

Summary
Host genetic factors are a major contributing factor to the inter-individual variation observed in response to human immunodeficiency virus (HIV) infection and are linked to resistance to HIV infection among exposed individuals, as well as rate of disease progression and the likelihood of viral transmission. Of the genetic variants that have been shown to affect the natural history of HIV infection, the human leukocyte antigen (HLA) class I genes exhibit the strongest and most consistent association, underscoring a central role for CD8+ T cells in resistance to the virus. HLA proteins play important roles in T-cell-mediated adaptive immunity by presenting immunodominant HIV epitopes to cytotoxic T lymphocytes (CTLs) and CD4+ T cells. Genetic and functional data also indicate a function for HLA in natural killer (NK) cell-mediated innate immunity against HIV by interacting with killer cell immunoglobulin-like receptors (KIR). We review the HLA and KIR associations with HIV disease and discuss the mechanisms underlying these associations.
Keywords: HLA, KIR, CTL, NK cells, GWAS, host genetic variation
Introduction
Human immunodeficiency virus (HIV) infection continues to be a major global public health issue with more than 25 million deaths over the past three decades and approximately 34 million people living with HIV (http://www.who.int/mediacentre/factsheets/fs360/en/index.html). These statistics have stimulated intensive research and substantial progress in understanding HIV pathogenesis. Despite the lack of a cure or an effective vaccine, these efforts have led to a plethora of information about HIV and its interactions with the host, strengthening our potential to devise effective therapeutics for controlling the virus.
Multiple longitudinal studies have provided ample evidence that the clinical outcome of untreated HIV-1 infection is highly variable. Some individuals will progress to acquired immunodeficiency syndrome (AIDS) within a year of seroconversion, while others are able to control the virus without drugs for more than two decades. The vast majority of these individuals have very low levels of viremia, in some cases below 50 copies of virus per ml of plasma (the latter are referred to as elite controllers) (1). A small fraction of individuals are resistant to HIV infection, even upon repeated exposure. Variation in response to HIV across individuals is likely due to complex interactions between virus, host, and environment. The importance of host genetic variation in HIV pathogenesis has shown progress over the last decade, in part because of high-throughput technologies that allow testing of millions of single nucleotide polymorphisms (SNPs) across the human genome. Defining host genetic factors that influence the response to HIV may be useful in predicting rates of disease progression and help to delineate modes of pathogenesis that can lead to the development of therapeutics and vaccines. Most of the genetic effects that have been conclusively identified involve genes encoding receptors for viral entry into cells and molecules that participate in innate or acquired immune responses. The human leukocyte antigen (HLA) class I loci clearly have shown the strongest and most consistent effects on HIV outcomes in general. This is probably not surprising, since these data affirm the importance of cytotoxic T-cell responses in controlling HIV. In addition to their role in acquired immunity, HLA class I molecules also serve as ligands for killer cell immunoglobulin-like receptors (KIR), a polymorphic set of molecules that modulate natural killer (NK) cell activity (and some T cells) (2), and therefore play an important part in the innate immune response. In this review, we focus primarily on the influence of variation at the HLA locus and the HLA/KIR loci combined on HIV-1 disease outcome.
HLA and KIR genetics and function
The HLA class I and class II molecules are encoded by genes located within the human major histocompatibility complex (MHC) on chromosome 6p21.3. The classical HLA class I (HLA-A, -B and -C) and class II (HLA-DR, -DQ and -DP) genes within the MHC are the most diverse loci in the human genome with the number of alleles varying from 31 for DQA1 to >2000 for HLA-B (http://www.ebi.ac.uk/imgt/hla). This variation is concentrated within the regions encoding the peptide binding groove, and historically, the exons encoding this region have been the main focus of study in terms of determining HLA effects on disease susceptibility/pathogenesis. Recent data, however, suggest that variants in noncoding regions, which may affect the level of transcription, translation, and splicing, may also be important. The HLA class I genes encode molecules that are expressed on the surface of virtually all nucleated cells. They bind peptide epitopes that are derived from self-proteins under normal conditions, but upon infection with intracellular pathogens, they bind antigenic peptides derived from the pathogen or self-stress proteins, and present them to CD8+ T cells, thereby initiating a cytotoxic T-cell (CTL) response. Class I molecules also regulate NK cell activity via interactions with NK cell receptors. The class II loci encode molecules that are expressed on the surface of antigen-presenting cells. They bind peptides that are primarily extracellularly derived and present them to CD4+ T cells, generally resulting in the production of cytokines that help other immune cells to respond.
The killer cell immunoglobulin-like receptor (KIR) genes are arranged in a ‘head-to-tail’ cluster on human chromosome 19q13.4 within the leukocyte receptor complex (LRC) and encode molecules that belong to the immunoglobulin (Ig) superfamily of receptors (3). They consist of a group of regulatory molecules that are expressed on natural killer (NK) cells and a subset of T cells. KIR2DL and KIR3DL genes encode molecules with long cytoplasmic tails and are inhibitory by virtue of the immunoreceptor tyrosine-based inhibition motifs (ITIMs) present in their cytoplasmic domains. KIR2DS and KIR3DS genes encode molecules with short cytoplasmic tails that transmit activating signals through their interaction with the adapter molecule DAP-12 (DNAX activation protein of 12 kDa), which contains an immunoreceptor tyrosine-based activation motif (ITAM) (4). There is extensive diversity of KIR haplotypes as a consequence of non-allelic homologous recombination (NAHR), but two basic groups of KIR haplotypes termed A and B have been described (5). Haplotype A is uniform in terms of gene content and is composed of nine genes that predominantly encode inhibitory receptors. The B group of haplotypes, on the other hand, contain variable numbers of genes encoding activating and inhibitory receptors ranging from 4 to 17 (6–8) (Fig. 1). Multiple alleles also exist for each KIR gene (http://www.ebi.ac.uk/ipd/kir/alleles.html), encoding products that can vary in expression level or functional capacity. Another notable feature of the KIR locus is that there is variegated expression of KIR on NK cell clones, such that a given KIR gene is expressed in some but not all NK cell clones in a given individual (9).
Fig. 1. Schematic representation of KIR haplotypes A and B.
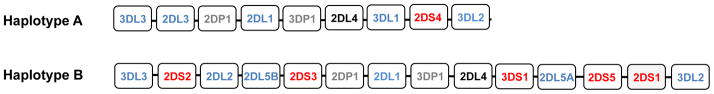
Genes encoding activating KIRs are shown in red, inhibitory KIRs are shown in blue and pseudogenes in grey. KIR2DL4, which has features of both an activating and an inhibitory receptor is shown in black.
Thus far, only HLA class I allotypes have been identified as ligands for KIR (Fig. 2). KIR3DL1 recognizes HLA-B molecules and a subset of HLA-A molecules that have the serologically defined Bw4 motif (determined by amino acid positions 77–83). Some KIR3DL1 subtypes exhibit a stronger inhibitory effect in the presence of HLA-B Bw4 subtypes that have isoleucine at position 80 (Bw4-80I) as opposed to threonine at the same position (Bw4-80T) (10, 11). HLA-B Bw6 allotypes on the other hand do not serve as ligands for KIR, so homozygotes for HLA-B Bw6 alleles serve as a very appropriate negative control grouping when studying disease effects of KIR3DL1/S1 in combination with HLA-B Bw4 alleles, as it does not. The activating KIR3DS1 and inhibitory KIR3DL1 segregate as alleles of the same locus and they share >97% similarity in their extracellular domains. Despite this similarity, there is no direct evidence of interactions between KIR3DS1 and Bw4 allotypes, although indirect evidence from genetic epidemiological (12, 13), functional (14), and population genetic studies (15) imply that some form of interaction, either direct or indirect, occurs between them. All HLA-C alleles can be divided into two distinct allotypic groups, group 1 and group 2 based on dimorphisms in the α1 domain (Ser77/Asn80 and Asn77/Lys80, respectively). The inhibitory KIR2DL1 interacts with group 2 allotypes, while KIR2DL2 and KIR2DL3, which segregate as alleles of the same locus, interact with group 1 allotypes (16–18). KIR3DL2 binds HLA-A*03 and HLA-A*11 (19, 20), KIR2DL4 binds the non-classical class I molecule HLA-G (21), and KIR2DS4 binds to HLA-A*11 as well as subsets of HLA-C groups 1 and 2 (22). The activating KIR2DS1 demonstrates weak binding to HLA-C group 2 (23), but with low affinity and its high affinity ligand has not been identified. The ligands for the activating KIR2DS2, KIR2DS3 and KIR2DS5 and the inhibitory KIR2DL5 and 3DL3 have not been identified.
Fig. 2. KIR and their HLA ligands.
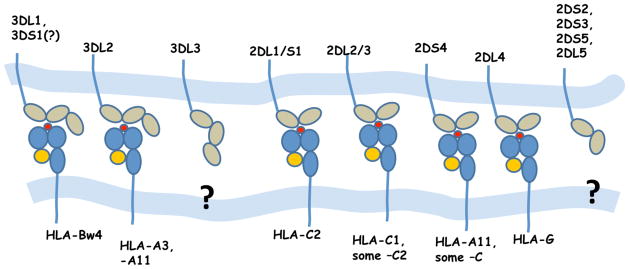
HLA-C2 = HLA-C allotypes with Asn77/Lys80. HLA-C1 = HLA-C allotypes with Ser77/Asn80. Bw4 = HLA allotypes with the serologically defined Bw4 motif.
Due to their spontaneous killing of targets that lack self MHC (missing self hypothesis), NK cells were initially thought to be non-MHC restricted (24), but it is now clear that inhibitory KIR are involved in restraining NK cell cytotoxicity. More recent data suggest that during the developmental process of NK cell education, interactions between inhibitory receptors expressed on the NK cell surface and self MHC class I molecules are important in determining functional potency to respond to and eliminate abnormal cells expressing aberrant levels of MHC (25). While inhibitory KIRs are relatively well studied, the function of the activating KIRs remains somewhat obscure. The activating KIRs have evolved from their inhibitory counterparts such that they bind HLA class I ligands with substantially lower affinity despite having nearly identical sequences in their extracellular domains. The biological relevance of low-affinity interactions between activating KIRs and class I molecules is not clear. It has also been difficult to study the function of these receptors since their high affinity ligands are not known. There could be specific conditions that are required for high affinity interactions, such as modification of the HLA molecule by a cofactor or recognition of HLA loaded with a disease-specific or stress peptide. It is also possible that activating KIR may recognize non-HLA ligands that are aberrantly expressed on abnormal cells. A recent report suggests an alternative role for the peptide in NK cell activation through inhibitory KIR. Peptide:MHC complexes that bind weakly to inhibitory KIR may antagonize the inhibition (referred to as peptide antagonism) and cause NK cell activation. Low affinity peptide:MHC complexes disrupt the inhibitory synapse either directly or by antagonizing the effect of high affinity peptide:MHC complexes (26, 27). Thus, for example, particular viral peptides may bind with lower affinity to an inhibitory KIR, thereby removing dominant inhibitory signals and resulting in NK cell activation. Ultimately however, NK cell activity will depend on a balance between inhibition and activation delivered through a variety of NK cell receptors, including the polymorphic KIR.
HLA and HIV
Over 100 diseases have been associated with the HLA loci, including autoimmune diseases, malignancies, and infectious diseases. HIV/AIDS is one of only a few infectious diseases showing a clear-cut and consistent HLA association. The primary influence of HLA loci on the HIV disease relative to all other single human genetic variants has now been confirmed by several genome-wide association studies (GWAS) (see below). The GWAS data supported numerous genetic epidemiological studies reported previously, which had implicated many HLA alleles in various aspects of HIV disease (Table 1). Thus, all the available evidence point to HLA as the most significant locus in differential control of HIV across humans.
Table 1.
HLA associations with HIV-1 disease
| HLA genotype | Epidemiological effect | Suggested mechanism |
|---|---|---|
| HLA class I homozygosity | Accelerated disease progression | Narrow CTL response(30, 32). Viral escape from common alleles (33, 34). |
| B*57 | Slow disease progression | Broad reactivity across multiple conserved gag epitopes and reduced viral fitness of escape variants (44, 55, 56, 191). |
| B*27 | Slow disease progression | Presents a conserved immunodominant gag epitope that requires a complex pattern of mutation for escape (46, 64–66). |
| B*35-Px | Accelerated disease progression | Peptide binding specificity (71). Interaction with ILT4 resulting in dendritic cell dysfunction (78). |
| Bw4 | Slow disease progression | More effective HIV-1-specific responses (84). |
| B*51 | Slow disease progression | Strong CTL response to Gag and Pol epitopes (192) |
| B*58:01 | Slow disease progression | Strong CTL response to Gag epitope (35) |
| B*58:02 | Accelerated disease progression | Ineffective CTL response to Env epitope (90) |
| B*13 | Slow disease progression | Targeting of a conserved nef epitope (86). Broad Gag-specific CD8+ response (87). |
| B*81:01 | Slow disease progression | Lower replication capacity of escape variants (89). |
| DR13 supertype & DRB1*13- DQB1*06 haplotype | Slow disease progression | Selection of a highly conserved Th1 gag epitope (102) |
| DRB1*01 | Protection against infection | Unknown (98, 99) |
| DRB1*15:03 | Increased susceptibility to infection | Unknown (98, 99) |
| DRB1*13:03 | Reduced viral load levels | Unknown (101) |
HLA heterozygote advantage
HLA class I and II allelic frequencies are more evenly distributed than expected by chance, with an excess of intermediate frequency alleles, pointing to the force of balancing selection through interaction between the host and infectious pathogens acting on these loci (28, 29). Because of the extensive polymorphism and fairly even distribution of most alleles, homozygosity at the class I loci is not very common, and few individuals are homozygous at all three loci. The primary function of MHC molecules is to present foreign antigens to elicit T-cell responses, so the number of distinct HLA allotypes expressed on the cell surface is directly related to the range of foreign antigens the host can present to T cells. Thus, individuals who are heterozygous at the HLA locus will be able to present a broader repertoire of antigenic peptides to T cells as compared to homozygotes, thereby exerting greater pressure on the pathogen to escape the CTL responses that may in turn affect pathogen fitness. Under this model, we would expect that HLA homozygous individuals would progress more rapidly to AIDS than HLA heterozygous individuals after HIV infection. Indeed, using a large cohort of HIV-infected individuals with known dates of seroconversion, we demonstrated a highly significant association of HLA class I homozygosity with rapid progression to AIDS in both Caucasians and African Americans. All three class I loci contributed independently to the association, and the effect was most pronounced in individuals who were homozygous at two or three loci (30). Similar results were observed for homozygosity at HLA-A and HLA-B in another study (31). Data from cynomolgus monkeys, which have a restricted set of MHC haplotypes, also support the protective effect of heterozygosity in that MHC heterozygous animals had lower chronic simian immunodeficiency virus (SIV) viral loads as compared to homozygous animals (32). Overall, the data suggest that a broader range of HIV-1 peptides are recognized by HLA heterozygous individuals resulting in more efficient specific CTL responses against the pathogen. It may also take the virus a longer time to accumulate escape mutations in HLA heterozygous individuals relative to homozygous individuals.
