

Abstract
We previously adapted the β-elimination/Michael addition chemistry to solid-phase derivatization on reversed-phase supports, and demonstrated the utility of this reaction format to prepare phosphoseryl peptides in unfractionated protein digests for mass spectrometric identification and facile phosphorylation-site determination. Here, we have expanded the use of this technique to β-N-acetylglucosamine peptides, modified at serine/threonine, phosphothreonyl peptides, and phosphoseryl/phosphothreonyl peptides, followed in sequence by proline. The consecutive β-elimination with Michael addition was adapted to optimize the solid-phase reaction conditions for throughput and completeness of derivatization. The analyte remained intact during derivatization and was recovered efficiently from the silica-based, reversed-phase support with minimal sample loss. The general use of the solid-phase approach for enzymatic dephosphorylation was demonstrated with phosphoseryl and phosphothreonyl peptides and was used as an orthogonal method to confirm the identity of phosphopeptides in proteolytic mixtures. The solid-phase approach proved highly suitable to prepare substrates from low-level amounts of protein digests for phosphorylation-site determination by chemical-targeted proteolysis. The solid-phase protocol provides for a simple, robust, and efficient tool to prepare samples for phosphopeptide identification in MALDI mass maps of unfractionated protein digests, using standard equipment available in most biological laboratories. The use of a solid-phase analytical platform is expected to be readily expanded to prepare digest from O-glycosylated- and O-sulfonated proteins for mass spectrometry-based structural characterization.
Keywords: phosphoprotein characterization, serial solid-phase derivatization, ZipTipC18 pipette tips, O-GlcNAcylation, alkaline phosphatase, chemical-targeted proteolysis
INTRODUCTION
Protein phosphorylation plays a major role in modulation of cellular processes, including cellular signaling, cell-cycle progression, and differentiation.1 Existing approaches to map this important post-translational modification often rely on mass spectrometry (MS) methods to identify and sequence the peptide of interest.2–5 MALDI-TOF-MS has been an early method to detect phosphopeptides in protein digests by monitoring the characteristic mass shift after alkaline phosphatase (AP) treatment.5 However, these methods are often challenged by the ionization inefficiency of the phosphopeptides and the limited information content of the tandem MS (MS/MS) spectra as a result of the intrinsic lability of the phosphate group upon collisionally induced dissociation (CID). Titanium dioxide chromatography is a frequently reported technique to ameliorate these problems by phosphopeptide enrichment, but this method exhibits a bias toward selection of monophosphorylated species.6 Additionally, peptides containing multiple basic residues are poorly retained by this support, rendering this method less suitable to prepare samples for structural characterization of peptides from basophilic kinase substrates. Although the above strategies afford improved phosphopeptide detection, issues still persist with regard to neutral loss of phosphate. Modified fragmentation regimes have been introduced to alleviate this problem. Neutral loss-triggered MS/MS, designated as MS,3 has been shown to generate more backbone cleavages that may improve the peptide sequence information content and thus, aid in phosphorylation-site determination.7 Electron transfer dissociation (ETD) has shown promise, as the phosphate group remains relatively stable during fragmentation. ETD has been informative for large, multiply protonated peptides and is often used in combination with CID in a complementary fashion.8 Furthermore, the more recent use of the higher-energy collisionally dissociation (HCD) cells, exhibiting lower levels of neutral loss, has facilitated phosphorylation-site localization.9
The substitution of the phosphate by a nucleophile via the Michael addition has emerged as a viable chemical strategy to preclude neutral loss as the primary path of low-energy, collisionally induced fragmentation.10,11 One approach advocates the use of ethanethiol as nucleophile for concurrent β-elimination/Michael addition.10 The derivatization proved to enhance ionization of the modified peptides during liquid chromatography (LC)/MS analysis and afforded improved MS/MS spectral information. As applied to digests of PKA, this advantage has been exploited to map comprehensively the phosphorylation sites of the protein in a single LC-MS/MS experiment.11 Subsequent studies focused on efforts to make this chemistry compatible with high-sensitivity proteomics applications, using barium hydroxide as elimination reagent and 2-aminoethanethiol as nucleophile, allowing the reactions to proceed under mild conditions in pure aqueous solutions. The S-2-aminoethylcysteine derivatives sequenced upon CID appreciably more informative than their native counterparts, thus enabling facile phosphorylation-site determination.12 Furthermore, the Michael addition products