

Abstract
Epithelial-mesenchymal transition (EMT) is a critical process for embryogenesis but is abnormally activated during cancer metastasis and recurrence. This process enables epithelial cancer cells to acquire mobility and traits associated with stemness. It is unknown whether epithelial stem cells or epithelial cancer stem cells are able to undergo EMT, and what molecular mechanism regulates this process in these specific cell types. We found that Epithelial Ovarian Cancer Stem cells (EOC stem cells) are the source of metastatic progenitor cells through a differentiation process involving EMT and Mesenchymal-Epithelial Transition (MET). We demonstrate both in vivo and in vitro the differentiation of EOC stem cells into mesenchymal spheroid-forming cells (MSFCs) and their capacity to initiate an active carcinomatosis. Furthermore, we demonstrate that human EOC stem cells injected i.p in mice are able to form ovarian tumors, suggesting that the EOC stem cells have the ability to “home” to the ovaries and establish tumors. Most interestingly, we found that TWIST1 is constitutively degraded in EOC stem cells, and that the acquisition of TWIST1 requires additional signals that will trigger the differentiation process. These findings are relevant for understanding the differentiation and metastasis process in EOC stem cells.
Introduction
Epithelial ovarian cancer (EOC) is the leading cause of gynecologic cancer deaths with a 5-year survival rate of only 15%. Although 80–90% of patients initially respond to first-line chemotherapy agents carboplatin and paclitaxel, less than 15% remain in complete remission and most patients recur within 5 years (1) (2, 3) (4). When the disease recurs, it usually presents as a carcinomatosis, or widespread metastatic disease, which is often not amenable to surgical debulking (5–7). In these patients, the only other option is the administration of chemotherapy. Unfortunately, recurrent ovarian cancer is extremely chemoresistant to currently available agents (8). This profile makes recurrent ovarian cancer very difficult to treat, with most patients succumbing to the disease. As such, recurrence and metastasis are the major causes of mortality in ovarian cancer (7, 9).
The establishment of metastatic disease involves multiple steps. In vessel-dependent metastasis, as seen in breast cancer, the process involves local invasion of cancer cells, intravasation, cancer cell survival in the circulation, extravasation, and colonization (10) (11). Ovarian cancer metastasis however, is not vessel-dependent. Metastatic ovarian cancer presents as localized intra-abdominal carcinomatosis and rarely spreads to distant sites. The main route of metastasis formation in ovarian cancer is direct dissemination to the peritoneal cavity and/or lymphatic dissemination (2). Unfortunately, the cellular and molecular processes required for the formation of ovarian cancer metastasis are not clearly understood.
The process of epithelial-mesenchymal transition (EMT) has been extensively utilized to explain how an epithelial cancer cell is able to acquire the capacity to migrate and metastasize. EMT was originally described as a developmental process in which epithelial cells break cell-cell contact and cell-extracellular matrix connections, which allows movement to other locations in the body during critical stages of embryonic development (12); (13). It has also been shown that through EMT, epithelial cancer cells can undergo a phenotypic switch that allows these polarized and immobile epithelial cells to become motile mesenchymal cells (14); (15, 16) . After migration to distant sites, the process can be reversed and the migratory mesenchymal cells can undergo mesenchymal-epithelial transition (MET) and revert back to an epithelial phenotype, thereby establishing a similar epithelial cancer in secondary sites. (17).
Several studies have provided convincing evidence that EMT plays an essential role in modulating the motility and invasiveness of ovarian cancer cells. However, given the heterogeneity of ovarian cancer tumors, it is not known if all or only certain and specific cancer cell populations have the plasticity to undergo EMT. The demonstration that metastatic sites have heterogeneous histological characteristics suggests that the cells capable of undergoing this process have the ability to self-renew as well as differentiate – properties that are unique to cancer stem cells (CSCs). Indeed, EMT has been linked to the ability of self -renewal and generation of multiple lineages (15).
Studies of neoplastic tissues have provided evidence of the existence of CSCs, which have the ability to recreate the heterogeneity of the original tumor in mice (18). CSCs, which are able to self-renew and differentiate, similar to normal stem cells, are not only the potential origin of the tumor, but also the possible source of recurrence and chemoresistance. Recent studies have shown that EMT can induce mammary epithelial cells or breast epithelial cancer cells to enter into a stem-like stage (19),(15),(20). However, whether epithelial stem cells or epithelial cancer stem cells are able to undergo EMT is unknown.
We and others have reported the presence of ovarian cancer stem cells using different cell surface markers (21–29). Although, the initial studies were done using cells lines, more recent reports confirmed their presence in different types of ovarian cancer tissues (22). In our studies, we reported the identification of a unique population of EOC cells with characteristics of CSCs (21, 30–32). These cells are characterized by the expression of CD44 and MyD88 (CD44+MyD88+ EOC stem cells). Additional characteristics associated with CSCs present in CD44+MyD88+ EOC stem cells includes tumorigenicity, capacity to recapitulate the heterogeneity of the original tumor in mice, ability to self-renew, expression of pleuripotency markers (CD44, MyD88, b-catenin, Oct-4, and SSEA-4 (32), chemoresistance, and capacity to differentiate into multiple cell types (21). CD44+/MyD88+ EOC stem cells give origin to CD44-MyD88- EOC cells (mature EOC cells), which have lost the capacity to self-renew, are more sensitive to chemotherapy, and are terminally differentiated (21, 31). These two cell populations can be identified and isolated from ovarian cancer tumors (22). Furthermore, pure populations of CD44+/MyD88+ EOC stem cells can differentiate, in vivo and in vitro, into CD44-MyD88- mature EOC cells (21).
Depending on the environment, CD44+/MyD88+ EOC stem cells can also differentiate into endothelial cells and therefore serve as tumor vascular progenitors (33). CD44+/MyD88+ EOC stem cells are different from their more differentiated progeny, CD44-/MyD88- mature EOC cells. Although both types are malignant cells, only CD44+/MyD88+ EOC stem cells have the capacity to self renew and to recapitulate the heterogeneity of the original tumor (21, 22). However, it is unknown whether CD44+/MyD88+ EOC stem cells have the capacity to give origin to cells with metastatic capacity. A process that would require CD44+/MyD88+ EOC stem cells to undergo EMT.
Although the acquisition of mesenchymal traits by the epithelial cancer cells appears to enable the dissemination of cancer cells from the primary site, the mechanisms that initiate and control these processes have been difficult to elucidate. TWIST1 is a basic helix–loop–helix (bHLH) transcription factor that regulates gastrulation and mesoderm differentiation during embryonic development. Moreover, it regulates genes that are essential for morphogenesis and cell migration. Recent data suggest that TWIST1 plays an important role in cancer metastasis by inducing EMT (34–37), and that hypoxia can stabilize hypoxia-inducible factor-1α (HIF-1α) leading to increased TWIST expression both at the mRNA and protein levels (36, 38). However, the mechanisms that regulate TWIST1 expression and function during EOC stem cell differentiation and during the process of metastasis are not well defined.
In the present study, we demonstrate that CD44+/MyD88+ EOC stem cells have the capacity to undergo EMT and in doing so generate mesenchymal spheroid-forming cells (MSFCs) with metastatic potential. The MSFCs are capable of forming tumors in vivo, not only in the peritoneum, but also in mice ovaries. Furthermore, we demonstrate that TWIST1 is constitutively degraded in EOC stem cells, and inhibition of TWIST1 expression prevents EOC stem cell differentiation. This may explain how EOC stem cells are able to keep their stemness through symmetric division even in regular cell culture conditions without adding any additional growth factors. These findings also suggest that CD44+/MyD88+ EOC stem cells are the possible source of metastatic ovarian cancer and highlight the central role of TWIST1 in regulating EMT in CD44+/MyD88+ EOC stem cells. The identification of the cellular source of metastatic ovarian cancer and the key players involved is fundamental not only for the better understanding of the origin of metastasis but also for developing new therapies.
Results
CD44+/MyD88+ EOC stem cells generate mesenchymal cells with migratory and tumorigenic capacity
A pure population of CD44+/MyD88+ EOC stem cells is able to form subcutaneous tumors in mice and generate a xenograft composed of almost 90% CD44-/MyD88- mature EOC cells (21). The capacity of CD44+/MyD88+ EOC stem cells to give origin to CD44-/MyD88- mature EOC cells is further demonstrated by monitoring the loss of these markers in a culture of 100% CD44+/GFP+ EOC stem cells. As shown in Supp. Figure 1, CD44+/GFP+ EOC stem cells are able to differentiate both in vitro and in vivo into 100% CD44-/GFP+ cells (22). The demonstration that the cells remained GFP+ while losing CD44 suggests that the CD44+ EOC stem cells can indeed generate CD44-mature EOC cells. Since CD44+/MyD88+ EOC stem cells have lower doubling time than CD44-/MyD88- mature EOC cells, the process of differentiation allows local tumor expansion by generating daughter epithelial cells with faster growth kinetics (21, 39, 40).
Metastasis represents another form of tumor expansion but requires the generation of cells with migratory and invasive capacity. We previously reported that freshly trypsinized CD44+/MyD88+ EOC stem cells can survive in low-attachment conditions and form viable compact spheroids (21, 41), suggesting another form of differentiation. To determine if this differentiation provided CD44+/MyD88+ EOC stem cells with invasive advantages, we used trans-well inserts coated with high-density Matrigel. CD44+/MyD88+ EOC stem cells or the derived spheroids were placed in the upper compartment of the chamber and media were placed in the lower compartment. Our results showed that the derived spheroids have enhanced invasion capacity compared to the progenitor CD44+/MyD88+ EOC stem cells (Supp. Fig. 2). CD44-/MyD88- mature EOC cells did not show any migratory capacity.
We next determined whether the CD44+/MyD88+ EOC stem cells could spontaneously differentiate in vitro from epithelial cells into spheroid-forming cells. We hypothesized that when grown in very high confluence, a monolayer of CD44+/MyD88+ EOC stem cells will spontaneously undergo differentiation. Thus, CD44+/MyD88+ EOC stem cells were maintained in high confluence (Fig. 1A, i) and low serum condition (1% FBS) in vitro, resembling the in vivo conditions of rapidly growing solid tumors. After about 2–4 weeks, we observed several foci of cells acquiring a differentiated phenotype based on their morphologic characteristics (Fig. 1A, ii-v). In each of these foci, we observed epithelial cells in the distal periphery; cells that lost their epithelial morphology and look more like fibroblast cells “ migrating” away from the epithelial cells; and a centrally located cluster of round cells (Fig. 1A, ii-iv). (Supp. Fig. 3A, B). When followed for the next few weeks, the clusters of round cells eventually detached from the tissue culture flask and formed compact spheroids (Fig. 1A, v). The viability of the spheroids was maintained when transferred to low- attachment tissue culture plates (Fig. 1A, vi). Interestingly, spheroids derived from CD44+/MyD88+ EOC stem cells show morphologic similarities to malignant ascites freshly isolated from ovarian cancer patients (Supp. Fig. 3C).
Figure 1. In vitro differentiation of CD44+/MyD88+ EOC stem cells into mesenchymal like spheroid-forming cells (MSFCs).
