

Summary
Accumulation of neurotoxic amyloid peptides (Aβ) in the brain, generated by β-site proteolytic processing of the amyloid precursor protein (APP), is the hallmark pathophysiologic feature of Alzheimer's disease. The plasmin-activating cascade, in which urokinase (uPA) and tissue-type (tPA) plasminogen activators convert plasminogen to the broad-spectrum protease plasmin, appears to serve a protective, Aβ-clearing, role in the central nervous system. Plasmin degrades Aβ and catalyzes α- site APP proteolysis generating nontoxic peptides. Plasmin activation in the brain is negatively regulated by the fast-acting clade E serine protease inhibitor (SERPIN) plasminogen activator inhibitor type-1 (PAI-1; SERPINE1) resulting in Aβ accumulation. PAI-1 and its major physiological inducer TGF-β1, moreover, are both increased in Alzheimer's disease models and implicated in the etiology and progression of human neurodegenerative disorders. Current findings support the hypothesis that targeting of PAI-1 function (by small molecule drugs) and/or gene expression (by histone deacetylase inhibitors) may constitute a clinically-relevant molecular approach to the therapy of neurodegenerative diseases associated with increased PAI-1 levels.
Plasmin-Activating System in Alzheimer's Disease
Aggregated β-amyloid peptide plaques accumulate in specific areas of the brain in patients with Alzheimer's disease (AD) by proteolytic processing of the single-pass transmembrane APP [1]. These deposits trigger prolonged inflammation, neuronal death, and progressive cognitive decline [2]. Aβ peptides are produced by aspartic protease (BACE)-induced β-site cleavage of APP creating a membrane-bound COOH-terminal C99 fragment followed by proteolysis (involving presenilin and nicastrin) at the C99 transmembrane-localized γ position [3–5]. There is also an alternative APP processing pathway in which a membrane-proximal (α-site) cleavage, by matrix metalloproteinases (TACE, ADAM 10), replaces β position utilization producing a membrane-anchored C83 fragment. Subsequent γ processing of the C83 product generates the nontoxic p3 peptide [3,6].
Among other targets, the broad-spectrum protease plasmin also degrades Aβ [7–9] and plasmin activation decreases Aβ peptide levels [10]. Plasmin-mediated proteolysis of APP, moreover, involves the α-site (either as a direct or indirect target) resulting in decreased Aβ production, suggesting a protective role for the plasmin cascade in the central nervous system. Indeed, plasmin levels in the brains of AD patients are considerably reduced [10] supporting a causal relationship between deficient activity of the plasmin-generating proteolytic system and accumulation of Aβ in the progression of AD (Figure 1).
Figure 1.
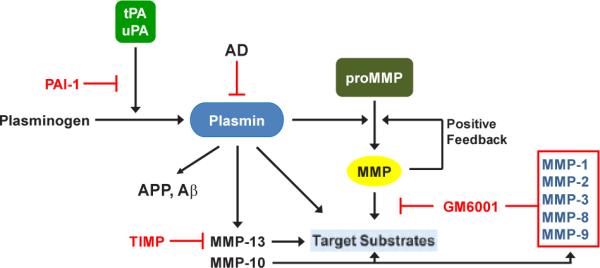
tPA and uPA convert plasminogen to the active protease plasmin that, in turn, degrades target substrates (e.g., APP, Aβ) directly as well as indirectly through downstream activation of matrix metalloproteinases (MMPs). Inhibition of MMP activity with tissue inhibitor of metalloproteinases (TIMP) or GM6001 has confirmed their participation in plasmin-initiated proteolysis. This cascade is effectively attenuated by expression of PAI-1 which blocks tPA and uPA catalysis inhibiting plasmin generation.
