

Abstract

Nucleic acid probes are used for diverse applications in vitro, in situ, and in vivo. In any setting, their power is limited by imperfect selectivity (binding of undesired targets) and incomplete affinity (binding is reversible, and not all desired targets bound). These difficulties are fundamental, stemming from reliance on base pairing to provide both selectivity and affinity. Shielded covalent (SC) probes eliminate the longstanding trade-off between selectivity and durable target capture, achieving selectivity via programmable base pairing and molecular conformation change, and durable target capture via activatable covalent cross-linking. In pure and mixed samples, SC probes covalently capture complementary DNA or RNA oligo targets and reject two-nucleotide mismatched targets with near-quantitative yields at room temperature, achieving discrimination ratios of 2–3 orders of magnitude. Semiquantitative studies with full-length mRNA targets demonstrate selective covalent capture comparable to that for RNA oligo targets. Single-nucleotide DNA or RNA mismatches, including nearly isoenergetic RNA wobble pairs, can be efficiently rejected with discrimination ratios of 1–2 orders of magnitude. Covalent capture yields appear consistent with the thermodynamics of probe/target hybridization, facilitating rational probe design. If desired, cross-links can be reversed to release the target after capture. In contrast to existing probe chemistries, SC probes achieve the high sequence selectivity of a structured probe, yet durably retain their targets even under denaturing conditions. This previously incompatible combination of properties suggests diverse applications based on selective and stable binding of nucleic acid targets under conditions where base-pairing is disrupted (e.g., by stringent washes in vitro or in situ, or by enzymes in vivo).
Introduction
Nucleic acids play myriad essential roles in the cell, including storage of genetic information, regulation of genetic expression, and catalysis of chemical reactions. Synthetic nucleic acid probes are at the heart of many of the techniques used to study how the parts and circuits encoded by the genome build and sustain life, relying on the programmable chemistry of base-pairing to recognize their endogenous nucleic acid targets. In vitro and in situ, nucleic acid probes are used to identify species within a community and genotypes within a species, to detect nucleic acid–protein interactions, and to measure gene expression patterns in time and space.1−4 In vivo, synthetic nucleic acid probes enable gene knockdown, providing essential research tools for the study of endogenous genetic circuitry5,6 and suggesting the potential for powerful therapeutic interventions.7−10
An ideal nucleic acid probe would fulfill three criteria: high affinity (complementary targets are bound durably and with high yield), high selectivity (mismatched targets are not bound), and robustness (insensitivity to environmental perturbations, enabling consistent measurements across laboratories). Failure on any of these counts will lead to false negatives, false positives, or irreproducible results that can confound interpretation of data and harm patients.
Existing probe technologies are unable to meet these goals simultaneously. A crucial difficulty is the reliance on base-pairing to provide both affinity and selectivity. Increasing probe length increases affinity for both complementary and mismatched targets, thus reducing selectivity. The handling of this fundamental affinity/selectivity trade-off is central to the design and performance of nucleic acid probes.3,11 Using DNA or RNA probes, single nucleotide mismatches are not sufficiently destabilizing to enable equilibrium binding of complementary targets with high yield and mismatched targets with low yield (section S2.1 in Supporting Information [SI]).12,13 Faced with this affinity/selectivity trade-off, simultaneous pursuit of quantitative on- and off-target binding yields is abandoned, and emphasis is placed on maximizing the ‘discrimination ratio’ (the ratio of on- and off-target binding yields). The discrimination ratio has more to gain from a small denominator than from a large numerator, so selectivity is prioritized over affinity. After several decades of optimization, two different probe concepts are widely used: (1) unstructured probes are fully complementary to their targets and achieve selectivity by destabilizing the probe/complement duplex using stringent hybridization conditions, while (2) structured probes achieve selectivity by destabilizing the probe/complement duplex using internal probe base pairs (Figure 1). Both approaches have significant conceptual limitations that undermine their performance in applications.
Figure 1.

Temperature-dependent selectivity/affinity trade-off for unstructured and structured nucleic acid probes. (a) Probe and target sequences used for calculations of panels b–d. Nucleotides contributing net base pairs in green. Mismatches in orange. (b) At room temperature, the unstructured probe is not selective, binding both the complementary target (C) and the 2-nt mismatch target (M) with high affinity. The structured hairpin probe exploits internal probe base pairs to shift the binding free energies along the yield curve and exploits the large discrimination energy gap (ΔΔG) to bind C with high affinity while selectively rejecting M. (c) At elevated temperature, both probes are selective, but ΔΔG is small, requiring precise control over temperature and reducing the achievable discrimination ratio (complement yield/mismatch yield). Chemical denaturants will have a qualitatively similar effect as they similarly reduce free energy benefit per base pair. (d) The discrimination energy gap, ΔΔG, between complementary and mismatched targets decreases monotonically with increasing temperature. Free energy calculations performed using NUPACK29 ([probe] = 3 μM, [target] = 1.8 μM, [Na+] = 195 mM).
Long unstructured probes (20–500 nt) are the most common approach for chromatographic applications and for profiling gene expression on microarrays or in fixed specimens. Selectivity is achieved using elevated temperature and/or stringent buffer conditions to operate near the melting temperature of the probe/complement duplex (Figure 1b,c).2,3,14 While this approach can be effective, there are serious drawbacks. Stringent hybridization conditions intentionally weaken base pairing but also reduce the energetic penalty for mismatches, shrinking the energy gap that is the basis for discrimination between complementary and mismatched targets (Figure 1c,d). As a result, precise control over temperature and buffer conditions are required in order to avoid deviation from the melting temperature, and the maximum achievable discrimination ratio is reduced. Significantly, multiplexed experiments require the design of multiple probes that all bind marginally to their complements at the same temperature. In practice, these requirements are difficult to meet, leading to inconsistent and irreproducible results that limit the utility of microarray and in situ hybridization assays for quantitative expression profiling.3,15−20 Furthermore, use of elevated temperature or stringent buffer conditions also precludes in vivo applications.