exhibited minimal peak splitting (as a result of diastereomer formation) during LC-MS/MS applications.13 An alternative approach to phosphorylation-site determination exploits the finding that S-2-aminoethylcysteine and β-methyl- S-2-aminoethylcysteine derivatives are recognized by lysine endopeptidase C (Lys-C) or trypsin as structural analogs of lysine. In this strategy, termed chemical targeted proteolysis, the phosphorylation site is determined by in silico mass, matching the predicted cleavage products to the known protein sequence.14,15 With these developments, the chemistry became amenable to solid-phase adaptation, as demonstrated earlier, using ZipTipC18 pipette tips as reaction vessels for the in situ derivatization.16,17 This technique exploits the advantages inherent in ZipTipC18 reversed-phase purification, a technique used widely in proteomics research for sample clean-up of in-gel protein digests, including simplicity of operation, significantly improved mass detection, and amenability to automation.18,19 We showed that the chemical strategy afforded improved phosphoseryl peptide detection in unfractionated digest from model proteins and experimental samples and demonstrated that immobilization on solid support provides for enhanced reaction efficiency, improved throughput, and efficient use of dilute samples, owing to the preconcentration effect and minimal sample handling during derivatization. The enhanced spectral MS/MS information content of the derivatives greatly facilitated mapping the site of phosphorylation. Since its introduction to phosphoprotein characterization, the solid-phase derivatization format has evolved into a valuable tool for improved de novo MS/MS spectra interpretation.20,21 We have demonstrated recently the use of ZipTipC18 pipette tips for peptide in situ permethylation.22 It should be noted here that current analytical sample-preparation techniques are based predominantly on solid-phase derivatization and have found widespread use for trace analysis of bioorganic compounds.23 Sensitivity improvement by a factor of 50 and higher over solution-phase reactions has been demonstrated by this sample-handling format. On microchip platforms, preconcentration factors as high as 1000 have been demonstrated with this sample-handling technique.24
A significant portion (>50%) of cellular proteins are targeted by proline-directed kinases, of which the cyclic- and calmodulin-dependent kinases collectively phosphorylate these substrates at hundreds of potential sites.25 A subset of these proteins containing the phosphoseryl/phosphothreonyl-proline sequence motif has attracted considerable interest, as they are substrates for prolyl isomerase Pin 1, an enzyme that plays a pivotal role in the regulation of a wide range of cellular processes, including cell-cycle progression, immune response, and neural functions.26 Reversible phosphorylation at threonine, although relatively infrequent in occurrence, is involved in diverse cellular reactions, such as the regulation of cell adhesion, cell attachment, and cell migration.27
The notable resistance of phosphothreonine, particularly when positioned adjacent to proline, to the barium hydroxide catalyzed β-elimination/Michael addition, using 2-aminoethanethiol or other thiols, is well-recognized, and efficient conversion of these phosphopeptides has, thus far, been met with limited success.12,14,15,28 In this report, we have expanded successfully our solid-phase derivatization approach to this subset of phosphopeptides to provide the requisite chemical basis for their detection in mixtures, subsequent phosphorylation-site determination, and for our ongoing efforts to exploit the thiol adducts for phosphopeptide affinity purification. We note that conversion of a phosphothreonyl-proline peptide has been demonstrated earlier, using propanedithiol as a nucleophile in an aprotic solvent mixture.29 However, as much as one nanomole of peptide was needed to ensure for completeness of derivatization.
O-linked β-N-acetylglucosamine (O-GlcNAc) is a dynamic protein modification that has gained considerable interest because of its global and often site-specific relationship with phosphorylation in many cellular responses.30 Analogous to phosphorylation-site determination, site mapping of the GlcNAc modification proved subject to ambiguity as a result of neutral loss of the glycan under the conditions of CID. Nucleophilic replacement of the O-glycan via elimination/Michael addition has been shown to generate thiol adducts whose CID spectra contain substantially more information than their native counterparts.30 We therefore examined the potential of the solid-phase format to generate S-2-aminoethanethiol conjugates from this class of peptides to facilitate their detection in MALDI-MS mass maps of unfractionated protein digests. AP treatment of phosphoprotein digests has been an early method to identify candidate phosphopeptides in MALDI-MS by the resultant mass shift of 80 Da/released phosphate.5 As shown by our study, the enzymatic dephosphorylation could be carried out readily on peptides adsorbed onto a ZipTipC18-ZipTipμ-C18 pipette tip.