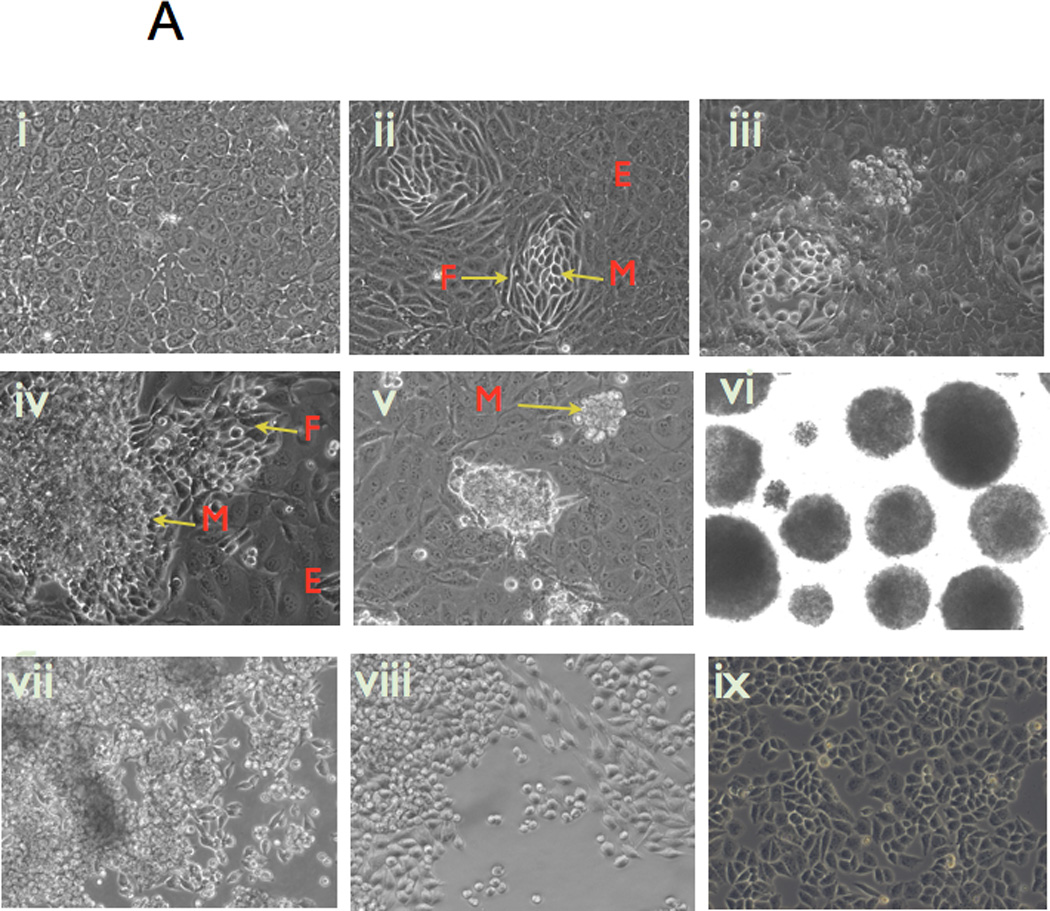
A) Over-confluent cultures of CD44+/MyD88+ EOC stem cells (i) form foci consisting of cells with fibroblast-like characteristics (ii-iv); these transformed cells eventually lose attachment and form mesenchymal compact spheroids (v-vi); spheroids can re-attach and re-form an epithelial monolayer when transferred to tissue culture plates (vii-ix);
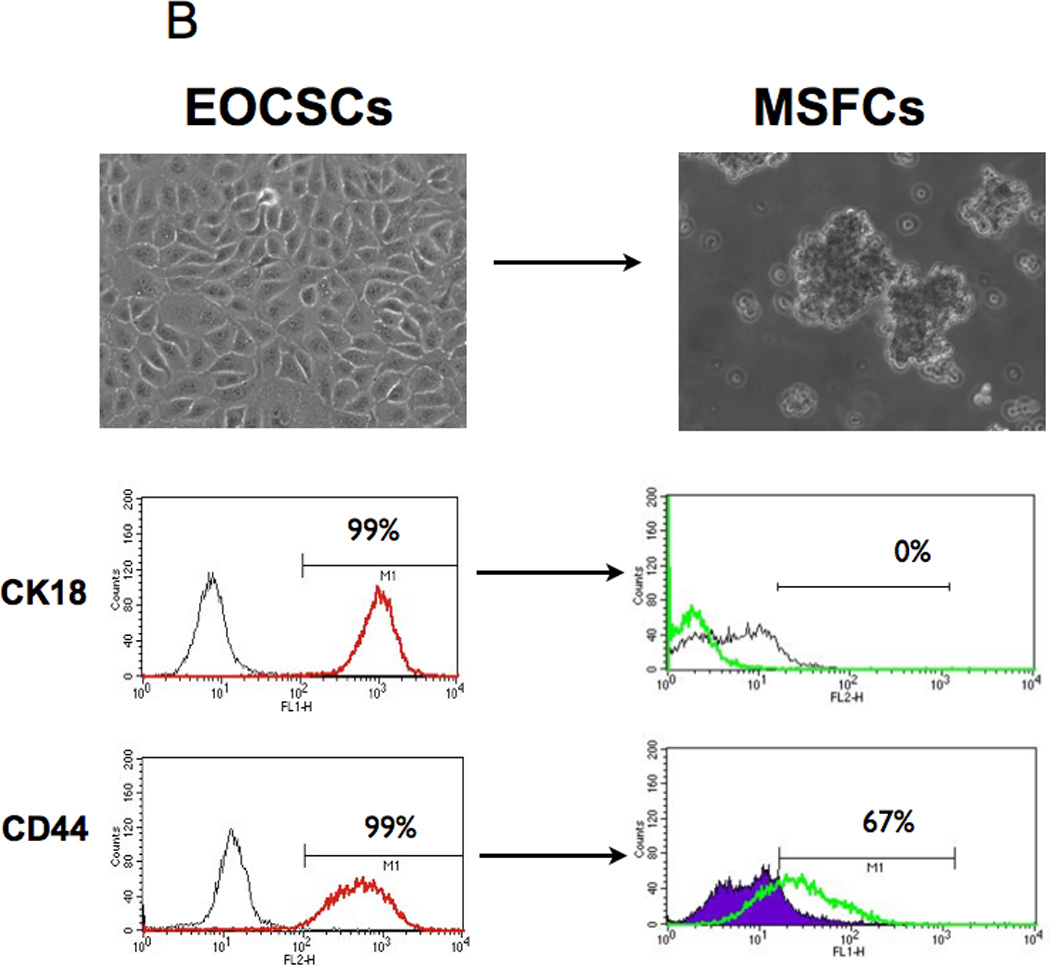
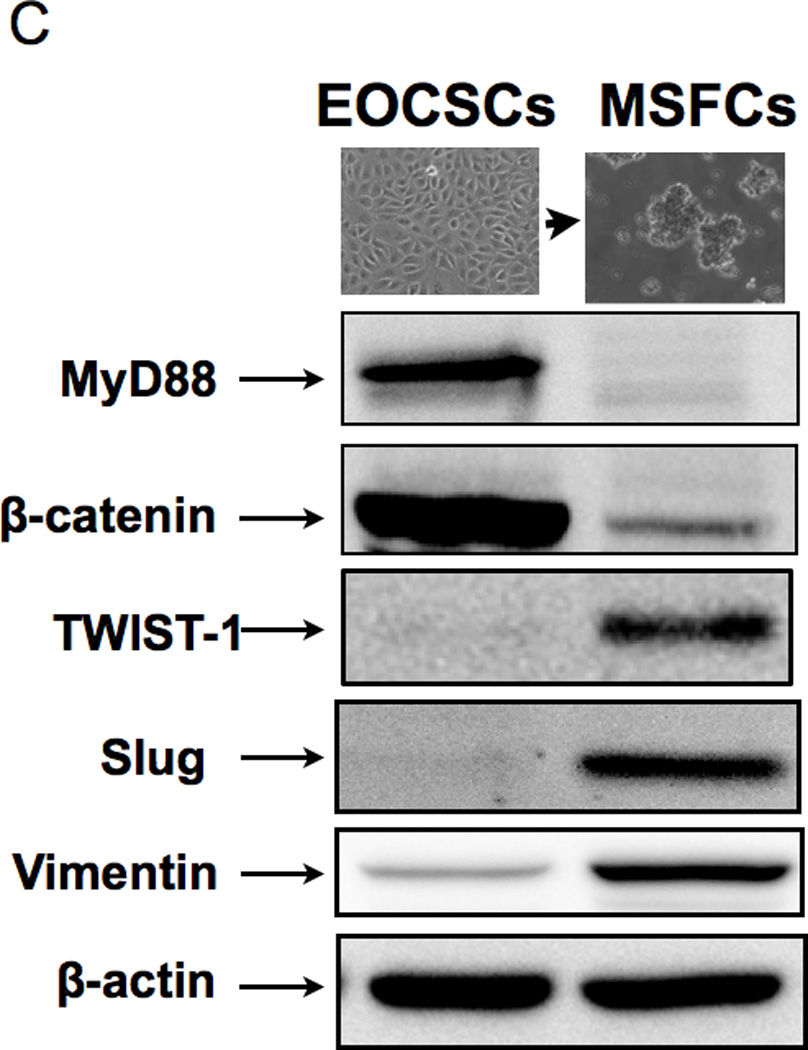
(B-C Upon spheroid formation, CD44+/MyD88+ EOC stem cells have lower levels of the epithelial marker Ck18 and the cancer stem cell markers, CD44 and B-catenin; but gain mesenchymal markers TWIST-1, Slug, and Vimentin. E, epithelial; F, fibroblast-like; M, mesenchymal-like; EOCSC - CD44+/MyD88+ EOC stem cells; MSFC - mesenchymal spheroids forming cells. Figures are representative of three clones done in independent experiments. Every experiment was repeated at least three times.
To further demonstrate that the mesenchymal spheroid-forming cells (MSFCs) originated from the CD44+/MyD88+ EOC stem cells, we repeated the same experiments using GFP-labeled CD44+/MyD88+ EOC stem cells. As shown in Supp. Fig 4, a monolayer of GFP+ CD44+/MyD88+ EOC stem cells is able to generate GFP+ MSFCs in vitro. Interestingly, when transferred to tissue culture plates, the GFP+ MSFCs are able to re-form a monolayer culture of GFP+ cancer cells, which morphologically resemble epithelial ovarian cancer cells (Fig 1A, vii-ix)(Supp. Fig. 4). These results suggest that MSFCs can revert back to an epithelial phenotype depending on culture conditions. None of the changes described above were observed when CD44-/MyD88- EOC cells were cultured in similar conditions. This suggests that the capacity to undergo these changes are unique to CD44+/MyD88+ EOC stem cells.
The observed cellular differentiation (acquisition of anchorage-independent growth, enhancement of migratory capacity, and re-acquisition of epithelial morphology), resemble processes that are associated with EMT and MET. Therefore, our next objective was to characterize the molecular phenotype of the cells during the differentiation process described above. CD44 and CK18 are highly expressed in CD44+/MyD88+ EOC stem cells (21, 24); therefore we used them as initial markers to monitor differentiation. While CD44+/MyD88+ EOC stem cells are 99.9% CD44+/ CK18+, the derived MSFCs are negative for the epithelial marker CK18 and are only 67% CD44+ (Fig. 1B). Further evaluation by Western blot showed that the MSFCs also lost MyD88 and B-catenin - two proteins that are highly expressed in the CD44+/MyD88+ EOC stem cells (Fig. 1C). Interestingly, the MSFCs acquired TWIST1, SLUG, and VIMENTIN, which are proteins associated with the process of EMT (Fig. 1C) suggesting that the process of differentiation from EOC stem cells into MSFCs involves EMT.