An alternative interpretation of the data is based on a model of frequency-dependent selection (or rare allele advantage), which argues that HIV adapts to HLA alleles that are common in the population. It is these alleles that are most likely to be observed in HLA homozygotes. HLA heterozygous individuals on the other hand are more likely to carry rare alleles (in combination with common alleles), to which the virus has not adapted as well, and therefore individuals with these alleles are better able to contain the virus. In support of a rare allele advantage, investigators have demonstrated a positive correlation between viral load and the frequency of HLA class I supertypes (33). Further, a recent study involving HIV cohorts from five continents demonstrated a strong positive correlation between the frequency of escape mutations of a given class I epitope and prevalence of the restricting HLA class I alleles (34), suggesting the occurrence of viral adaptation at the population level. It is plausible that both heterozygote advantage and rare allele advantage apply to HIV-infected individuals.
HLA class I variation
Distinct HLA allotypes may mediate differential responses to HIV infection based on their capacity to contain HIV replication. As a general rule, HLA-B appears to have the most obvious differential effects on HIV outcome, perhaps as a consequence of its extensive diversity. The HLA-B locus is the most polymorphic of all the HLA loci (http://www.ebi.ac.uk/imgt/hla). Functional data also support the dominant role of HLA-B in control of HIV. A large study of HIV-infected patients showed that most CD8+ T-cell responses to HIV were HLA-B restricted and that variation in viral load was predominantly associated with variation at the HLA-B locus (35).
Among the protective HLA effects identified to date, one of the most consistent associations has been with B*57 (35–39). Nearly 40% of elite controllers carry B*57 compared to only 9% in non-controllers, which is about the same frequency as that observed in the general population (38, 39). B*57+ carriers are also less likely to present symptoms of acute HIV-1 infection (40). Multiple GWAS have also demonstrated that SNPs in LD with B*57 showed strong association with low HIV-1 viral load set point (see below).
Perhaps because of the dominant effect of B*57 on HIV, the CTL responses restricted by B*57 molecules in HIV infection have been well studied and provide some insight into the mechanism behind this protection. The B*57 molecule binds a number of immunodominant epitopes located in conserved regions of Gag (41–45) with the most frequently targeted epitope being Gag TW10 (46), and CTL responses to these highly conserved HIV epitopes may sufficiently maintain a low viral load. The Caucasian B*57:01 and the closely related African B*57:03 molecules both bind the same Gag peptide even though they utilize mutually exclusive T-cell receptor (TCR) repertoires (47, 48). There is strong evidence that CD8+ T cells exert extreme selection pressure on HIV sequences over the course of infection (44, 49–54). Escape mutations may compromise the CD8+ T-cell response by disrupting binding of a viral peptide to HLA class I or by impairing recognition by the T-cell receptor, or they may result in significantly reduced viral fitness due to the escape variants. Indeed, reduced viral fitness of escape variants from B*57+ individuals has been demonstrated (44, 55, 56). When escape mutants are transmitted to a B*57-negative individual, the B*57-imprinted mutants tend to revert to the wildtype (44), which is an indication of their detrimental effect on viral fitness. CTL responses restricted by B*57 molecules also appear to involve a larger pool of naive T cells with greater cross-reactivity as compared to other HLA alleles, which may contribute to the control of viral replication and may also prevent viral escape to some extent (57). Interestingly, small differences between B*57 alleles (referred to as micropolymorphisms) can have a significant impact on HIV control as suggested in a study of individuals infected with clade C virus in which B*57:03 associated with lower viral load set points than B*57:02 and the closely related B*58:01 (58). These three alleles were shown to bind unique HIV epitopes and exerted differences in selection pressure on the virus, thus highlighting the effect of small changes (B*57:02 and B*57:03 differ by a single amino acid at residue 156) on the ability to control the virus. In addition, a recent report demonstrated that CD8+ T cells restricted by the protective B*57 and B*27 are resistant to the suppressive effects of Tregs because they express very low levels of Tim-3 (59). B*57 and B*27-restricted CTLs were also shown to directly kill Tregs. Tim-3 negatively regulates the immune response upon interaction with its ligand Gal-9 (60), which is constitutively expressed by Tregs (61). The overall effect is that CTLs restricted by these alleles are not suppressed, thus allowing them to function maximally during chronic infection. Taken together, these characteristics of the B*57-restricted CTL response may explain its protective role in HIV infection. Additionally, B*57 is involved in regulating NK cell function via its interaction with KIR3DL1/S1 (see below), which may also contribute to the B*57 protection against AIDS progression.
HLA-B*27 is also associated with slower progression to AIDS and control of viral load levels (36, 37, 39, 62, 63). Like B*57, B*27 recognizes a highly conserved immunodominant gag epitope, but unlike B*57, its restriction is focused on a single gag epitope KK10 (46, 64, 65). Restriction of KK10 efficiently suppresses HIV replication and delays the onset of AIDS. The KK10 peptide is conserved epitope and can tolerate little variation. Consequently, under B*27 pressure, the virus must undergo a complex pattern of mutations in order to escape recognition, but once the virus fully escapes, it is able to replicate at wild-type levels (66), resulting in rapid disease progression (64, 65, 67). In contrast to B*57 escape mutations which tend to revert to wildtype virus in B*57 subjects, B*27 escape mutations seem to be stable upon transmission to B*27 subjects (68). Furthermore, transmission of the escape variant to B*27+ subjects results in failure of the recipient to control viral replication (69). An alternate mechanism of viral evasion has also been suggested. A recent study found that a CTL escape variant of the KK10 epitope with a leucine to methionine amino acid substitution at position 6 (L6M) was capable of eliciting a secondary de novo CD+ T-cell response with an alternative TCR repertoire, but also substantially enhanced the binding of the B*27-KK10 L6M complex to the inhibitory ILT4 receptor (now referred to as LILRB2) which is expressed on myelomonocytic cells (70). This interaction resulted in a tolerogenic phenotype of these cells characterized by lower surface expression of dendritic cell (DC) maturation markers and costimulatory molecules. DC dysfunction in turn may result in functional impairment of CD8+ T cells.
Unlike B*57 and B*27, HLA-B*35 alleles consistently associate with susceptibility to HIV outcomes (30, 71–74). The B*35 effect appears to be more pronounced in people of European descent than in those of African descent, raising the question of whether specific B*35 are responsible for this effect. Analysis of B*35 subtypes indicated that more rapid progression to AIDS among B*35 positive subjects is due to the less common B*35 subtypes, B*35:02 and B*35:03, and not the most common B*35:01 (71). B*35:01 is the predominant B*35 subtype in individuals of African descent, while B*3502 and B*3503 are only sporadically detected, which likely explains the lack of B*35 association with AIDS progression in African Americans. This dichotomy of B*35 subtypes effects may be based on their peptide-binding specificity. The common B*35:01 allele preferably binds epitopes with proline at position 2 and tyrosine at position 9 (referred to as B*35PY). B*35:02/03 and the related B*53:01 is more broadly reactive and binds epitopes with proline at position 2 but accepts several amino acids at position 9, although amino acids with aromatic side chains including tyrosine are disfavored (referred to as B*35Px) (75–77). The major difference between B*35Px and PY subtypes is at position 116 (tyrosine or phenylalanine for Px vs. Serine for PY), which forms the floor of the peptide anchoring F pocket and directly interacts with residue P9 of the bound peptide (77). These results indicate that minor variations in the class I MHC can have a significant impact on AIDS pathogenesis. The peptide binding specificity may not explain the difference between B35 subtypes in HIV disease, however, as in vitro data do not support clear differences in peptide-binding specificity between B*35Px and B*35PY (78, 79). Functional assays demonstrated that the level of the gag-specific CTL is inversely correlated with HIV viral loads in B*35PY but not B*35Px individuals, suggesting a difference in the quality and/or the quantity, of HIV-specific CTL activity between B*35Px and B*35PY positive individuals (80).
A novel view of the susceptibility effect of B*35 was recently suggested that involves dendritic cells (DCs) (78), which are crucial in regulating CTL responses. DC activity is regulated by a number of inhibitory and activating receptors, one of which is the inhibitory Ig-like transcript 4 (ILT4) now referred to LILRB2 (81, 82). B*35Px tetramers were shown to bind to ILT4 with greater affinity than did B*35PY tetramers folded with the same HIV-1 peptides, potentially leading to DC dysfunction and accelerated disease progression (78). Interestingly, ILT4 is upregulated during HIV infection (83).
Although the three alleles B*27, B*57, and B*35Px consistently associate with altered rates of AIDS progression, their influence appears to occur during distinct intervals after HIV infection. Using a cohort of >2,600 individuals, the effects of these alleles on progression at distinct intervals after infection were determined: seroconversion to CD4<200; CD4<200 to an AIDS-defining illness; and AIDS-defining illness to death (36). None of the associations appears to last throughout the entire disease course; rather, each allele targets a distinct interval of AIDS progression. The B*35Px effect is short-lived during the earlier stage of CD4+ T-cell decline. The B*57-mediated protection also occurs early after infection, but once the CD4+ T-cell counts have fallen to 200 cells/ml blood, the protective effect of B*57 begins to subside. In contrast, B*27 shows no significant protection in the early stages before CD4+ T-cell counts have fallen, but significantly delays progression to an AIDS-defining illness after CD4 depletion. The discrete timing of HLA allele influence on HIV suggests that the HLA effect is not necessarily uniform and may depend on the nature of the antigenic epitopes recognized by these allotypes.
Apart from the major associations described above, other class I associations with HIV outcomes have also been described (see Table 1). HLA-B alleles with the Bw4 public epitope (as defined by amino acids 77–83) have been correlated with AIDS protection in a number of studies (84, 85). When treated as a group, the Bw4 protection is typically weaker and less consistent compared to B*27 or B*57 both of which carry the Bw4 epitope. However, when HLA alleles are ranked by relative hazard there is a clear trend for Bw4+ alleles to cluster on the protective side (Fig. 3). Other alleles that associate with HIV outcomes include the protective B*13 (86, 87), B*51:01 (37), B*58:01 (33, 35), and B*81:01 alleles (88, 89), and the susceptible B*58:02 (35, 90). Some of these alleles have frequency distributions that differ across populations, which may explain their somewhat inconsistent associations in different cohort studies.
Fig. 3. Effect of HLA-B alleles on CD4+ T-cell counts and viral load levels over time.

Average change in the square root CD4+ T-cell counts (left side) and log viral load measurements (right side) over time and 95% confidence intervals were determined using a linear mixed effects regression. HLA-B alleles are ordered from most protective (top) to least protective (bottom). The vertical line in the middle of each plot represents the sample average. Confidence intervals for alleles deviating significantly from this value do not cross the line. HLA-B Bw4 alleles are shown in green (189).
HLA class II variation
CD4+ T cells play a critical role in the immune response and are also the principal targets for HIV infection. Although most HIV specific CD4+ T cells are not infected (91) and are therefore capable of contributing to the antiviral response, there is significant impairment in their proliferative capacity in viremic patients (92, 93). CD4+ T cells are also required for the development and maintenance of memory and cytotoxic CD8+ T cells (94–97). Despite the role of CD4+ T cells in the antiviral response, the data regarding the effect of HLA class II genetic variation on HIV disease outcomes are not as convincing as those for class I. Nevertheless, there is some evidence from several studies indicating an effect of specific class II alleles/haplotypes on disease outcome. A protective effect for HLA-DRB1*01 and a susceptible effect of DRB1*15:03 have been described among African populations (98–100). DRB1*13:03 associates with reduced viral load levels in both B and C clade populations (101), and the DRB1*13-DQB1*06 haplotype has shown an association with viral control after ART interruption (102). DRB1*13:01 carriers were also reported to be less likely to transmit virus to their seronegative partners, despite the lack of an effect on viral load (100).
The most successful antiviral vaccines are those that elicit neutralizing antibodies (103, 104). However, for highly variable viruses such as HIV, vaccine design can be challenging due to the difficulties associated with eliciting broadly neutralizing antibodies that will be effective against all isolates. Interestingly, a small percentage of HIV-infected individuals develop broadly neutralizing protective antibodies (105–108), and it will be important to determine if HLA class II variation plays a role in the quality and quantity of anti-HIV antibodies.