MATERIALS AND METHODS
TFA, formic acid, N-hydroxysulfosuccinimide ester of acetic acid (sulfo-NHS acetate), hydroxylamine hydrochloride, and Gel Code Blue Stain reagent were purchased from Pierce (Rockford, IL, USA). 2-Aminoethanethiol hydrochloride and barium hydroxide octahydrate were from Sigma-Aldrich (St. Louis, MO, USA). O-Methylisourea hemisulfate (94%) was from Acros Organics (Morris Plains, NJ, USA). Ammonium hydroxide, American Chemical Society reagent grade, 14.8 N, and ammonium hydrogen carbonate were purchased from Fluka (Ronkonkoma, NJ, USA). AP from calf intestine (20 U/μl) and N-octyl glucoside (OGS) were obtained from Roche Diagnostics (Indianapolis, IN, USA). Methanol and acetonitrile were from Burdick & Jackson (Muskegon, MI, USA). Tris (hydroxymethyl) aminomethane, EDTA, sodium carbonate, iodoacetamide, acetyl chloride (99%), anhydrous methanol, anhydrous deuterated methanol (CD3OD), the monophosphorylated fragment T6 of β-casein, HSA, horse heart myoglobin, β-lactoglobulin from bovine milk, carbonic anhydrase from bovine erythrocytes, OVA, angiotensin I human acetate salt hydrate [DRVYIHPFHL, mass:charge ratio (m/z) 1296.5], and human fibrinopeptide B (pyrGVNDNEEGFFSAR, m/z 1552.5) were purchased from Sigma-Aldrich. Bis(2-mercaptoethylsulfone) (BMS) was obtained from Calbiochem (La Jolla, CA, USA). Polyacrylamide gels (Criterion Precast gel, 1 mm, 10%) were from Bio-Rad (Hercules, CA, USA). ZipTipC18 pipette tips (0.6 μl bed volume) and ZipTipμ-C18 pipette tips (0.2 μl bed volume) were purchased from Millipore (Billerica, MA, USA). Trypsin (modified sequencing grade) was purchased from Promega (Madison, WI, USA). α-Cyano-4-hydroxycinnamic acid (α-CHCA) was obtained from Agilent Technologies (Palo Alto, CA, USA). The O-GlcNAc-modified peptides [SVES(O-GlcNAc)GSADAK, m/z 1152.2; and SVET(O-GlcNAc)GSADAK, m/z 1166.2] were custom-synthesized by Sussex Research Laboratories (Ottawa, Ontario, Canada). The phosphorylated cholecystokinin fragment, residues 10–20 (IKNLQpSLDPSH, m/z 1331.4), and the phosphorylated calcitonin fragment, residues 15–29 (DFNKFHpTFPQTAIGV, m/z 1801.9), were obtained from Protea Biosciences (Morgantown, WV, USA). The following phosphopeptides were purchased from AnaSpec (San Jose, CA, USA): UOM9, PKC substrate-3 phosphopeptide (KRPpSQRHGSKY amide, m/z 1422.5); CaM-dependent protein kinase-2 (CaMK2) substrate phosphopeptide (KKALRRQEpTVDA, m/z 1608.8); PKC substrate-5 phosphopeptide (RRGRpTGRGRRGIFR, m/z 1782.0); EGFR phosphopeptide (KRELVEPLpTPSGEAPNQALLR amide, m/z 2398.7); and myelin basic protein (MBP) phosphopeptide (APRpTPGGR, m/z 1048.1). The following phosphopeptides were synthesized in-house: P1 (RGApSPVE, m/z 795.5); P2 (RRApSPVA, m/z 835.5); P3 [p62 (dok); SHNSALYpSQVQK, m/z 1441.2]; P4 [p62 (dok); GIKSHNpSALYSQVQK, m/z 1739.3]; and UOM11, PKC substrate phosphopeptide (KRpTIRR, m/z 909.5).