To achieve selectivity in permissive hybridization conditions where the discrimination energy gap is maximized, it is desirable to decrease the net number of base pairs gained during probe/complement hybridization in order to lower the melting temperature of the resulting duplex. In principle, this can be accomplished by using a short unstructured probe (7–15 nt), but this approach has the fundamental flaw that the recognition sequence is then too short to uniquely address genes in the context of a genome.3,11,21 One approach to increasing the length of the recognition sequence while maintaining selectivity is to use the target to template reactions between two unstructured probes.21 Alternatively, structured probes (of which molecular beacons are the most familiar example) exploit internal base-pairing to compete with probe/complement hybridization, making it possible to adjust the trade-off between affinity and selectivity at a temperature of choice while maintaining a large recognition sequence (Figure 1b,c).22−28 Structured probes can be designed to achieve selective detection of single-nucleotide mismatches at room or physiological temperatures22,23,27,28 and are suitable for in vivo applications.24,25 However, because structured probes achieve selectivity by operating near the melting temperature of the probe/complement duplex, they are unable to stably capture their targets, precluding the use of washes which are critical to applications in vitro and in situ (e.g., removing unbound targets on a microarray or unbound probes within a fixed embryo).
A parallel thread of research has focused on the development of antisense and antigene agents based on covalent binding of an unstructured nucleic acid probe to a complementary RNA or DNA target, inhibiting expression by steric blockade. A variety of reactive groups have been developed to achieve durable covalent capture of nucleic acid targets.30−39 Stringent hybridization conditions are unavailable in vivo, but selective reactivity of the cross-linker (e.g., reactivity only with pyrimidines) can be used to augment the poor hybridization selectivity of unstructured probes.31,34,36,40 This approach applies only to mismatches located at the cross-linker’s target base and is susceptible to inadvertent covalent cross-linking of off-targets that hybridize transiently to a portion of the probe. Given the potential harm resulting from covalent capture of off-targets, the poor selectivity of unstructured probes is a major drawback for these efforts.
This article describes shielded covalent (SC) probes, a new class of probes that eliminates the longstanding trade-off between selectivity and durable target capture, combining the sequence selectivity of a structured probe with the durable target capture of a covalent probe. Figure 2a shows a schematic of the SC probe concept. One or more activatable cross-linkers are shielded within the duplex stem of a nucleic acid hairpin probe (DNA, RNA, or artificial variants). The single-stranded toehold rapidly hybridizes to potential targets, mediating a competitive branch migration process41,42 in which probe/probe base pairs are isoenergetically replaced by probe/target base pairs if the target is fully complementary to the probe sequence. Any mismatches destabilize the probe/target duplex and introduce a kinetic barrier to branch migration due to the need to break a probe/probe pair without formation of a corresponding probe/target pair. Completion of the branch migration process shields the cross-linker within the probe/target duplex and opens the hairpin loop, providing an entropic anchor to maintain binding. Activation of the cross-linker produces a covalent bond between the probe and target. Notably, the cross-linker is shielded within a duplex both before and after target hybridization, limiting the opportunity for covalent capture of biomolecules that are not hybridized to the probe. Hence, base pairing and molecular conformation change contribute to both hybridization selectivity and to cross-linking selectivity.
Figure 2.

Shielded covalent (SC) probes achieve high sequence selectivity and stable target capture at a temperature of choice. (a) Concept. High sequence selectivity is achieved at a temperature of choice via competition between internal probe base pairs and probe/target base pairs. Durable target capture is achieved via activation of one or more covalent cross-linkers which are shielded within a duplex both before and after target hybridization, limiting side reactions. Covalent bonds are stable even when base pairing is disrupted, enabling diverse applications. (b) Photoactivated cross-linker used in the current study. The vinyl bond (blue) of the 3-cyanovinylcarbazole (CNVK) nucleoside analogue undergoes [2 + 2] cycloaddition to the double bond (red) in an opposite-strand pyrimidine (T depicted) when exposed to 365 nm UV light, forming a stable photoadduct.43,44 If desired, the target can be recovered by reversing the cross-link in denaturing conditions with 311 nm UV light.
The SC probe concept is suitable for use with diverse cross-linker chemistries. The cross-linker employed in the current work is 3-cyanovinylcarbazole (CNVK; Figure 2b), a photoactive nucleoside analogue developed by Yoshimura et al.43,44 that can be incorporated into nucleic acids using standard solid-phase synthesis protocols. CNVK can be activated rapidly using low-cost UV-A light sources, and the resulting cross-links are durable and, if desired, can be reversed with UV-B light. The only sequence requirement for CNVK cross-linking is a single opposite-strand pyrimidine, allowing a wide choice of target sequences.43,44
Like conventional structured probes, SC probes can be designed to achieve high selectivity at a temperature of choice (including room temperature), but with the crucial difference that SC probes capture targets covalently so that binding persists even when base pairing is disrupted (e.g., by stringent washes in vitro or in situ, or by enzymes in vivo). Like conventional unstructured covalent probes, SC probes provide durable target capture, but with the crucial difference that SC probes employ molecular conformation change to achieve hybridization selectivity at a temperature of choice and to shield the cross-linker from undesired side reactions. Combining the advantages of structured probes and covalent cross-linking yields profound conceptual and practical advantages over existing probe chemistries for many applications.
Results
To characterize the performance of the SC probe concept, we designed a battery of experiments in which DNA or RNA probes were hybridized with complementary or mismatched DNA or RNA targets (including mixtures), and then cross-linked using UV-A light. Reactions are analyzed by denaturing gel electrophoresis to demonstrate covalent target capture. Oligonucleotide targets are fluorophore-labeled to enable quantification of capture yields by comparison of band intensities within a lane.
Near-Quantitative Discrimination of 2-Nucleotide Substitutions
Figure 3 demonstrates the performance of SC probes in discriminating complementary targets and mismatched targets containing 2-nt substitutions. For both DNA and RNA targets, SC probes with 5-nt toeholds achieve two key goals: (1) near-quantitative covalent capture of the complementary target, (2) near-quantitative rejection of the mismatched targets. As a result, discrimination ratios of 2–3 orders of magnitude are achieved. Mismatches are detected at different positions along the probe stem and for disruption of either strong (G·C) or weak (A·T/U) base pairs. By contrast, an unstructured DNA probe complementary to the same target is completely unable to discriminate the mismatches (Figure S14, SI). As expected, the probes are found in one of two states after irradiation: cross-linked to the target or internally cross-linked (section S6 in SI). The cross-links are durable in highly stringent conditions (e.g., 50% formamide at 95 °C; see section S4.1 in SI).
