antibodies against Snail (green) and the podocyte marker, WT1 (red), in frozen renal sections of untreated diabetic patients. The colocalization signal in yellow is indicated (arrows). Scale bars are indicated.
References
- 1.Wild S., Roglic G., Green A., Sicree R., King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 2.Gall M.A., Borch-Johnsen K., Hougaard P., Nielsen F.S., Parving H.H. Albuminuria and poor glycemic control predict mortality in NIDDM. Diabetes. 1995;44:1303–1309. doi: 10.2337/diab.44.11.1303. [DOI] [PubMed] [Google Scholar]
- 3.Remuzzi G., Schieppati A., Ruggenenti P. Clinical practice: nephropathy in patients with type 2 diabetes. N Engl J Med. 2002;346:1145–1151. doi: 10.1056/NEJMcp011773. [DOI] [PubMed] [Google Scholar]
- 4.Zatz R., Dunn B.R., Meyer T.W., Anderson S., Rennke H.G., Brenner B.M. Prevention of diabetic glomerulopathy by pharmacological amelioration of glomerular capillary hypertension. J Clin Invest. 1986;77:1925–1930. doi: 10.1172/JCI112521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gagliardini E., Conti S., Benigni A., Remuzzi G., Remuzzi A. Imaging of the porous ultrastructure of the glomerular epithelial filtration slit. J Am Soc Nephrol. 2010;21:2081–2089. doi: 10.1681/ASN.2010020199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Remuzzi A., Perico N., Amuchastegui C.S., Malanchini B., Mazerska M., Battaglia C., Bertani T., Remuzzi G. Short- and long-term effect of angiotensin II receptor blockade in rats with experimental diabetes. J Am Soc Nephrol. 1993;4:40–49. doi: 10.1681/ASN.V4140. [DOI] [PubMed] [Google Scholar]
- 7.Durvasula R.V., Petermann A.T., Hiromura K., Blonski M., Pippin J., Mundel P., Pichler R., Griffin S., Couser W.G., Shankland S.J. Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int. 2004;65:30–39. doi: 10.1111/j.1523-1755.2004.00362.x. [DOI] [PubMed] [Google Scholar]
- 8.Kriz W., Gretz N., Lemley K.V. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int. 1998;54:687–697. doi: 10.1046/j.1523-1755.1998.00044.x. [DOI] [PubMed] [Google Scholar]
- 9.Pavenstadt H., Kriz W., Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- 10.Benigni A., Gagliardini E., Remuzzi G. Changes in glomerular perm-selectivity induced by angiotensin II imply podocyte dysfunction and slit diaphragm protein rearrangement. Semin Nephrol. 2004;24:131–140. doi: 10.1016/j.semnephrol.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 11.Doublier S., Salvidio G., Lupia E., Ruotsalainen V., Verzola D., Deferrari G., Camussi G. Nephrin expression is reduced in human diabetic nephropathy: evidence for a distinct role for glycated albumin and angiotensin II. Diabetes. 2003;52:1023–1030. doi: 10.2337/diabetes.52.4.1023. [DOI] [PubMed] [Google Scholar]
- 12.Welsh G.I., Saleem M.A. The podocyte cytoskeleton: key to a functioning glomerulus in health and disease. Nat Rev Nephrol. 2012;8:14–21. doi: 10.1038/nrneph.2011.151. [DOI] [PubMed] [Google Scholar]
- 13.Gagliardini E., Corna D., Zoja C., Sangalli F., Carrara F., Rossi M., Conti S., Rottoli D., Longaretti L., Remuzzi A., Remuzzi G., Benigni A. Unlike each drug alone, lisinopril if combined with avosentan promotes regression of renal lesions in experimental diabetes. Am J Physiol Renal Physiol. 2009;297:F1448–F1456. doi: 10.1152/ajprenal.00340.2009. [DOI] [PubMed] [Google Scholar]
- 14.Fukuda A., Wickman L.T., Venkatareddy M.P., Sato Y., Chowdhury M.A., Wang S.Q., Shedden K.A., Dysko R.C., Wiggins J.E., Wiggins R.C. Angiotensin II-dependent persistent podocyte loss from destabilized glomeruli causes progression of end stage kidney disease. Kidney Int. 2012;81:40–55. doi: 10.1038/ki.2011.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ilagan M.X., Kopan R. SnapShot: notch signaling pathway. Cell. 2007;128:1246. doi: 10.1016/j.cell.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 16.Murea M., Park J.K., Sharma S., Kato H., Gruenwald A., Niranjan T., Si H., Thomas D.B., Pullman J.M., Melamed M.L., Susztak K. Expression of Notch pathway proteins correlates with albuminuria, glomerulosclerosis, and renal function. Kidney Int. 2010;78:514–522. doi: 10.1038/ki.2010.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niranjan T., Bielesz B., Gruenwald A., Ponda M.P., Kopp J.B., Thomas D.B., Susztak K. The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat Med. 2008;14:290–298. doi: 10.1038/nm1731. [DOI] [PubMed] [Google Scholar]
