

Abstract
Lecithin:cholesterol acyltransferase (LCAT), a key enzyme in HDL metabolism, has been proposed to have atheroprotective properties, by promoting reverse cholesterol transport. Over expression of LCAT in various animal models, however, has led to conflicting results on its overall effect on lipoproteins and atherosclerosis. In this study, the effect of overexpression of LCAT in non-human primates on lipoprotein metabolism is examined. Human LCAT was expressed with adenovirus in squirrel monkeys (n=8), resulting on day 4 in a 22-fold increase of LCAT activity (257±23 vs. 5618±799 nmol/mL/h; p<0.0001). At its peak, LCAT was found to nearly double the level of HDL-C from baseline (113±7 vs. 260±24 mg/dL; p<0.01). HDL formed after treatment with the adenovirus was larger in size, as assessed by FPLC analysis. By kinetic studies, it was determined that there was a decrease in apoA-I resident time (0.373±0.027 vs. 0.685±0.045 d−1; p<0.0001), and almost a doubling in the apoA-I synthetic rate (22±2 vs. 41±3 mg/kgxd, p<0.0001) but no overall change in apoA-I levels. In addition, increased expression of LCAT was associated with a 37% reduction of apoB levels (12±1 vs. 19±1 mg/dL; p<0.05), due to increased LDL catabolism (FCR = 1.7±0.1 in controls vs. 4.2±0.3 d−1 in LCAT treated group; p<0.05). In summary, overexpression of LCAT in non-human primates leads to an anti-atherogenic lipoprotein profile, by increasing HDL-C and lowering apoB, thus making LCAT a potential drug target for reducing atherosclerosis.
Keywords: Adenovirus, apolipoprotein A-I, apolipoprotein B, LCAT, HDL, LDL, non-human primate, cholesterol
INTRODUCTION
Lecithin:cholesterol acyltransferse (LCAT), a plasma enzyme produced by the liver, catalyzes the conversion of cholesterol to cholesteryl esters on lipoproteins, by the transacylation of fatty acid from the sn-2 position of phosphatidylcholine to the 3-hydroxyl group on the A-ring of cholesterol1. The majority of LCAT activity is found on HDL but approximately 30% is also on apoB-containing lipoproteins2. Because of the increased hydrophobicity of cholesteryl ester compared to cholesterol, cholesteryl ester formed by LCAT on the surface of lipoprotein particles partitions into the neutral lipid core of lipoproteins. This has a profound effect on lipoprotein structure, particularly HDL; it converts the nascent discoidal shaped HDL to the mature spherical shaped alpha-migrating form of HDL.
The physiologic consequences of LCAT on atherosclerosis, however, have not been definitively established. A long standing hypothesis is that LCAT is anti-atherogenic, because it promotes the reverse cholesterol transport pathway, the pathway by which excess cellular cholesterol is returned to the liver for excretion 3. This potentially occurs by two mechanisms. First, LCAT increases the level of HDL, which in itself may increase the flux of cholesterol from cells by increasing the amount of extracellular acceptors of cholesterol. Secondly, the esterification of cholesterol by LCAT on HDL would be predicted to limit the spontaneous back exchange of cholesterol from HDL to cells and instead promote the net delivery of cholesterol to HDL and then to the liver. In support of this model, the level of LCAT activity, in some studies, have been shown to be positively correlated with HDL-C and appears to be inversely related to the risk of coronary heart disease (CHD) 4. In addition, patients with a genetic deficiency of LCAT, have a profound decrease in HDL-C, but paradoxically these patients do not appear to have a significant increase risk of CHD 5;6. This may be due, at least in part, to the fact that LCAT deficiency also leads to a decrease in LDL-C 7, possibly as a consequence of the decreased formation of cholesteryl esters on HDL, which is normally transferred from HDL to LDL and other apoB containing lipoproteins by the Cholesteryl Ester Transfer Protein (CETP).
Various animal models of both the absence and overexpression of LCAT have been described 4;8–13, but the effect of LCAT on lipoproteins and atherosclerosis varies, depending on the animal model. In mice, overexpression of LCAT results in an increase in the level of a large lipid-rich form of HDL and accelerated atherosclerosis. In contrast, increased LCAT expression in rabbits also raises HDL-C but markedly decreases atherosclerosis43. Mice, unlike rabbits and humans, lack CETP, which causes excess cholesteryl esters to accumulate on HDL in LCAT transgenic mice14. This results in a large dysfunctional form of HDL, with decreased ability to deliver cholesterol to the liver 14. In contrast, when LCAT was expressed in transgenic mice that also express CETP, LCAT was found line in rabbits8,43 to decrease atherosclerosis. Interestingly, LCAT K/O mice also have a decrease propensity for atherosclerosis15.
In this study, we examined the effect of LCAT in non-human primates, namely squirrel monkeys. Because of the conflicting animal and genetic studies, it is important to establish the effect of LCAT in another animal model, which more closely resembles humans in their lipoprotein metabolism. Squirrel monkeys have been commonly used to investigate lipoprotein metabolism, as well as the impact of various drug therapies on plasma lipids60,61. Squirrel monkeys like humans also contain CETP, but they do not have the very high levels found in rabbits16 and contain, like humans, apoA-II, which is not present in rabbits17. Furthermore, squirrel monkeys among New World monkeys have the greatest propensity to develop diet-induced atherosclerosis and have also been found to spontaneously develop atherosclerosis 35. In this study, human LCAT was overexpressed in squirrel monkeys, using adenovirus, which was found to raise HDL-C and lower apoB. Results from this study raise the possibility of LCAT as a target for the development of new drugs for modulating lipoprotein metabolism and reducing atherosclerosis.
MATERIAL AND METHODS
ANIMALS
Sixteen adult male squirrel monkeys (Saimiri sciureus), ranging in weight from 850 to 1200g, were fed with Fiber-Plus Monkey Diet # 5049 (PMI Nutrition Int’l Inc., St. Louis, MO). The animals used in these studies were maintained in accordance with the NIH Guide for the CARE of Laboratory Animals, protocol H-0059/H-0059R1.