Figure 3.

Near-quantitative capture of complementary targets and rejection of 2-nt mismatched targets for DNA and RNA. (a) Denaturing polyacrylamide gels. Fluorescent channels: Cy5 (red) and SYBR Gold poststain (green). ssDNA ladder at left. (b) Probe, complementary target (C), and mismatched target (M1, M2, M3) sequences. Probe: 5-nt toehold in green, CNVK cross-linker in red, 6-nt hairpin loop. Targets: Mismatches in orange, thymidine cross-link partner in blue (uracil for RNA targets), Cy5 fluorophore label at the 5′-end of a 6-nt linker. (c) Cross-linking yield (mean ± standard deviation, N = 3) determined as the ratio of Cy5 fluorescence in the cross-link band to the lane total. Complementary targets are cross-linked in high yield, and mismatched targets are cross-linked in low yield, resulting in large discrimination ratios (complement yield/mismatch yield).
Efficient Discrimination of Single-Nucleotide Substitutions
Single-nucleotide polymorphisms (SNPs) are the most common type of genetic variation in the human genome and are associated with numerous disease phenotypes.45,46 They are also the most challenging mutation to discriminate with hybridization assays because they impose the smallest energetic penalties on probe/target duplex formation.3,11,28 To achieve a high discrimination ratio for SNPs, the SC probe toehold can be shortened to simultaneously achieve moderate yield in capturing complementary targets and low yield in capturing SNPs. Figure 4 shows that DNA SC probes with 3-nt toeholds efficiently reject six SNP mismatches that disrupt either strong or weak base pair stacks, while covalently capturing the complementary DNA target with ∼35% yield. The resulting discrimination ratios are 1–2 orders of magnitude (median 90). Due to the greater energetic benefit of RNA/RNA base pairs, the same 5-nt toehold RNA probe used to achieve near-quantitative discrimination of 2-nt mismatches (Figure 3) also discriminates RNA SNPs efficiently (median 20), with the exception of A-to-G substitutions. This exception is not surprising, as the resulting G·U wobble pair is predicted to be nearly isoenergetic with the A·U pair it replaces.12 By reducing the probe toehold to 4 nt (section S6.4 in SI), it is possible to efficiently reject even these SNPs, at the cost of reduced capture yield for the complementary target (∼12%). With this probe, the resulting discrimination ratios for seven RNA SNPs range from 7 to 49 (median 31; Figure S16 in SI). Notably, two of these mismatched targets contain only RNA wobble SNPs.
Figure 4.

Efficient discrimination of single-nucleotide substitutions for DNA and RNA. (a) Denaturing polyacrylamide gels. Fluorescent channels: Cy5 (red) and SYBR Gold poststain (green). ssDNA ladder at left. (b) Probe, complementary target (C), and mismatched target (S1, S2, S3, ...) sequences, color coded as in Figure 3. (c) Cross-linking yields (mean ± standard deviation, N = 3). See section S6.4 in SI for enhanced discrimination of RNA wobble mismatches (S5 and S6) with a 4-nt toehold probe.
Selective Capture in a Large Pool of Mismatched Targets
For most applications, SC probes must selectively capture the complementary target within a large and diverse pool of mismatched targets. Figure 5 demonstrates SC probe performance in capturing a complementary DNA or RNA target within a pool of 96 mismatched targets (24 1-nt mismatches and 72 2-nt mismatches) when all of the targets are at approximately the same concentration. In this needle-in-a-haystack situation, where the complementary target is outnumbered by ∼100:1, SC probes covalently capture the complementary target with high yield and efficiently reject mismatched targets.
Figure 5.

Selective capture of a complementary DNA or RNA target in a pool containing 24 1-nt and 72 2-nt mismatched targets. (a) Denaturing polyacrylamide gel. Fluorescent channels: 6-FAM (blue) and Cy5 (red). Probe at 3 μM, each target at ∼30 nM. See section S6.5 in SI for additional probe toehold length studies and examination of the unexpected band splitting observed for the complementary RNA target. (b) Probe, complementary target (C), and mismatched target (P1–P8) sequences, color coded as in Figure 3 (4-nt toehold probe for DNA, 5-nt toehold probe for RNA). Targets: fluorophore label at the 5′-end of a 6-nt linker (6-FAM for complement, Cy5 for mismatches), IUB nucleotide codes (N = A, C, G, or T; D = A, G or T; H = A, C or T; V = A, C or G). (c) Cross-linking yields (mean ± standard deviation, N = 3).
Kinetic vs Thermodynamic Discrimination
We wished to determine the physical basis for the high discrimination ratios observed in these studies. One hypothesis is that discrimination is kinetic, with the mismatch creating an energetic barrier that blocks completion of branch migration, preventing the cross-linker from coming into contact with the target. If this hypothesis is correct, mismatch discrimination should be independent of probe toehold length, provided that the toehold is long enough for initial binding to occur. Accordingly, we synthesized a series of DNA probes with different toehold lengths and measured their cross-linking yield with both complementary and 2-nt mismatch DNA targets. As shown in Figure 6a, the data appear inconsistent with the kinetic discrimination hypothesis: mismatch targets are cross-linked with substantial yield by probes with long toeholds. Efficient rejection of mismatches on the loop side of the cross-linker (Figure 3, target M3; Figure 5, target pool P7) provides additional evidence that appears inconsistent with the kinetic barrier hypothesis.
Figure 6.
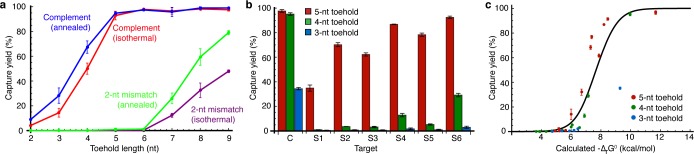
Kinetic vs thermodynamic discrimination for DNA probes and targets. Capture yields are mean ± standard deviation, N = 3. (a) Cross-linking yield vs probe toehold length for complementary and 2-nt mismatch targets (C and M1 from Figure 3). When the toehold becomes sufficiently long, the mismatch is cross-linked with substantial yield, indicating that this 2-nt substitution does not produce a prohibitive kinetic barrier to branch migration. (b) Cross-linking yield for complementary and SNP targets (from Figure 4) using SC probes with 3-, 4-, and 5-nt toeholds. The pattern of yields is the same across toehold lengths, and the mismatch yields fall more rapidly than the complement yield as the toehold is shortened. (c) Cross-linking yield vs calculated reaction free energies for probe/target hybridization. Probes and targets from Figure 6b plus five 2-nt mismatched targets with 4- and 5-nt toehold probes (section S6.6 in SI). Data cluster around the predicted yield curve for two-state equilibrium (black line).