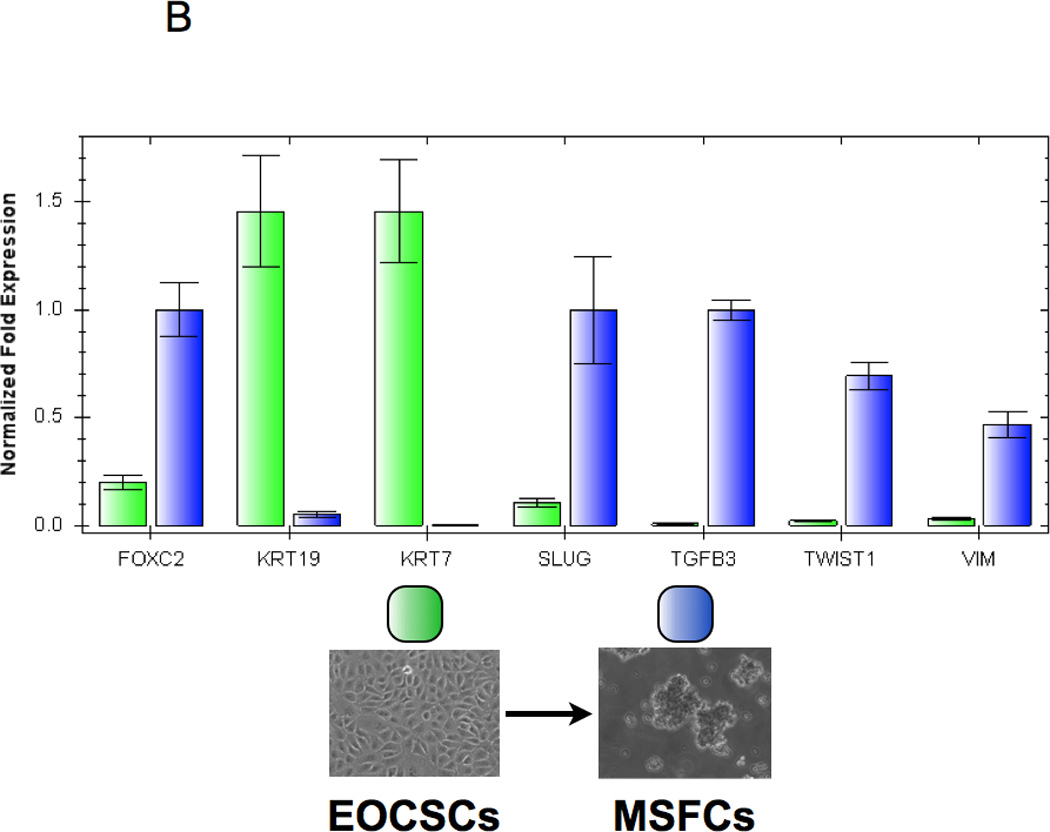
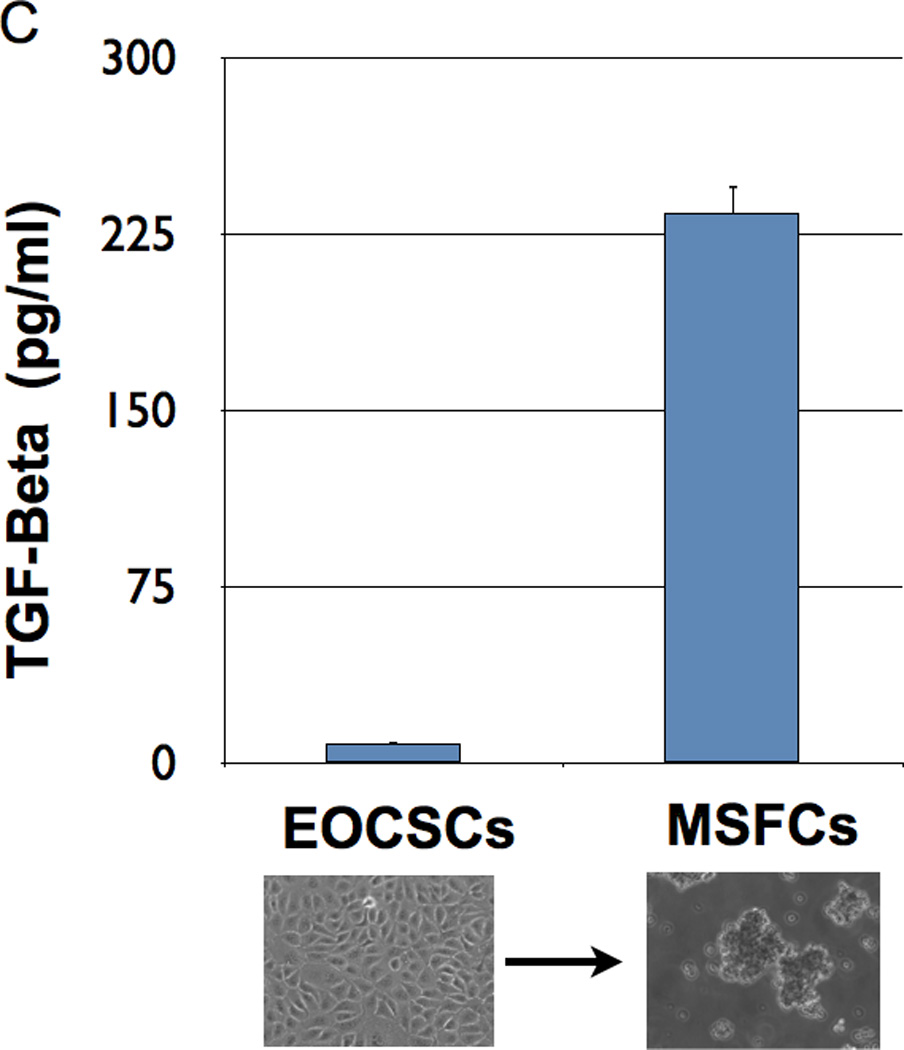
To further confirm the occurrence of EMT, we used two clones of CD44+/MyD88+ EOC stem cells and their derived MSFCs for the Human EMT RT2 Profiler PCR Array, which profiles the expression of 84 key genes. Fifty-one of these genes are directly associated with the EMT/MET process, while the rest are associated with cell growth, proliferation, and cytoskeleton organization. The results of the array showed that 31 out of the 51 genes associated with EMT/MET were differentially expressed between the CD44+/MyD88+ EOC stem cells and the MSFCs (Fig. 2A). To validate these results, we used 5 different clones of CD44+/MyD88+ cells and 5 clones of derived MSFCs cells, and we evaluated 9 of these genes at the mRNA as well as at the protein level. We tested the mesenchymal marker VIMENTIN (42); epithelial markers CK19/KRT19, (43) and CK7/KRT7 (44); transcription factors FOXC2 (15, 45), TWIST1, TCF4/E2-2 (46) and SLUG; and EMT-associated cytokine, TGFβ. Real-time PCR results showed that the EMT inducers and mesenchymal markers FOXC2, SLUG, TWIST1, and VIMENTIN were increased and that the epithelial markers KRT19 and KRT7 were decreased during the differentiation process from CD44+/MyD88+ EOC stem cells to MSFCs (Fig. 2B). Western blot analysis further confirmed the induction of mesenchymal markers VIMENTIN, FOXC2, SLUG, and TWIST1 (Fig. 2B). Finally, the induction of TGFβ during differentiation was confirmed both at the mRNA and secreted protein level (Fig. 2B–C)
Figure 2. Spheroid formation is an EMT process.

(A EMT array showed that the transition from CD44+/MyD88+ EOC stem cells to MSFCs is characterized by upregulation of genes associated with EMT. Array was done using two clones for each stage of differentiation;
(B Down-regulation of epithelial genes (CK7 and CK19) and up-regulation of mesenchymal genes (FOXC2, TWIST1, TGFB3, SLUG, VIMENTIN) during spheroid formation were validated by Real-time PCR;
(C TGFβ secretion was significantly increased after spheroid formation; EOCSC - EOC stem cells; MSFC - mesenchymal spheroids forming cells. . Each experiment was done in triplicate using three different cell clones.
CD44+/MyD88+ EOC stem cells undergo EMT in vivo to form metastatic tumors
Epithelial cancer cells, if injected in mouse peritoneum, mostly undergo apoptosis. This is due to the lack of cell-to-cell and/or surface contact (anoiksis), limited nutrients, as well as limited oxygen (47). We previously showed that CD44+/MyD88+ EOC stem cells injected subcutaneously (s.c.) in the presence of high-density Matrigel could develop solid s.c. tumors (21). In the present study, we evaluated the outcome of intra-peritoneally (i.p) injected CD44+/MyD88+ EOC stem cells. Thus, EOC stem cells (3 × 106) were injected i.p. in nude mice in the absence of Matrigel. After 25 days, we observed multiple small implants, which progressed to carcinomatosis within 35 days post-injection (Fig. 3A–B). Analysis of the mice ovaries showed prominent ovarian tumors, (Fig. 3B, C, Di). Analysis of tumor implants from other organs (i.e. small intestines, colon, liver) showed superficial attachment but not invasion (Fig. 3A, B). Thus, invasion was only observed in the ovaries. All the animals injected with CD44+/MyD88+ EOC stem cells developed ovarian tumors at least on one side. No tumors were observed when CD44-/MyD88- mature EOC cells were injected under similar conditions (Fig. 3Dii). Furthermore, when we injected CD44+/MyD88+ EOC stem cells in the fallopian tube of the mice, we were able to observe ovarian tumors and also metastatic lesions in the peritoneum (Data not shown). This suggests that the capacity to survive and to establish carcinomatosis is limited to the CD44+/MyD88 EOC stem cells.
Figure 3. CD44+/MyD88+ EOC stem cells form ovarian tumors and carcinomatosis in nude mice.
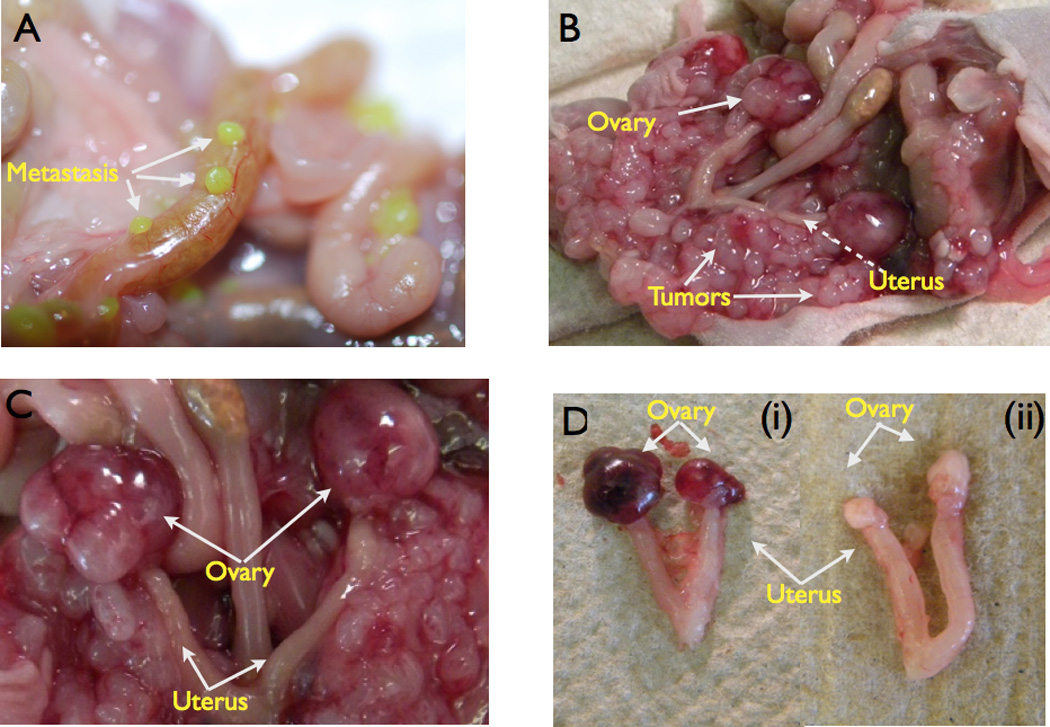
(A 3 × 106 GFP+/CD44+/MyD88+ EOC stem cells were injected i.p. in nude mice. Note the intestinal metastases;
(B-C 3 × 106 CD44+/MyD88+ EOC stem cells were injected i.p formed ovarian tumors and extensive carcinomatosis.