GENERATION OF RECOMBINANT ADENOVIRUS
A full-length human wild type LCAT, and luciferase cDNA’s were subcloned into a shuttle vector (pAdl2HL), containing a CMV promoter/enhancer fragment [HindII-Pbsl, −601 bp to f52 bp] promoter elements, as well as the SV40 polyadenylation signal. Recombinant adenovirus encoding luciferase (rLucif-AdV) and LCAT (rLCAT-AdV) were generated after co-transfection of pAdl2LCAT and pJM17 (Ad5 genome) in 293 cells, as previously described18, propagated in 293 cells, and purified by cesium chloride density ultracentrifugation. The purified virus was then tittered and diluted in 0.2% albumin (Sigma Chemical Co., St. Louis, MO) before infusion into the animals. 2×1010 pfu of either rLCAT-AdV or rLucif-AdV was infused in each monkey by tail venipuncture.
ANALYSIS OF LIPIDS AND APOLIPOPROTEINS
Blood was collected after overnight fasting either by tail venipuncture or from an indwelling catheter into tubes containing 0.1M EDTA and centrifuged for 15 min and stored at 4°C before analysis. Total cholesterol, triglycerides, phospholipids, free cholesterol, and ApoA-I and ApoB were determined, using enzymatic or immuno-turbidometric kits from Sigma Diagnostics and Wako Chemicals. HDL cholesterol was measured after dextran sulfate (Ciba-Corning) precipitation 19. Plasma lipoproteins were further analyzed by FPLC by gel filtration chromatography, using two Superose 6 HR 10/30 columns connected in series (Pharmacia Biotech Inc.). Lipoproteins were eluted at a constant flow rate of 0.3 ml/min with PBS buffer, containing 1mM EDTA and 0.02% sodium azide.
LCAT ACTIVITY ASSAY
LCAT activity was determined by the formation of cholesteryl esters, using proteoliposomes containing apoA-I as substrate20.
ISOLATION OF LIPOPROTEINS
HDL (d=1.063–1.21) and LDL (d=1.030–1.050 g/mL) were isolated from monkey plasma by sequential ultracentrifugation, followed by dialysis at 4 °C against PBS, containing 0.01% (wt/vol) EDTA. Agarose gel electrophoresis and FPLC analysis were used to confirm the purity of each lipoprotein fraction.
IN VIVO METABOLIC STUDIES
131I-apoA-I HDL was prepared by a modification of the iodine monochloride method21. Lyophilized purified human apoA-I was dissolved in a 6M guanidine-HCl, 1M glycine (pH 8.5) buffer containing one mCi of Na131I and iodinated after adding 0. 33 mM iodine monochloride and then dialyzed. 131I-labeled apoA-I was then reassociated with HDL from either untreated squirrel monkey plasma or rAdv-LCAT modified plasma for 30 min at 37°C and extensively dialyzed at 4 °C against PBS, containing 0.01% (wt/vol) EDTA. All radiolabeled HDL preparations were analyzed by FPLC and native agarose gel to confirm the purity and integrity.
LDL isolated by ultracentrifugation from squirrel monkeys was dialyzed against 1M glycine (pH 10.0) before iodination, using a modification of the iodine monochloride method21. Briefly, 1 mCi of 125I was added to LDL rapidly followed by 0.33 mM iodine monochloride with no vortexing. After dialysis, the final LDL preparations was then analyzed by FPLC and agarose gel electrophoresis to assess the integrity and purity of the labeled LDL.
All radiolabeled lipoproteins were filter sterilized through a 0.22 micrometer filter before injection into the femoral artery. Sequential blood samples of less than 50 uL were obtained for the kinetic analysis at the indicated times from an indwelling catheter or by tail vein venipuncture. Multi exponential functions were fit to the plasma decay curves, with the use of WINSAAM program. Residence times (RT) were obtained from the areas under the plasma decay curves for the first 72 h, and the fractional catabolic rate (FCR) was calculated as the reciprocal of the residence time.
STATISTICAL ANALYSIS
Unless otherwise indicated, all values are expressed as mean±SEM. Comparisons between groups of mice were made, using 2-tailed Student’s t-test for independent samples.
RESULTS
Plasma LCAT activity levels
Four days following injection of adenovirus, containing human LCAT transgene (rLCAT-Adv), into squirrel monkeys, a marked increase in LCAT activity was observed (Table 1). Compared to a control group treated with an adenovirus encoding Luciferase (rLucif-Adv), there was approximately a 22-fold increase in LCAT activity by day 4, which returned to baseline by day 8.
Table 1.
Plasma levels of LCAT, lipids and lipoproteins.
| LCAT act | TC | TG | PL | FC | CE | Apo AI | HDL | Apo B | non-HDL | |
|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||
| (nmol/ml/h) | (mg/dL) | |||||||||
| LCAT d0 (n=11) | 257±23 | 178±11 | 67±12 | 245±21 | 42±3 | 136±8 | 134±8 | 113±7 | 19±1 | 65±7 |
| LCAT d4 (n=11) | 5618±799* | 325±20* | 35±6* | 196±9+ | 57±5* | 268±18* | 136±6 | 260±24* | 12±1* | 65±13 |
| Luc d0 (n=9) | 251±13 | 166±10 | 41±4 | 199±10 | 35±2 | 130±8 | 135±6 | 103±9 | 20±2 | 63±11 |
| Luc d4 (n=9) | 257±12 | 171±10 | 43±3 | 210±11 | 39±3 | 132±8 | 132±4 | 113±13 | 20±2 | 57±8 |
= INCREASED where p < 0.00001 copared to day 0;
= DECREASED where p < 0.05 compared to day 0 LCAT activity (LCAT act), total cholesterol (TC), triglycerides (TG). phospholipids (PL), free cholesterol (FC). cholesteryl ester (CE), apolipoprotein A-I (ApoA-I). HDL cholesterol (HDL), apolipoprotein B (ApoB) and non-HDL cholesterol (defined as TC-HDL) before the study (d0) or at the peak expression day, at a steady state level (d4).