If 2-nt mismatches do not produce a large kinetic barrier to branch migration, an alternative hypothesis is that the probe/target interaction rapidly reaches equilibrium, and that the yield is therefore determined by the thermodynamics of probe/target hybridization. The toehold data of Figure 6a appear consistent with this hypothesis: longer toeholds compensate for the mismatch to allow high-yield cross-linking. Our DNA SNP detection data (Figure 6b) are also consistent with the thermodynamic hypothesis: discrimination improves as the toehold is shortened, with the pattern of yields remaining constant across SNPs. Figure 6c plots measured cross-linking yields vs calculated reaction free energy changes based on nearest-neighbor thermodynamic parameters.13,29CNVK is expected to destabilize hybridization,43,44 but the sequence context is similar in the SC probe stem and in the probe/target duplex, and therefore this destabilization is not expected to significantly alter the hybridization yield. Data are shown for DNA SC probes detecting complementary and mismatched targets with 1- and 2-nt substitutions. The data cluster around the sigmoidal curve predicted for two-state equilibrium (section S2.1 in SI), suggesting that for these sequences and toehold lengths, mismatch discrimination is dominated by the thermodynamics of probe/target hybridization, rather than by a kinetic barrier to branch migration.
Annealing (heating followed by slow cooling) is commonly used to promote equilibration of nucleic acid systems.47,48 Anneals involving hairpins must be interpreted with caution because intramolecular base pairing can cause kinetic traps to form during cooling, preventing equilibration.49,50 To test for equilibration of probe/target hybridization, we annealed probes and targets together prior to cross-linking. Across a range of toehold lengths, and for both complementary and 2-nt mismatch targets, annealing produced yield patterns similar to the isothermal case (Figure 6a), though with somewhat higher capture yields for data that were not already saturated at high yield. These results may indicate that probe/target hybridization has not fully equilibrated in the isothermal experiments, or alternatively, that annealing does not relax the system to equilibrium. Further study is warranted.
Selective Target Capture from a Mixture of Closely Related Fluorescent Protein Sequences
The results of Figures 3 and 6c suggest that, for probe concentrations in the micromolar range, high-yield cross-linking of the complementary DNA target and low-yield cross-linking of 2-nt (and larger) mismatches can be achieved simultaneously by designing probes to bind the complementary target with a calculated reaction free energy change of approximately −12 kcal/mol (section S2.3 in SI). We tested the utility of this design criterion by designing a pair of DNA probes (section S7 in SI) to selectively hybridize to one of a pair of targets drawn from the DsRed2 and mCherry red fluorescent protein sequences;51,52 these targets differ by only a 2-nt substitution. Probe sequences and target sequences are shown in Figure 7, together with results demonstrating selective capture of one target or the other from a mixture of the two targets. Each probe captures its complementary target and rejects its mismatched target with near-quantitative yield, achieving a discrimination ratio of approximately 3 orders of magnitude for both DNA and RNA targets.
Figure 7.
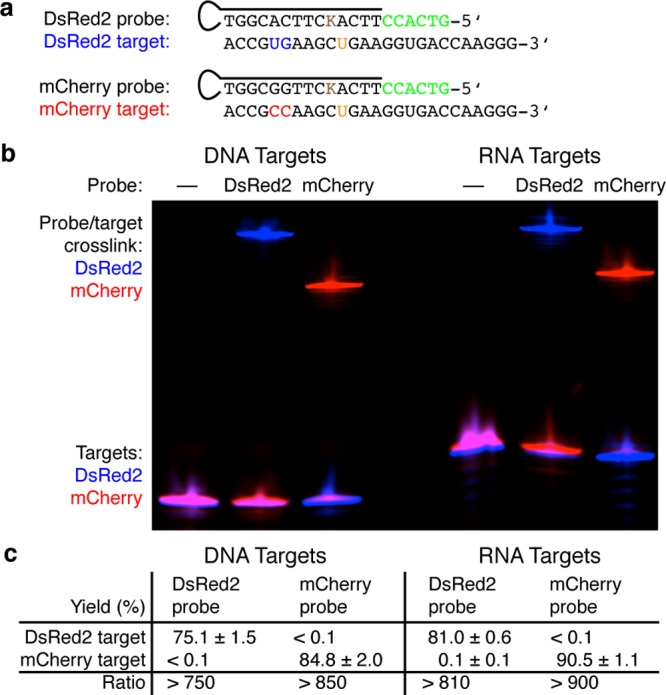
Near-quantitative selective target capture from a mixture of closely related fluorescent protein sequences. (a) Probe and target sequences. Probes: toehold in green, CNVK cross-linker in brown, DNA probes used for both DNA and RNA targets. Targets: mismatches in blue and red, cross-link partner in orange, 5′-fluorophore-labeled (6-FAM for DsRed2, Cy5 for mCherry). (b) Denaturing polyacrylamide gel. Fluorescent channels: 6-FAM (blue) and Cy5 (red). (c) Cross-linking yields (mean ± standard deviation, N = 3). See sections S7.4 and S7.5 in SI for additional demonstrations of selective target capture in mixtures of fluorescent protein sequences.