(D 3 × 106 CD44+/MyD88+ EOC stem cells or CD44-/MyD88- EOC cells were injected i.p.. Ovarian tumors were observed only in CD44+/MyD88+ EOC stem cells (i) and not in CD44-/MyD88- EOC cells (ii).
We next determined if the in vitro-derived MSFCs behave similarly in vivo as the CD44+/MyD88+ EOC stem cells. Thus, CD44+/MyD88+ EOC stem cells were induced to undergo in vitro differentiation into MSFCs as described above. Once the cells formed compact spheroids, they were collected and injected i.p. in nude mice and tumor formation evaluated after 30 days. The MSFCs were able to form ovarian tumors and carcinomatosis, similar to those observed with CD44+/MyD88+ EOC stem cells. More importantly, comparative analysis of tumor histology showed that tumors formed by the CD44+/MyD88+ EOC stem cells and the MSFCs have identical epithelial morphology (Supp Fig. 5). These results suggest that in order to generate metastatic tumors, CD44+/MyD88+ EOC stem cells undergo EMT to produce generate MSFCs with metastatic capacity. CD44-/MyD88- mature EOC cells do not form spheroids nor do they form tumors when injected in the peritoneum or fallopian tubes.
Differentiation of CD44+/MyD88+ EOC stem cells into mesenchymal cells is associated with TWIST1 expression
TWIST1 is a major regulator of EMT during embryogenesis and is over-expressed in metastatic tumors. As mentioned above, EMT gene array results showed acquisition of TWIST1 in the process of differentiation from EOC stem cells to MSFCs. Therefore we evaluated TWIST1 expression and its association with CD44+/MyD88+ EOC stem cell differentiation using immunofluorescence. TWIST1+ cells were observed in cell cultures of CD44+/MyD88+ EOC stem cells that displayed foci of cells with fibroblast morphology (Fig. 4A, ii). TWIST1 was not observed in the surrounding CD44+/MyD88+ EOC stem cells, which displayed the classic epithelial morphology. Furthermore, TWIST1 expression is high in the MSFCs (Fig. 4A, iii) and its expression is mainly nuclear (Fig. 4A, iv). This suggests an early role for TWIST1 during the differentiation process.
Figure 4. TWIST-1 expression during EMT.
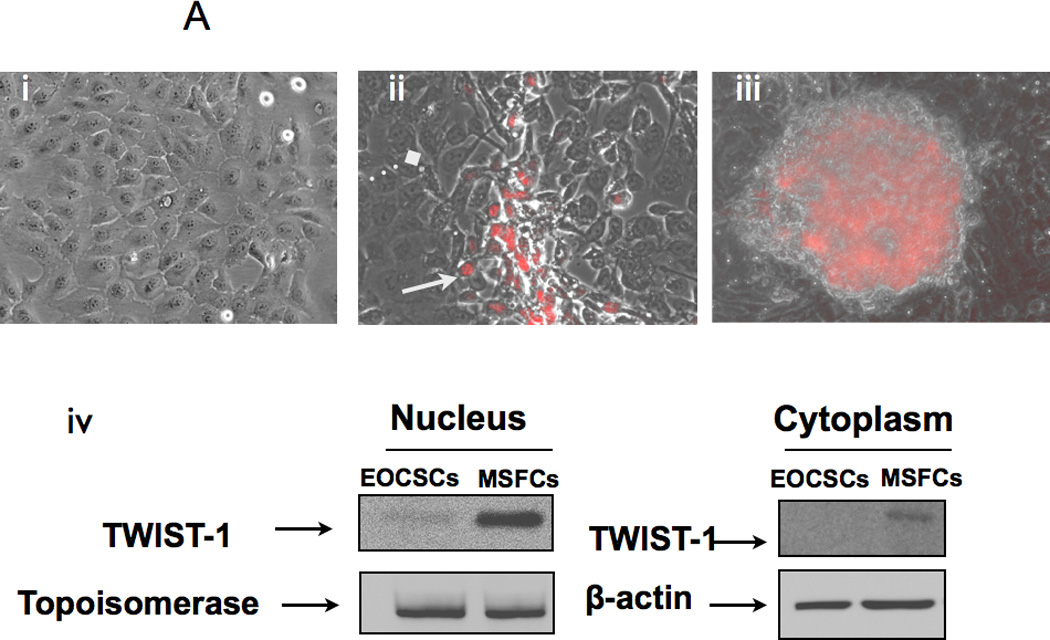
(A Immunofluorescence show specific expression of TWIST-1 only in cells undergoing EMT; (i) CD44+/MyD88+ EOC stem cell monolayer is negative for TWIST-1; (ii) TWIST-1 is expressed in cells with fibroblast/mesenchymal morphology; (iii) TWIST-1 is highly expressed in cells forming spheroids; (iv) Western blot analysis showing TWIST-1 expression is mainly nuclear. EOCSC - CD44+/MyD88+ EOC stem cells; MSFC - mesenchymal-like spheroid forming cells.
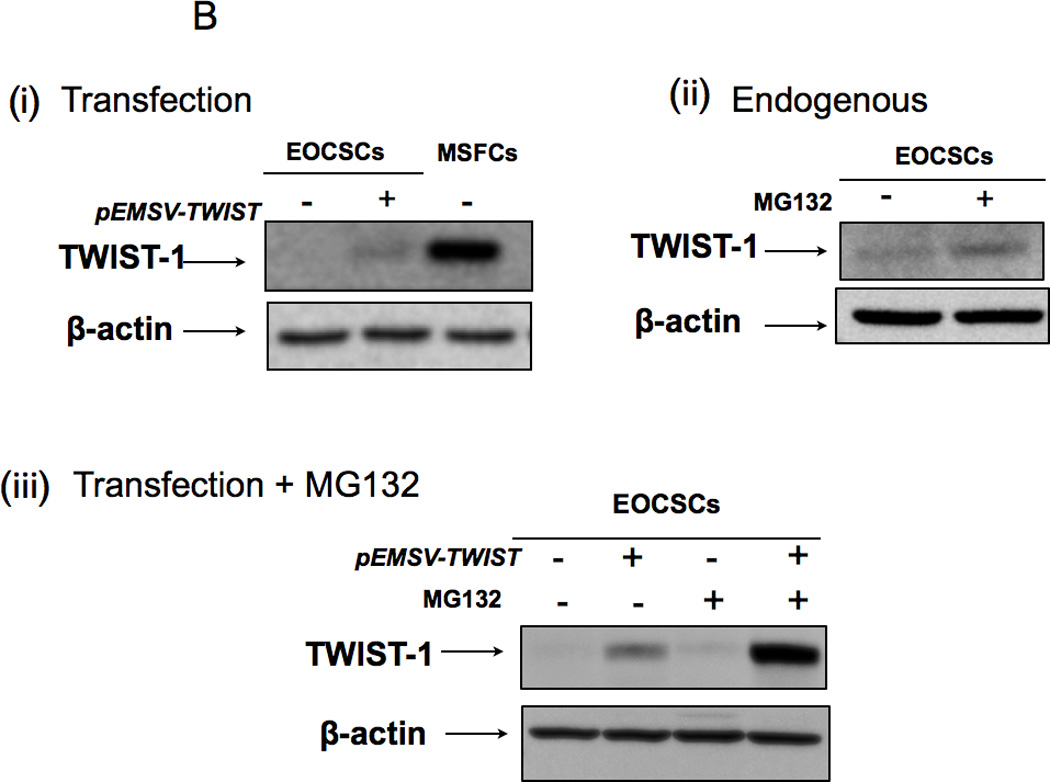
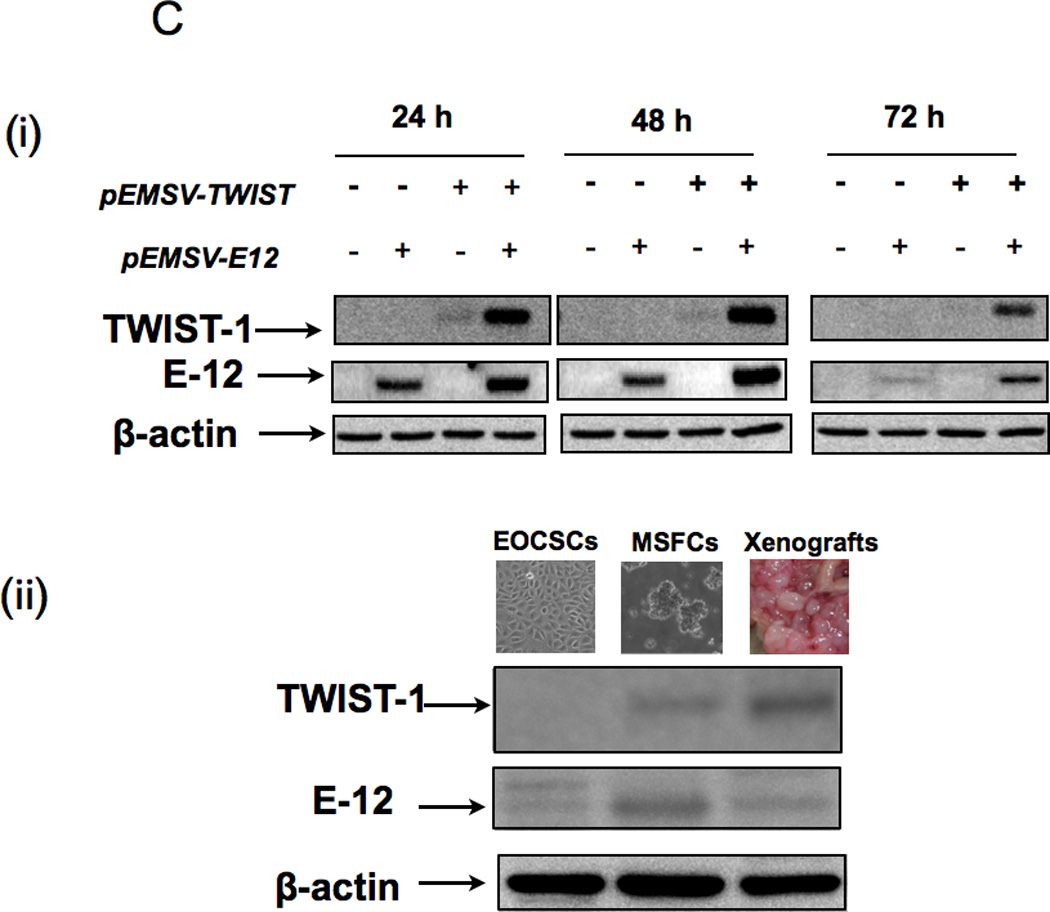
(B (i) Transfection of pEMSV-TWIST1 into CD44+/MyD88+ EOC stem cells is not able to induce expression of TWIST-1 protein; (ii-iii) the proteasome inhibitor, MG132 is able to increase endogenous and ectopically expressed TWIST-1.