- 18.Sahlgren C., Gustafsson M.V., Jin S., Poellinger L., Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci U S A. 2008;105:6392–6397. doi: 10.1073/pnas.0802047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsui I., Ito T., Kurihara H., Imai E., Ogihara T., Hori M. Snail, a transcriptional regulator, represses nephrin expression in glomerular epithelial cells of nephrotic rats. Lab Invest. 2007;87:273–283. doi: 10.1038/labinvest.3700518. [DOI] [PubMed] [Google Scholar]
- 20.Saleem M.A., O'Hare M.J., Reiser J., Coward R.J., Inward C.D., Farren T., Xing C.Y., Ni L., Mathieson P.W., Mundel P. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol. 2002;13:630–638. doi: 10.1681/ASN.V133630. [DOI] [PubMed] [Google Scholar]
- 21.Lapinski R., Perico N., Remuzzi A., Sangalli F., Benigni A., Remuzzi G. Angiotensin II modulates glomerular capillary permselectivity in rat isolated perfused kidney. J Am Soc Nephrol. 1996;7:653–660. doi: 10.1681/ASN.V75653. [DOI] [PubMed] [Google Scholar]
- 22.Macconi D., Abbate M., Morigi M., Angioletti S., Mister M., Buelli S., Bonomelli M., Mundel P., Endlich K., Remuzzi A., Remuzzi G. Permselective dysfunction of podocyte-podocyte contact upon angiotensin II unravels the molecular target for renoprotective intervention. Am J Pathol. 2006;168:1073–1085. doi: 10.2353/ajpath.2006.050701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruggenenti P., Cravedi P., Sghirlanzoni M.C., Gagliardini E., Conti S., Gaspari F., Marchetti G., Abbate M., Remuzzi G. Effects of rituximab on morphofunctional abnormalities of membranous glomerulopathy. Clin J Am Soc Nephrol. 2008;3:1652–1659. doi: 10.2215/CJN.01730408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weibel E.R. Academic Press Inc.; London: 1979. Practical Methods for Biological Morphometry. pp 40–116. [Google Scholar]
- 25.Macconi D., Sangalli F., Bonomelli M., Conti S., Condorelli L., Gagliardini E., Remuzzi G., Remuzzi A. Podocyte repopulation contributes to regression of glomerular injury induced by ACE inhibition. Am J Pathol. 2009;174:797–807. doi: 10.2353/ajpath.2009.080227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyata K., Ohashi N., Suzaki Y., Katsurada A., Kobori H. Sequential activation of the reactive oxygen species/angiotensinogen/renin-angiotensin system axis in renal injury of type 2 diabetic rats. Clin Exp Pharmacol Physiol. 2008;35:922–927. doi: 10.1111/j.1440-1681.2008.04938.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zoja C., Cattaneo S., Fiordaliso F., Lionetti V., Zambelli V., Salio M., Corna D., Pagani C., Rottoli D., Bisighini C., Remuzzi G., Benigni A. Distinct cardiac and renal effects of ETA receptor antagonist and ACE inhibitor in experimental type 2 diabetes. Am J Physiol Renal Physiol. 2011;301:F1114–F1123. doi: 10.1152/ajprenal.00122.2011. [DOI] [PubMed] [Google Scholar]
- 28.Welsh G.I., Saleem M.A. Nephrin-signature molecule of the glomerular podocyte? J Pathol. 2010;220:328–337. doi: 10.1002/path.2661. [DOI] [PubMed] [Google Scholar]
- 29.Vooijs M., Ong C.T., Hadland B., Huppert S., Liu Z., Korving J., van den Born M., Stappenbeck T., Wu Y., Clevers H., Kopan R. Mapping the consequence of Notch1 proteolysis in vivo with NIP-CRE. Development. 2007;134:535–544. doi: 10.1242/dev.02733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Purow B. Notch inhibitors as a new tool in the war on cancer: a pathway to watch. Curr Pharm Biotechnol. 2009;10:154–160. doi: 10.2174/138920109787315060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balla T., Varnai P., Tian Y., Smith R.D. Signaling events activated by angiotensin II receptors: what goes before and after the calcium signals. Endocr Res. 1998;24:335–344. doi: 10.3109/07435809809032613. [DOI] [PubMed] [Google Scholar]