Selective Cross-Linking of Full-Length Fluorescent Protein mRNAs
We wished next to examine whether these SC probes could selectively capture the corresponding full-length mRNA targets. The switch to long targets required three changes. First, because cross-linking to a long target does not cause a discernible gel mobility shift, the target-labeling scheme that we employed for short targets in Figures 3–7 is not applicable here. Instead, we monitor probe/target cross-linking by observing depletion of the cross-linked probe band after irradiation in the presence of an excess of target mRNA. Second, to reduce the quantity of mRNA needed for these studies, we operated at lower probe concentrations than for Figure 7, necessitating a 1-nt increase in the length of the SC probe toehold. Third, to make the target sites accessible for binding, it was necessary to include auxiliary oligonucleotides (so-called ‘helper strands’) that hybridize to the regions flanking the target sites. Figure 8 demonstrates that DsRed2 and mCherry DNA SC probes targeting either short RNAs or full-length mRNAs selectively capture their cognate targets with high yield, while efficiently rejecting their noncognate targets containing 2-nt mismatches. Within the context of this semiquantitative assay (a property following from the need to compare bands between lanes rather than within each lane; see section S5 in SI), this selective capture property is indistinguishable for full-length mRNA targets and the short RNA targets that were studied quantitatively in Figure 7.
Figure 8.
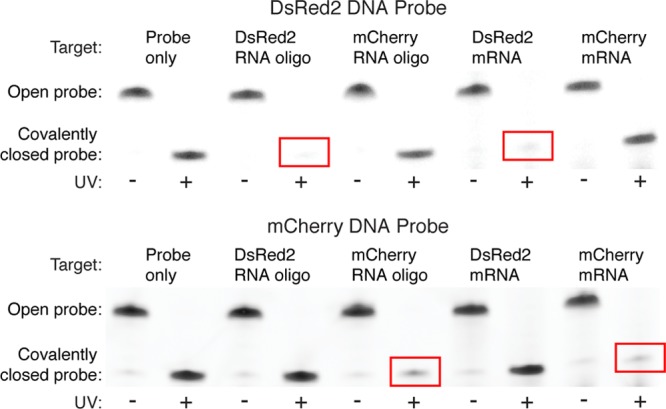
Selective cross-linking of full-length mRNA targets. DsRed2 and mCherry DNA SC probes target full-length DsRed2 and mCherry mRNAs at sites differing by only a 2-nt substitution (same target sites as Figure 7a, 7-nt toehold probes). Denaturing polyacrylamide gel poststained with SYBR Gold. Probes are open in (−UV) lanes and covalently closed in (+UV) lanes. The covalently closed probe band is depleted only for complementary targets (red boxes). To combat native secondary structure in mRNA targets, each SC probe is accompanied by RNA helper strands complementary to the sequences flanking the intended target site.
Sensitive and Selective Capture As Target Concentration Is Decreased
For many applications, target concentration will not be under experimental control and will often be significantly lower than the probe concentration. Hence, sensitivity is an important aspect of probe performance. Ideally, the capture yields for complementary and mismatched targets would be independent of target concentration. Because the data of Figure 6 suggest that our covalent capture yields are consistent with the thermodynamics of probe/target hybridization, we examine equilibrium theory in setting expectations for probe sensitivity. Equilibrium hybridization calculations predict that for any given probe/target pair, the hybridization yield asymptotes to a constant from below as the target concentration is decreased at fixed probe concentration (section S2.2 in SI). To test this prediction, we measured the cross-link yields for the complementary and 2-nt mismatched DNA targets of Figure 3 across 3 orders of magnitude in target concentration (section S6.10 in SI). Consistent with equilibrium theory, there is no observed degradation in SC probe performance as the target concentration decreases, with near-quantitative capture of the complement and rejection of the mismatch observed down to 1 nM (the sensitivity limit of our gel-based assay using fluorophore-labeled targets). To read out the signal at lower target concentrations, selective target capture with an SC probe could be followed by postcapture signal amplification (e.g., using PCR, catalytic reporter deposition,14 or HCR53).
Photoreversal of Probe/Target Cross-Links
Many applications of nucleic acid probes require that targets are first captured and then released once other materials have been washed away. For covalent probes, target release requires reversal of the probe/target cross-link. Yoshimura et al. have shown that CNVK cross-links between single-stranded oligonucleotides can be reversed by high-intensity UV-B irradiation.43,44 We observe efficient reversal of SC probe/target cross-links by low-intensity UV-B irradiation (99% after a 20 min exposure with 8 mW/cm2 at 311 nm; Figure S7 in SI). Interestingly, we find that efficient target recovery requires irradiation in denaturing conditions, and that the SC probes are found in a mix of covalently closed and open states after exposure, implying that irradiation at this wavelength drives both the forward (cross-linking) and reverse reactions (section S4.4 in SI). Subsequent investigations revealed that 254 and 365 nm light also drive both the forward and reverse reactions, with the forward reaction more favored at longer wavelengths (section S4.5 in SI). This phenomenon, which is not observed for psoralen,54 a widely used [2 + 2] photoactive cross-linker, has significant implications for applications utilizing CNVK, and to our knowledge has not been reported in the literature.
Discussion
Shielded covalent probes combine the three key ingredients of programmable base pairing, molecular conformation change, and activatable covalent cross-linking to simultaneously achieve high sequence selectivity and durable target capture at a temperature of choice. Existing probe concepts lack at least one of these three key ingredients, undermining the selectivity, stability, or robustness of target binding.
The data presented here show that SC probes covalently capture their complementary DNA or RNA targets with near-quantitative yield (75–97%) while achieving near-quantitative rejection of mismatched targets containing 2-nt substitutions (<1%). The resulting discrimination ratios are 2–3 orders of magnitude. These results are obtained for mismatches at a variety of locations along the probe stem that disrupt either weak or strong base pairs, eliminating the need to design probes for specific mismatches.
SC probes are also capable of discriminating single-nucleotide substitutions, the most difficult type of mutations to detect, at the cost of a modest reduction in capture yield for the complementary target. For either DNA or RNA targets, SNPs disrupting either strong or weak base pairs (including those leading to G·U wobbles) are efficiently discriminated at different locations along the probe stem. For DNA targets, discrimination ratios of 1–2 orders of magnitude are achieved (median 90). This is roughly an order of magnitude better than unstructured allele-specific microarray probes18,20,55 and compares favorably with the discrimination ratios of recent structured probe designs (ref (27): median 13; ref (28): median 26). Crucially, unlike these earlier methods, SC probes capture their targets covalently, achieving binding that is not only selective but also stable.