(C (i) Co-transfection of E12 and TWIST-1 significantly increased TWIST-1 expression in the CD44+/MyD88+ EOC stem cells; ii) parallel increase in E12 and TWIST-1 was observed in the MSFCs and in mouse xenograft. EOCSC - CD44+/MyD88+ EOC stem cells; MSFCs - mesenchymal spheroids forming cells.
TWIST1 protein is constitutively degraded in the CD44+/MyD88+ EOC stem cells
To further characterize the role of TWIST1 in the EMT process observed in our system, we transfected CD44+/MyD88+ EOC stem cells with a plasmid containing full length TWIST1 (pEMSV-TWIST). Although we saw a significant increase in TWIST1 mRNA after transfection (data not shown), this was not associated with a corresponding increase in TWIST1 protein (Fig. 4B, i). This suggests that CD44+/MyD88+ EOC stem cells have an active mechanism to prevent the accumulation of TWIST1 protein even in the presence of its mRNA. This regulation could be at the RNA level (via microRNAs) or at the protein level (via degradation). Evaluation of the involvement of the ubiquitin–proteasome system was determined using the proteasome inhibitor, MG132. Thus, wild type or pEMSV-TWIST-transfected CD44+/MyD88+ EOC stem cells were treated with MG132, and TWIST1 expression were determined by Western blot. MG132 was able to increase the levels of endogenous TWIST1 (Fig. 4B, ii) as well as the ectopically expressed TWIST1 (Fig.4B, iii). Thus, blocking proteasome activity allowed the accumulation of both the endogenous, as well as the ectopically expressed TWIST1 in the CD44+/MyD88+ EOC stem cells. These data suggest that TWIST1 expression in CD44+/MyD88+ EOC stem cells is regulated at the protein level.
E12 stabilizes TWIST1 protein expression in CD44+/MyD88+ EOC stem cells
TWIST1 is a Class II basic-helix-loop-helix (bHLH) transcription factor and can form heterodimers with Class I bHLH proteins (E proteins). Previous studies have shown that binding of Class II transcription factors to Class I proteins can enhance the activity of the Class II transcription factors (48, 49). We hypothesize that this binding may also be required to stabilize Class II proteins such as TWIST1. We evaluated the role of the Class I E protein, E12, which is expressed in MSFCs but not in CD44+/MyD88+ EOC stem cells (Fig. 4C, ii). Thus, pEMSV-TWIST and pEMSV-E12 were co-transfected in CD44+/MyD88+ EOC stem cells. As shown in Figure 4C, co-expression with E12 is able to stabilize TWIST1 levels in CD44+/MyD88+ EOC stem cells (Fig. 4C, i).
We then evaluated whether the acquisition of TWIST1 and E12 occurs in CD44+/MyD88+ EOC stem cells in vivo. Thus, we determined the expression of these proteins in the tumors obtained when CD44+/MyD88+ EOC stem cells were injected i.p. in mice. While CD44+/MyD88+ EOC stem cells in culture do not express either TWIST1 or E12, the resulting mouse xenografts expressed high levels of both proteins (Fig. 4C, ii).
Hypoxia/HIF-1 induces TWIST1 expression in CD44+/MyD88+ EOC stem cells
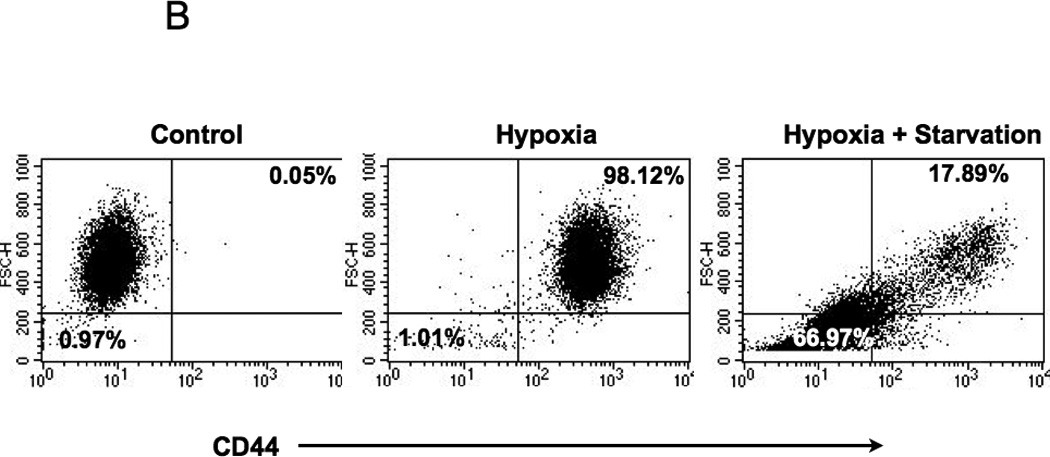
Next, we looked at the potential signal(s) that would trigger TWIST1 expression and initiate the differentiation process. A common factor found in our different models (peritoneum and tumor confluence) is low oxygen (hypoxia) and low nutrients. The peritoneal environment is characterized in part by hypoxia, which is a stimulus for the induction of HIF-1. Since HIF-1 is known to be upregulated in metastatic lesions, we hypothesized that hypoxic conditions may induce HIF-1 expression and promote differentiation by enhancing TWIST1 expression in CD44+/MyD88+ EOC stem cells. To test this hypothesis we treated CD44+/MyD88+ EOC stem cells with the hypoxia mimetic, cobalt chloride hexahydrate (CCH) and determined the expression of TWIST1 and E12. As shown in Figure 5A, CCH induced HIF-1 expression in a time-dependent manner. The induction of HIF-1 expression in CD44+/MyD88+ EOC stem cells correlates with the induction of TWIST1 and E12 expression. This was further confirmed when we evaluated the expression of CD44 in CD44+/MyD88+ EOC stem cells cultured under hypoxic and “starvation” conditions (1% O2 and supplemented with 1% FBS). We found a significant decrease in CD44 expression (98% vs 17%) and also a significant change in their morphology and cell size (1% vs 66% small cells) suggestive of differentiation (Fig. 5B). These results imply that hypoxia and starvation are potent stimuli for CD44+/MyD88+ EOC stem cells to undergo EMT and give origin to MSFCs with metastatic potential.
Figure 5. Effect of hypoxia on EOC stem cell differentiation and TWIST-1 expression.
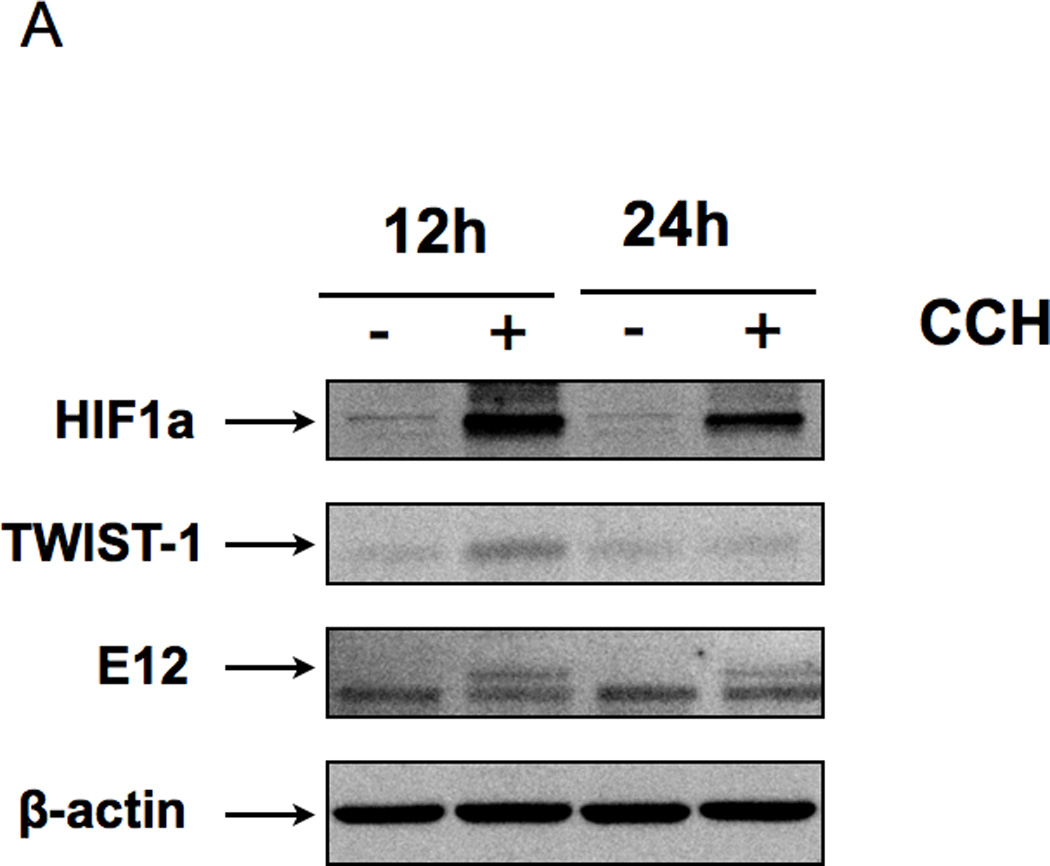
(A CD44+/MyD88+ EOC stem cells treated with Cobalt chloride hexahydrate (CCH) showed a significant increase in HIF1a, TWIST-1, and E12 in a time-dependent manner;
(B 5 days in hypoxic and starvation conditions (as described in the Materials and Methods section) promote CD44+/MyD88+ EOC stem cell differentiation as detected by loss of CD44;
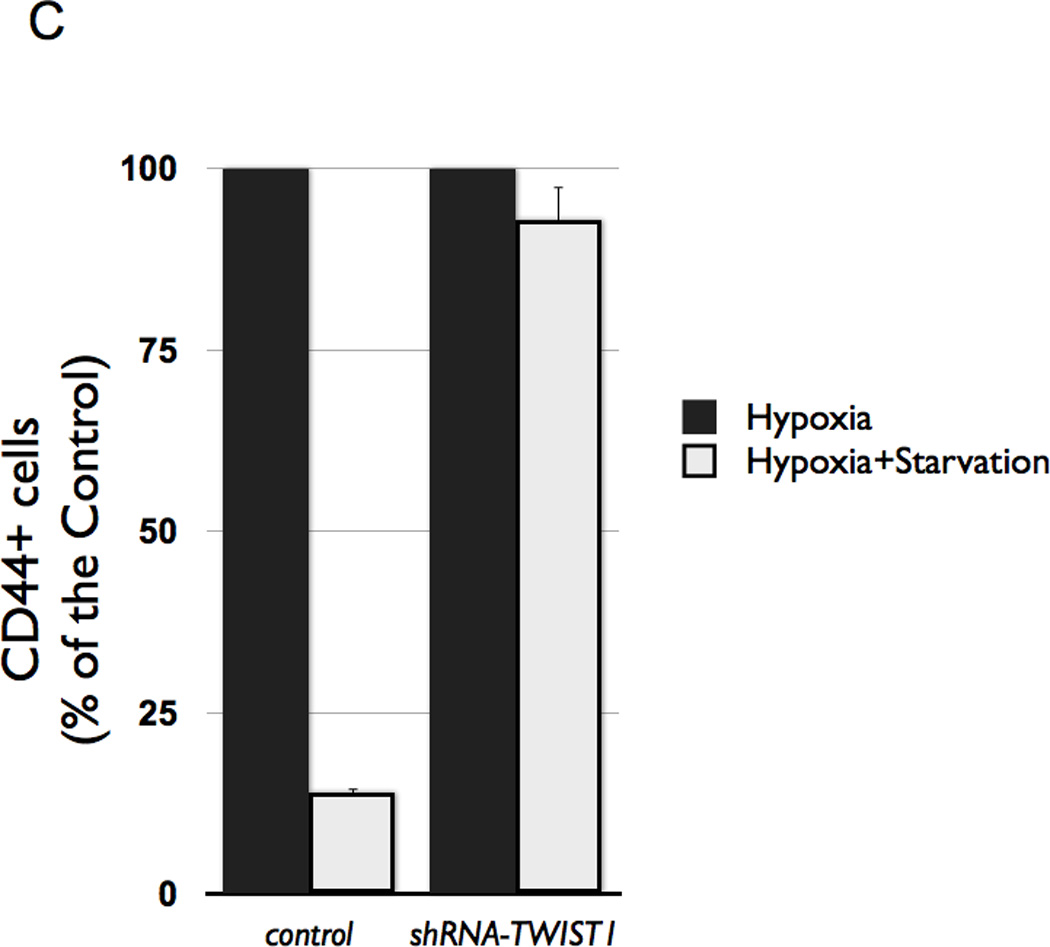
(C Stable transfection of shRNA TWIST1 in CD44+/MyD88+ EOC stem cells prevented loss of CD44 when exposed to conditions in B.