Effect of LCAT on plasma lipids and lipoproteins
The increase of LCAT activity in the rLCAT-Adv treated group was associated with a significant increase in total plasma cholesterol, HDL-C, as well as a reduction apoB (Table 1). No significant changes in lipids and lipoproteins were observed in the control group treated with rLucif-Adv, indicating that adenoviral infection in itself did not significantly alter lipoprotein metabolism by non-specific liver damage. Compared to baseline results on day 0, LCAT expression significantly increased total cholesterol from 178±11 to 325±20 mg/dL by day 4 (p<0.001). Approximately 90% of the increase in total cholesterol was due to an increase in cholesteryl esters, which nearly doubled by day 4 (p<0.001). Almost all of the increase in total cholesterol was also associated with HDL, which increased 2.3 fold on day 4 (p<0.001). No significant change was observed in the level of non-HDL-C. No change was also observed in the plasma concentration of apoA-I, but interestingly the level of plasma apoB decreased by approximately by 37% on day 4 (p=0.001). As shown in Fig. 1, the effect of the adenoviral expression of LCAT on plasma cholesterol reached a plateau on day 4 and started to decrease by day 7–8.
Figure 1. Effect of LCAT on plasma total cholesterol.

rLCAT-Adv (◆) or rLucif-Adv (■) were injected squirrel monkeys and plasma total cholesterol was measured from baseline to 8 days post-injection. Results are expressed as mean± SEM (n=8 per group) and (* p<0.01 compared to day 0).
Increased LCAT activity was also associated with a change in the size of HDL, as assessed by FPLC analysis (Fig. 2). A significant shift of HDL toward larger size particles was observed (peak elution shifted from 30 mL elution volume to 27 mL). Consistent with the LCAT induced increase of HDL-C observed after dextran sulfate precipitation (Table 1), there was also an overall increase in cholesterol from the FPLC fractions for HDL from the rLCAT-Adv treated monkeys. A relatively small LDL peak was observed from both the control and experimental group, but the rLCAT-Adv treated monkeys appeared to have slightly lower cholesterol content, although no difference was observed for non-HDL cholesterol levels (Table 1).
Figure 2. Elution profiles of lipoproteins on gel permeation chromatography.

Plasma pool from squirrel monkeys (n=4) that received rLCAT-Adv were fractionated, by using two Superose 6 HR 10/30 columns connected in series. Each fraction was analyzed for the total cholesterol content (ug/mL). The dark line ( ) represents the pre-treatment value, whereas the lighter line (
) represents the pre-treatment value, whereas the lighter line ( ) represents the result from plasma collected on day 4 after injection of adenovirus. Arrows indicate elution position for major lipoproteins. Results are expressed as mean± SEM.
) represents the result from plasma collected on day 4 after injection of adenovirus. Arrows indicate elution position for major lipoproteins. Results are expressed as mean± SEM.
Effect of LCAT on Apolipoprotein Kinetics
To determine the mechanism for the effect of LCAT on lipid and lipoprotein changes, a kinetic analysis of radioiodinated apoA-I associated with HDL and radioiodinated apoB on LDL was performed (Fig. 3). We first established that potential liver damage from the adenoviral infection did not significantly alter apolipoprotein kinetic parameters. As can be seen in Fig. 3A, treatment of squirrel monkeys with luciferase by rLucif-Adv did not change the catabolism of radioiodinated apoA-I on HDL compared to a control group not treated with adenovirus over the 8 days of the study (FCR= 0.373±0.027 pools/day). Next, the effect of LCAT expression on apoA-I catabolism was assessed (Figure 3B). This was done on day 4 after treatment with rLCAT-Adv, because the effect of LCAT on lipoproteins appeared to peak at that time and to be relatively stable for at least the following 3 days, as shown by the total plasma cholesterol levels (Fig. 1). The LCAT treated group was divided in two arms; one arm received HDL isolated from control monkeys that had not received any adenovirus, whereas the other arm received HDL isolated from monkeys previously treated 4 days earlier with rLCAT-Adv. Before injection, the two different HDL preparations were associated with radioiodinated apoA-I. The objective of this experiment was to test if apoA-I from either the native control HDL or HDL that had been modified by LCAT would have a different catabolic or production rate. Compared to rLucif-Adv treated monkeys that had received native control HDL (FCR=0.373±0.027 pools/day), monkeys treated with rLCAT-Adv showed a similar increased rate of catabolism for both the native control HDL (FCR=0.685±0.045 pools/day) and the LCAT modified HDL (FCR=0.731±0.077 pools/day). Analysis of the estimated production rate (Table 2) that was calculated 72 h after injection of the labeled HDL indicated that there was also a significant increase (1.9-fold) in the apoA-I production rate for both groups. The rLucif-Adv treated monkeys that had received the control native HDL had an increased PR of 41±3 mg/kg/d, which was similar to the group receiving LCAT modified HDL (PR=39±5 mg/kg/d) and much higher compared to the rLucif-Adv control group (PR=22±2mg/kg/d; p<0.05). The overall plasma level of apoA-I, however, did not change after LCAT treatment (Table 1), so the increased catabolism of apoA-I from the LCAT treatment was fully compensated by an increase in the production rate of apoA-I.
Figure 3. Kinetic analysis of apoA-I and apoB in non-human primates.
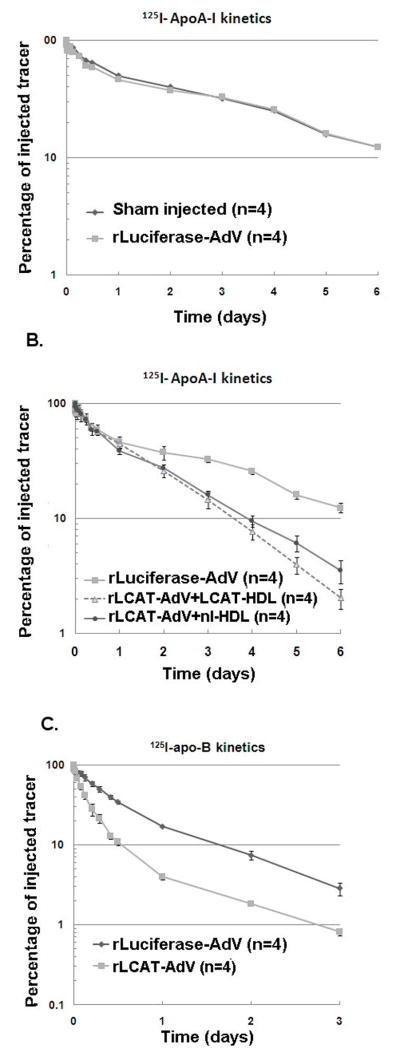
Panel 3A illustrates the apoA-I kinetic decay curve of control monkeys () versus rLucif-Adv injected monkeys (). Panel 3B illustrates the kinetic decay curve of apoA-I in monkeys (n= 4 per group) injected with either rLuc-Adv () or with LCAT-Adv (△ and  ). Within the group treated with rLCAT-Adv, 4 monkeys received LCAT modified HDL (△), and four monkeys received control native HDL (). Panel 3C illustrates apoB kinetics in monkeys (n=4) treated with either rLCAT-Adv () or with rLucif-Adv (n=4) (). Results are expressed as mean± SEM.