Historically, a great deal of emphasis has been placed on discriminating SNPs, leading to diverse probe technologies that produce nonquantitative yields for complementary targets.18−20,26−28,34,37,55,56 For quantitative expression profiling studies focused on discriminating genes within a genome, where 1-nt resolution is not necessarily needed, we believe it is highly significant that SC probes enable robust, near-quantitative capture of complementary targets and near-quantitative rejection of 2-nt mismatches, providing a powerful framework for genome-wide analysis. Studies with large pools of DNA or RNA mismatched targets demonstrate that SC probes efficiently capture complementary targets and reject mismatched targets when the complementary targets are greatly outnumbered.
It was not clear a priori whether SC probes would discriminate mismatches based on the kinetics or thermodynamics of probe/target hybridization. Although 1-nt mismatches have been shown to introduce a substantial barrier to 4-way DNA branch migration in Holliday junctions,57,58 the literature is conflicted as to their effect on the kinetics of 3-way branch migration (which is central to SC probe/target hybridization). Studies have reported that 1-nt mismatches effectively block 3-way DNA branch migration,41,56 have little effect on 3-way DNA branch migration kinetics,57 or sometimes slow 3-way DNA branch migration.23 Our data suggest that even 2-nt mismatches do not produce a large kinetic barrier to 3-way DNA branch migration. Given the importance of this process to nucleic acid nanotechnology,59 further study of the effect of mismatches on 3-way branch migration kinetics is warranted; by trapping kinetically accessible states, SC probes provide a convenient tool for examining this phenomenon.
For our probe and target sequences, mismatch discrimination appears to be governed primarily by the thermodynamics of probe/target hybridization. The nearest-neighbor model of DNA secondary structure thermodynamics13,29 usefully predicted capture yields, providing a basis for rational SC probe design, as demonstrated for two pairs of target sequences for closely related fluorescent proteins. The corresponding RNA model12 should also provide a useful guide for design, though salt corrections are needed for direct comparison to experimental data. The length of the toehold (or, likely, the size of the loop) can be adjusted to overcome native secondary structure or to optimize yield or sequence selectivity. For long targets, helper strands complementary to the regions flanking the target site can be used to combat native secondary structure in the desired target. Because helper strands will usually base pair imperfectly to off-targets, they also have the potential to augment SC probe selectivity.
SC probes are unique in providing both high sequence selectivity and covalent target capture. This combination of properties is desirable for numerous applications, including profiling genetic expression in vitro, mapping genetic expression in situ, and regulating genetic expression in vivo. The ability to form covalent bonds with targets is particularly valuable in the common situation in which unwanted material including surplus probes or targets must be washed away while preserving genuine probe/target interactions. Currently, washes are often performed by adjusting stringency, but without a covalent link between the probe and target, this process remains subject to the same affinity/selectivity trade-off as the initial detection step. The covalent link afforded by SC probes should dramatically improve and simplify such assays. The ability to photoreverse the cross-link and recover the target after washing will be valuable for chromatographic applications such as target enrichment for high-throughput sequencing60,61 or pulldown of specific RNAs or RNA/protein complexes.4 To date, nucleic acid cross-linkers developed for antisense and antigene applications30,32,33,37−39 have been studied in the context of unstructured probes that lack the sequence selectivity of structured probes. SC probes offer a promising conceptual framework for covalent inhibition of genetic expression while minimizing off-target effects. While SC probes employing photoactivated cross-linkers are conceptually suited for use in photoaccessible tissues (e.g., cultured cells, small model organisms, or shallow tissue in larger organisms), the capture of targets deep within opaque tissue would require development of SC probes that employ conformation-activated cross-linkers. SC probes may prove useful in multiple areas of nucleic acid nanotechnology, including as (optionally reversible) covalent elements for structural engineering62 and strand displacement cascades.59
This article has introduced the concept of shielded covalent probes and explored their performance using the photoactivated CNVK cross-linker. Significant work will be required to realize SC probes’ full potential. Depending on the application, probes may need to be immobilized on solid surfaces or introduced into fixed samples or living cells. It also may be desirable to include materials other than DNA or RNA (e.g., 2′-OMe RNA, LNA, PNA) in the probes to improve their performance further, or to employ different cross-linker chemistries. On the basis of these initial studies, it appears that the shielded covalent probe concept offers a powerful new tool for exploring the many essential functions of nucleic acids in biology.
Methods Summary
Target and Probe Sequences
Sequences of the oligonucleotides used in this article are listed in section S1 in SI, as well as a discussion of how they were designed.
Oligonucleotide Synthesis and Preparation
HPLC-purified oligonucleotide targets were purchased from Integrated DNA Technologies and used without further purification. SC probes were synthesized using standard solid-phase protocols and purified by RP-HPLC (see section S3 in SI for details). After quantification by UV absorbance, the probes were stored in opaque microtubes to prevent activation by ambient light.
mRNA Transcription
mRNA targets were produced by in vitro transcription using MegaScript kits (Invitrogen) as directed by the manufacturer. The DsRed2 template was created by linearizing pTNT-DsRed2 plasmid with NotI and transcribed with T7 polymerase. The mCherry template was created by linearizing pCS2+mCherry:H2B (a plasmid containing an mCherry-human histone H2B fusion) with KpnI and transcribed with SP6 polymerase.
Probe Hybridization and Cross-Linking
Hybridization and cross-linking assays were performed in SSC buffer (150 mM NaCl, 15 mM trisodium citrate, pH 7.0) with probes at 3 μM and the concentration of each target as follows: 1.8 μM for the single-target studies of Figures 3, 4, and 6; ∼30 nM for the pool studies of Figure 5; 1.5 μM for the mixture studies of Figure 7. For all experiments except the anneals in Figure 6a, probes in buffer were annealed separately (5 min at 95 °C for DNA probes, 10 min at 70 °C for RNA, followed by cooling to RT over 35 min in the dark). Targets were then added and the reactions incubated at 22 °C for 35 min. For the anneal experiments, the probes and targets were annealed together as above, and the incubation step was omitted. A portion of each reaction was individually irradiated for 1 min with a 365 nm LED (LED-100, Electro-Lite) at 4 °C in a clear 96-well microplate. Alternative activation procedures are detailed in section S4 in SI. For mRNA targets (Figure 8), SC probes (0.5 μM) were heated (65 °C, 15 min) with targets (1 μM) and 30-nt RNA helper strands (2 μM; complementary to the regions flanking the SC probe binding site) in SSC buffer and incubated at RT for 3 h, then irradiated on glass coverslips for 1 min.