Finally, we wanted to test whether inhibition of TWIST1 expression could alter the effect of hypoxia on CD44+/MyD88+ EOC stem cell differentiation. Thus, CD44+/MyD88+ EOC stem cells were transfected with shRNA for TWIST1 (in order to prevent its expression during differentiation) and then cultured under the hypoxic and starvation conditions described above. As shown in Figure 5C, inhibition of TWIST1 expression prevented CD44+/MyD88+ EOC stem cell differentiation. Taken together, these results support the requirement for TWIST1 in CD44+/MyD88+ EOC stem cell differentiation into MSFCs.
Discussion
The present data suggest that CD44+/MyD88+ EOC stem cells could be a source of ovarian cancer metastasis by generating MSFCs with enhanced migratory capacity and ability to recreate an epithelial ovarian cancer tumor in a secondary site. This process is the result of EMT and is regulated by TWIST1.
In spite of debulking and chemotherapy, 60–80% of patients with ovarian cancer will present with recurrence and carcinomatosis in less than five years. The source of metastatic ovarian cells and how EOC cells acquire migratory properties and survive without cell-to-cell contact has not been elucidated. Previously, we reported that the CD44+/MyD88+ EOC stem cells could differentiate into a CD44-/MyD88- epithelial culture, which has lost stemness markers. In addition, we have shown that CD44+/MyD88+ EOC stem cells can give origin to different cell types including endothelial-like cells (21, 33). In this present study, we report a third type of differentiation. Under specific conditions, CD44+/MyD88+ EOC stem cells can form MSFCs with enhanced migratory/metastatic potential.
Ovarian cancer metastasis typically presents on local pelvic and abdominal organs and rarely involves metastasis to distant sites. Therefore, in contrast to other types of cancer, no anatomical barrier exists between the primary ovarian tumor and the metastatic sites. In ovarian cancer, small clusters of cells shed by the primary ovarian tumor can implant on peritoneal surface and form metastatic nodules. In order to initiate the shedding of cancer cells, the epithelial cancer cells comprising the solid tumor need to undergo a transformation into mesenchymal-like cells to acquire the capacity to migrate. Once the solid tumor has shed, then metastatic sites can be established. We were able to demonstrate this process both in vitro and in vivo and showed that the generation of MSFCs from CD44+/MyD88+ EOC stem cells is associated with migratory potential, which is required for the process of metastasis formation. Interestingly, the MSFCs present unique characteristics, which differ from the CD44+/MyD88+ EOC stem cells from which they are derived. None of these characteristics are observed in CD44-/MyD88- mature EOC cells.
EMT is a complex process by which epithelial cells lose cell-cell adhesion and apical-basal polarity but acquire mesenchymal features such as motility, invasiveness, and resistance to apoptosis (50–52). In our in vitro system we demonstrated these changes during the formation of MSFCs from CD44+/MyD88+ EOC stem cells. We showed morphological changes from epithelial to fibroblast-like mesenchymal cells, loss of cell-cell contact, capacity to form viable spheroids, and acquisition of migratory properties. We also showed that the formation of MSFCs from CD44+/MyD88+ EOC stem cells is associated with increased expression of mesenchymal markers (vimentin, thrombospondin, vitronectin) and decreased expression of epithelial markers (CK19, CK18, occluding, desmoplakin and Mucin-1) typically observed during EMT. In addition, we demonstrated increased expression of extracellular matrix components (collagen IV and fibronectin), altered expression of transcription factors (β-catenin, Snail, Slug, TWIST-1, Sox 10 and NFκB), and activation of specific kinases (ERK, AKT) (52). These changes were observed in vitro during the formation of MSFCs from CD44+/MyD88+ EOC stem cells and also in vivo when CD44+/MyD88+ EOC stem cells were injected i.p. or in the fallopian tubes of nude mice and formed carcinomatosis.
The general understanding has been that the process of EMT is associated with the generation of CSCs. Our data suggest that, at least in ovarian cancer, epithelial cancer stem cells can also undergo EMT, a transdifferentiation process. During this differentiation process from CD44+/MyD88+ EOC stem cells to MSFCs, there is a significant change in gene expression associated with the observed morphologic and functional changes. We observed a decrease in a few of the stemness genes such as CD44, MyD88, and CTNNB1 (gene of β-CATENIN). Instead the cells acquired mesenchymal markers as mentioned above. However, the MSFCs still conserve some stemness properties and high degree of plasticity since they can create a monolayer culture again once they are placed on tissue culture plates. The newly-formed monolayer culture obtained from the MSFCs have morphologic characteristics of epithelial cells, but lack stemness markers such as CD44, MyD88. The observed process resembles MET although the signals associated with this process need further investigation.
An important characteristic of the MSFCs is their capacity to migrate. This capacity was observed both in vitro as well as in vivo. When injected i.p. or intra uterine in mice, these cells are able to survive and establish xenografts in adjacent organs, including the mouse ovary. In the ovaries we observed three processes; 1) attachment to the surface of the ovary, 2) invasion of the ovarian stroma; and 3) differentiation into fast dividing epithelial ovarian cancer cells and expansion of the tumor. The changes in gene expression observed during EMT provide the biological basis for these processes. For adhesion, we observed the expression of adhesion molecules such as CTNNB1, COL1A2, COL3A1, COL5A2, ERBB3, and FOXC2. For the process of migration and motility we observed the increase in the expression of CALD1, NODAL, PDGFRB, TGFB1, VIM, MMP2, MMP9, TGFB1, TGFB2, and TIMP1; and finally for differentiation we found increased expression of BMP7, COL3A1, COL5A2, CTNNB1, ERBB3, FOXC2, FZD7, GSC, KRT14, NODAL, SNAI2, SOX10, TGFB2, TGFB3, TWIST1, WNT1. (Supplementary Table.2)
TWIST-1 is temporally expressed during normal development, but constitutively expressed in some forms of cancer (53); (36, 54). Indeed, we found that TWIST-1 is highly expressed in the MSFCs but not in the CD44+/MyD88+ EOC stem cells. Furthermore, we demonstrate that the differentiation of EOC stem cells to MSFCs depends on TWIST-1 expression.
Previous studies have shown that over-expression of TWIST-1 in normal or malignant mammary epithelial cells sufficiently leads to EMT and can generate mesenchymal cells (15, 36). In EOC cells however, we demonstrate that the expression of TWIST-1 is highly regulated. While inhibition of TWIST-1 expression prevents EMT, the over-expression of TWIST-1 alone in CD44+/MyD88+ EOC stem cells is not enough to induce EMT. These data suggest a different mechanism of TWIST-1 regulation in differentiated epithelial cells and epithelial cancer stem cells. In CD44+/MyD88+ EOC stem cells, the induction of EMT requires additional signals that will trigger the differentiation process. One of the mechanisms regulating TWIST-1 expression in CD44+/MyD88+ EOC stem cells involves the proteasome. Our data suggest that maintenance of an epithelial phenotype is a primordial objective of the CSCs and is achieved by mechanisms that actively prevent the expression of TWIST-1. Under stress conditions, such as low nutrients or/and hypoxia, the factors produced (TGFβ or HIF-1α) can reverse the inhibition on TWIST-1 expression leading to EMT. This is further supported by our findings, which show that the increase in TWIST-1 during EMT is accompanied by an increase in E12 expression.
E12 is a class I HLH transcription factor, and has emerged as a key regulator of both B and T lymphocyte differentiation (55, 56). E proteins continue to be essential at subsequent stages of development. Recent studies have reported that TWIST-1 requires E12 to be functional transcription factor (55). Therefore, we propose that TWIST-1 and E12 are essential factors to trigger the process of transition from CD44+/MyD88+ EOC stem cells to MSFCs, and therefore formation of metastasis.
Most epithelial tissues maintain their structure and characteristics throughout adult life due to the presence of multipotent epithelial stem cells (57). Despite some common characteristics, there are significant morphological and functional differences among epithelial cells; however, it is not very clear what are the molecular mechanisms controlling epithelial stem cell maintenance, activation, lineage determination, and differentiation in each of the different epithelial tissue. Our finding that TWIST-1 plays a central role in EOC stem cell differentiation may contribute in the understanding of ovarian epithelial stem cell maintenance and differentiation process.
A striking finding in the animal studies was the presence of mouse ovarian tumors following the i.p. or intra-uterus injection of CD44+/MyD88+ EOC stem cells or MSFCs. The demonstration that these cells are able to reach the ovaries in a very specific manner indicates that these cells have the ability to “home” to the ovaries and that the ovary has the optimum environment to support their growth. These data advocate the possibility that ovarian cancer may originate from transformed cells outside of the ovaries (i.e. fallopian tubes as some studies have suggested) but still present clinically as an “in situ” ovarian cancer (58, 59).
In summary, we demonstrated that CD44+/MyD88+ EOC stem cells could be a source of ovarian cancer metastasis by generating MSFCs through EMT. Regulation of TWIST-1 expression and function is a critical step in this process and therefore it represents a potential marker and target for monitoring and preventing ovarian cancer metastasis.
Materials and methods
Reagents and antibodies
Cobalt (II) chloride hexahydrate (sc-203004) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). MG132 (#C2211) was purchased from Sigma (St. Louis, MO, USA). Beta-actin antibody was purchased from Sungene Biotech, (Tianjin, China) clone KM9001. Additional antibodies are listed in supplemental material.
Cell culture and culture conditions
Cells used in these studies were isolated from either ovarian cancer tissues or ovarian ascites and grown as previously described (60). The isolation and characterization of the CD44+/MyD88+ EOC stem cells has been previously reported (21, 31). All patients signed consent forms, and the use of patient samples was approved under the Yale University’s Human Investigations Committee (HIC no. 10425).
In short, CD44+/MyD88+ EOC stem cells are maintained in 60–80% confluence in RPMI (GIBCO, Cat. 2340-021)+10%FBS (Gemini Bio-products, Woodland, CA, Cat. 100–106) with out grow factors and hormones. The confluence of the culture is critical to prevent differentiation. Differentiation is induced by maintaining the cells at 100% confluence for 5–10 days. Media is changed every four days.
The primary cultures are maintained at low number of passages (5–10). We have immortalized two CD44+/MyD88+ EOC stem cells lines and two derived CD44-/MyD88- EOC cells. These clones have been used for establishing GFP- cells. The experiments described in this study are from passages 25–40. We are extremely careful to maintain low passage number for the cells in order to prevent changes associated with long-term cultures.