Protein In-Gel Proteolytic Digestion
The protocol, as described by Speicher et al.,31 was used, except that BMS was used as a reductant. GelCode Blue-stained bands from 10 to 20 pmol OVA and α-S1 casein loaded onto the gels were destained twice with 200 μl 25 mM ammonium bicarbonate in 50% aqueous acetonitrile for 30 min at 37°C. GelCode Blue-stained bands from yeast Maf1 (20 pmol), which had been phosphorylated in vitro as described in a previous report,17 were processed in the same manner. Bands were dried briefly in a SpeedVac and then incubated for 15 min at 37°C in 100 μl 2 mM BMS/25 mM ammonium bicarbonate; the supernatant was removed. The gel bands were incubated in 100 μl 20 mM iodoacetamide in 25 mM ammonium bicarbonate for 30 min at 37°C; the supernatant was discarded. The gel bands were washed three times with 200 μl 25 mM ammonium bicarbonate for 15 min and subsequently dried. Bands were dehydrated for 10 min in 100 μl acetonitrile and then dried briefly in a SpeedVac. Dried gel bands were reswollen at room temperature in 20 μl 25 mM ammonium bicarbonate/0.01% OGS, supplemented with Promega-modified trypsin to an enzyme:substrate ratio of 1:10. After 20 min, 40 μl 25mM ammonium bicarbonate was added, and the digestion continued for 18 h at 37°C. After incubation, 50 μl 0.1% TFA was added. The supernatant was removed, and another 50 μl 0.1% TFA was added. The gel bands were incubated for 30 min 37°C. The combined extracts were reduced in volume to 35 μl and acidified by addition of 5 μl 10% TFA before sample immobilization.
Protein In-Solution Proteolytic Digestion
In-solution tryptic digestion was performed in 40 μl 25 mM ammonium bicarbonate/0.01% OGS, typically at an enzyme:substrate ratio of 1:100. After 18 h incubation at 37°C, the digests were acidified with 5 μl 10% TFA before solid-phase immobilization, as described below.
Sample Immobilization
ZipTipC18 pipette tips/ZipTipμ-C18 pipette tips were wetted six times with 10 μl methanol, followed by six washes with 0.1% TFA. Model peptides (0.2–20 pmol) prepared in 0.2–1% TFA/0.01% OGS solutions were placed in 10 μl aliquots into 0.5 ml microfuge tubes and subjected to 10 sample-loading/dispense cycles. The immobilized test peptides were then washed three times with 10 μl 0.1% TFA and once with 10 μl water. For high-volume sample enrichment (>10 μl), peptides and the in-gel proteolytic digests were loaded sequentially in 10-μl aliquots onto ZipTipC18 pipette tips and dispensed into a 0.5-ml microfuge tube. The sample was then transferred back in this step-wise mode to the original collection tube. This alternating load/dispense-enrichment cycle was repeated five times. The ZipTip pipette tips were then washed five times with 10 μl 0.1% TFA. Optionally, 200 μl disposable pipette tips can be press-fitted into ZipTipμ-C18 pipette tips or ZipTipC18 pipette tips, as suggested by the manufacturer and operated with a 200-μl pipettor to pass the sample slowly 10 times over the resin. Up to 200 μl sample can be processed for binding in this manner (www.millipore.com/publications).
General Procedure for On-Column Peptide Derivatization
The reagents (10 μl), specified in the solid-phase protocols described below, were aspirated three times onto the ZipTipC18 pipette tips or onto ZipTipμ-C18 pipette tips and dispensed to waste. The reagents (10 μl) were then loaded onto the tips from 60 μl that had been placed into 0.5 ml microfuge tubes. Alternatively, the incubation was carried out in 1 ml-capped microcentrifuge tubes, which had been loaded with 80 μl reagents. During incubation, the tips were left immersed in the reagents. Unless stated otherwise, the samples were desalted by passing 100 μl 0.1% TFA in 10 μl aliquots over the resin. The material was desorbed from ZipTipC18 pipette tips in 5–10 μl 50% acetonitrile/0.1% TFA/0.01% OGS, of which 1 μl was used for MALDI-MS analysis. ZipTipμ-C18 pipette tips were eluted in 0.5–1 μl matrix containing 0.1% TFA/0.01% OGS onto the MALDI plate. The droplet was aspirated carefully and dispensed on the target three times and then allowed to dry.