Genome-wide association studies: the importance of agnosticism
A genome-wide association study (GWAS) of HIV disease was much anticipated by the time the first of such studies came to fruition (109). It may have been disappointing to some that the most significant variant genome-wide was one that marked the HLA-B*57:01 allele (rs2395029 in the HCP5 gene), but to those studying genetic effects of HLA class I in HIV disease outcomes, it was a testimony to the importance of this locus in determining outcome after HIV infection. Since that time, several GWAS have identified the MHC region as the dominant locus in terms of its influence on outcome to HIV infection, including the two largest studies (109, 110) (Table 2). Indeed, the only locus identified in these studies was the HLA class I region, especially in the region of HLA-B and HLA-C. A number of other variants near interesting genes outside of the MHC, such as CXCR6 and PARD3, have also shown genome-wide significance, but these have not been replicated in a second study. Most of the GWAS designed to study outcomes after HIV infection have involved Caucasian samples with the exception of Pelak et al. (111) and Pereyra et al. (110). Both studies pointed to HLA-B*57:03 as having the greatest effect, and this allele is well known to associate with protection in people of African descent. Table 2 lists all of the GWAS performed to date in cohorts involving various outcomes of HIV infection all in the absence of therapy, and three of which studied HIV acquisition. The lack of any clear association in HIV infection identified through GWAS has raised the possibility that rare genetic variants may protect from HIV infection but that these are difficult to uncover given the power of GWAS (112, 113).
Table 2.
Genome wide association studies in HIV/AIDS
| Reference | HIV-1 Outcome tested | Sample Size | Genes/Loci with GW association | Chr. | Platform |
|---|---|---|---|---|---|
| (109) | Viral setpoint | 486 infected | HCP5, HLA-C | 6 | Illumina HumanHap550 |
| Time to CD4<350 | 337 seroconverters | no GW significance | |||
| (114) | Early plasma HIV-RNA | 605 seroconverters | HLA-B, HLA-C | 6 | Illumina HumanHap300 |
| cellular HIV-DNA | 590 seroconverters | no GW significance | |||
| (193) | AIDS non-progression | 275 LTNP, 1352 seronegative controls | HCP5 | 6 | Illumina HumanHap300 |
| (194) | AIDS rapid progression | 85 rapid progressors | no GW significance | Illumina HumanHap300 | |
| 1352 seronegative controls | |||||
| (119) | HIV-1 viral setpoint | 2362 | HCP5, HLA-C | 6 | Illumina HumanHap550, 1M |
| Time to CD4<350 | 1071 seroconverters | HCP5, HLA-C, RNF39, ZNRD1 | 6 | ||
| (111) | Viral setpoint | 515 infected | no GW significance; lowest p-val - SNP linked to B*5703 | Illumina HumanHap550, 1M, 1M-Duo | |
| B*5703 in 285 HLA-typed p=5.6 × 10−10 | |||||
| (195) | Mother-to-child transmission | 100 infant cases | no GW significance | Illumina HumanHap650Y | |
| 126 infant controls | |||||
| (110) | Viral control | 974 controllers | HLA-C, MICA, HCP5, HLA-B, PSORS1C3, HCG22 | 6 | Illumina HumanHap650Y, 1M-Duo |
| 2648 progressors | |||||
| (196) | AIDS non-progression | 186 LTNP (31+59 -2nd) | no GW significance | 3 | Illumina HumanHap300, Affymetrix 500K (MACS) |
| 697 seronegative controls | CXCR6 after the second stage meta-analysis p=2.1×10−8 | ||||
| (197) | AIDS progression | 755 seroconverters | PARD3B | 2 | Affymetrix array 6.0 |
| (198)} | HIV-1 acquisition | 848 high-risk seronegative | no GW significance | Illumina 1M, 1M-Duo | |
| 531 seropositive | |||||
| (199) | AIDS non-progression with low-freq. SNPs | 365 LTNP | HCP5, C6orf48, NOTCH4 | 6 | Illumina HumanHap300 |
| 1394 seronegative controls | RICH2 | 17 | Affymetrix 500K | ||
| AIDS rapid progression with low-freq. SNPs | 147 rapid progressors | no GW significance | |||
| 1394 seronegative controls | |||||
| (200) | AIDS progression | 404 serocoverters | no GW significance | Illumina HumanHap300 | |
| (201) | HIV-1 acquisition | 302 high-risk seronegative | no GW significance | Illumina 1M-Duo | |
| 496 seropositive | |||||
| HIV-1 viral setpoint | 403 infected | no GW significance | |||
| (202) | HIV-1 replication in macrophages in vitro | 96 low replication donors | no GW significance | Illumina 610 Quad | |
| 96 high replication donors | |||||
| (203) | AIDS non-progression | 144 LTNP | HCP5, HLA-B, C6orf48, MICA | 6 | Illumina HumanHap300 |
| 605 seroconverters | |||||
| (204) | HIV-1 acquisition | 764 seropositive (975 - 2nd) | no GW significance; | Illumina HumanHap300 | |
| 1073 seronegative (324 -2nd) | CYP7B1 after the second stage meta-analysis p=7.8 × 10−8 | 8 | |||
| (205) | HIV-1 acquisition | 60 infection resistant | no GW significance | Affymetrix 50K | |
| 48 infected | |||||
| (206) | Development of neutralizing antibodies | 282 infected MSM | no GW significance | Illumina HumanHap300, 370CNV | |
| (207) | HIV-1 acquisition | 431 uninfected hemophiliacs | no GW significance | Illumina 1M, 1M-Duo | |
| 765 infected controls |
The variant rs9264942 located 35 Kb upstream of the HLA-C coding region was the second most significant, independent hit in the Euro-CHAVI study (109) and the top hit in the International HIV Controller Study (110). Although this SNP was absent on the earlier Illumina chip versions utilized by some studies, another variant, rs10484554, which is located in close proximity to rs9264942, was identified as genome-wide significant in the analysis of early HIV viral load in the PRIMO/ANRS01 study (114). It is likely that this SNP is marking the same effect as rs9264942 through linkage disequilibrium (D′=1, R2=0.31 based on the 1000 genome data). More detailed discussion of the rs9264942 variant is provided in the subsequent section.
Pereyra et al. (110) took their analysis of the GWAS data a step further by analyzing individual amino acid residues within the HLA loci after HLA typing or imputation of the HLA types. These data showed that the SNP associations derived from the GWAS data were primarily due to polymorphisms in amino acids comprising the HLA-B peptide binding groove (reviewed in 115) and in particular positions 67, 70 and 97 in HLA-B, which are all highly polymorphic, involved in direct interaction with peptide, and in strong LD with the other variants involved in peptide binding. There is no known function of these amino acid positions in HIV control beyond their participation in peptide binding, with the exception of a dimorphism at position 245 in the α-3 domain of the class I molecule, which also showed a significant association with HIV control and is known to affect the strength of the interaction between the HLA allotype and the CD8 molecule (116, 117).
Heterozygosity for CCR5Δ32, a variant that is not covered by any SNP on the chips, has been reported many times to have a modest effect on HIV outcomes prior to GWAS (118), but this variant did not show genome-wide significance until it was interrogated with very large numbers (109, 119). By most accounts, the candidate gene approach that was prevalent prior to the GWAS era produced a fair number of false positive results.
Differential HLA-C expression: beyond peptide binding
Growing evidence points to the HLA-C locus as a key player in control of HIV ever since the identification of a SNP 35 Kb upstream of the coding region for HLA-C (rs9264942) as the one of the two most significant, independent variants genome-wide in HIV control (109, 110). In general, a number of features distinguish HLA-C from the other classical class I loci, HLA-A and HLA-B, including its more limited polymorphism (http://www.ebi.ac.uk/imgt/hla) (120), lower expression on the cell surface (121–126), more extensive ligand-receptor interactions with killer cell immunoglobulin-like receptors (KIR) (127), and with respect to HIV, resistance to Nef-mediated downregulation (128). The decreased diversity of HLA-C relative to HLA-B in exons encoding the peptide-binding region (PBR) is evident when plotting nucleotide diversity (π) by nucleotide position across 4.6 Kb of 45 HLA-C and 19 HLA-B aligned sequences (Fig. 4). The lower overall expression of HLA-C compared to HLA-A and –B is due to a number of post-translational events, such as their poor assembly with β2 microglobulin (122, 123, 125), retention in the endoplasmic reticulum where they are degraded to some extent (123), and the presence of an internalization and lysosomal targeting signal in the cytoplasmic domain of the HLA-C molecule that further regulates its surface expression (124). Given the distinctions characterizing HLA-C relative to HLA-A and HLA–B, HLA-C may have unique functions as well. It is also possible that variation outside of the PBR confers a different type of diversity to the HLA-C locus, compensating for the more limited overall repertoire of peptides restricted by this locus.
Fig. 4. Nucleotide diversity (Π) in HLA-B and -C genes.

The vertical axis shows Π (%) measured in 100 bp windows across 4.6 kb of aligned sequences consisting of 45 HLA-C (red line) and 19 HLA-B (blue line) sequences. The horizontal axis indicates the nucleotide position in the alignment. The approximate positions and sizes of protein-coding regions are indicated by horizontal dark lines (190).
The rs9264942 variant was also shown to correlate with levels of HLA-C mRNA transcripts (129) and cell surface expression (130) in people of European ancestry, where the genotype associating with higher expression also associates with protection against HIV as well (109, 130). These data led to the proposal that the higher the HLA-C expression levels as a continuum (based on the three rs9264942 genotypes), the greater the protection against HIV (130). Another study concluded that differences in HLA-C cell surface expression levels as a function of rs9264942 genotypes could be attributed to the low expression HLA-C*07 lineage of subtypes alone (131). These authors proposed that the associations between rs9264942 genotype and HIV outcomes reported previously (109, 130) were actually due to specific HLA-B alleles (and perhaps a weak C*07 susceptibility effect) that are in significant linkage disequilibrium (LD) with the rs9264942 SNP. Resolving the question as to whether HLA-C expression levels have a direct on HIV control or are simply marking the effects of individual HLA-B alleles first requires an understanding of HLA-C expression: do the alleles differ significantly in their expression levels and what mechanism is controlling their expression?
Two studies, one of 50 European Americans (EA) and a second of 200 African Americans (AA), show definitely that HLA-C alleles differ significantly in expression levels as a function of allele type [Fig. 5, p (ANOVA) = 0.002 for EA and 8 × 10−21 for AA]. Most individuals are heterozygous, and therefore the differential expression of the two allotypes present in a given individual will necessarily affect their total mean fluorescent intensity (MFI), causing variation in the expression level of a given allotype across individuals. It is also possible that other modulators of HLA-C expression may themselves be polymorphic, differentially regulating expression of the same allele to some extent, though none have been identified. In spite of these confounding factors, the significant differences in HLA-C expression as a function of HLA-C allotype are remarkable. The relative expression levels of the HLA-C allotypes that are present in both African Americans and European Americans are consistent, indicating that the primary regulatory mechanisms of HLA-C expression are common to these two ethnic groups.
Fig. 5. HLA-C allotype expression levels in European and African Americans.
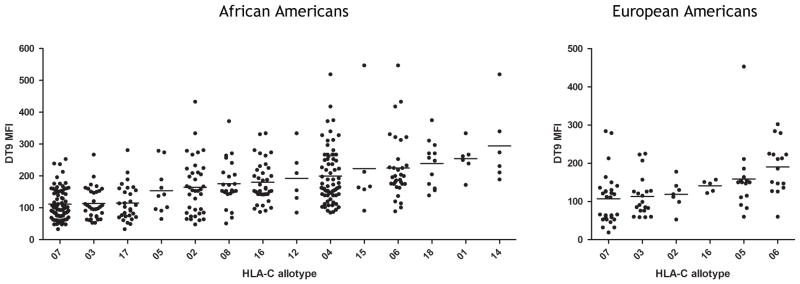
CD3+ cells from 200 African American (left plot) and 50 European American (right plot) normal donors were stained by flow cytometry for HLA-C expression level using the monoclonal antibody DT9. Median fluorescence intensity (MFI) of HLA-C staining is plotted twice for each donor (i.e. once for each HLA-C allele present).
The genetic variants and mechanisms that regulate differential HLA-C allotype expression are beginning to be addressed. We can, with fair certainty, rule out the possibility that rs9264942 has any direct effect on HLA-C expression. It is not located in any canonical transcription factor binding site, nor are there polymorphisms in known HLA-C cis-acting regulatory elements (132) that are in linkage disequilibrium (LD) with rs9264942 or that show better association with viral load control (130). Importantly, the rs9264942 SNP is in poor linage disequilibrium with HLA-C alleles in people of African descent and neither mark HLA-C expression levels nor associate with HIV control in African Americans (109–111). The mechanisms by which high levels of HLA-C protect against HIV would be expected to be consistent across ethnic groups. More efficient presentation of HIV epitopes to cytotoxic T-lymphocytes (CTL) and/or more effective regulation of natural killer (NK) cells through KIR recognition, resulting in beneficial effector cell responses against infected targets, could readily explain why high HLA-C expression associates with protection. Given the general applicability across all humans of these most obvious explanatory mechanisms, it is difficult to imagine that the rs9264942 effect could be direct. For these reasons, other polymorphisms within or near to the HLA-C region that may directly regulate its expression were considered.