Analysis
Cross-linking reactions were analyzed using two methods. All reactions were analyzed by denaturing polyacrylamide gel electrophoresis and imaged with a Fujifilm FLA-5100 fluorescence scanner. Additionally, some reactions were analyzed by HPLC in order to confirm the values obtained from the gels. In all cases, the HPLC and gel cross-linking yields agreed closely. Experimental details for both methods and discussion of uncertainties can be found in section S5 in SI.
Photoreversal of Cross-Links
For the photoreversal experiments, 1.2 and 1 nmol of probe and complementary target were hybridized and cross-linked as described above. Acetonitrile and urea were then added to final concentrations of 50% and 2 M. The reaction was irradiated at RT in a clear microplate using a small lamp fitted with two Philips PL-S 9 W narrowband UVB bulbs. The nominal output of these bulbs is 1.2 W with a narrow spectrum centered at 311 nm; we measured an irradiance of 8 mW/cm2 at the reaction position using a UVX-31 photometer (UVP). Aliquots were removed at various time points, and the yields were determined as for the cross-linking reactions.
Acknowledgments
We thank Dr. Peng Yin for discussions, Victoria Hsiao for performing additional characterizations of photoreversal conditions, Dr. Joshua Day for synthesis of alternative photocross-linkers, Dr. Le Trinh for providing plasmid pCS2+mCherry:H2B and Maayan Schwarzkopf for providing plasmid pTNT-DsRed2. This work was funded by NIH 5R01CA140759, NIH P50 HG004071, the Caltech Innovation Initiative, the Caltech Programmable Molecular Technology Initiative via Grant GBMF2809 from the Gordon and Betty Moore Foundation, an NSF Graduate Research Fellowship (H.M.N.), and a Ford Foundation Predoctoral Fellowship (H.M.N.).
Supporting Information Available
Additional details on modeling, materials, methods, and data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Stoughton R. B. Annu. Rev. Biochem. 2005, 74, 53–82. [DOI] [PubMed] [Google Scholar]
- Kreil D. P.; Russell R. R.; Russell S. Methods Enzymol. 2006, 410, 73–98. [DOI] [PubMed] [Google Scholar]
- Silverman A. P.; Kool E. T. Adv. Clin. Chem. 2007, 43, 79–115. [PubMed] [Google Scholar]
- Hegarat N.; Francois J. C.; Praseuth D. Biochimie 2008, 90, 1265–1272. [DOI] [PubMed] [Google Scholar]
- Voorhoeve P.; Agami R. Trends Biotechnol. 2003, 21, 2–4. [DOI] [PubMed] [Google Scholar]
- Kim D. H.; Rossi J. J. BioTechniques 2008, 44, 613–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. H.; Rossi J. J. Nat. Rev. Genet. 2007, 8, 173–184. [DOI] [PubMed] [Google Scholar]
- Aagaard L.; Rossi J. J. Adv. Drug Delivery Rev. 2007, 59, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanotto D.; Rossi J. J. Nature 2009, 457, 426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett C. F.; Swayze E. E. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [DOI] [PubMed] [Google Scholar]
- Demidov V.; Frank-Kamenetskii M. Trends Biochem. Sci. 2004, 29, 62–71. [DOI] [PubMed] [Google Scholar]
- Mathews D. H.; Sabina J.; Zuker M.; Turner D. H. J. Mol. Biol. 1999, 288, 911–940. [DOI] [PubMed] [Google Scholar]
- SantaLucia J. Jr.; Hicks D. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 415–440. [DOI] [PubMed] [Google Scholar]
- Qian X.; Jin L.; Lloyd R. V. J. Histotechnol. 2004, 27, 53–67. [Google Scholar]
- Marshall E. Science 2004, 306, 630–631. [DOI] [PubMed] [Google Scholar]
- Yilmaz L. S.; Noguera D. R. Appl. Environ. Microbiol. 2004, 70, 7126–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salit M. In DNA Microarrays, Part B: Databases and Statistics; Kimmel A., Oliver B., Eds.; Methods in Enzymology, Vol. 411; 2006; pp 63–78. [DOI] [PubMed] [Google Scholar]
- Suzuki S.; Ono N.; Furusawa C.; Kashiwagi A.; Yomo T. BMC Genomics 2007, 8, 373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder H.; Fasold M.; Glomb T. PLoS One 2009, 4, e7862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan F.; Pauley M. A.; Spindel E. R.; Zhang L.; Norgren R. B. Jr. BioData Mining 2010, 3, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman A. P.; Kool E. T. Chem. Rev. 2006, 106, 3775–3789. [DOI] [PubMed] [Google Scholar]
- Bonnet G.; Tyagi S.; Libchaber A.; Kramer F. R. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 6171–6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q. Q.; Luan G. Y.; Guo Q. P.; Liang J. X. Nucleic Acids Res. 2002, 30, e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratu D. P.; Cha B. J.; Mhlanga M. M.; Kramer F. R.; Tyagi S. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 13308–13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santangelo P.; Nitin N.; Bao G. Ann. Biomed. Eng. 2006, 34, 39–50. [DOI] [PubMed] [Google Scholar]