On-Column β-Elimination with Concurrent Michael Addition
The reagent mixture was prepared by mixing an aqueous 100-mM barium hydroxide solution with an aqueous 100-mM 2-aminoethanethiol hydrochloride in a 2:1 ratio to give a final concentration of 66 mM barium hydroxide and 33 mM 2-aminoethanethiol (pH 12.2). Barium hydroxide was finely ground to facilitate dissolution by vigorously vortexing for 1.5 min. The stock solution (31.54 mg/ml) was then centrifuged for 1 min at 13,000 rpm to remove carbonate precipitates. The barium hydroxide and the 2-aminothanethiol hydrochloride stock solution (11.36 mg/ml) were prepared fresh for daily use. The ZipTipC18 pipette tips were loaded with 10 μl of the reagent mixture and incubated typically for 1 h at 37°C. In some experiments, the incubation was carried out at 55°C for 1 h or at the same temperature extended for 1 additional hour. After incubation, the tips were washed with 100 μl 0.1% TFA, passed over the resin in 10 μl aliquots, and prepared for MALDI-MS analysis as described above.
On-Column β-Elimination with Consecutive Michael Addition
The β-elimination reaction was carried in 10 μl 50 mM barium hydroxide (pH 12.6) for 30 min at 55°C. ZipTipC18 pipette tips were subsequently flushed three times to waste with 10 μl of the Michael addition reagent prepared by mixing 100 mM aqueous barium hydroxide with 400 mM aqueous 2-aminoethanethiol hydrochloride (45.44 mg/ml) in a 3:1 ratio (Reagent A, 100 mM 2-aminoethanethiol/75 mM barium hydroxide, pH 10.6). The Michael addition medium (10 μl) was loaded onto the ZipTipC18 pipette tips, which were then incubated at 55°C for 1, 2, or 3 h. The tips were desalted by passing 100 μl 0.1% TFA in 10 μl aliquots over the support and prepared for MALDI-MS analysis as described above. The additional nucleophile reagents examined during chemistry optimization were prepared as follows: 100 mM aqueous barium hydroxide was mixed in a 3:1 ratio with 200 mM aqueous 2-aminoethanethiol (Reagent B, 50 mM 2-aminoethanethiol/75 mM barium hydroxide, pH 11.8) or with 100 mM aqueous 2-aminoethanethiol (Reagent C, 25 mM 2-aminoethanethiol/75 mM barium hydroxide, pH 12.2). Aqueous barium hydroxide (50 mM) was mixed in a 3:1 ratio with 400 mM aqueous 2-aminothanethiol (Reagent D, 100 mM 2-aminoethanethiol/37.50 mM barium hydroxide, pH 8.6). Aqueous barium hydroxide (25 mM) was mixed in a 3:1 ratio with 400 mM 2-aminoethanethiol (Reagent E, 100 mM 2-aminoethanethiol/18.75 barium hydroxide, pH 7.6). The various additional reagents were prepared freshly for daily use.
In a variation of the protocol, the β-elimination reaction was allowed to proceed in 10 μl of 50 mM barium hydroxide, typically for 2 h at 37°C. The β-eliminated samples were washed five times with 10 μl 0.1% TFA and then flushed five times to waste with 10 μl of a 200-mM 2-aminoethanethiol hydrochloride solution that had been adjusted in a pH meter with solid Tris base to pH 8.6. The nucleophilic addition was allowed to proceed for 2 h at 55°C in 10 μl of the Michael addition reagent. The ZipTipC18 pipette tips were desalted and processed for MALDI-MS analysis as described above.
ZipTipC18 Pipette Tip Alkali-Compatibility Evaluation
The relative efficiency of peptide recovery from the silica-based reversed-phase support, before and after the alkali treatment, was assessed using the isotope-dilution procedure, as described previously, with minor modifications.17 Efficiency and reproducibility of relative peptide recovery from the support were assessed as follows: aliquots of angiotensin I solutions in 0.2% TFA/0.01% OGS, containing 50 pmol peptide, were bound in replicates to ZipTipC18 pipette tips that were then washed three times with 10 μl 0.1% TFA to remove unbound material and eluted twice with 10 μl 50% acetonitrile/0.1% TFA/0.01% OGS. Eluates were dried in a SpeedVac, dissolved in 25 μl of the isotopically labeled methanolic HCl, prepared as described previously,17 and incubated for 90 min at 12°C and subsequently dried. The methyl-d3-esterified peptides were then dissolved in 10 μl 50% acetonitrile/0.1% TFA, and 5 μl of the peptide solutions was mixed with 5 μl of the methyl-d0-esterified reference peptide prepared as described previously,17 followed by addition of 10 μl MALDI matrix. The mixture (1 μl) was applied to the MALDI target for relative quantitation of the eluted peptides.