CD44-/MYD88- EOC cells are derived in vitro or in vivo from CD44+/MyD88+ Type EOC stem cells and have lost their tumor initiating capacity but conserves the epithelial phenotype and have a high growth rate (21).
Details for the conditions on the hypoxia culture conditions are described in the supplementary material.
Mouse xenograft studies
CD44+/MyD88+ EOC stem cells (3×106 cells) or MSFCs (3×106 cells) were re-suspended in 200 µl total volume of RPMI 10%FBS. Uterus from nude mice was exposed by lateral incision and 25 µl were injected into the fallopian tube of each side using a 30 G needle. Intra-peritoneal injection in nude mice was done using 26G needle. Control groups were injected with the same amount of CD44-/MyD88- EOC cells using similar conditions as described for CD44+/MyD88+ EOC stem cells. Tumor samples were collect and snap frozen in liquid nitrogen or fixed in 4% paraformaldehyde for further studies. For each experiment we used a n=6
Plasmids
pEMSV-TWIST1 and pEMSV-E12 were given by Dr. Ernst-Martin Fuchtbauer, University of Aarhus, Denmark, pFUGW was given by Dr. Wange Lu, University of Southern California, USA. Lentivirus vector of shRNA of TWIST1, TWIST1-siRNA3, was purchased from Addgene (#1784). The full length open reading frame (ORF) of the cDNA encoding TWIST1 was obtained by PCR amplification from the pEMSV-TWIST1, and was cloned into pFUGW, which was used to generate lentivirus. The pFUGW and pFUGW-TWIST1 can express GFP since it contains a GFP coding sequence. The primers of TWIST1 for vector construction is shown in Supplementary Table 1.
Plasmid transfection
Ovarian cancer stem cells (5*105) were mixed with 10 µg of the indicated expression vectors, and transfection process was performed by Gene Pulser X-cell (Bio-Rad) according to Gene Pulser Electroprotocal. Twenty-four to 72 hours after transfection, cells were trypsinized and collected for RNA purification or protein sample preparation.
Flow cytometry
Cells were stained with CD44 (eBioscience, #11-0441, 1:100 dilution) or CK18 (Cell Signaling, #4548, 1:1000 dilution) as previously described (33, 61). Data was acquired using BD LSR II System and analyzed using FloJo FACS analysis software (Tree Star, Inc., Ashland, OR).
Protein preparation and cellular fractionation
Protein extraction was carried out as previously described (62). For separation of the cytoplasmic/nuclear fractions, cell pellets were processed using NE- PER Nuclear and Cytoplasmic Extraction kit (Pierce Biotechnology, Inc., Rockford, IL). Proteins concentration was determined by BCA Protein Assay (Pierce Biotechnology, Rockford, IL, USA) and proteins were stored at −80° until further use.
SDS–polyacrylamide gel electrophoresis and Western blots
SDS-PAGE performance and the antibody dilutions are listed in the supplemental material.
Cytokine profiling
Levels of TGFbeta1,2,3 were measured from cell-free supernatants using the MILLIPLEX MAP TGFß 3-Plex Assay (#TGFB-64K-03) purchased from Millipore (Billerica, MA, USA). Data were acquired using the Bioplex system (Biorad) and analysis was carried out using the Bioplex software as previously described (63, 64).
Lentiviral Transduction and Stable Cell Line Generation
Lentiviral vector and packaging vectors were transfected into the packaging cell line 293T (ATCC) using the FuGENE6 Transfection Reagent (Roche Applied Science). The medium was changed 8 h post-transfection, and the medium containing lentivirus was collected 48 h later. Ovarian cancer stem cells were infected with lentivirus in the presence of 8 ng/ml Polybrene (Sigma). For stable transfection cell line selection, GFP fluorescence was used as a sorting method for FUGW-TWIST, and the antibiotic puromycin was used to select for TWIST-siRNA3-lentivirus (49).
Immunohistonchemistry and Immunofluorescence Staining
The immunohistochemistry and immunocytochemistry experiments are described in the supplementary material. GFP fluorescence was captured by a Zeiss fluorescence microscope and pictures obtained with Velocity Image analysis system (PerkinElmer (Waltham, MA).
Transwell migration and Matrigel Invasion Assays
100ul Matrigel (BD Biosciences) was coated onto the lower side of a transwell filter with 8-mm pores (Costar, Lowell, MA, USA), and incubated at 37° for 30min. Cells were plated at a concentration of 1×105 in 500ul RPMI-10% FBS to the upper side of the filter on a 24-well plate. 500ul of OPTIMEM (about 1% FBS) was added to the lower chamber. After 24 h, the filters were removed and the cells on the bottom of the well were counted. Images were captured using a Zeiss (Melville, NY, USA) microscope system. For each experiment, the number of cells in nine random fields on the underside of the filter was counted and three independent filters were analyzed using the Velocity Image analysis system (PerkinElmer (Waltham, MA).
EMT array
Total RNA was prepared from two clones of CD44+/MyD88 EOC stem cell lines, two clones of derived spheroids and CD44-/MyD88- EOC cells using the RNeasy Mini kit (Qiagen, Valencia, CA, USA). Total RNA isolated from each samples was then used as a template for cDNA synthesis, prepared with a RT2 first strand kit (SABioscience, no.C-03). Total cDNA was used as template for EMT array using RT2 Profiler™ PCR Array Human Epithelial to Mesenchymal Transition (EMT) plate (SABioscience, no. PAHS-090A). This plate was pre-coated with 84 pairs of primers for 84 kinds of genes (included in the Supplementary Table 2). The expression levels of various genes were assessed by real-time PCR amplification (95 °C for 10 min; (95 °C for 15 s, 60 °C for 1 min; 40 cycles), with PCR Master Mix RT2 Real-Time SYBR Green/ROX (SABioscience, no. PA-012) using the ABI 7500 Real-Time Standard Cycler (Applied Biosystems, Foster City, CA, USA). Validation of the gene array was done in five cell cultures for each subtype of cells (n=15)
mRNA quantitative RT–PCR
RT-qPCR procedure is described in supplementary material. The primer sets used in this study are listed in Supp. Table 1. All PCR reactions were carried out in triplicate and validated by the presence of a single peak in the melt curve analysis. Changes in gene expression were calculated relative to GAPDH using the 2−ΔCt method (65).
Statistical analysis
Data are expressed as mean ± standard error for the in vitro studies and median ± first or third quartiles for the in vivo studies. Statistical significance (p<0.05) was determined using either two tailed unpaired t-tests or Mann-Whitney U test for non-parametric data. Unless stated otherwise, all experiments were performed in duplicate.
Supplementary Material
Acknowledgements
This study was supported in part by grants from NCI/NIH RO1CA127913, RO1CA118678, The Janet Burros Memorial Foundation, The Sands Family Foundation and the Discovery To Cure Research Program. GY was supported by the Brozman Foundation.
Special thanks to Dr. Ernst-Martin Fuchtbauer for the plasmids, pEMSV, pEMSV-TWIST1, and pEMSV-E12; and to Dr. Wange Lu for the plasmid pFUIGW.
Footnotes
Conflict of interest. The authors declare no conflict of interest.
References
- 1.Alison MR, Murphy G, Leedham S. Stem cells and cancer: a deadly mix. Cell Tissue Res. 2008 Jan;331(1):109–124. doi: 10.1007/s00441-007-0510-7. [DOI] [PubMed] [Google Scholar]
- 2.Vergara D, Merlot B, Lucot JP, Collinet P, Vinatier D, Fournier I, et al. Epithelial-mesenchymal transition in ovarian cancer. Cancer Lett. 2010 May 1;291(1):59–66. doi: 10.1016/j.canlet.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 3.Espey DK, Wu XC, Swan J, Wiggins C, Jim MA, Ward E, et al. Annual report to the nation on the status of cancer, 1975–2004, featuring cancer in American Indians and Alaska Natives. Cancer. 2007 Nov 15;110(10):2119–2152. doi: 10.1002/cncr.23044. [DOI] [PubMed] [Google Scholar]
- 4.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: a cancer journal for clinicians. 2011 Mar-Apr;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 5.Mutch D. Surgical manegement of ovarian cancer. Semin Oncol. 2002;29:3–8. doi: 10.1053/sonc.2002.31589. [DOI] [PubMed] [Google Scholar]
- 6.Cannistra SA, Bast RC, Jr., Berek JS, Bookman MA, Crum CP, DePriest PD, et al. Progress in the management of gynecologic cancer: consensus summary statement. J Clin Oncol. 2003 May 15;21(10 Suppl):129–132. doi: 10.1200/JCO.2003.04.003. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz PE. Current diagnosis and treatment modalities for ovarian cancer. Cancer Treat Res. 2002;107:99–118. doi: 10.1007/978-1-4757-3587-1_4. [DOI] [PubMed] [Google Scholar]
- 8.Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer. 2003 Jul;3(7):502–516. doi: 10.1038/nrc1123. [DOI] [PubMed] [Google Scholar]
- 9.Clarke-Pearson DL. Clinical practice. Screening for ovarian cancer. N Engl J Med. 2009 Jul 9;361(2):170–177. doi: 10.1056/NEJMcp0901926. [DOI] [PubMed] [Google Scholar]
- 10.Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S, et al. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005 Oct 15;65(20):9328–9337. doi: 10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- 11.Chambers JT, Schwartz PE. Treatment of persistent or recurrent ovarian carcinoma with sequential methotrexate and 5-fluorouracil. Gynecol Oncol. 1986;23(3):346–349. doi: 10.1016/0090-8258(86)90136-8. [DOI] [PubMed] [Google Scholar]
- 12.Yang M, Mailhot G, Birnbaum MJ, MacKay CA, Mason-Savas A, Odgren PR. Expression of and role for ovarian cancer G-protein-coupled receptor 1 (OGR1) during osteoclastogenesis. J Biol Chem. 2006 Aug 18;281(33):23598–23605. doi: 10.1074/jbc.M602191200. [DOI] [PubMed] [Google Scholar]
- 13.Radisky DC, Przybylo JA. Matrix metalloproteinase-induced fibrosis and malignancy in breast and lung. Proc Am Thorac Soc. 2008 Apr 15;5(3):316–322. doi: 10.1513/pats.200711-166DR. [DOI] [PubMed] [Google Scholar]
- 14.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006 Feb;7(2):131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 15.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008 May 16;133(4):704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003 Dec;15(6):740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 17.Chaffer CL, Dopheide B, Savagner P, Thompson EW, Williams ED. Aberrant fibroblast growth factor receptor signaling in bladder and other cancers. Differentiation. 2007 Nov;75(9):831–842. doi: 10.1111/j.1432-0436.2007.00210.x. [DOI] [PubMed] [Google Scholar]
- 18.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006 Oct 1;66(19):9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 19.Radisky DC, LaBarge MA. Epithelial-mesenchymal transition and the stem cell phenotype. Cell Stem Cell. 2008 Jun 5;2(6):511–512. doi: 10.1016/j.stem.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 20.Hollier BG, Evans K, Mani SA. The epithelial-to-mesenchymal transition and cancer stem cells: a coalition against cancer therapies. J Mammary Gland Biol Neoplasia. 2009 Mar;14(1):29–43. doi: 10.1007/s10911-009-9110-3. [DOI] [PubMed] [Google Scholar]