MicroRNAs (miRNA) are a class of small, non-protein coding RNAs that regulate 30% or more of all genes in animals (133) by binding to specific sites, generally in the 3′UTR of the gene, resulting in posttranscriptional repression, cleavage or destabilization (134–136). Fig. 4 illustrates the extensive polymorphism in the 3′UTR of the HLA-C gene relative to HLA-B; indeed, the highest peak of variation between all pairs of distinct HLA-C gene sequences (including flanking regions) occurs in the 3′UTR. A recent scan of variation in the 3′UTR of HLA-C identified an intact binding site for the microRNA, miR-148a, that is present in the sequence of some HLA-C alleles, but missing in others due to polymorphism in the region that includes an insertion/deletion polymorphism in the seed region of miR-148a at position 263 of the 3′UTR (263D/I) (137). The variation in the miR-148a binding site was shown to regulate levels of expression of a reporter construct, as well as endogenous HLA-C cell surface levels, accounting in part for the varied levels of HLA-C expression on the cell surface that was reported previously to associate with the rs9264942 variant (137). The 263D/I variant is in strong linkage disequilibrium with the rs9264942 variant in people of European descent, but not those of African descent. On the other hand, the coding region of the HLA-C gene is in perfect LD with the 3′UTR, such that certain HLA-C alleles always contains an intact miR-148a binding site across individuals carrying those alleles and others never do (137). That is, there is no evidence that recombination has ever occurred within the short distance (just over 250 bp) between the HLA-C coding region and the miR-148a binding site. These data begin to address the identification of the causal variant for differential HLA-C expression levels, and they also provide the mechanism by which this variant regulates HLA-C expression. While the direct effect of miR-148a on level of HLA-C expression does not account completely for the varied expression of HLA-C, the miR-148a binding site, which is in very strong LD with rs9264942 in European Americans, probably accounts for the majority of the previous association between rs9264942 and HLA-C expression.
The second question addresses whether levels of HLA-C have a direct influence on control of HIV or whether the effect of HLA-C expression (as marked by genetic variants), is largely/completely attributable to individual alleles at the HLA-B locus, as suggested previously (131). Stepwise multivariate analyses that included as covariables all HLA class I alleles with frequencies of 5% or greater (n = 63) along with the 3′UTR variant (263D/I) were performed to determine whether the 3′UTR variant has an effect on HIV control that is independent of individual HLA class I alleles. B*57:01, B*57:03, B*58:01, B*27:05, C*14:01, and 263D/I were the only variables to remain in the model, indicating that each of these had a significant effect that was independent of the others. Given the strong LD between rs9264942 and 263D/I in the miR-148a binding site of HLA-C among European Americans, it is not surprising that the 263D/I variant associates significantly with control of HIV, just as rs9264942 does in this ethnic group (137). Thus, variation in the miR-148a binding site of the HLA-C 3′UTR associates significantly with HIV control, potentially because of its direct effect on HLA-C expression. However, it is particularly difficult to eliminate the possibility that individual alleles of the HLA-B locus, some of which are well known to associate strongly with HIV outcomes, are still confounding our ability to assign genetic causation.
The 3′UTR insertion/deletion variant is clearly not the sole determinant of HLA-C expression. Thus, a more direct approach to determining whether HLA-C expression has an effect on HIV control is to use HLA-C expression levels as a continuous variable rather than coarsely dividing individuals into three groups based on a genotype. The cell surface expression of sixteen common HLA-C allotypes that were determined in 200 African Americans accurately predict the expression levels of HLA-C that were determined previously in the 50 European Americans (138). The mean allotypic expression levels determined in the 200 African Americans were therefore used to assign the sum of HLA-C expression level based on the two alleles present in each of >5,000 HIV infected individuals. The continuous distribution of HLA-C allele expression levels correlated significantly with HIV outcomes in both European and African Americans. The consistency of effects across ethnic groups is strong evidence for a direct effect of HLA-C expression level on HIV control, rather than an indirect effect due to LD with HLA-B alleles, because there are extensive differences in HLA allelic frequencies and LD patterns across these two ethnic groups. The effect of HLA-C expression in this study was independent of all individual HLA allelic effects in a model that included all class I alleles (≥2%), including protection conferred by B*57.
Assigning causation for a genetic association with disease outcome is dependent on functional data to support the genetic findings. An analysis of viral sequences from 1,800 HIV infected showed that there was greater selection pressure on the virus with increasing expression of HLA-C level, based on 22 viral escape mutations in 12 HLA-C restricted epitopes. Interestingly, a similar conclusion was drawn from a previous study in Han Chinese patients, in which six HLA-C associated viral mutations occurred more commonly among individuals homozygous for high HLA-C expression alleles as compared to those homozygous for low expression HLA-C alleles (based on the rs9264942 genotype) (139). These data were the first to suggest a functional mechanism for protection conferred by high expression alleles (i.e. a better CTL response with higher expression), and they support the idea that such protection is ubiquitous across ethnic groups.
Perhaps most telling was an analysis of CTL responses to overlapping peptides spanning the HIV proteome in 1,010 African patients, which strongly supported a protective effect conferred through high expression HLA-C alleles by demonstrating a significantly greater likelihood of responding to the corresponding epitopes with increasing expression of HLA-C alleles (138). Taken together, the data support an independent, direct effect of HLA-C expression on HIV control. It is noteworthy that GWAS identified for the first time an effect of HLA class I variation on HIV disease that does not directly involve the peptide binding region, but rather another characteristic of HLA that participates in its overall function.
NK cells in HIV infection
Natural killer (NK) cells make up 5–15% of peripheral blood lymphocytes and are an important component of the innate immune system (140). They play a role in early responses against infected or transformed cells without prior sensitization through production of cytokines and direct cytotoxicity (141, 142) NK cells are also involved in bridging the innate and adaptive immune systems by the production of cytokines and chemokines that mediate activation of effector cells involved in the adaptive immune response (143).
Two subsets of NK cells in have been described depending on the level of cell surface expression of CD56 and CD16 (144–146). Although they were initially thought to be distinct cell lineages, recent data suggest that they may represent different stages of NK cell maturation (147, 148). The majority of circulating NK cells (~90%) express low levels of CD56 (CD56dim) and high levels of CD16 (CD16bright), while the rest are CD56bright and CD16dim/−. The CD56dim subset express KIR at high levels compared to the CD56bright cells. Functional studies indicate that the CD56dim subset is responsible for natural cytoxicity and contain large quantities of perforin and granzyme but produce only moderate amounts of cytokines (146). The CD56bright subset is the primary source of NK-cell-derived immunoregulatory cytokines, including IFN-γ, TNF-β, IL-10, IL-13, and GM-CSF (149).
Following HIV infection, NK cells rapidly expand in response to a variety of cytokines before the expansion of CD8+ T cells. This expansion occurs predominantly in the cytolytic CD56dim NK cell population (14, 150). However, HIV has devised strategies to evade recognition by NK cells, as is the case for other viruses. HIV downregulates HLA class I expression on the surface of infected cells in an effort to escape CD8+ T-cell lysis, rendering the cells vulnerable to NK cell lysis. The HIV-1 Nef protein selectively downregulates HLA-A and HLA-B by triggering their retention in the golgi (151), while sparing HLA-C, the dominant ligand for inhibitory KIR2D NK cell receptors, and the non-classical class I HLA-E, the ligand for the inhibitory NK cell receptor CD94/NKG2A (128, 152). Nef also downregulates stress induced molecules, such as the MHC class I-related chain-A (MIC-A) and UL-16 binding proteins-1 and -2 (ULBP-1/2), some of which serve as ligands for the activating C-type lectin NK cell receptor NKG2D (153). Thus, Nef may allow the virus to overcome both CD8+ T-cell and NK-cell-mediated recognition by selectively downregulating the major contributors to the CTL response (HLA-A and –B) but leaving expression levels of the major contributors to NK cell inhibition (HLA-C and HLA-E) intact.
Several studies have also shown that NK cell phenotype and function are altered during the course of HIV-1 infection (154–157). Acute HIV-1 infection is characterized by elevated NK cell numbers, specifically the CD56dim CD16+ cells, and a concomitant depletion of CD56brightCD16− NK cells. As infection progresses, depletion of CD56dimCD16+ cells occurs along with an increase in the functionally anergic CD56−CD16+ cells, which express significantly higher levels of inhibitory receptors and lower levels of natural cytotoxicity receptors compared with that of CD56+ NK cells (156, 158). CD56−CD16+ NK cells are rare in healthy individuals. Additional defects in NK cell function, receptor expression, and effects on other immunomodulatory cells have also been described (159–161). Interestingly, a correlation between NK cell activity and resistance to HIV infection has also been demonstrated. NK cells from Vietnamese intravascular drug users (IDUs) who remained HIV-1 negative despite many years of high risk exposure exhibited [exposed uninfected IDU (EU IDU)] significantly augmented cytolytic activity against various target cell lines compared with seronegative controls and seroconverters before or after seroconversion (162). NK cell cytokine production was also increased in EU IDUs, suggesting that enhanced NK cell function contributed to the protection against HIV-1 infection. Taken together, the data suggest that NK cells confer some level of protection against HIV and that their anergy in chronic infection may contribute to disease progression.
The KIR3DL1/S1 locus in HIV/AIDS
The KIR3DL1/S1 gene is unique in terms of its diversity and expression patterns. It is the only KIR locus that encodes both inhibitory (KIR3DL1) and activating (KIR3DS1) allotypes. It also appears to play a significant role in the control of HIV infection. Inhibitory KIR3DL1 allotypes have specificity for HLA-B (and sometimes HLA-A) molecules with the Bw4 serological motif. Although the ligands for the activating KIR3DS1 allotype have not been defined by binding studies, both population and disease association data as well as functional studies, suggest that it may interact directly or indirectly with the subset of HLA-B Bw4 molecules that have Isoleucine at position 80 (Bw4-80I). We previously reported a protective effect of the specific combination of the activating receptor KIR3DS1 with HLA-B Bw4-80I against AIDS progression using a large multicenter cohort of ART naive seroconverters composed of European American and African American individuals (12). This study was the first to describe an epistatic interaction between KIR and HLA in disease association and indicated the possibility that activating KIR may have biological significance in viral infection. Subsequent analyses showed that the KIR3DS1/HLA-B Bw4-80I compound genotype also correlated with lower viral load and protection from opportunistic infections but not AIDS associated malignancies (13). In vitro studies have demonstrated that NK cells expressing KIR3DS1 degranulate more potently in response to HIV-infected Bw4−80I+ CD4+ T cells and suppress viral replication (14). KIR3DS1+ NK cells have been shown to expand during acute HIV-1 infection, persist at elevated levels in subjects with Bw4-80I, and are able to effectively suppress HIV-1 replication in Bw-80I+target cells in vitro (150). Taken together, these epidemiological and functional data support some sort of interaction between KIR3DS1 and HLA-B Bw4-80I in the NK cell response to HIV infection.
Other studies have failed to replicate the synergistic protective effect of KIR3DS1+HLA-B Bw4 80I. A study of treatment -naive individuals with early HIV-1 infection showed that NK cells derived from individuals with KIR3DS1 responded more potently to HLA class I negative target cells than NK cells from KIR3DS1 negative subjects (163). However, this effect was only partially dependent on carrying both Bw4-80I and KIR3DS1. NK cell responses were strongest in individuals with both KIR3DS1 and HLA-Bw4-80I but the presence of KIR3DS1 alone was sufficient for producing an enhanced NK cell effector function. Gaudieri et al. (164) found that individuals with KIR3DS1 and Bw4-80I actually exhibited an accelerated progression to AIDS, as did individuals with haplotype B in general (KIR3DS1 is present on B haplotypes). O’Connell et al. (165) also found that in a small cohort of African American elite controllers, there was no correlation with KIR/HLA genotype and control of HIV replication by NK cells in vitro. However, of the eight individuals tested in vitro, only one had KIR3DS1 + Bw4 80I (B*57:03). These contradictory findings are puzzling but may be due in part to differences in the characteristics of the cohorts as well as differences in analytical methodologies. Nevertheless, failure to detect any direct binding of KIR3DS1 to HLA-B Bw4-80I does question whether these two sets of variants are simply marking the true causative genotypes through linkage disequilibrium. Several scenarios could account for this paradox. For example, a specific viral or stress peptide generated during infection and presented by Bw4-80I may be necessary to alter the affinity of KIR3DS1 for Bw4-80I or an accessory molecule might be required to generate a bridging interaction.
KIR3DS1 may also play a role in acquisition of HIV. NK cell activity in a group of highly exposed uninfected (EU) intravenous drug users (IDU) was found to be significantly greater than that in HIV-infected patients and unexposed individuals (162). This was subsequently shown to be due to the presence of elevated KIR3DS1 transcripts in the EU individuals, suggesting that KIR3DS1 may also be involved in protection from infection (166). Further, two studies have shown that homozygosity for KIR3DS1 is enriched in EU (167, 168).
KIR3DS1 may also be important in the antibody response to HIV. Findings from a recent study suggest that NK cells expressing KIR3DS1 significantly influence antibody-mediated inhibition of HIV-1, where more potent neutralization was observed when PBMCs from 3DS1+ donors were used in a neutralization assay (169). These data may have implications for identification of additional correlates of protection for future HIV-1 vaccine trials.