To assess the potential impact of silica alkali exposure on peptide recovery, 50 pmol peptide was immobilized in replicates as described above. The ZipTipC18 pipette tips were rinsed three times with 10 μl 0.1% TFA, then loaded with 10 μl 50 mM barium hydroxide, and incubated for 30 min at 55°C. The elimination base was then exchanged in situ for 75 mM barium hydroxide/100 mM 2-aminoethanethiol (pH 10.6), and the incubation continued in 10 μl of the addition medium for 2 h at 55°C. The ZipTips were washed by passing 100 μl 0.1% TFA in 10 μl aliquots over the resin and eluted twice with 10 μl 50% acetonitrile/0.1% TFA/0.01% OGS. The eluates were dried and methyl-d3-esterified under the conditions described above. The methyl-d3-esterified peptides were then dissolved in 10 μl 50% acetonitrile/0.1% TFA, and 5 μl of the peptide solutions was mixed with 5 μl of the methyl-d0-esterified reference peptide, followed by addition of 10 μl MALDI matrix. The mixture (1 μl) was applied to the MALDI target for relative quantitation of the eluted peptides. Potential alkali-induced sample loss from the reversed-phase support was determined by measuring the ratio of the relative abundance of the methyl-d3 and methyl-d0-esterified peptides in the mass spectra.
On-Column Guanidination
The reaction mixture (pH 10.6), described by Beardsley and Reilly32 for in-solution derivatization, was prepared by diluting 110 μl 7 N ammonium hydroxide solutions with 100 μl water. The guanidination stock solution (30 μl) was then added and prepared by vigorously mixing 50 mg O-methylisourea hemisulfate with 51 μl water. Freshly prepared O-methylisourea hemisulfate stock solution was used for each reaction. The ZipTipC18 pipette tips were flushed twice to waste with 10 μl of the reagent solution, which was then loaded onto the tips. After incubation for 10–15 min at 65°C, the tips were washed 10 times with 10 μl 0.1% TFA and then three times with 10 μl water. The reaction products were eluted with 5–10 μl 50% acetonitrile/0.1% TFA/0.01% OGS, of which 1 μl was used for MALDI-MS analysis. In some experiments, ZipTipμ-C18 pipette tips were used and eluted in matrix onto the target.
On-Column Sequential Derivatization of O-GlcNAc-Modified Peptides
ZipTipC18 pipette tip-bound, O-GlcNAc-modified peptides were subjected to on-column guanidination, as described above. The postreaction, desalting step was omitted. Instead, the tips were flushed three times with 66 mM barium hydroxide/33 mM 2-aminoethanethiol and then loaded with 10 μl of the reagent mixture. The O-GlcNAc serine peptide was incubated for 1 h at 37°C. The O-GlcNAc threonine peptide was allowed to react for 1 h at 55°C. The tips were washed 10 times with 10 μl 0.1% TFA and prepared for MALDI-MS analysis, as described above. In some experiments, the peptides were subjected to β-elimination with consecutive Michael addition. The addition reaction was allowed to proceed for up to 2 h.
On-Column Acetylation
A 20-mM sodium phosphate buffer (pH 8.0) was supplemented with 20 mM sulfo-NHS acetate. ZipTipC18 pipette tip-bound protein digests (2–20 pmol) were flushed three times with the reagent, incubated in 10 μl reagent for 20 min at 55°C, and then washed 10 times with 10 μl 0.1% TFA. Sample processing for MALDI-MS was as described above.
On-Column Deacylation
A 1-M sodium carbonate solution was supplemented with 2% hydroxylamine hydrochloride (pH 9.4). ZipTipC18 pipette tips containing the acetylated samples were flushed three times with 10 μl of the reagent. The reagent (10 μl) was loaded onto the supports and allowed to react for 15 min at 37°C. To terminate the reaction, the ZipTips were washed 10 times with 10 μl 0.1% TFA and five times with 10 μl water. Sample processing for MALDI-MS was as described above.