Therapeutic Approaches
Several members of the SERPIN superfamily exhibit cell-type neurotrophic, neuroprotective, or neuropathophysiologic activities [11]. These include SERPINF1, SERPINI1 (neuroserpin), SERPINE1 (PAI-1), SERPINE2 (nexin-1), and SERPINA3 [11]. PAI-1 (SERPINE1), in particular, has multifunctional roles in the central nervous system as it both maintains neuronal cellular structure and initiates signaling through the mitogen-activated protein kinase pathway [12]. Significantly increased PAI-1 immunoreactivity in the central nervous system of AD patients was associated with development of senile plaques and ghost tangle structures [13] consistent with the earlier colocalization of plasminogen activators and PAI-1 in plaque structures [14] which are sites of sustained inflammation [15]. Tg2576 and TgCRN8 transgenic mice, engineered to express brain-targeted Swedish mutant Aβ and the double Swedish/V717F mutant Aβ, respectively, exhibit age-dependent Aβ plaque development as well as cognitive deficiencies [16]. tPA activity in these mice was significantly decreased specifically in the hippocampus and amygdala corelating with corresponding regional increases in brain PAI-1 expression [17]. Since direct Aβ peptide injection increased PAI-1 expression and Aβ removal from the hippocampal region required both tPA and plasminogen, a functional tPA-plasmin axis appears required for Aβ clearance [17]. While PAI-1 may be neuroprotective in specific acute injury settings (e.g., tPA-triggered neuronal apoptosis) [18,19], chronically elevated PAI-1 levels likely promote Aβ accumulation by inhibiting plasmin-dependent degradation. Genetic evidence clearly indicates that brain PAI-1 expression is increased in Aβ precursor protein presenilin 1 (APP/PS1) mice as well as in AD patients [20] while PAI-1-deficiency in an APP/PS1 transgenic background reduces amyloid accumulation. These findings have therapeutic implications as a small molecule inhibitor of PAI-1 activity (PAZ-417) partially blocks amyloid deposition in a mouse model of AD. The mechanism, perhaps as expected, suggests that PAI-1 inhibition stimulates tPA/plasmin activity, decreasing brain Aβ levels and reverses cognitive deficits [21]. The development of pharmacologic strategies to prevent and therapeutically manage patients at risk for, and who present with, AD by inhibiting the function of a key contributor (PAI-1) to disease progression has significant translational relevance. Indeed, histone deacetylase inhibitors (HDACi) have potential promise as a therapy for neurodegenerative disease [22]. Sodium butyrate (NaB), a broad-spectrum HDACi, attenuated streptozotocin-induced endothelial dysfunction and improved learning and memory (Morris water maze test) in rats [23]. Butyrate localizes to the cerebral cortex in KCl-induced spreading cortical depression [24] while NaB (as well as TSA and valproic acid) are neuroprotective in the ischemic brain [25]. Most importantly, several HDACi (NaB, TSA, SAHA and to a lesser extent sirtinol) effectively reduced TGF-β1-induced PAI-1 expression [19] (Figure 2). This has translational implications as brain TGF-β1 levels are elevated in the onset and progression of Parkinson's disease, AD and stroke [19]. Increased expression of TGF-β1 correlates with Aβ angiopathy and transgenic mice that overexpress TGF-β in astrocytes exhibit early onset Aβ deposition [26]. TGF-β1, moreover, induces astrocyte APP expression through a TGF- β1-responsive element in the APP promoter while Aβ production was enhanced by TGF-β1 signaling [27]. The coordinate overexpression of PAI-1 and increased Aβ generation in response to elevated TGF-β1 in AD patients is likely to predispose to disease progression [28]. Collectively, these findings raise the possibility that targeting TGF-β1-inducible genes (e.g., PAI-1, APP) will provide a therapeutic benefit in the setting of AD. HDACi coupled with a small molecule central nervous system-accessible PAI-1 functional inhibitor may have efficacy as an approach to reverse the ongoing accumulation of amyloid deposits even after disease development.
Figure 2.