The inhibitory KIR3DL1 shows extensive polymorphism, and its variation has functional significance in terms of cell surface expression levels and inhibitory capacity. Staining with the KIR3DL1-specific monoclonal antibody DX9 revealed that KIR3DL1 subtypes are expressed at different levels (high, low, and null) on the NK cell surface, a phenomenon due at least in part to variation in the extracellular domains (170–172). As a general rule, the higher expressing alleles also exhibit stronger inhibitory capacity (172). Ironically, the strongly inhibitory KIR3DL1 alleles + Bw4-80I are also very protective against HIV. Using a sample size of over 1500 HIV+ individuals, we showed that multiple distinct allelic combinations of the KIR3DL1 and HLA-B loci significantly and strongly influence AIDS progression (173). In this study, the high expression/inhibition allotypes included KIR3DL1*001, *002, *008, *015, and *009, while the low expression/inhibition alleles were comprised of KIR3DL1*005 and *007. KIR3DL1*004 is not expressed on the cell surface. A trend of relatively stronger protection through higher expression KIR3DL1 alleles (referred to as 3DL1*h/y, where h = a high expressing allele and y = a high expressing allele or the null allele 3DL1*004) in combination with HLA-B Bw4-80I alleles was observed. A weaker protective effect was observed among subjects with lower expression KIR3DL1 alleles (referred to as 3DL1*l/x, where l = a low expressing allele and x = a low or high expressing allele or *004) in combination with HLA-B Bw4-80T alleles (i.e. those that have threonine at position 80 of the Bw4 motif). Notably, the most protective HLA-B allele, B*57 belongs to the Bw4-80I group and individuals carrying both 3DL1*h/y and B*57 had the strongest protective effect (Fig. 6). A related study observed that the presence of KIR3DL1 in combination with the HLA-B*57 supertype (made up of B*57 and B*58 alleles) had a highly protective effect against progression to AIDS in a Zambian cohort (174).
Fig. 6. KIR3DL1 + HLA-B Bw4 continuum of HIV viral load control.

Genotypes are ordered by degree of protection in terms of (A) disease progression and (B) viral load control. (A) Relative hazard ranges (bars) for two AIDS outcomes (CD4+ T-cell count >200 cells/mm3 and AIDS1987) relative to the Bw6/Bw6 control group. (B) Odds ratios (dots) of genotypes relative to the Bw6/Bw6 control group in terms of their distributions in the <2,000 vs. the >10,000 HIV RNA mean viral load groupings (173)
That both the activating KIR3DS1 and the inhibitory KIR3DL1 alleles confer protection against HIV seems perplexing, but the mechanism may involve NK cell activation in both cases. Protection conferred by an activating receptor (KIR3DS1) is instinctively obvious, but how does protection associating with an inhibitory KIR translate to activation? In 2005, a breakthrough was achieved in our understanding of NK cell development and function. Studies in mice indicated that NK cells with self-specific Ly49 inhibitory receptors were functionally competent while their counterparts lacking such receptors were not. The data suggested that direct interaction of self-MHC class I with inhibitory receptors on NK cells during their maturation resulted in the acquisition of NK cell functional competence that allows them to be triggered through their activating receptors when exposed to non-self or altered self, a process referred to as ‘licensing’ (25, 175, 176). Studies on human NK cells also support this concept (177, 178). Thus, it is conceivable that higher expression of KIR3DL1 on a developing NK cell in the presence of its ligand may result in the generation of an NK cell population with more vigorous activating potential under conditions where the ligand for that inhibitory receptor is missing, such as HIV infection.
KIR3DL1/S1 copy number variation
The initial focus of genome-wide association studies was primarily on SNPs, but there is a significant amount of structural variation in the human genome, including deletions, duplications, and large-scale copy-number variants, as well as insertions, inversions, and translocations. These structural variants also contribute to human diversity as well as disease susceptibility.
Copy number variation (CNV) is defined as a segment of DNA that is 1kb or larger that is present at a variable copy number in comparison with the reference genome (179). Because of their homology and tandem arrangement on chromosome 19q13.4, KIR genes frequently undergo non-allelic homologous recombination (NAHR), which causes deletions or duplications of KIR genes resulting in the extensive haplotypic variation that characterizes the locus. A recent genome-wide screen in a large HIV cohort identified a CNV in the KIR region that associates with HIV control in individuals of European ancestry (180). This CNV was found to encompass the KIR3DL1-KIR3DS1 locus. The vast majority of KIR haplotypes has a single copy of either KIR3DL1 or KIR3DS1, which segregate as alleles of a single locus. A unique 14kb sequence upstream of KIR2DL4 (2DL4 is immediately upstream of 3DL1/S1) is a frequent site of unequal crossing over, which may account for the not uncommon CNV observed in this region (7). Subsequent quantification of KIR3DS1 and KIR3DL1 copy numbers by quantitative PCR showed that increased KIR3DS1 CNV associated with lower viral load set point in the presence of its putative ligand HLA-B Bw4 80I (referred to as effective KIR3DS1 count), and regardless of the presence or absence of KIR3DL1. On the other hand, an increase in effective KIR3DL1 count (i.e. KIR3DL1 + HLA-B Bw4) was protective only when at least one effective copy of KIR3DS1 (i.e. KIR3DS1+Bw4 80I) was also present (180). Functional studies demonstrated that NK cells from individuals with a single effective copy of KIR3DL1 and KIR3DS1 had an increased capacity to inhibit HIV replication in vitro relative to NK cells from individuals who did not, which was even more pronounced in individuals with multiple copies of KIR3DL1. Moreover, there was a significant expansion in the frequency of KIR3DS1+ NK cells, which was also more pronounced in individuals with multiple copies of KIR3DL1. Overall, these data suggest that there is a beneficial interaction between KIR3DL1 and KIR3DS1, which may reflect more robust NK cell licensing in the presence of KIR3DL1. Individuals with multiple copies of KIR3DL1 in the presence of KIR3DS1 may harbor an expanded pool of KIR3DS1+ cells, which could contribute to greater antiviral activity upon infection. Another study reported stronger NK cell responses to HLA deficient K562 cells among HIV infected slow progressor individuals with KIR3DL1 and HLA-Bw4 as compared to individuals without this receptor/ligand combination (181), further supporting a contribution of an NK cell licensing effect. All individuals in this study were homozygous for KIR3DL1. Interestingly, increasing copy numbers of the activating KIR3DH also associates with control of SIV replication in rhesus macaques during primary infection (182).
KIR-mediated viral evolution
CD8+ T cells are well known to exert strong selection pressure on HIV-1 over the course of infection (183). Most of these changes arise within the HLA class I-restricted CD8+ T-cell epitopes, where they disrupt binding of viral peptide to HLA class I or impair recognition by the T-cell receptor as a means of viral escape. They may also involve amino acids upstream of the T-cell epitope that are important in antigen processing (184). Recently, efforts have turned to determining whether NK cells also place pressure on the virus, given the growing evidence that NK cells have an important role in the control of HIV-1 infection. Indeed, an HLA-Cw4 tetramer folded with an escape variant of the Cw4-restricted HIV-1 gp120 epitope SF9 (referred to as SL9) was shown to bind to KIR2DL1 with higher affinity than Cw4-SF9, resulting in inhibition of NK cell function (185). A recent report described several amino acid polymorphisms within the HIV-1 sequence that were significantly associated with the presence of specific KIR genes. Polymorphisms in a region encoding the carboxy-terminal end of Vpu and the amino-terminal end of Env were found to be significantly higher in individuals with the inhibitory receptor KIR2DL2 (186). Functionally, these ‘escape’ variants reduced the anti-viral activity of KIR2DL2+ NK cells in vitro. This represents a novel approach by which HIV can evade the host immune response.
Common HIV-1 peptide variants have also been shown to mediate differential binding of KIR3DL1 to HLA-Bw4. Point mutations that are frequently selected by CD8+ T cells within dominant HLA-B*57-restricted HIV-1 epitopes were shown to abrogate binding of KIR3DL1 to HLA-B*5701, including one within the well-characterized TW10 immunodominant epitope (T242N) (187). These results are in line with another study showing that an early emerging mutation in the TW10 epitope (G9E) that was identified in two HLA-B*5703+ patients acutely infected with HIV-1 was not associated with strong escape from CD8+ T-cell recognition, rather it was found to abrogate KIR3DL1 binding (188). Thus, viral escape from CTL recognition may also result in abrogation of inhibitory KIR-HLA interactions, potentially resulting in disinhibition of NK cells and promoting NK cell lysis of infected cells. Interestingly, the G9E variant is rare (188), suggesting some degree of KIR3DL1 mediated immune pressure on the virus.
Concluding remarks
The outcome of HIV infection in humans is largely the result of the complex interactions between virus and host immune system. Because of the extensive genetic variability of HIV, it presents an extreme challenge to the host immune system. Nevertheless, it is becoming apparent that host genetic variation, particularly variation at the HLA class I loci, is exerting significant pressure on the virus. Thus, genetic associations warrant functional studies to further investigate the molecular basis of their impact on AIDS pathogenesis and help dissect the relative contribution of the innate and adaptive immune responses, which may lead to the development of better therapies and vaccines. Advances in nucleic acid sequencing will likely drive further studies in this field as well.
Acknowledgments
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This Research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. We thank Arman Bashirova for helpful comments.
References
- 1.Deeks SG, Walker BD. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity. 2007;27:406–416. doi: 10.1016/j.immuni.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 2.Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 3.Wilson MJ, et al. Plasticity in the organization and sequences of human KIR/ILT gene families. Proc Natl Acad Sci USA. 2000;97:4778–4783. doi: 10.1073/pnas.080588597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lanier LL, Corliss BC, Wu J, Leong C, Phillips JH. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature. 1998;391:703–707. doi: 10.1038/35642. [DOI] [PubMed] [Google Scholar]
- 5.Uhrberg M, et al. Human diversity in killer cell inhibitory receptor genes. Immunity. 1997;7:753–763. doi: 10.1016/s1074-7613(00)80394-5. [DOI] [PubMed] [Google Scholar]
- 6.Martin MP, Single RM, Wilson MJ, Trowsdale J, Carrington M. KIR haplotypes defined by segregation analysis in 59 Centre d’Etude Polymorphisme Humain (CEPH) families. Immunogenetics. 2008;60:767–774. doi: 10.1007/s00251-008-0334-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pyo CW, et al. Recombinant structures expand and contract inter and intragenic diversification at the KIR locus. BMC Genomics. 2013;14:89. doi: 10.1186/1471-2164-14-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Traherne JA, et al. Mechanisms of copy number variation and hybrid gene formation in the KIR immune gene complex. Hum Mol Genet. 2010;19:737–751. doi: 10.1093/hmg/ddp538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valiante NM, et al. Functionally and structurally distinct NK cell receptor repertoires in the peripheral blood of two human donors. Immunity. 1997;7:739–751. doi: 10.1016/s1074-7613(00)80393-3. [DOI] [PubMed] [Google Scholar]
- 10.Cella M, Longo A, Ferrara GB, Strominger JL, Colonna M. NK3-specific natural killer cells are selectively inhibited by Bw4- positive HLA alleles with isoleucine 80. J Exp Med. 1994;180:1235–1242. doi: 10.1084/jem.180.4.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gumperz JE, Litwin V, Phillips JH, Lanier LL, Parham P. The Bw4 public epitope of HLA-B molecules confers reactivity with natural killer cell clones that express NKB1, a putative HLA receptor. J Exp Med. 1995;181:1133–1144. doi: 10.1084/jem.181.3.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin MP, et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet. 2002;31:429–434. doi: 10.1038/ng934. [DOI] [PubMed] [Google Scholar]
- 13.Qi Y, et al. KIR/HLA pleiotropism: Protection against both HIV and opportunistic infections. PLoS Pathog. 2006;2:e79. doi: 10.1371/journal.ppat.0020079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alter G, et al. Differential natural killer cell-mediated inhibition of HIV-1 replication based on distinct KIR/HLA subtypes. J Exp Med. 2007;204:3027–3036. doi: 10.1084/jem.20070695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Single RM, et al. Global diversity and evidence for coevolution of KIR and HLA. Nat Genet. 2007;39:1114–1119. doi: 10.1038/ng2077. [DOI] [PubMed] [Google Scholar]