). Within the group treated with rLCAT-Adv, 4 monkeys received LCAT modified HDL (△), and four monkeys received control native HDL (). Panel 3C illustrates apoB kinetics in monkeys (n=4) treated with either rLCAT-Adv () or with rLucif-Adv (n=4) (). Results are expressed as mean± SEM.
Table 2.
Kinetic analysis of apoA-I and apoB.
Kinetic parameters of HDL and LDL metabolism in non-human primates
| AdV | ApoA-I mg/d | FCR d−1 | PR mg/kg.d |
|---|---|---|---|
| rLCAT-AdV (n=4) | 134±8 | 0.685±0.045* | 41±3* |
| rLucif-AdV (n=4) | 132±4 | 0.373±0.027 | 22±2 |
|
| |||
| AdV | ApoB mg/d | FCR d−1 | PR mg/kg.d |
|
| |||
| rLCAT-AdV (n=4) | 12±2* | 4.2±0.3* | 22±3 |
| rLucif-AdV (n=4) | 20±2 | 1.7±0.1 | 16±2 |
p<0.05 compared to control; rLucif-rAdV= control group; rLCAT-rAdv= LCAT adenovirus injected monkeys.
Because we observed a decrease in plasma apoB plasma levels (Table 1), we also investigated the effects of increased LCAT activity on LDL kinetics (Fig. 3C). Compared to the rLucif-Adv treatment group (FCR=1.7±0.1 pools/day), monkeys treated with rLCAT-Adv showed a significant increase in the catabolism of apoB (FCR=4.2±0.3 pools/day; p<0.01). The ApoB production rate was also slightly increased (1.4-fold) compared to the control group, which apparently was negated by the much larger relative increase in LDL catabolism, resulting in lower apoB plasma levels (Table 1).
To gain further insight into the effect of LCAT on the catabolism of HDL and LDL, samples collected at various time points after injection with rLCAT-Adv or Lucif-Adv were analyzed by FPLC (Fig. 4). For the Lucif-Adv treated group, there was no apparent change in the size distribution of the HDL or LDL tracer over time (data not shown). In other words, there was a gradual decrease in the total radioactive counts of the radiolabeled lipoproteins with no shift in the lipoprotein size distribution with time. The same was also true for radiolabeled LDL given to the rLCAT-Adv treated group (Fig. 4A). In contrast, in the case of radiolabeled HDL given to the rLCAT-Adv treated monkeys, the majority of the apoA-I tracer on HDL shifted after 30 min to a larger size HDL particle, as assessed by FPLC analysis (Fig. 4B). Thereafter, the tracer remained on this larger HDL particle and then gradually decreased over time, which is consistent with a direct catabolism of the larger form of HDL produced by the increased expression of LCAT.
Figure 4. Elution profiles of radiolabeled lipoproteins on gel permeation chromatography.
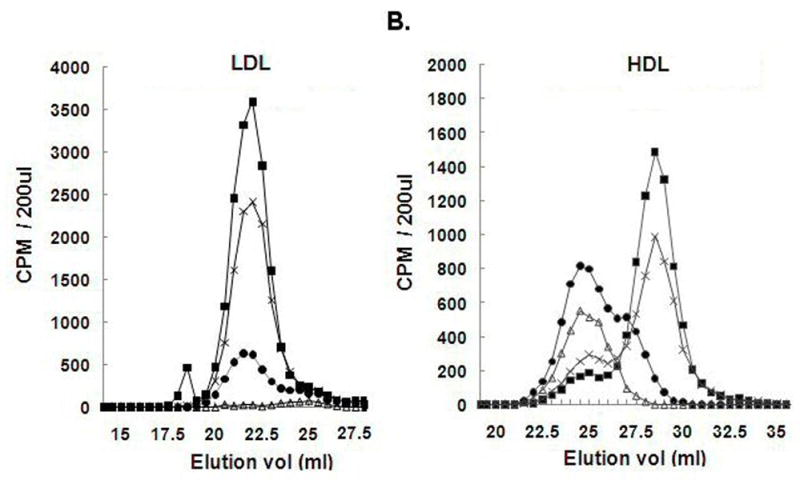
A plasma pool from 4 monkeys that received either radiolabeled LDL (Panel 4A) or HDL (Panel 4B) four days following treatment with rLCAT-Adv were fractionated, by using two Superose 6 HR 10/30 columns connected in series, and radioactive counts were determined. Samples were analyzed at 2 min (■), 30 min (X), 7 hours (●) and 24 hours (△) after injection of the tracer. Results are expressed as mean± SEM.
DISCUSSION
There is growing evidence that increasing the level of HDL will reduce the risk for developing for CHD 22;23. The main mechanism that has been proposed for the atheroprotective effect of HDL is its ability to increase the reverse cholesterol transport pathway 24, although HDL has also been reported to have other beneficial effects on atherosclerosis25;26. Unfortunately, there are a limited number of drugs for raising HDL and not all approaches that increase HDL may be anti-atherogenic 27. Interfering with the hepatic uptake of cholesterol from HDL, for example, will markedly raise HDL-C in SR-BI K/O mice but leads to increased atherosclerosis28. Similarly, there is concern that drug strategies based on the inhibition of CETP, which also raise HDL-C, may not be atheroprotective, because of decreased delivery of cholesterol to the liver by HDL29, 59. In contrast, LCAT is believed to raise HDL by enhancing its formation rather than decreasing its catabolism and thus would be predicted to increase overall reverse cholesterol transport. Evidence against a possible atheroprotective role of LCAT is the fact that patients with a genetic defect in LCAT do not appear to have a marked increase risk of CHD30 and in mice, increased expression of LCAT leads to increase atherosclerosis but this may be due to their lack of CETP31. One advantage of the present study is that LCAT was expressed in squirrel monkeys, which because they have CETP are likely to be a better animal model than mice for human atherosclerosis35. Another strength of this study is that not only the level of HDL was examined but also the effect of LCAT on the catabolism and production rate of apoA-I and apoB was assessed.