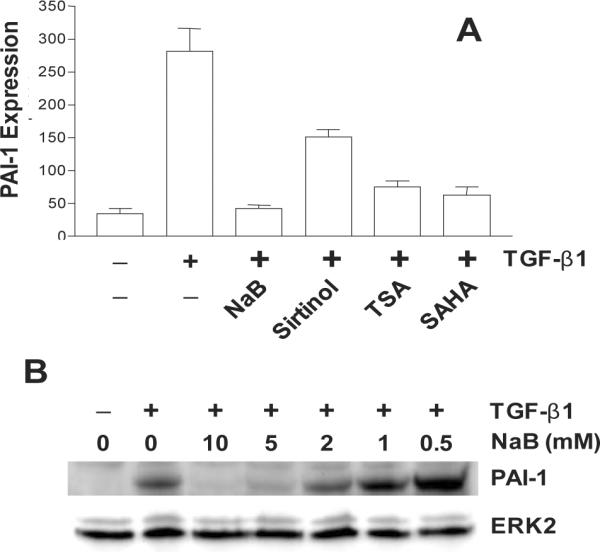
Effect of the histone deacetylase inhibitors sodium butyrate (NaB), sirtinol, trichostatin A (TSA) and suberoylanilide hydroxamic acid (SAHA) on TGF-β1-induced PAI-1 expression (A). Data plotted is a summary of western blot analyses. Preincubation with NaB concentrations of 5–10 mM completely ablates TGF–β1-stimulated PAI-1 expression (B).
Acknowledgements
Supported by NIH grant GM057242 to PJH.
References
- 1.Glenner GG, Wong CW. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature. 1999;399 doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- 3.Parvathy S, Hussain I, Karran EH, Turner AJ, Hooper NM. Cleavage of Alzheimer's amyloid precursor protein by α-secretase occurs at the surface of neuronal cells. Biochemistry. 1999;38:9728–9734. doi: 10.1021/bi9906827. [DOI] [PubMed] [Google Scholar]
- 4.Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, et al. β-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 5.Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and βAPP processing. Nature. 2000;407:48–54. doi: 10.1038/35024009. [DOI] [PubMed] [Google Scholar]
- 6.Periz G, Fortini ME. Proteolysis in Alzheimer's disease. Can plasmin tip the balance? EMBO Rep. 2000;1:477–478. doi: 10.1093/embo-reports/kvd124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tucker HM, Kihiko M, Caldwell JN, Wright S, Kawarabayashi T, et al. The plasmin system is induced by and degrades amyloid-β aggregates. J Neurosci. 2000;20:3937–3946. doi: 10.1523/JNEUROSCI.20-11-03937.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Nostrand WE, Porter M. Plasmin cleavage of the amyloid β-protein: alteration of secondary structure and stimulation of tissue plasminogen activator activity. Biochemistry. 1999;38:11570–11576. doi: 10.1021/bi990610f. [DOI] [PubMed] [Google Scholar]
- 9.Wnendt S, Wetzels I, Gunzler WA. Amyloid β peptides stimulate tissue-type plasminogen activator but not recombinant prourokinase. Thromb Res. 1997;85:217–224. doi: 10.1016/s0049-3848(97)00006-6. [DOI] [PubMed] [Google Scholar]
- 10.Ledesma MD, Da Silva JS, Crassaerts K, Delacourte A, De Strooper B, et al. Brain plasmin enhances APP α-cleavage and Aβ degradation and is reduced in Alzheimer's disease brains. EMBO Reports. 2000;1:530–535. doi: 10.1093/embo-reports/kvd107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Silverman GA, Bird PI, Carrell RW, Church FC, Coughlin PB, et al. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions and a revised nomenclature. J Biol Chem. 2001;276:33293–33296. doi: 10.1074/jbc.R100016200. [DOI] [PubMed] [Google Scholar]
- 12.Vivien D, Buisson A. Serine protease inhibitors: novel therapeutic targets for stroke? J Cereb Blood Flow Metabol. 2000;20:755–764. doi: 10.1097/00004647-200005000-00001. [DOI] [PubMed] [Google Scholar]
- 13.Hino H, Akiyama H, Iseki E, Kato M, Kondo H, et al. Immunohistochemical localization of plasminogen activator inhibitor-1 in rat and human brain tissues. Neurosci Lett. 2001;297:105–108. doi: 10.1016/s0304-3940(00)01679-7. [DOI] [PubMed] [Google Scholar]
- 14.Rebeck GW, Harr SD, Strickland DK, Hyman BT. Multiple, diverse senile plaque-associated proteins are ligands of an apolipoprotein e receptor, the α2-macroglobulin receptor/ low-density-lipoprotein receptor - related protein. Ann Neurol. 1995;37:211–217. doi: 10.1002/ana.410370212. [DOI] [PubMed] [Google Scholar]