- 16.Biassoni R, et al. Amino acid substitutions can influence the natural killer (NK)-mediated recognition of HLA-C molecules. Role of serine-77 and lysine-80 in the target cell protection from lysis mediated by “group 2” or “group 1” NK clones. J Exp Med. 1995;182:605–609. doi: 10.1084/jem.182.2.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Colonna M, Borsellino G, Falco M, Ferrara GB, Strominger JL. HLA-C is the inhibitory ligand that determines dominant resistance to lysis by NK1- and NK2-specific natural killer cells. Proc Natl Acad Sci USA. 1993;90:12000–12004. doi: 10.1073/pnas.90.24.12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Winter CC, Gumperz JE, Parham P, Long EO, Wagtmann N. Direct binding and functional transfer of NK cell inhibitory receptors reveal novel patterns of HLA-C allotype recognition. J Immunol. 1998;161:571–577. [PubMed] [Google Scholar]
- 19.Dohring C, Scheidegger D, Samaridis J, Cella M, Colonna M. A human killer inhibitory receptor specific for HLA-A1. J Immunol. 1996;156:3098–3101. [PubMed] [Google Scholar]
- 20.Pende D, et al. The natural killer cell receptor specific for HLA-A allotypes: a novel member of the p58/p70 family of inhibitory receptors that is characterized by three immunoglobulin-like domains and is expressed as a 140-kD disulphide-linked dimer. J Exp Med. 1996;184:505–518. doi: 10.1084/jem.184.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rajagopalan S, Long EO. A human histocompatibility leukocyte antigen (HLA)-G-specific receptor expressed on all natural killer cells. J Exp Med. 1999;189:1093–1100. doi: 10.1084/jem.189.7.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Graef T, et al. KIR2DS4 is a product of gene conversion with KIR3DL2 that introduced specificity for HLA-A*11 while diminishing avidity for HLA-C. J Exp Med. 2009;206:2557–2572. doi: 10.1084/jem.20091010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biassoni R, et al. Role of amino acid position 70 in the binding affinity of p50.1 and p58. 1 receptors for HLA-Cw4 molecules. Eur J Immunol. 1997;27:3095–3099. doi: 10.1002/eji.1830271203. [DOI] [PubMed] [Google Scholar]
- 24.Karre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. 1986;319:675–678. doi: 10.1038/319675a0. [DOI] [PubMed] [Google Scholar]
- 25.Jonsson AH, Yokoyama WM. Natural killer cell tolerance licensing and other mechanisms. Adv Immunol. 2009;101:27–79. doi: 10.1016/S0065-2776(08)01002-X. [DOI] [PubMed] [Google Scholar]
- 26.Borhis G, et al. A Peptide Antagonist Disrupts NK Cell Inhibitory Synapse Formation. J Immunol. 2013 doi: 10.4049/jimmunol.1201032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fadda L, et al. Peptide antagonism as a mechanism for NK cell activation. Proc Natl Acad Sci U S A. 2010;107:10160–10165. doi: 10.1073/pnas.0913745107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hughes AL, Yeager M. Natural selection at major histocompatibility complex loci of vertebrates. Annu Rev Genet. 1998;32:415–435. doi: 10.1146/annurev.genet.32.1.415. [DOI] [PubMed] [Google Scholar]
- 29.Parham P, Ohta T. Population biology of antigen presentation by MHC class I molecules. Science. 1996;272:67–74. doi: 10.1126/science.272.5258.67. [DOI] [PubMed] [Google Scholar]
- 30.Carrington M, et al. HLA and HIV-1: Heterozygote advantage and B*35-Cw*04 disadvantage. Science. 1999;283:1748–1752. doi: 10.1126/science.283.5408.1748. [DOI] [PubMed] [Google Scholar]
- 31.Tang J, et al. HLA class I homozygosity accelerates disease progression in human immunodeficiency virus type 1 infection. AIDS Res Hum Retroviruses. 1999;15:317–324. doi: 10.1089/088922299311277. [DOI] [PubMed] [Google Scholar]
- 32.O’Connor SL, et al. MHC heterozygote advantage in simian immunodeficiency virus-infected Mauritian cynomolgus macaques. Science translational medicine. 2010;2:22ra18. doi: 10.1126/scitranslmed.3000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trachtenberg E, et al. Advantage of rare HLA supertype in HIV disease progression. Nat Med. 2003;9:928–935. doi: 10.1038/nm893. [DOI] [PubMed] [Google Scholar]
- 34.Kawashima Y, et al. Adaptation of HIV-1 to human leukocyte antigen class I. Nature. 2009;458:641–645. doi: 10.1038/nature07746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kiepiela P, et al. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature. 2004;432:769–775. doi: 10.1038/nature03113. [DOI] [PubMed] [Google Scholar]
- 36.Gao X, et al. AIDS restriction HLA allotypes target distinct intervals of HIV-1 pathogenesis. Nature Med. 2005;11:1290–1292. doi: 10.1038/nm1333. [DOI] [PubMed] [Google Scholar]
- 37.Kaslow RA, et al. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat Med. 1996;2:405–411. doi: 10.1038/nm0496-405. [DOI] [PubMed] [Google Scholar]
- 38.Migueles SA, et al. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc Natl Acad Sci USA. 2000;97:2709–2714. doi: 10.1073/pnas.050567397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pereyra F, et al. Genetic and immunologic heterogeneity among persons who control HIV infection in the absence of therapy. J Infect Dis. 2008;197:563–571. doi: 10.1086/526786. [DOI] [PubMed] [Google Scholar]
- 40.Altfeld M, et al. Influence of HLA-B57 on clinical presentation and viral control during acute HIV-1 infection. AIDS. 2003;17:2581–2591. doi: 10.1097/00002030-200312050-00005. [DOI] [PubMed] [Google Scholar]
- 41.Brumme ZL, et al. Marked epitope- and allele-specific differences in rates of mutation in human immunodeficiency type 1 (HIV-1) Gag, Pol, and Nef cytotoxic T-lymphocyte epitopes in acute/early HIV-1 infection. J Virol. 2008;82:9216–9227. doi: 10.1128/JVI.01041-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crawford H, et al. Evolution of HLA-B*5703 HIV-1 escape mutations in HLA-B*5703-positive individuals and their transmission recipients. J Exp Med. 2009;206:909–921. doi: 10.1084/jem.20081984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klein MR, et al. Characterization of HLA-B57-restricted human immunodeficiency virus type 1 Gag- and RT-specific cytotoxic T lymphocyte responses. J Gen Virol. 1998;79:2191–2201. doi: 10.1099/0022-1317-79-9-2191. [DOI] [PubMed] [Google Scholar]
- 44.Leslie AJ, et al. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med. 2004;10:282–289. doi: 10.1038/nm992. [DOI] [PubMed] [Google Scholar]
- 45.Migueles SA, et al. The differential ability of HLA B*5701+ long-term nonprogressors and progressors to restrict human immunodeficiency virus replication is not caused by loss of recognition of autologous viral gag sequences. J Virol. 2003;77:6889–6898. doi: 10.1128/JVI.77.12.6889-6898.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Altfeld M, et al. HLA alleles associated with delayed progression to AIDS contribute strongly to the initial CD8(+) T cell response against HIV-1. PLoS Med. 2006;3:e403. doi: 10.1371/journal.pmed.0030403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gillespie GM, et al. Cross-reactive cytotoxic T lymphocytes against a HIV-1 p24 epitope in slow progressors with B*57. AIDS. 2002;16:961–972. doi: 10.1097/00002030-200205030-00002. [DOI] [PubMed] [Google Scholar]
- 48.Yu XG, et al. Mutually exclusive T-cell receptor induction and differential susceptibility to human immunodeficiency virus type 1 mutational escape associated with a two-amino-acid difference between HLA class I subtypes. J Virol. 2007;81:1619–1631. doi: 10.1128/JVI.01580-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Allen TM, et al. Selective escape from CD8+ T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV-1) sequence diversity and reveals constraints on HIV-1 evolution. J Virol. 2005;79:13239–13249. doi: 10.1128/JVI.79.21.13239-13249.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Borrow P, et al. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med. 1997;3:205–211. doi: 10.1038/nm0297-205. [DOI] [PubMed] [Google Scholar]
- 51.Cao J, McNevin J, Malhotra U, McElrath MJ. Evolution of CD8+ T cell immunity and viral escape following acute HIV-1 infection. J Immunol. 2003;171:3837–3846. doi: 10.4049/jimmunol.171.7.3837. [DOI] [PubMed] [Google Scholar]
- 52.Goonetilleke N, et al. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med. 2009;206:1253–1272. doi: 10.1084/jem.20090365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Y, et al. Selection on the human immunodeficiency virus type 1 proteome following primary infection. J Virol. 2006;80:9519–9529. doi: 10.1128/JVI.00575-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Price DA, et al. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc Natl Acad Sci USA. 1997;94:1890–1895. doi: 10.1073/pnas.94.5.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brockman MA, et al. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. J Virol. 2007;81:12608–12618. doi: 10.1128/JVI.01369-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martinez-Picado J, et al. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J Virol. 2006;80:3617–3623. doi: 10.1128/JVI.80.7.3617-3623.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kosmrlj A, et al. Effects of thymic selection of the T-cell repertoire on HLA class I-associated control of HIV infection. Nature. 2010;465:350–354. doi: 10.1038/nature08997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kloverpris HN, et al. HLA-B*57 Micropolymorphism shapes HLA allele-specific epitope immunogenicity, selection pressure, and HIV immune control. J Virol. 2012;86:919–929. doi: 10.1128/JVI.06150-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Elahi S, et al. Protective HIV-specific CD8+ T cells evade Treg cell suppression. Nat Med. 2011;17:989–995. doi: 10.1038/nm.2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu C, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245–1252. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 61.Li XC, Turka LA. An update on regulatory T cells in transplant tolerance and rejection. Nat Rev Nephrol. 2010;6:577–583. doi: 10.1038/nrneph.2010.101. [DOI] [PubMed] [Google Scholar]
- 62.Hendel H, et al. New class I and II HLA alleles strongly associated with opposite patterns of progression to AIDS. J Immunol. 1999;162:6942–6946. [PubMed] [Google Scholar]
- 63.Magierowska M, et al. Combined genotypes of CCR5, CCR2, SDF1, and HLA genes can predict the long-term nonprogressor status in human immunodeficiency virus-1- infected individuals. Blood. 1999;93:936–941. [PubMed] [Google Scholar]
- 64.Goulder PJ, et al. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat Med. 1997;3:212–217. doi: 10.1038/nm0297-212. [DOI] [PubMed] [Google Scholar]
- 65.Kelleher AD, et al. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J Exp Med. 2001;193:375–386. doi: 10.1084/jem.193.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schneidewind A, et al. Escape from the dominant HLA-B27-restricted cytotoxic T-lymphocyte response in Gag is associated with a dramatic reduction in human immunodeficiency virus type 1 replication. J Virol. 2007;81:12382–12393. doi: 10.1128/JVI.01543-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Feeney ME, et al. Immune escape precedes breakthrough human immunodeficiency virus type 1 viremia and broadening of the cytotoxic T-lymphocyte response in an HLA-B27-positive long-term-nonprogressing child. J Virol. 2004;78:8927–8930. doi: 10.1128/JVI.78.16.8927-8930.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schneidewind A, et al. Transmission and long-term stability of compensated CD8 escape mutations. J Virol. 2009;83:3993–3997. doi: 10.1128/JVI.01108-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goulder PJ, et al. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature. 2001;412:334–338. doi: 10.1038/35085576. [DOI] [PubMed] [Google Scholar]
- 70.Lichterfeld M, et al. A viral CTL escape mutation leading to immunoglobulin-like transcript 4-mediated functional inhibition of myelomonocytic cells. J Exp Med. 2007;204:2813–2824. doi: 10.1084/jem.20061865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gao X, et al. Effect of a single amino acid change in MHC class I molecules on the rate of progression to AIDS. N Engl J Med. 2001;344:1668–1675. doi: 10.1056/NEJM200105313442203. [DOI] [PubMed] [Google Scholar]
- 72.Itescu S, et al. HLA-B35 is associated with accelerated progression to AIDS. J Acquir Immune Defic Syndr. 1992;5:37–45. [PubMed] [Google Scholar]
- 73.Sahmoud T, Laurian Y, Gazengel C, Sultan Y, Gautreau C, Costagliola D. Progression to AIDS in French haemophiliacs: association with HLA-B35. AIDS. 1993;7:497–500. doi: 10.1097/00002030-199304000-00007. [DOI] [PubMed] [Google Scholar]
- 74.Scorza Smeraldi R, Fabio G, Lazzarin A, Eisera NB, Moroni M, Zanussi C. HLA-associated susceptibility to acquired immunodeficiency syndrome in Italian patients with human-immunodeficiency-virus infection. Lancet. 1986;2:1187–1189. doi: 10.1016/s0140-6736(86)92197-5. [DOI] [PubMed] [Google Scholar]
- 75.Falk K, et al. Peptide motifs of HLA-B35 and -B37 molecules. Immunogenetics. 1993;38:161–162. doi: 10.1007/BF00190906. [DOI] [PubMed] [Google Scholar]
- 76.Hill AV, et al. Molecular analysis of the association of HLA-B53 and resistance to severe malaria. Nature. 1992;360:434–439. doi: 10.1038/360434a0. [DOI] [PubMed] [Google Scholar]
- 77.Steinle A, et al. Motif of HLA-B*3503 peptide ligands. Immunogenetics. 1996;43:105–107. doi: 10.1007/BF00186615. [DOI] [PubMed] [Google Scholar]
- 78.Huang J, et al. HLA-B*35-Px-mediated acceleration of HIV-1 infection by increased inhibitory immunoregulatory impulses. J Exp Med. 2009;206:2959–2966. doi: 10.1084/jem.20091386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Streeck H, et al. Recognition of a Defined Region within p24 Gag by CD8+ T Cells during Primary Human Immunodeficiency Virus Type 1 Infection in Individuals Expressing Protective HLA Class I Alleles. J Virol. 2007;81:7725–7731. doi: 10.1128/JVI.00708-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jin X, et al. Human immunodeficiency virus type 1 (HIV-1)-specific CD8+-T-cell responses for groups of HIV-1-infected individuals with different HLA-B*35 genotypes. J Virol. 2002;76:12603–12610. doi: 10.1128/JVI.76.24.12603-12610.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Colonna M, Nakajima H, Navarro F, Lopez-Botet M. A novel family of Ig-like receptors for HLA class I molecules that modulate function of lymphoid and myeloid cells. J Leukoc Biol. 1999;66:375–381. doi: 10.1002/jlb.66.3.375. [DOI] [PubMed] [Google Scholar]