- Narita A.; Ogawa K.; Sando S.; Aoyama Y. Nat. Protoc. 2007, 2, 1105–1116. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Plakos K. J. I.; Lou X. H.; White R. J.; Qian J. R.; Plaxco K. W.; Soh H. T. Angew. Chem., Int. Ed. 2009, 48, 4354–4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D. Y.; Chen S. X.; Yin P. Nat. Chem. 2012, 4, 208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadeh J. N.; Steenberg C. D.; Bois J. S.; Wolfe B. R.; Pierce M. B.; Khan A. R.; Dirks R. M.; Pierce N. A. J. Comput. Chem. 2011, 32, 170–173. [DOI] [PubMed] [Google Scholar]
- Knorre D. G.; Vlassov V. V. Prog. Nucleic Acid Res. Mol. Biol. 1985, 32, 291–320. [DOI] [PubMed] [Google Scholar]
- Coleman R. S.; Pires R. M. Nucleic Acids Res. 1997, 25, 4771–4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrer O. D. ChemBioChem 2005, 6, 27–32. [DOI] [PubMed] [Google Scholar]
- Sasaki S.; Nagatsugi F. Curr. Opin. Chem. Biol. 2006, 10, 615–621. [DOI] [PubMed] [Google Scholar]
- Peng X. H.; Greenberg M. M. Nucleic Acids Res. 2008, 36, e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M. E.; Christian E. L. Methods Enzymol. 2009, 468, 127–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori K.; Hirohama T.; Imoto S.; Kusano S.; Nagatsugi F. Chem. Commun. 2009, 6463–6465. [DOI] [PubMed] [Google Scholar]
- Higuchi M.; Kobori A.; Yamayoshi A.; Murakami A. Bioorg. Med. Chem. 2009, 17, 475–483. [DOI] [PubMed] [Google Scholar]
- Stevens K.; Madder A. Nucleic Acids Res. 2009, 37, 1555–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatsugi F.; Imoto S. Org. Biomol. Chem. 2011, 9, 2579–2585. [DOI] [PubMed] [Google Scholar]
- Shigeno A.; Sakamoto T.; Yoshimura Y.; Fujimoto K. Org. Biomol. Chem. 2012, 10, 7820–7825. [DOI] [PubMed] [Google Scholar]
- Quartin R. S.; Plewinska M.; Wetmur J. G. Biochemistry 1989, 28, 8676–8682. [DOI] [PubMed] [Google Scholar]
- Yurke B.; Turberfield A. J.; Mills A. P. Jr.; Simmel F. C.; Neumann J. L. Nature 2000, 406, 605–608. [DOI] [PubMed] [Google Scholar]
- Yoshimura Y.; Fujimoto K. Org. Lett. 2008, 10, 3227–3230. [DOI] [PubMed] [Google Scholar]
- Yoshimura Y.; Ohtake T.; Okada H.; Fujimoto K. ChemBioChem 2009, 10, 1473–1476. [DOI] [PubMed] [Google Scholar]
- Donnelly P. Nature 2008, 456, 728–731. [DOI] [PubMed] [Google Scholar]
- Altshuler D. M.; Gibbs R. A.; Peltonen L.; Dermitzakis E.; Schaffner S. F.; Yu F. L.; Bonnen P. E.; de Bakker P. I. W.; Deloukas P.; Gabriel S. B.; Gwilliam R.; Hunt S.; Inouye M.; Jia X. M.; Palotie A.; Parkin M.; Whittaker P.; Chang K.; Hawes A.; Lewis L. R.; Ren Y. R.; Wheeler D.; Muzny D. M.; Barnes C.; Darvishi K.; Hurles M.; Korn J. M.; Kristiansson K.; Lee C.; McCarroll S. A.; Nemesh J.; Keinan A.; Montgomery S. B.; Pollack S.; Price A. L.; Soranzo N.; Gonzaga-Jauregui C.; Anttila V.; Brodeur W.; Daly M. J.; Leslie S.; McVean G.; Moutsianas L.; Nguyen H.; Zhang Q. R.; Ghori M. J. R.; McGinnis R.; McLaren W.; Takeuchi F.; Grossman S. R.; Shlyakhter I.; Hostetter E. B.; Sabeti P. C.; Adebamowo C. A.; Foster M. W.; Gordon D. R.; Licinio J.; Manca M. C.; Marshall P. A.; Matsuda I.; Ngare D.; Wang V. O.; Reddy D.; Rotimi C. N.; Royal C. D.; Sharp R. R.; Zeng C. Q.; Brooks L. D.; McEwen J. E. Nature 2010, 467, 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih W. M.; Quispe J. D.; Joyce G. F. Nature 2004, 427, 618–621. [DOI] [PubMed] [Google Scholar]
- Rothemund P. W. K. Nature 2006, 440, 297–302. [DOI] [PubMed] [Google Scholar]
- Bois J. S.Analysis of Interacting Nucleic Acids in Dilute Solutions. Ph.D. Thesis, California Institute of Technology, 2007. [Google Scholar]
- Yin P.; Choi H. M. T.; Calvert C. R.; Pierce N. A. Nature 2008, 451, 318–322. [DOI] [PubMed] [Google Scholar]
- Matz M. V.; Fradkov A. F.; Labas Y. A.; Savitsky A. P.; Zaraisky A. G.; Markelov M. L.; Lukyanov S. A. Nat. Biotechnol. 1999, 17, 969–973. [DOI] [PubMed] [Google Scholar]
- Shaner N. C.; Campbell R. E.; Steinbach P. A.; Giepmans B. N. G.; Palmer A. E.; Tsien R. Y. Nat. Biotechnol. 2004, 22, 1567–1572. [DOI] [PubMed] [Google Scholar]
- Choi H. M. T.; Chang J.; Trinh L. A.; Padilla J.; Fraser S. E.; Pierce N. A. Nat. Biotechnol. 2010, 28, 1208–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearst J. Annu. Rev. Biophys. Bioeng. 1981, 10, 69–86. [DOI] [PubMed] [Google Scholar]
- Naiser T.; Ehler O.; Kayser J.; Mai T.; Michel W.; Ott A. BMC Biotechnol. 2008, 8, 48–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian H. K. K.; Chakraborty B.; Sha R.; Seeman N. C. Nano Lett. 2011, 11, 910–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panyutin I. G.; Hsieh P. J. Mol. Biol. 1993, 230, 413–424. [DOI] [PubMed] [Google Scholar]
- Biswas I.; Yamamoto A.; Hsieh P. J. Mol. Biol. 1998, 279, 795–806. [DOI] [PubMed] [Google Scholar]
- Zhang D. Y.; Seelig G. Nat. Chem. 2011, 3, 103–113. [DOI] [PubMed] [Google Scholar]
- Summerer D. Genomics 2009, 94, 363–368. [DOI] [PubMed] [Google Scholar]
- Mamanova L.; Coffey A. J.; Scott C. E.; Kozarewa I.; Turner E. H.; Kumar A.; Howard E.; Shendure J.; Turner D. J. Nat. Methods 2010, 7, 111–118. [DOI] [PubMed] [Google Scholar]
- Pinheiro A. V.; Han D. R.; Shih W. M.; Yan H. Nat. Nanotechnol. 2011, 6, 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.