- 15.McGeer PL, McGeer EG. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev. 1995;21:195–218. doi: 10.1016/0165-0173(95)00011-9. [DOI] [PubMed] [Google Scholar]
- 16.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, et al. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 17.Melchor JP, Pawlak R, Strickland S. The tissue plasminogen activator-plasminogen proteolytic cascade accelerates amyloid-β (Aβ) degradation and inhibits Aβ-induced neurodegeneration. J Neurosci. 2003;23:8867–8871. doi: 10.1523/JNEUROSCI.23-26-08867.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flavin MP, Zhao G, Ho LT. Microglial tissue plasminogen activator (tPA) triggers neuronal apoptosis in vitro. Glia. 2000;29:347–354. [PubMed] [Google Scholar]
- 19.Higgins PJ. The TGF-β1/upstream stimulatory factor-regulated PAI-1 gene: potential involvement and a therapeutic target in Alzheimer's disease. J Biomed Biotechnol. 2006;2006:15792. doi: 10.1155/JBB/2006/15792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu RM, van Groen T, Katre A, Cao D, Kadisha I, et al. Knockout of plasminogen activator inhibitor 1 gene reduces amyloid beta peptide burden in a mouse model of Alzheimer's disease. Neurobiol Aging. 2011;32:1079–1089. doi: 10.1016/j.neurobiolaging.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacobsen JS, Comery TA, Martone RL, Elokdah H, Crandall DL, et al. Enhanced clearance of Abeta in brain by sustaining the plasmin proteolysis cascade. Proc Natl Acad Sci USA. 2008;105:8754–8759. doi: 10.1073/pnas.0710823105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodeling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- 23.Sarma B, Singh N. Attenuation of vascular dementia by sodium butyrate in streptozotocin diabetic rats. Psychopharmacology (Berl) 2011;215:677–687. doi: 10.1007/s00213-011-2164-0. [DOI] [PubMed] [Google Scholar]
- 24.Dienel GA, Liu K, Cruz NF. Local uptake of 14C-labeled acetate and buyrate in rat brain in vivo during spreading cortical depression. J Neuroscience Res. 2001;66:812–820. doi: 10.1002/jnr.10063. [DOI] [PubMed] [Google Scholar]
- 25.Kim HJ, Leeds P, Chuang DM. The HDAC inhibitor, sodium butyrate, stimulates neurogenesis in the ischemic brain. J Neurochem. 2009;110:1226–1240. doi: 10.1111/j.1471-4159.2009.06212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wyss-Coray T, lin C, Sanan DA, Mucke L, Mashiah E. Chronic overproduction of transforming growth factor-β1 by astrocytes promotes Alzheimer's disease-like microvascular degeneration in transgenic mice. Am J Pathol. 2000;156:139–150. doi: 10.1016/s0002-9440(10)64713-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lesné S, Docagne F, Gabriel C, Liot G, Lahiri DK, et al. Transforming growth factor-β1 potentiates amyloid-β generation in astrocytes and in transgenic mice. J Biol Chem. 2003;278:18408–18418. doi: 10.1074/jbc.M300819200. [DOI] [PubMed] [Google Scholar]
- 28.Lesné S, Blanchet S, Docagne F, Liot G, Plawinski L, et al. Transforming growth factor-β1-modulated cerebral gene expression. J Cereb Blood Flow Metabol. 2002;22:1114–1123. doi: 10.1097/00004647-200209000-00009. [DOI] [PubMed] [Google Scholar]