- 82.Colonna M, et al. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J Exp Med. 1997;186:1809–1818. doi: 10.1084/jem.186.11.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huang J, et al. Dendritic cell dysfunction during primary HIV-1 infection. The Journal of infectious diseases. 2011;204:1557–1562. doi: 10.1093/infdis/jir616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Flores-Villanueva PO, et al. Control of HIV-1 viremia and protection from AIDS are associated with HLA-Bw4 homozygosity. Proc Natl Acad Sci USA. 2001;98:5140–5145. doi: 10.1073/pnas.071548198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Welzel TM, et al. HLA-B Bw4 alleles and HIV-1 transmission in heterosexual couples. AIDS. 2007;21:225–229. doi: 10.1097/QAD.0b013e3280123840. [DOI] [PubMed] [Google Scholar]
- 86.Harrer EG, et al. A conserved HLA B13-restricted cytotoxic T lymphocyte epitope in Nef is a dominant epitope in HLA B13-positive HIV-1-infected patients. AIDS. 2005;19:734–735. doi: 10.1097/01.aids.0000166099.36638.56. [DOI] [PubMed] [Google Scholar]
- 87.Honeyborne I, et al. Control of human immunodeficiency virus type 1 is associated with HLA-B*13 and targeting of multiple gag-specific CD8+ T-cell epitopes. J Virol. 2007;81:3667–3672. doi: 10.1128/JVI.02689-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McLaren PJ, et al. Fine-mapping classical HLA variation associated with durable host control of HIV-1 infection in African Americans. Hum Mol Genet. 2012;21:4334–4347. doi: 10.1093/hmg/dds226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wright JK, et al. Gag-protease-mediated replication capacity in HIV-1 subtype C chronic infection: associations with HLA type and clinical parameters. J Virol. 2010;84:10820–10831. doi: 10.1128/JVI.01084-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ngumbela KC, et al. Targeting of a CD8 T cell env epitope presented by HLA-B*5802 is associated with markers of HIV disease progression and lack of selection pressure. AIDS Res Hum Retroviruses. 2008;24:72–82. doi: 10.1089/aid.2007.0124. [DOI] [PubMed] [Google Scholar]
- 91.Douek DC, et al. HIV preferentially infects HIV-specific CD4+ T cells. Nature. 2002;417:95–98. doi: 10.1038/417095a. [DOI] [PubMed] [Google Scholar]
- 92.McNeil AC, et al. High-level HIV-1 viremia suppresses viral antigen-specific CD4(+) T cell proliferation. Proc Natl Acad Sci USA. 2001;98:13878–13883. doi: 10.1073/pnas.251539598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Palmer BE, Boritz E, Blyveis N, Wilson CC. Discordance between frequency of human immunodeficiency virus type 1 (HIV-1)-specific gamma interferon-producing CD4(+) T cells and HIV-1-specific lymphoproliferation in HIV-1-infected subjects with active viral replication. J Virol. 2002;76:5925–5936. doi: 10.1128/JVI.76.12.5925-5936.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–856. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 95.Kalams SA, Walker BD. The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J Exp Med. 1998;188:2199–2204. doi: 10.1084/jem.188.12.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat Immunol. 2004;5:927–933. doi: 10.1038/ni1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lacap PA, et al. Associations of human leukocyte antigen DRB with resistance or susceptibility to HIV-1 infection in the Pumwani Sex Worker Cohort. AIDS. 2008;22:1029–1038. doi: 10.1097/QAD.0b013e3282ffb3db. [DOI] [PubMed] [Google Scholar]
- 99.Ndung’u T, et al. Major histocompatibility complex class II (HLA-DRB and -DQB) allele frequencies in Botswana: association with human immunodeficiency virus type 1 infection. Clin Diagn Lab Immunol. 2005;12:1020–1028. doi: 10.1128/CDLI.12.9.1020-1028.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tang J, Penman-Aguilar A, Lobashevsky E, Allen S, Kaslow RA. HLA-DRB1 and -DQB1 alleles and haplotypes in Zambian couples and their associations with heterosexual transmission of HIV type 1. J Infect Dis. 2004;189:1696–1704. doi: 10.1086/383280. [DOI] [PubMed] [Google Scholar]
- 101.Julg B, et al. Possession of HLA class II DRB1*1303 associates with reduced viral loads in chronic HIV-1 clade C and B infection. J Infect Dis. 2011;203:803–809. doi: 10.1093/infdis/jiq122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Malhotra U, et al. Role for HLA class II molecules in HIV-1 suppression and cellular immunity following antiretroviral treatment. J Clin Invest. 2001;107:505–517. doi: 10.1172/JCI11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Amanna IJ, Messaoudi I, Slifka MK. Protective immunity following vaccination: how is it defined? Human vaccines. 2008;4:316–319. doi: 10.4161/hv.4.4.5751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Plotkin SA. Immunologic correlates of protection induced by vaccination. The Pediatric infectious disease journal. 2001;20:63–75. doi: 10.1097/00006454-200101000-00013. [DOI] [PubMed] [Google Scholar]
- 105.Stamatatos L, Morris L, Burton DR, Mascola JR. Neutralizing antibodies generated during natural HIV-1 infection: good news for an HIV-1 vaccine? Nat Med. 2009;15:866–870. doi: 10.1038/nm.1949. [DOI] [PubMed] [Google Scholar]
- 106.Trkola A, et al. Cross-clade neutralization of primary isolates of human immunodeficiency virus type 1 by human monoclonal antibodies and tetrameric CD4-IgG. J Virol. 1995;69:6609–6617. doi: 10.1128/jvi.69.11.6609-6617.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Walker LM, et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009;326:285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wu X, et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science. 2010;329:856–861. doi: 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fellay J, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pereyra F, et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330:1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pelak K, et al. Host determinants of HIV-1 control in African Americans. J Infect Dis. 2010;201:1141–1149. doi: 10.1086/651382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 113.Shea PR, Shianna KV, Carrington M, Goldstein DB. Host Genetics of HIV Acquisition and Viral Control. Annu Rev Med. 2013;64:203–217. doi: 10.1146/annurev-med-052511-135400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dalmasso C, et al. Distinct genetic loci control plasma HIV-RNA and cellular HIV-DNA levels in HIV-1 infection: the ANRS Genome Wide Association 01 study. PLoS ONE. 2008;3:e3907. doi: 10.1371/journal.pone.0003907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Carrington M, Walker BD. Immunogenetics of spontaneous control of HIV. Annu Rev Med. 2012;63:131–145. doi: 10.1146/annurev-med-062909-130018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gao GF, et al. Classical and nonclassical class I major histocompatibility complex molecules exhibit subtle conformational differences that affect binding to CD8alphaalpha. The Journal of biological chemistry. 2000;275:15232–15238. doi: 10.1074/jbc.275.20.15232. [DOI] [PubMed] [Google Scholar]
- 117.Martinez-Naves E, et al. Interactions of HLA-B*4801 with peptide and CD8. Tissue Antigens. 1997;50:258–264. doi: 10.1111/j.1399-0039.1997.tb02869.x. [DOI] [PubMed] [Google Scholar]
- 118.Carrington M, Dean M, Martin MP, O’Brien SJ. Genetics of HIV-1 infection: chemokine receptor CCR5 polymorphism and its consequences. Hum Mol Genet. 1999;8:1939–1945. doi: 10.1093/hmg/8.10.1939. [DOI] [PubMed] [Google Scholar]
- 119.Fellay J, et al. Common genetic variation and the control of HIV-1 in humans. PLoS Genet. 2009;5:e1000791. doi: 10.1371/journal.pgen.1000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zemmour J, Parham P. Distinctive polymorphism at the HLA-C locus: implications for the expression of HLA-C. The Journal of experimental medicine. 1992;176:937–950. doi: 10.1084/jem.176.4.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.McCutcheon JA, Gumperz J, Smith KD, Lutz CT, Parham P. Low HLA-C expression at cell surfaces correlates with increased turnover of heavy chain mRNA. J Exp Med. 1995;181:2085–2095. doi: 10.1084/jem.181.6.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Neefjes JJ, Ploegh HL. Allele and locus-specific differences in cell surface expression and the association of HLA class I heavy chain with beta 2-microglobulin: differential effects of inhibition of glycosylation on class I subunit association. Eur J Immunol. 1988;18:801–810. doi: 10.1002/eji.1830180522. [DOI] [PubMed] [Google Scholar]
- 123.Neisig A, Melief CJ, Neefjes J. Reduced cell surface expression of HLA-C molecules correlates with restricted peptide binding and stable TAP interaction. J Immunol. 1998;160:171–179. [PubMed] [Google Scholar]
- 124.Schaefer MR, Williams M, Kulpa DA, Blakely PK, Yaffee AQ, Collins KL. A novel trafficking signal within the HLA-C cytoplasmic tail allows regulated expression upon differentiation of macrophages. J Immunol. 2008;180:7804–7817. doi: 10.4049/jimmunol.180.12.7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Setini A, et al. Distinctive features of the alpha 1-domain alpha helix of HLA-C heavy chains free of beta 2-microglobulin. Hum Immunol. 1996;46:69–81. doi: 10.1016/0198-8859(96)00011-0. [DOI] [PubMed] [Google Scholar]
- 126.Snary D, Barnstable CJ, Bodmer WF, Crumpton MJ. Molecular structure of human histocompatibility antigens: the HLA-C series. Eur J Immunol. 1977;7:580–585. doi: 10.1002/eji.1830070816. [DOI] [PubMed] [Google Scholar]
- 127.Bashirova AA, Martin MP, McVicar DW, Carrington M. The killer immunoglobulin-like receptor gene cluster: Tuning the genome for defense. Annu Rev Genomics Hum Genet. 2006;7:277–300. doi: 10.1146/annurev.genom.7.080505.115726. [DOI] [PubMed] [Google Scholar]
- 128.Cohen GB, et al. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity. 1999;10:661–671. doi: 10.1016/s1074-7613(00)80065-5. [DOI] [PubMed] [Google Scholar]
- 129.Stranger BE, et al. Genome-wide associations of gene expression variation in humans. PLoS Genet. 2005;1:e78. doi: 10.1371/journal.pgen.0010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Thomas R, et al. HLA-C cell surface expression and control of HIV/AIDS correlate with a variant upstream of HLA-C. Nat Genet. 2009;41:1290–1294. doi: 10.1038/ng.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Corrah TW, et al. Reappraisal of the relationship between the HIV-1-protective single-nucleotide polymorphism 35 kilobases upstream of the HLA-C gene and surface HLA-C expression. J Virol. 2011;85:3367–3374. doi: 10.1128/JVI.02276-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yao Z, Volgger A, Scholz S, Albert E. Polymorphism of the HLA-C promotor region. Immunogenetics. 1997;45:428–431. doi: 10.1007/s002510050225. [DOI] [PubMed] [Google Scholar]
- 133.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 134.Giraldez AJ, et al. Zebrafish MiR-430 promotes deadenylation and clearance of maternal mRNAs. Science. 2006;312:75–79. doi: 10.1126/science.1122689. [DOI] [PubMed] [Google Scholar]
- 135.Lim LP, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 136.Wu L, Fan J, Belasco JG. MicroRNAs direct rapid deadenylation of mRNA. Proc Natl Acad Sci USA. 2006;103:4034–4039. doi: 10.1073/pnas.0510928103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kulkarni S, et al. Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature. 2011;472:495–498. doi: 10.1038/nature09914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Apps R, et al. Influence of HLA-C expression level on HIV control. Science. 2013 doi: 10.1126/science.1232685. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Blais ME, et al. High frequency of HIV mutations associated with HLA-C suggests enhanced HLA-C-restricted CTL selective pressure associated with an AIDS-protective polymorphism. J Immunol. 2012;188:4663–4670. doi: 10.4049/jimmunol.1103472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Trinchieri G. Biology of natural killer cells. Adv Immunol. 1989;47:187–376. doi: 10.1016/S0065-2776(08)60664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bancroft GJ. The role of natural killer cells in innate resistance to infection. Curr Opin Immunol. 1993;5:503–510. doi: 10.1016/0952-7915(93)90030-v. [DOI] [PubMed] [Google Scholar]
- 142.Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: Function and regulation by innate cytokines. Annu Rev Immunol. 1999;17:189–220. doi: 10.1146/annurev.immunol.17.1.189. [DOI] [PubMed] [Google Scholar]
- 143.Vivier E, et al. Innate or adaptive immunity? The example of natural killer cells Science. 2011;331:44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Caligiuri MA, Zmuidzinas A, Manley TJ, Levine H, Smith KA, Ritz J. Functional consequences of interleukin 2 receptor expression on resting human lymphocytes. Identification of a novel natural killer cell subset with high affinity receptors. J Exp Med. 1990;171:1509–1526. doi: 10.1084/jem.171.5.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cooper MA, et al. Human natural killer cells: a unique innate immunoregulatory role for the CD56(bright) subset. Blood. 2001;97:3146–3151. doi: 10.1182/blood.v97.10.3146. [DOI] [PubMed] [Google Scholar]
- 146.Nagler A, Lanier LL, Cwirla S, Phillips JH. Comparative studies of human FcRIII-positive and negative natural killer cells. J Immunol. 1989;143:3183–3191. [PubMed] [Google Scholar]
- 147.Romagnani C, et al. CD56brightCD16- killer Ig-like receptor- NK cells display longer telomeres and acquire features of CD56dim NK cells upon activation. J Immunol. 2007;178:4947–4955. doi: 10.4049/jimmunol.178.8.4947. [DOI] [PubMed] [Google Scholar]
- 148.Yu J, et al. CD94 surface density identifies a functional intermediary between the CD56bright and CD56dim human NK-cell subsets. Blood. 2010;115:274–281. doi: 10.1182/blood-2009-04-215491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22:633–640. doi: 10.1016/s1471-4906(01)02060-9. [DOI] [PubMed] [Google Scholar]