Consistent with what has been observed in all other animals models studied to date32–34, increased LCAT expression in monkeys led to a significant increase in HDL-C; the level of HDL-C more than doubled from 113 mg/dL to 260 mg/dL (Table 1). ApoA-I, the main protein component of HDL, however, did not significantly change, which would indicate that increased LCAT activity promotes the formation of a lipid enriched form of HDL. This is consistent with the observed increase in the size of HDL by FPLC (Fig. 2 and 4). As would be expected, a large increase in cholesteryl esters was also observed, which was found mostly associated with HDL (Table 1). Similar changes in HDL from humans has been shown to be atheroprotective36;37. For example, large lipid rich HDL, such as the HDL2 sub fraction, has been shown in numerous epidemiologic studies to correlate better with CHD risk reduction than total HDL-C36;37. The effect of LCAT on lipoproteins still partially persisted by day 8 even after LCAT returned to baseline most likely because of the lag in time needed for the catabolism of HDL and the clearance of cholesteryl esters formed by LCAT.
Based on previous studies in other animal models38;39, the effect of LCAT on apoB concentration appears to better correlate with atherosclerosis than changes in the level of HDL-C. In mice fed a pro-atherogenic diet, over expression of LCAT led to an increase in apoB and atherosclerosis40. When LCAT was overexpressed in CETP transgenic mice, there was reduced levels of apoB and reduced atherosclerosis41. Over expression of LCAT in rabbits, which have CETP, also resulted in reduced apoB and reduced atherosclerosis 42;43. Overexpression of LCAT in rabbits that lack LDL receptor, however, did not alter apoB levels and was not atheroprotective44, indicating that the LDL receptor is necessary for LCAT to lower apoB levels, most likely through its ability to remove LDL that has been enriched in cholesteryl esters by LCAT and CETP. In this study, LCAT was also found to significantly lower apoB levels in squirrel monkeys, but did not significantly change non-HDL-C levels (Table 1). Because squirrel monkeys normally contain relatively low levels of apoB compared to humans, the effect of the marked increase of LCAT on decreasing apoB may, however, may not be generalized to humans. LCAT over expression was also found to reduce total triglycerides by almost 50% (Table 1), which may be the consequence of the exchange of cholesteryl esters for triglycerides on LDL by CETP.
Another potential consideration in predicting the effect of LCAT over expression on atherosclerosis is the mechanism behind any change in lipoprotein pool size45. The pool of HDL and LDL can be altered by either a change in the rate of production or catabolism or both. Because adenoviral constructs only transiently express proteins, the kinetic parameters of apoA-I and apoB may be altered, if the lipoprotein changes induced by LCAT are not in steady state. To minimize this potential problem, the kinetic analysis on apoA-I and apoB were done from day 4 to day 7 when the effect of LCAT on total cholesterol appeared to peak and remained relatively stable (Fig. 1). During this time period, LCAT over expression was associated with an increase in both the production and catabolism of apoA-I, which balanced each other out (Table 2), resulting in no change in the plasma concentration of apoA-I, although HDL-C increased. The mechanism for the change in the catabolism of apoA-I by LCAT is not known but is likely related to the HDL lipid compositional differences (Table 1) caused by LCAT46. The effect of LCAT over expression on the production of apoA-I is likely an indirect effect, such as an alteration in the hepatic expression of genes like apoA-I and ABCA1 that are involved in the initial secretion of nascent HDL. In addition, the increased esterification of cholesterol by LCAT could also potentially facilitate the extracellular formation and stabilization of HDL.
Several studies support the notion that increased production of apoA-I or increased formation of HDL-C, as was observed in this study, should be anti-atherogenic. Transgenic mice and rabbits that over express the gene for apoA-I are protected against atherosclerosis47;48. Similarly, increased hepatic expression of ABCA1, which increases the formation of HDL by promoting the initial lipidation of apoA-I is atheroprotective 49–51. Infusion of HDL or apoA-I mimetic peptides, which would simulate increased production of apoA-I is also atheroprotective52;53.
The effect of increasing the catabolism of HDL or apoA-I on atherosclerosis is not as clear and appears to depend on other factors. Increased catabolism of HDL, leading to a decrease in HDL pool size as occurs in patients with hypertriglyceridemia54, is associated with increased CHD risk. Delaying HDL catabolism, however, such as when SR-B1 gene is deleted in the liver, leads to a marked increase in HDL-C and increased atherosclerosis, because it interferes with one of the last steps in the reverse cholesterol transport pathway, the hepatic uptake of cholesterol55. Mice over expressing LCAT in the absence of CETP, which have increased atherosclerosis, were found to have delayed catabolism of apoA-I, because the altered lipid composition of HDL interferes with its hepatic uptake14. In this study, LCAT increased HDL-C even though it also increased apoA-I catabolism, presumably because the CETP-mediated transfer of cholesteryl esters from HDL to apoB containing lipoproteins prevented the formation of a dysfunctional lipid rich form of HDL that was found in mice14. Additional studies will have to be performed to fully understand the impact of apoA-I kinetic parameters on atherosclerosis, but overall these findings are consistent with a model whereby increased catabolism of apoA-I may not necessarily promote atherosclerosis, so long as the overall HDL-C pool size is not significantly decreased. In this situation, it would be predicted that the net flux of cholesterol from peripheral cells to HDL and then to the liver by the reverse cholesterol transport pathway may in fact be increased.
LCAT was also found in this study to lower apoB levels by markedly increasing its catabolism (Table 2), which would be expected to be atheroprotective. Statins, currently our most effective drug for treating atherosclerosis, decrease LDL largely by increasing its catabolism56;57. The mechanism for how LCAT modulates apoB metabolism is not known, but one possible mechanism may be related to its ability to alter the lipid composition of LDL. Besides the CETP-mediated transfer of cholesteryl esters produced by LCAT from HDL to LDL, a significant fraction of cholesteryl esters can also be directly made by LCAT on LDL58.