- 21.Alvero AB, Chen R, Fu HH, Montagna M, Schwartz PE, Rutherford T, et al. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle. 2009 Jan 1;8(1):158–166. doi: 10.4161/cc.8.1.7533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steffensen KD, Alvero AB, Yang Y, Waldstrom M, Hui P, Holmberg JC, et al. Prevalence of epithelial ovarian cancer stem cells correlates with recurrence in early-stage ovarian cancer. J Oncol. 2011;2011:620523. doi: 10.1155/2011/620523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao Q, Geng L, Kvalheim G, Gaudernack G, Suo Z. Identification of cancer stem-like side population cells in ovarian cancer cell line OVCAR-3. Ultrastruct Pathol. 2009 Jul-Aug;33(4):175–181. doi: 10.1080/01913120903086072. [Research Support, Non-U.S Gov't] [DOI] [PubMed] [Google Scholar]
- 24.Bapat SA, Mali AM, Koppikar CB, Kurrey NK. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005 Apr 15;65(8):3025–3029. doi: 10.1158/0008-5472.CAN-04-3931. [DOI] [PubMed] [Google Scholar]
- 25.Moserle L, Indraccolo S, Ghisi M, Frasson C, Fortunato E, Canevari S, et al. The side population of ovarian cancer cells is a primary target of IFN-alpha antitumor effects. Cancer Res. 2008 Jul 15;68(14):5658–5668. doi: 10.1158/0008-5472.CAN-07-6341. [Research Support, Non-U.S Gov't] [DOI] [PubMed] [Google Scholar]
- 26.Szotek PP, Pieretti-Vanmarcke R, Masiakos PT, Dinulescu DM, Connolly D, Foster R, et al. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proceedings of the National Academy of Sciences of the United States of America. 2006 Jul 25;103(30):11154–11159. doi: 10.1073/pnas.0603672103. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curley MD, Garrett LA, Schorge JO, Foster R, Rueda BR. Evidence for cancer stem cells contributing to the pathogenesis of ovarian cancer. Front Biosci. 2011;16:368–392. doi: 10.2741/3693. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't Review] [DOI] [PubMed] [Google Scholar]
- 28.Curley MD, Therrien VA, Cummings CL, Sergent PA, Koulouris CR, Friel AM, et al. CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells. 2009 Dec;27(12):2875–2883. doi: 10.1002/stem.236. [DOI] [PubMed] [Google Scholar]
- 29.Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008 Jun 1;68(11):4311–4320. doi: 10.1158/0008-5472.CAN-08-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen R, Alvero AB, Silasi DA, Mor G. Inflammation, cancer and chemoresistance: taking advantage of the toll-like receptor signaling pathway. Am J Reprod Immunol. 2007 Feb;57(2):93–107. doi: 10.1111/j.1600-0897.2006.00441.x. [DOI] [PubMed] [Google Scholar]
- 31.Chen R, Alvero AB, Silasi DA, Kelly MG, Fest S, Visintin I, et al. Regulation of IKKbeta by miR-199a affects NF-kappaB activity in ovarian cancer cells. Oncogene. 2008 Aug 7;27(34):4712–4723. doi: 10.1038/onc.2008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mor G, Yin G, Chefetz I, Yang Y, Alvero A. Ovarian cancer stem cells and inflammation. Cancer Biol Ther. 2011 Apr 15;11(8) doi: 10.4161/cbt.11.8.14967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alvero AB, Fu HH, Holmberg J, Visintin I, Mor L, Marquina CC, et al. Stem-like ovarian cancer cells can serve as tumor vascular progenitors. Stem Cells. 2009 Oct;27(10):2405–2413. doi: 10.1002/stem.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharabi AB, Aldrich M, Sosic D, Olson EN, Friedman AD, Lee SH, et al. Twist-2 controls myeloid lineage development and function. PLoS Biol. 2008 Dec 16;6(12):e316. doi: 10.1371/journal.pbio.0060316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watanabe O, Imamura H, Shimizu T, Kinoshita J, Okabe T, Hirano A, et al. Expression of twist and wnt in human breast cancer. Anticancer Res. 2004 Nov-Dec;24(6):3851–3856. [PubMed] [Google Scholar]
- 36.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004 Jun 25;117(7):927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 37.Cheng GZ, Zhang WZ, Sun M, Wang Q, Coppola D, Mansour M, et al. Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. J Biol Chem. 2008 May 23;283(21):14665–14673. doi: 10.1074/jbc.M707429200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gort EH, van Haaften G, Verlaan I, Groot AJ, Plasterk RH, Shvarts A, et al. The TWIST1 oncogene is a direct target of hypoxia-inducible factor-2alpha. Oncogene. 2008 Mar 6;27(11):1501–1510. doi: 10.1038/sj.onc.1210795. [Research Support, Non-U.S Gov't] [DOI] [PubMed] [Google Scholar]
- 39.Alvero AB, Montagna MK, Holmberg JC, Craveiro V, Brown D, Mor G. Targeting the mitochondria activates two independent cell death pathways in ovarian cancer stem cells. Mol Cancer Ther. 2011 Aug;10(8):1385–1393. doi: 10.1158/1535-7163.MCT-11-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chefetz I, Holmberg JC, Alvero AB, Visintin I, Mor G. Inhibition of Aurora-A kinase induces cell cycle arrest in epithelial ovarian cancer stem cells by affecting NFkB pathway. Cell Cycle. 2011 Jul 1;10(13):2206–2214. doi: 10.4161/cc.10.13.16348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mor G, Yin G, Chefetz I, Yang Y, Alvero A. Ovarian cancer stem cells and inflammation. Cancer Biol Ther. 2011 Apr 15;11(8):708–713. doi: 10.4161/cbt.11.8.14967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009 Jun;119(6):1429–1437. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alix-Panabieres C, Vendrell JP, Slijper M, Pelle O, Barbotte E, Mercier G, et al. Full-length cytokeratin-19 is released by human tumor cells: a potential role in metastatic progression of breast cancer. Breast Cancer Res. 2009;11(3):R39. doi: 10.1186/bcr2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carambula SF, Matikainen T, Lynch MP, Flavell RA, Goncalves PB, Tilly JL, et al. Caspase-3 is a pivotal mediator of apoptosis during regression of the ovarian corpus luteum. Endocrinology. 2002 Apr;143(4):1495–1501. doi: 10.1210/endo.143.4.8726. [DOI] [PubMed] [Google Scholar]
- 45.Hader C, Marlier A, Cantley L. Mesenchymal-epithelial transition in epithelial response to injury: the role of Foxc2. Oncogene. 2010 Feb 18;29(7):1031–1040. doi: 10.1038/onc.2009.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bechtel W, Zeisberg M. Twist: a new link from hypoxia to fibrosis. Kidney Int. 2009 Jun;75(12):1255–1256. doi: 10.1038/ki.2009.102. [DOI] [PubMed] [Google Scholar]
- 47.Horbinski C, Mojesky C, Kyprianou N. Live free or die: tales of homeless (cells) in cancer. Am J Pathol. 2010 Sep;177(3):1044–1052. doi: 10.2353/ajpath.2010.091270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee YB, Bantounas I, Lee DY, Phylactou L, Caldwell MA, Uney JB. Twist-1 regulates the miR-199a/214 cluster during development. Nucleic Acids Res. 2009 Jan;37(1):123–128. doi: 10.1093/nar/gkn920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laursen KB, Mielke E, Iannaccone P, Fuchtbauer EM. Mechanism of transcriptional activation by the proto-oncogene Twist1. J Biol Chem. 2007 Nov 30;282(48):34623–34633. doi: 10.1074/jbc.M707085200. [DOI] [PubMed] [Google Scholar]
- 50.Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001 Jun;7(6):1267–1278. doi: 10.1016/s1097-2765(01)00260-x. [DOI] [PubMed] [Google Scholar]
- 51.Hartwell KA, Muir B, Reinhardt F, Carpenter AE, Sgroi DC, Weinberg RA. The Spemann organizer gene, Goosecoid, promotes tumor metastasis. Proc Natl Acad Sci U S A. 2006 Dec 12;103(50):18969–18974. doi: 10.1073/pnas.0608636103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hay ED. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev Dyn. 2005 Jul;233(3):706–720. doi: 10.1002/dvdy.20345. [DOI] [PubMed] [Google Scholar]
- 53.Valsesia-Wittmann S, Magdeleine M, Dupasquier S, Garin E, Jallas AC, Combaret V, et al. Oncogenic cooperation between H-Twist and N-Myc overrides failsafe programs in cancer cells. Cancer Cell. 2004 Dec;6(6):625–630. doi: 10.1016/j.ccr.2004.09.033. [DOI] [PubMed] [Google Scholar]
- 54.Entz-Werle N, Stoetzel C, Berard-Marec P, Kalifa C, Brugiere L, Pacquement H, et al. Frequent genomic abnormalities at TWIST in human pediatric osteosarcomas. Int J Cancer. 2005 Nov 10;117(3):349–355. doi: 10.1002/ijc.21068. [DOI] [PubMed] [Google Scholar]
- 55.El Ghouzzi V, Legeai-Mallet L, Aresta S, Benoist C, Munnich A, de Gunzburg J, et al. Saethre-Chotzen mutations cause TWIST protein degradation or impaired nuclear location. Hum Mol Genet. 2000 Mar 22;9(5):813–819. doi: 10.1093/hmg/9.5.813. [DOI] [PubMed] [Google Scholar]
- 56.Shintani Y, Maeda M, Chaika N, Johnson KR, Wheelock MJ. Collagen I promotes epithelial-to-mesenchymal transition in lung cancer cells via transforming growth factor-beta signaling. Am J Respir Cell Mol Biol. 2008 Jan;38(1):95–104. doi: 10.1165/rcmb.2007-0071OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Blanpain C, Horsley V, Fuchs E. Epithelial stem cells: turning over new leaves. Cell. 2007 Feb 15;128(3):445–458. doi: 10.1016/j.cell.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kurman RJ, Shih Ie M. The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol. 2010 Mar;34(3):433–443. doi: 10.1097/PAS.0b013e3181cf3d79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shih Ie M, Chen L, Wang CC, Gu J, Davidson B, Cope L, et al. Distinct DNA methylation profiles in ovarian serous neoplasms and their implications in ovarian carcinogenesis. Am J Obstet Gynecol. 2010 Dec;203(6):e1–e22. doi: 10.1016/j.ajog.2010.08.003. 584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kamsteeg M, Rutherford T, Sapi E, Hanczaruk B, Shahabi S, Flick M, et al. Phenoxodiol--an isoflavone analog--induces apoptosis in chemoresistant ovarian cancer cells. Oncogene. 2003 May 1;22(17):2611–2620. doi: 10.1038/sj.onc.1206422. [DOI] [PubMed] [Google Scholar]
- 61.Abrahams V, Straszewski S, Kamsteeg M, Hanczaruk B, Schwartz P, Rutherford T, et al. Epithelial Ovarian Cancer secrete funcitonal Fas Ligand. Cancer Res. 2003;63:5573–5581. [PubMed] [Google Scholar]
- 62.Green JM, Alvero AB, Kohen F, Mor G. 7-(O)-Carboxymethyl daidzein conjugated to N-t-Boc-hexylenediamine: a novel compound capable of inducing cell death in epithelial ovarian cancer stem cells. Cancer Biol Ther. 2009 Sep;8(18):1747–1753. doi: 10.4161/cbt.8.18.9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koga K, Cardenas I, Aldo P, Abrahams VM, Peng B, Fill S, et al. Activation of TLR3 in the trophoblast is associated with preterm delivery. Am J Reprod Immunol. 2009 Mar;61(3):196–212. doi: 10.1111/j.1600-0897.2008.00682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Visintin I, Feng Z, Longton G, Ward DC, Alvero AB, Lai Y, et al. Diagnostic markers for early detection of ovarian cancer. Clin Cancer Res. 2008 Feb 15;14(4):1065–1072. doi: 10.1158/1078-0432.CCR-07-1569. [DOI] [PubMed] [Google Scholar]
- 65.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.