- 150.Alter G, et al. HLA class I subtype-dependent expansion of KIR3DS1+ and KIR3DL1+ NK cells during acute human immunodeficiency virus type 1 infection. J Virol. 2009;83:6798–6805. doi: 10.1128/JVI.00256-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Schwartz O, Marechal V, Le Gall S, Lemonnier F, Heard JM. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med. 1996;2:338–342. doi: 10.1038/nm0396-338. [DOI] [PubMed] [Google Scholar]
- 152.Le Gall S, et al. Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity. 1998;8:483–495. doi: 10.1016/s1074-7613(00)80553-1. [DOI] [PubMed] [Google Scholar]
- 153.Cerboni C, et al. Human immunodeficiency virus 1 Nef protein downmodulates the ligands of the activating receptor NKG2D and inhibits natural killer cell-mediated cytotoxicity. The Journal of general virology. 2007;88:242–250. doi: 10.1099/vir.0.82125-0. [DOI] [PubMed] [Google Scholar]
- 154.Bonaparte MI, Barker E. Inability of natural killer cells to destroy autologous HIV-infected T lymphocytes. AIDS. 2003;17:487–494. doi: 10.1097/00002030-200303070-00003. [DOI] [PubMed] [Google Scholar]
- 155.Kottilil S, et al. Innate immunity in human immunodeficiency virus infection: effect of viremia on natural killer cell function. J Infect Dis. 2003;187:1038–1045. doi: 10.1086/368222. [DOI] [PubMed] [Google Scholar]
- 156.Mavilio D, et al. Natural killer cells in HIV-1 infection: dichotomous effects of viremia on inhibitory and activating receptors and their functional correlates. Proc Natl Acad Sci USA. 2003;100:15011–15016. doi: 10.1073/pnas.2336091100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ullum H, Gotzsche PC, Victor J, Dickmeiss E, Skinhoj P, Pedersen BK. Defective natural immunity: an early manifestation of human immunodeficiency virus infection. J Exp Med. 1995;182:789–799. doi: 10.1084/jem.182.3.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mavilio D, et al. Characterization of CD56-/CD16+ natural killer (NK) cells: a highly dysfunctional NK subset expanded in HIV-infected viremic individuals. Proc Natl Acad Sci USA. 2005;102:2886–2891. doi: 10.1073/pnas.0409872102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Alter G, et al. IL-10 induces aberrant deletion of dendritic cells by natural killer cells in the context of HIV infection. J Clin Invest. 2010;120:1905–1913. doi: 10.1172/JCI40913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.De Maria A, et al. The impaired NK cell cytolytic function in viremic HIV-1 infection is associated with a reduced surface expression of natural cytotoxicity receptors (NKp46, NKp30 and NKp44) Eur J Immunol. 2003;33:2410–2418. doi: 10.1002/eji.200324141. [DOI] [PubMed] [Google Scholar]
- 161.Wong AH, et al. Alterations in natural killer cell receptor profiles during HIV type 1 disease progression among chronically infected South African adults. AIDS Res Hum Retroviruses. 2010;26:459–469. doi: 10.1089/aid.2009.0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Scott-Algara D, et al. Cutting edge: increased NK cell activity in HIV-1-exposed but uninfected Vietnamese intravascular drug users. J Immunol. 2003;171:5663–5667. doi: 10.4049/jimmunol.171.11.5663. [DOI] [PubMed] [Google Scholar]
- 163.Long BR, et al. Conferral of enhanced natural killer cell function by KIR3DS1 in early human immunodeficiency virus type 1 infection. J Virol. 2008;82:4785–4792. doi: 10.1128/JVI.02449-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gaudieri S, et al. Killer immunoglobulin-like receptors and HLA act both independently and synergistically to modify HIV disease progression. Genes Immun. 2005;6:683–690. doi: 10.1038/sj.gene.6364256. [DOI] [PubMed] [Google Scholar]
- 165.O’Connell KA, Han Y, Williams TM, Siliciano RF, Blankson JN. Role of natural killer cells in a cohort of elite suppressors: low frequency of the protective KIR3DS1 allele and limited inhibition of human immunodeficiency virus type 1 replication in vitro. J Virol. 2009;83:5028–5034. doi: 10.1128/JVI.02551-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ravet S, et al. Distinctive NK-cell receptor repertoires sustain high-level constitutive NK-cell activation in HIV-exposed uninfected individuals. Blood. 2007;109:4296–4305. doi: 10.1182/blood-2006-08-040238. [DOI] [PubMed] [Google Scholar]
- 167.Boulet S, et al. Increased proportion of KIR3DS1 homozygotes in HIV-exposed uninfected individuals. AIDS. 2008;22:595–599. doi: 10.1097/QAD.0b013e3282f56b23. [DOI] [PubMed] [Google Scholar]
- 168.Guerini FR, et al. Under representation of the inhibitory KIR3DL1 molecule and the KIR3DL1+/BW4+ complex in HIV exposed seronegative individuals. J infect Dis. 2011;203:1235–1239. doi: 10.1093/infdis/jir020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Brown BK, et al. The role of natural killer (NK) cells and NK cell receptor polymorphisms in the assessment of HIV-1 neutralization. PLoS ONE. 2012;7:e29454. doi: 10.1371/journal.pone.0029454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gardiner CM, et al. Different NK cell surface phenotypes defined by the DX9 antibody are due to KIR3DL1 gene polymorphism. J Immunol. 2001;166:2992–3001. doi: 10.4049/jimmunol.166.5.2992. [DOI] [PubMed] [Google Scholar]
- 171.Pando MJ, Gardiner CM, Gleimer M, McQueen KL, Parham P. The protein made from a common allele of KIR3DL1 (3DL1*004) is poorly expressed at cell surfaces due to substitution at positions 86 in Ig domain 0 and 182 in Ig domain 1. J Immunol. 2003;171:6640–6649. doi: 10.4049/jimmunol.171.12.6640. [DOI] [PubMed] [Google Scholar]
- 172.Yawata M, Yawata N, Draghi M, Little AM, Partheniou F, Parham P. Roles for HLA and KIR polymorphisms in natural killer cell repertoire selection and modulation of effector function. J Exp Med. 2006;203:633–645. doi: 10.1084/jem.20051884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Martin MP, et al. Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nat Genet. 2007;39:733–740. doi: 10.1038/ng2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lopez-Vazquez A, et al. Interaction between KIR3DL1 and HLA-B*57 supertype alleles influences the progression of HIV-1 infection in a Zambian population. Hum Immunol. 2005;66:285–289. doi: 10.1016/j.humimm.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 175.Fernandez NC, Treiner E, Vance RE, Jamieson AM, Lemieux S, Raulet DH. A subset of natural killer cells achieves self-tolerance without expressing inhibitory receptors specific for self-MHC molecules. Blood. 2005;105:4416–4423. doi: 10.1182/blood-2004-08-3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kim S, et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature. 2005;436:709–713. doi: 10.1038/nature03847. [DOI] [PubMed] [Google Scholar]
- 177.Anfossi N, et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity. 2006;25:331–342. doi: 10.1016/j.immuni.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 178.Kim S, et al. HLA alleles determine differences in human natural killer cell responsiveness and potency. Proc Natl Acad Sci U S A. 2008;105:3053–3058. doi: 10.1073/pnas.0712229105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- 180.Pelak K, et al. Copy number variation of KIR genes influences HIV-1 control. PLoS biology. 2011;9:e1001208. doi: 10.1371/journal.pbio.1001208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kamya P, et al. Inhibitory Killer Immunoglobulin-like Receptors to self HLA-B and HLA-C ligands contribute differentially to Natural Killer cell functional potential in HIV infected slow progressors. Clin Immunol. 2012;143:246–255. doi: 10.1016/j.clim.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 182.Hellmann I, Lim SY, Gelman RS, Letvin NL. Association of activating KIR copy number variation of NK cells with containment of SIV replication in rhesus monkeys. PLoS Pathogens. 2011;7:e1002436. doi: 10.1371/journal.ppat.1002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Carlson JM, Brumme ZL. HIV evolution in response to HLA-restricted CTL selection pressures: a population-based perspective. Microbes Infect. 2008;10:455–461. doi: 10.1016/j.micinf.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 184.Allen TM, et al. Selection, transmission, and reversion of an antigen-processing cytotoxic T-lymphocyte escape mutation in human immunodeficiency virus type 1 infection. J Virol. 2004;78:7069–7078. doi: 10.1128/JVI.78.13.7069-7078.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Thananchai H, et al. Reciprocal recognition of an HLA-Cw4-restricted HIV-1 gp120 epitope by CD8+ T cells and NK cells. AIDS. 2009;23:189–193. doi: 10.1097/QAD.0b013e32831fb55a. [DOI] [PubMed] [Google Scholar]
- 186.Alter G, et al. HIV-1 adaptation to NK-cell-mediated immune pressure. Nature. 2011;476:96–100. doi: 10.1038/nature10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Fadda L, et al. Common HIV-1 peptide variants mediate differential binding of KIR3DL1 to HLA-Bw4 molecules. J Virol. 2011;85:5970–5974. doi: 10.1128/JVI.00412-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Brackenridge S, et al. An early HIV mutation within an HLA-B*57-restricted T cell epitope abrogates binding to the killer inhibitory receptor 3DL1. J Virol. 2011;85:5415–5422. doi: 10.1128/JVI.00238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Baker BM, Block BL, Rothchild AC, Walker BD. Elite control of HIV infection: implications for vaccine design. Expert Opin Biol Ther. 2009;9:55–69. doi: 10.1517/14712590802571928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.O’Huigin C, et al. The molecular origin and consequences of escape from miRNA regulation by HLA-C alleles. Am J Hum Genet. 2011;89:424–431. doi: 10.1016/j.ajhg.2011.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Miura T, et al. HLA-B57/B*5801 human immunodeficiency virus type 1 elite controllers select for rare gag variants associated with reduced viral replication capacity and strong cytotoxic T-lymphocyte [corrected] recognition. J Virol. 2009;83:2743–2755. doi: 10.1128/JVI.02265-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhang Y, et al. Multilayered defense in HLA-B51-associated HIV viral control. J Immunol. 2011;187:684–691. doi: 10.4049/jimmunol.1100316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Limou S, et al. Genomewide association study of an AIDS-nonprogression cohort emphasizes the role played by HLA genes (ANRS Genomewide Association Study 02) J Infect Dis. 2009;199:419–426. doi: 10.1086/596067. [DOI] [PubMed] [Google Scholar]
- 194.Le Clerc S, et al. Genomewide association study of a rapid progression cohort identifies new susceptibility alleles for AIDS (ANRS Genomewide Association Study 03) J Infect Dis. 2009;200:1194–1201. doi: 10.1086/605892. [DOI] [PubMed] [Google Scholar]
- 195.Joubert BR, Lange EM, Franceschini N, Mwapasa V, North KE, Meshnick SR. A whole genome association study of mother-to-child transmission of HIV in Malawi. Genome Med. 2010;2:17. doi: 10.1186/gm138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Limou S, et al. Multiple-cohort genetic association study reveals CXCR6 as a new chemokine receptor involved in long-term nonprogression to AIDS. J Infect Dis. 2010;202:908–915. doi: 10.1086/655782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Troyer JL, et al. Genome-wide association study implicates PARD3B-based AIDS restriction. J Infect Dis. 2011;203:1491–1502. doi: 10.1093/infdis/jir046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Petrovski S, et al. Common human genetic variants and HIV-1 susceptibility: a genome-wide survey in a homogeneous African population. AIDS. 2011;25:513–518. doi: 10.1097/QAD.0b013e328343817b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Le Clerc S, et al. Screening low-frequency SNPS from genome-wide association study reveals a new risk allele for progression to AIDS. J Acquir Immune Defic Syndr. 2011;56:279–284. doi: 10.1097/QAI.0b013e318204982b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.van Manen D, et al. Genome-wide association scan in HIV-1-infected individuals identifying variants influencing disease course. PLoS ONE. 2011;6:e22208. doi: 10.1371/journal.pone.0022208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lingappa JR, et al. Genomewide association study for determinants of HIV-1 acquisition and viral set point in HIV-1 serodiscordant couples with quantified virus exposure. PLoS ONE. 2011;6:e28632. doi: 10.1371/journal.pone.0028632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Bol SM, et al. Genome-wide association study identifies single nucleotide polymorphism in DYRK1A associated with replication of HIV-1 in monocyte-derived macrophages. PLoS ONE. 2011;6:e17190. doi: 10.1371/journal.pone.0017190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Guergnon J, et al. Single-nucleotide polymorphism-defined class I and class III major histocompatibility complex genetic subregions contribute to natural long-term nonprogression in HIV infection. J Infect Dis. 2012;205:718–724. doi: 10.1093/infdis/jir833. [DOI] [PubMed] [Google Scholar]
- 204.Limou S, et al. Multicohort genomewide association study reveals a new signal of protection against HIV-1 acquisition. J Infect Dis. 2012;205:1155–1162. doi: 10.1093/infdis/jis028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Luo M, et al. A genetic polymorphism of FREM1 is associated with resistance against HIV infection in the Pumwani sex worker cohort. J Virol. 2012;86:11899–11905. doi: 10.1128/JVI.01499-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Euler Z, van Gils MJ, Boeser-Nunnink BD, Schuitemaker H, van Manen D. Genome-Wide Association Study on the Development of Cross-Reactive Neutralizing Antibodies in HIV-1 Infected Individuals. PLoS ONE. 2013;8:e54684. doi: 10.1371/journal.pone.0054684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lane J, et al. A genome-wide association study of resistance to HIV infection in highly exposed uninfected individuals with hemophilia A. Hum Mol Genet. 2013;22:1903–1910. doi: 10.1093/hmg/ddt033. [DOI] [PMC free article] [PubMed] [Google Scholar]