In summary, increased expression of human LCAT in squirrel monkeys by adenovirus was associated with increased HDL-C and decreased apoB levels. These results along with the potential beneficial changes in kinetic parameters for apoA-I and apoB suggest that, like in rabbits, increased expression of LCAT in an animal model that also expresses CETP is most likely anti-atherogenic. The results from this study, therefore, suggest that drug strategies to increase LCAT activity may be useful for reducing atherosclerosis because of its combined beneficial effect on HDL and LDL metabolism.
Acknowledgments
This work was supported in by the intramural funds from the NHLBI at the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Dobiasova M, Frohlich JJ. Advances in understanding of the role of lecithin cholesterol acyltransferase (LCAT) in cholesterol transport. Clin Chim Acta. 1999;286:257–271. doi: 10.1016/s0009-8981(99)00106-0. [DOI] [PubMed] [Google Scholar]
- 2.Rajaram OV, Barter PJ. Reactivity of human lipoproteins with purified lecithin: cholesterol acyltransferase during incubations in vitro. Biochim Biophys Acta. 1985;835:41–49. doi: 10.1016/0005-2760(85)90028-1. [DOI] [PubMed] [Google Scholar]
- 3.Glomset JA. The plasma lecithins:cholesterol acyltransferase reaction. J Lipid Res. 1968;9:155–167. [PubMed] [Google Scholar]
- 4.Hovingh GK, de Groot E, van der Steeg W, et al. Inherited disorders of HDL metabolism and atherosclerosis. Curr Opin Lipidol. 2005;16(2):139–45. doi: 10.1097/01.mol.0000162318.47172.ef. [DOI] [PubMed] [Google Scholar]
- 5.Peelman F, Verschelde JL, Vanloo B, et al. Effects of natural mutations in lecithin:cholesterol acyltransferase on the enzyme structure and activity. J Lipid Res. 1999;40:59–69. [PubMed] [Google Scholar]
- 6.Peelman F, Vanloo B, Verschelde JL, et al. Effect of mutations of N- and C-terminal charged residues on the activity of LCAT. J Lipid Res. 2001;42:471–479. [PubMed] [Google Scholar]
- 7.Miller NE, Rajput-Williams J, Nanjee MN, et al. Relationship of high density lipoprotein composition to plasma lecithin:cholesterol acyltransferase concentration in men. Atherosclerosis. 1988;69:123–129. doi: 10.1016/0021-9150(88)90005-6. [DOI] [PubMed] [Google Scholar]
- 8.Foger B, Chase M, Amar MJ, et al. Cholesteryl ester transfer protein corrects dysfunctional high density lipoproteins and reduces aortic atherosclerosis in lecithin cholesterol acyltransferase transgenic mice. J Biol Chem. 1999;274:36912–36920. doi: 10.1074/jbc.274.52.36912. [DOI] [PubMed] [Google Scholar]
- 9.Forte TM, Subbanagounder G, Berliner JA, et al. Altered activities of anti-atherogenic enzymes LCAT, paraoxonase, and platelet-activating factor acetylhydrolase in atherosclerosis-susceptible mice. J Lipid Res. 2002;43:477–485. [PubMed] [Google Scholar]
- 10.Furbee JW., Jr Parks JS Transgenic overexpression of human lecithin: cholesterol acyltransferase (LCAT) in mice does not increase aortic cholesterol deposition. Atherosclerosis. 2002;165:89–100. doi: 10.1016/s0021-9150(02)00201-0. [DOI] [PubMed] [Google Scholar]
- 11.Furbee JW, Jr, Francone O, Parks JS. In vivo contribution of LCAT to apolipoprotein B lipoprotein cholesteryl esters in LDL receptor and apolipoprotein E knockout mice. J Lipid Res. 2002;43:428–437. [PubMed] [Google Scholar]
- 12.Furbee JW, Jr, Sawyer JK, Parks JS. Lecithin:cholesterol acyltransferase deficiency increases atherosclerosis in the low density lipoprotein receptor and apolipoprotein E knockout mice. J Biol Chem. 2002;277:3511–3519. doi: 10.1074/jbc.M109883200. [DOI] [PubMed] [Google Scholar]
- 13.Nong Z, Gonzalez-Navarro H, Amar M, et al. Hepatic lipase expression in macrophages contributes to atherosclerosis in apoE-deficient and LCAT-transgenic mice. J Clin Invest. 2003;112:367–378. doi: 10.1172/JCI16484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berard AM, Foger B, Remaley A, et al. High plasma HDL concentrations associated with enhanced atherosclerosis in transgenic mice overexpressing lecithin-cholesteryl acyltransferase. Nat Med. 1997;3:744–749. doi: 10.1038/nm0797-744. [DOI] [PubMed] [Google Scholar]
- 15.Ng DS, Maguire GF, Wylie J, et al. Oxidative stress is markedly elevated in lecithin:cholesterol acyltransferase-deficient mice and is paradoxically reversed in the apolipoprotein E knockout background in association with a reduction in atherosclerosis. J Biol Chem. 2002;277:11715–11720. doi: 10.1074/jbc.M112320200. [DOI] [PubMed] [Google Scholar]
- 16.Son YS, Zilversmit DB. Increased lipid transfer activities in hyperlipidemic rabbit plasma. Arteriosclerosis. 1986;6:345–351. [PubMed] [Google Scholar]
- 17.Yanni AE. The laboratory rabbit: an animal model of atherosclerosis research. Laboratory Animals. 2004;38:246–256. doi: 10.1258/002367704323133628. [DOI] [PubMed] [Google Scholar]
- 18.McGrory WJ, Bautista DS, Graham FL. A simple technique for the rescue of early region I mutations into infectious human adenovirus type 5. Virology. 1988;163:614–617. doi: 10.1016/0042-6822(88)90302-9. [DOI] [PubMed] [Google Scholar]
- 19.Vaisman BL, Klein HG, Rouis M, et al. Overexpression of human lecithin cholesterol acyltransferase leads to hyperalphalipoproteinemia in transgenic mice. J Biol Chem. 1995;270:12269–12275. doi: 10.1074/jbc.270.20.12269. [DOI] [PubMed] [Google Scholar]