- 32.Saward L., Zahradka P. Angiotensin II activates phosphatidylinositol 3-kinase in vascular smooth muscle cells. Circ Res. 1997;81:249–257. doi: 10.1161/01.res.81.2.249. [DOI] [PubMed] [Google Scholar]
- 33.Liu X.C., Liu B.C., Zhang X.L., Li M.X., Zhang J.D. Role of ERK1/2 and PI3-K in the regulation of CTGF-induced ILK expression in HK-2 cells. Clin Chim Acta. 2007;382:89–94. doi: 10.1016/j.cca.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 34.Molloy C.J., Taylor D.S., Weber H. Angiotensin II stimulation of rapid protein tyrosine phosphorylation and protein kinase activation in rat aortic smooth muscle cells. J Biol Chem. 1993;268:7338–7345. [PubMed] [Google Scholar]
- 35.Sadoshima J., Qiu Z., Morgan J.P., Izumo S. Angiotensin II and other hypertrophic stimuli mediated by G protein-coupled receptors activate tyrosine kinase, mitogen-activated protein kinase, and 90-kD S6 kinase in cardiac myocytes: the critical role of Ca(2+)-dependent signaling. Circ Res. 1995;76:1–15. doi: 10.1161/01.res.76.1.1. [DOI] [PubMed] [Google Scholar]
- 36.Jin Y.H., Kim H., Oh M., Ki H., Kim K. Regulation of Notch1/NICD and Hes1 expressions by GSK-3alpha/beta. Mol Cells. 2009;27:15–19. doi: 10.1007/s10059-009-0001-7. [DOI] [PubMed] [Google Scholar]
- 37.Perico N., Amuchastegui S.C., Colosio V., Sonzogni G., Bertani T., Remuzzi G. Evidence that an angiotensin-converting enzyme inhibitor has a different effect on glomerular injury according to the different phase of the disease at which the treatment is started. J Am Soc Nephrol. 1994;5:1139–1146. doi: 10.1681/ASN.V541139. [DOI] [PubMed] [Google Scholar]
- 38.Parving H.H., Lehnert H., Brochner-Mortensen J., Gomis R., Andersen S., Arner P. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med. 2001;345:870–878. doi: 10.1056/NEJMoa011489. [DOI] [PubMed] [Google Scholar]
- 39.Brenner B.M., Cooper M.E., de Zeeuw D., Keane W.F., Mitch W.E., Parving H.H., Remuzzi G., Snapinn S.M., Zhang Z., Shahinfar S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 40.Ruggenenti P., Fassi A., Ilieva A.P., Iliev I.P., Chiurchiu C., Rubis N., Gherardi G., Ene-Iordache B., Gaspari F., Perna A., Cravedi P., Bossi A., Trevisan R., Motterlini N., Remuzzi G. Effects of verapamil added-on trandolapril therapy in hypertensive type 2 diabetes patients with microalbuminuria: the BENEDICT-B randomized trial. J Hypertens. 2011;29:207–216. doi: 10.1097/hjh.0b013e32834069bd. [DOI] [PubMed] [Google Scholar]
- 41.Ruggenenti P., Fassi A., Ilieva A.P., Bruno S., Iliev I.P., Brusegan V., Rubis N., Gherardi G., Arnoldi F., Ganeva M., Ene-Iordache B., Gaspari F., Perna A., Bossi A., Trevisan R., Dodesini A.R., Remuzzi G. Preventing microalbuminuria in type 2 diabetes. N Engl J Med. 2004;351:1941–1951. doi: 10.1056/NEJMoa042167. [DOI] [PubMed] [Google Scholar]
- 42.Ruggenenti P., Perticucci E., Cravedi P., Gambara V., Costantini M., Sharma S.K., Perna A., Remuzzi G. Role of remission clinics in the longitudinal treatment of CKD. J Am Soc Nephrol. 2008;19:1213–1224. doi: 10.1681/ASN.2007090970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The expression of ZO-1 and CD2AP in diabetic patients, either treated with ramipril or untreated, was comparable to controls. A: Immunoperoxidase staining for ZO-1. No specific signal was obtained, omitting the primary antibodies (minus). B: Immunoperoxidase staining for CD2AP. No signal was observed in the negative control (minus). Scale bars are indicated.
Snail shows specific podocyte localization in human diabetic glomeruli. Representative image of the double immunofluorescence with antibodies against Snail (green) and the podocyte marker, WT1 (red), in frozen renal sections of untreated diabetic patients. The colocalization signal in yellow is indicated (arrows). Scale bars are indicated.