- 20.Chen CH, Albers JJ. Characterization of proteoliposomes containing apoprotein A-I: a new substrate for the measurement of lecithin: cholesterol acyltransferase activity. J Lipid Res. 1982;23:680–691. [PubMed] [Google Scholar]
- 21.McFarlane AS. Labelling of plasma proteins with radioactive iodine. Biochem J. 1956;62:135–143. doi: 10.1042/bj0620135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nicholls SJ, Tuzcu EM, Sipahi I, et al. Statins, high-density lipoprotein cholesterol, and regression of coronary atherosclerosis. JAMA. 2007;297:499–508. doi: 10.1001/jama.297.5.499. [DOI] [PubMed] [Google Scholar]
- 23.Brown B Greg, Zhao Xue-Qiao, Cheung Marian C. Should both HDL-C and LDL-C be targets for lipid therapy? A review of current evidence. Journal of Clinical Lipidology. 2008;1:88–94. doi: 10.1016/j.jacl.2007.02.004. (GENERIC) Ref Type: Journal (Full) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lewis GF, Rader DJ. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ Res. 2005;96:1221–1232. doi: 10.1161/01.RES.0000170946.56981.5c. [DOI] [PubMed] [Google Scholar]
- 25.MNTRAC-DJNP Sviridov D. Antiatherogenic Functionality of High Density Lipoprotein: How Much versus How Good. Journal of Atherosclerosis and Thrombosis. 2008;15:52–56. doi: 10.5551/jat.e571. [DOI] [PubMed] [Google Scholar]
- 26.Feig JE, Shamir R, Fisher EA. Atheroprotective effects of HDL: Beyond reverse cholesterol transport. Current Drug Targets. 2008;9:196–203. doi: 10.2174/138945008783755557. [DOI] [PubMed] [Google Scholar]
- 27.Patel TN, Shishehbor MH, Bhatt DL. A review of high-dose statin therapy: targeting cholesterol and inflammation in atherosclerosis. European Heart Journal. 2007;28:664–672. doi: 10.1093/eurheartj/ehl445. [DOI] [PubMed] [Google Scholar]
- 28.Rigotti A, Trigatti BL, Penman M, et al. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc Natl Acad Sci USA. 1997;94:12610–12615. doi: 10.1073/pnas.94.23.12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tchoua U, D’Souza W, Mukhamedova N, et al. The effect of cholesteryl ester transfer protein overexpression and inhibition on reverse cholesterol transport. Cardiovasc Res. 2008;77:732–739. doi: 10.1093/cvr/cvm087. [DOI] [PubMed] [Google Scholar]
- 30.Santamarina-Fojo S, Lambert G, Hoeg JM, et al. Lecithin-cholesterol acyltransferase: role in lipoprotein metabolism, reverse cholesterol transport and atherosclerosis. Curr Opin Lipidol. 2000;11:267–275. doi: 10.1097/00041433-200006000-00007. [DOI] [PubMed] [Google Scholar]
- 31.Berard AM, Foger B, Remaley A, et al. High plasma HDL concentrations associated with enhanced atherosclerosis in transgenic mice overexpressing lecithin-cholesteryl acyltransferase. Nat Med. 1997;3:744–749. doi: 10.1038/nm0797-744. [DOI] [PubMed] [Google Scholar]
- 32.Dugi KA, Vaisman BL, Sakai N, et al. Adenovirus-mediated expression of hepatic lipase in LCAT transgenic mice. J Lipid Res. 1997;38:1822–1832. [PubMed] [Google Scholar]
- 33.Brousseau ME, Santamarina-Fojo S, Zech LA, et al. Hyperalphalipoproteinemia in human lecithin cholesterol acyltransferase transgenic rabbits In vivo apolipoprotein A-I catabolism is delayed in a gene dose-dependent manner. J Clin Invest. 1996;97:1844–1851. doi: 10.1172/JCI118614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoeg JM, Vaisman BL, Demosky SJ, Jr, et al. Lecithin:cholesterol acyltransferase overexpression generates hyperalpha-lipoproteinemia and a nonatherogenic lipoprotein pattern in transgenic rabbits. J Biol Chem. 1996;271:4396–4402. doi: 10.1074/jbc.271.8.4396. [DOI] [PubMed] [Google Scholar]
- 35.McCombs HL, Zook BC, et al. Fine structure of spontaneous atherosclerosis of the aorta in the squirrel monkey. Am J Pathol. 1969;55(2):235–252. [PMC free article] [PubMed] [Google Scholar]
- 36.Havel RJ. High-density lipoproteins, cholesterol transport and coronary heart disease. Circulation. 1979;60:1–3. doi: 10.1161/01.cir.60.1.1. [DOI] [PubMed] [Google Scholar]
- 37.Gordon T, Castelli WP, Hjortland MC, et al. High density lipoprotein as a protective factor against coronary heart disease The Framingham Study. Am J Med. 1977;62:707–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 38.Brousseau ME, Santamarina-Fojo S, Vaisman BL, et al. Overexpression of human lecithin:cholesterol acyltransferase in cholesterol-fed rabbits: LDL metabolism and HDL metabolism are affected in a gene dose-dependent manner. J Lipid Res. 1997;38:2537–2547. [PubMed] [Google Scholar]
- 39.Brousseau ME, Wang J, Demosky SJ, Jr, et al. Correction of hypoalphalipoproteinemia in LDL receptor-deficient rabbits by lecithin:cholesterol acyltransferase. J Lipid Res. 1998;39:1558–1567. [PubMed] [Google Scholar]
- 40.Berard AM, Foger B, Remaley A, et al. High plasma HDL concentrations associated with enhanced atherosclerosis in transgenic mice overexpressing lecithin-cholesteryl acyltransferase. Nat Med. 1997;3:744–749. doi: 10.1038/nm0797-744. [DOI] [PubMed] [Google Scholar]
- 41.Foger B, Chase M, Amar MJ, et al. Cholesteryl ester transfer protein corrects dysfunctional high density lipoproteins and reduces aortic atherosclerosis in lecithin cholesterol acyltransferase transgenic mice. J Biol Chem. 1999;274:36912–36920. doi: 10.1074/jbc.274.52.36912. [DOI] [PubMed] [Google Scholar]
- 42.Brousseau ME, Santamarina-Fojo S, Vaisman BL, et al. Overexpression of human lecithin:cholesterol acyltransferase in cholesterol-fed rabbits: LDL metabolism and HDL metabolism are affected in a gene dose-dependent manner. J Lipid Res. 1997;38:2537–2547. [PubMed] [Google Scholar]
- 43.Hoeg JM, Santamarina-Fojo S, Berard AM, et al. Overexpression of lecithin:cholesterol acyltransferase in transgenic rabbits prevents diet-induced atherosclerosis. Proc Natl Acad Sci USA. 1996;93:11448–11453. doi: 10.1073/pnas.93.21.11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brousseau ME, Kauffman RD, Herderick EE, et al. LCAT modulates atherogenic plasma lipoproteins and the extent of atherosclerosis only in the presence of normal LDL receptors in transgenic rabbits. Arterioscler Thromb Vasc Biol. 2000;20:450–458. doi: 10.1161/01.atv.20.2.450. [DOI] [PubMed] [Google Scholar]
- 45.Rashid S, Patterson BW, Lewis GF. What have we learned about HDL metabolism from kinetics studies in humans? Journal of Lipid Research. 2006;47:1631–1642. doi: 10.1194/jlr.R600008-JLR200. [DOI] [PubMed] [Google Scholar]
- 46.Vaisman BL, Klein HG, Rouis M, et al. Overexpression of human lecithin cholesterol acyltransferase leads to hyperalphalipoproteinemia in transgenic mice. J Biol Chem. 1995;270:12269–12275. doi: 10.1074/jbc.270.20.12269. [DOI] [PubMed] [Google Scholar]
- 47.Belalcazar LM, Merched A, Carr B, et al. Long-term stable expression of human apolipoprotein A-I mediated by helper-dependent adenovirus gene transfer inhibits atherosclerosis progression and remodels atherosclerotic plaques in a mouse model of familial hypercholesterolemia. Circulation. 2003;107:2726–2732. doi: 10.1161/01.CIR.0000066913.69844.B2. [DOI] [PubMed] [Google Scholar]
- 48.Duverger N, Kruth H, Emmanuel F, et al. Inhibition of atherosclerosis development in cholesterol-fed human apolipoprotein A-I-transgenic rabbits. Circulation. 1996;94:713–717. doi: 10.1161/01.cir.94.4.713. [DOI] [PubMed] [Google Scholar]
- 49.Joyce C, Freeman L, Brewer HB, Jr, et al. Study of ABCA1 function in transgenic mice. Arterioscler Thromb Vasc Biol. 2003;23:965–971. doi: 10.1161/01.ATV.0000055194.85073.FF. [DOI] [PubMed] [Google Scholar]
- 50.Joyce CW, Wagner EM, Basso F, et al. ABCA1 overexpression in the liver of LDLr-KO mice leads to accumulation of pro-atherogenic lipoproteins and enhanced atherosclerosis. J Biol Chem. 2006;281:33053–33065. doi: 10.1074/jbc.M604526200. [DOI] [PubMed] [Google Scholar]
- 51.Singaraja RR, Stahmer B, Brundert M, et al. Hepatic ATP-binding cassette transporter A1 is a key molecule in high-density lipoprotein cholesteryl ester metabolism in mice. Arterioscler Thromb Vasc Biol. 2006;26:1821–1827. doi: 10.1161/01.ATV.0000229219.13757.a2. [DOI] [PubMed] [Google Scholar]
- 52.Amar MJ, Basso F, Demosky S, et al. The ApoA-I peptide mimetic 5A protects against atherosclerosis in ApoE deficient mice. Circulation (Supplement) 2006;144:II–23. [Google Scholar]
- 53.Sethi AA, Amar M, Shamburek RD, et al. Apolipoprotein AI mimetic peptides: possible new agents for the treatment of atherosclerosis. Curr Opin Investig Drugs. 2007;8:201–212. [PubMed] [Google Scholar]
- 54.Brinton EA, Eisenberg S, Breslow JL. Increased apo A-I and apo A-II fractional catabolic rate in patients with low high density lipoprotein-cholesterol levels with or without hypertriglyceridemia. J Clin Invest. 1991;87:536–544. doi: 10.1172/JCI115028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rigotti A, Trigatti BL, Penman M, et al. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc Natl Acad Sci USA. 1997;94:12610–12615. doi: 10.1073/pnas.94.23.12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anonymous MRC/BHF Heart Protection Study of cholesterol-lowering therapy and of antioxidant vitamin supplementation in a wide range of patients at increased risk of coronary heart disease death: early safety and efficacy experience. Eur Heart J. 1999;20:725–741. doi: 10.1053/euhj.1998.1350. [DOI] [PubMed] [Google Scholar]
- 57.Cannon CP. Intensive versus moderate lipid lowering with statins after acute coronary syndromes (vol 350, pg 1495, 2004) New England Journal of Medicine. 2006;354:778. doi: 10.1056/NEJMoa040583. [DOI] [PubMed] [Google Scholar]
- 58.Dobiasova M, Frohlich J. Understanding the mechanism of LCAT reaction may help to explain the high predictive value of LDL/HDL cholesterol ratio. Physiological Research. 1998;47:387–397. [PubMed] [Google Scholar]
- 59.Tchoua U, D’Souza W, Mukhamedova N, Blum D, et al. The effect of cholesteryl ester transfer protein overexpression and inhibition on reverse cholesterol transport. Cardiovasc Res. 2007;77(4):732–9. doi: 10.1093/cvr/cvm087. [DOI] [PubMed] [Google Scholar]
- 60.Hojnacki J, Mulligan J, Cluette-Brown J, Igoe F, Osmolski T. Oral nicotine impairs clearance of plasma low density lipoproteins. Proc Soc Exp Biol Med. 1986;182(3):414–8. doi: 10.3181/00379727-182-3-rc2. [DOI] [PubMed] [Google Scholar]
- 61.Portman Oscar W, Alexander Manfred. Metabolism of sphingolipids by normal and atherosclerotic aorta of squirrel monkeys. Journal of Lipid Research. 1970;11:23–30. [PubMed] [Google Scholar]