Preparation of Substrates for Chemical-Targeted, Phosphorylation-Site Determination
ZipTipμ-C18 pipette tip-bound α-S1 protein digests (2 pmol) were subjected to on-column acetylation, as described above. The postreaction desalting step was omitted. Instead, the pipette tips were flushed three times with 10 μl of the 66-mM base/33-mM nucleophile solution, loaded with 10 μl of the reagent, and subjected to concurrent β-elimination with Michael addition for 1 h at 37°C. The ZipTipμ-C18 pipette tips were desalted by passing 100 μl 0.1% TFA in 10 μl aliquots over the resin and eluted in 0.5–1 μl matrix, supplemented with 0.1% TFA/0.01% OGS directly onto the MALDI plate.
On-Column Phosphopeptide Enzymatic Dephosphorylation
AP (20 U/μl) stock solution was diluted into dephosphorylation buffer (0.1 M Tris HCl/0.2 mM EDTA, pH 8.5) to final enzyme concentrations of 2, 4, and 8 U/μl. In-gel protein digests (20 pmol) and model peptides (5–20 pmol) were loaded onto ZipTipC18 pipette tips or ZipTipμ-C18 pipette tips from 0.2% formic acid or 0.2% TFA solutions and washed three times with 10 μl 0.1% TFA, three times with 10 μl water, and once with 3 μl dephosphorylation buffer. The enzyme solutions (20 μl) were then placed into 0.5 ml microfuge tubes and the tips flushed three times with 3 μl of the enzyme solutions while immersed in liquid. The tips were then loaded with 3 μl enzyme solution, immersed in liquid into an air-heated incubator, and set at 37°C. ZipTipμ-C18 pipette tips were incubated in 3 μl of the enzyme solution, aspirated from 10 μl enzyme solution that had been placed into 0.2 ml microfuge tubes. The 0.2-ml tubes were inserted into 0.5 ml tubes, and the space between tubes and tips was sealed with Parafilm to minimize solvent evaporation. After incubation for predetermined times, the tips were washed 10 times with 10 μl 0.1% TFA and desorbed in 5–10 μl 50% acetonitrile/0.1% TFA/0.01% OGS before MALDI-MS.
Differential Peptide Mass Mapping
In-gel digests prepared in replicates from equimolar amounts of protein loaded onto the gel, model peptides used for method optimization, and in-solution digests were split into equal fractions. Samples were immobilized on ZipTipC18 pipette tips or ZipTipμ-C18 pipette tips. One set of ZipTips was loaded with 10 μl 0.1% TFA, left immersed in solvent, and set aside as controls. The other set of ZipTips was processed for chemical or enzymatic treatment. Unmodified and derivatized material was eluted from the ZipTipC18 pipette tips, typically in 5–10 μl 50% acetonitrile/0.1% TFA/0.01% OGS, of which 1 μl was used for MALDI-MS. ZipTipμ-C18 pipette tips were deposited directly in 1 μl matrix containing 0.1% TFA onto the MALDI plate.
Optimal Use of Low-Level Samples
For efficient use of low-level samples, the analyte was recovered from MALDI spots, according to a previous procedure, with the following modifications.33 The protein digest (<2 pmol), enriched from dilute solutions onto a ZipTipμ-C18 pipette tip, was deposited in 1 μl α-CHCA matrix, supplemented with 0.1% TFA onto the MALDI plate. After MALDI-TOF analysis, the plate was removed from the instrument. TFA (0.5%; 4–5 μl) was placed onto the MALDI spot. The solubilized analyte was concentrated on a preconditioned ZipTipμ-C18 pipette tip directly from the droplet using 10 load/dispense cycles. The ZipTipμ-C18 pipette tip was washed 10 times with 10 μl 0.1% TFA to remove the matrix. The sample was then subjected to the chemical or enzymatic treatment, as described below, desalted, and deposited in the matrix onto the sample stage. In this manner, only a single sample needed to be committed to differential peptide mass mapping.
MS
A Voyager-DE STR (Applied Biosystems, Foster City, CA, USA) was used and operated in the reflector mode at an accelerating voltage of 20 kV, 67% grid voltage, and 250 ns extraction delay time. Laser intensity was typically set at 1400–1600, and spectra were acquired using 100 laser shots/spectrum. When operated in the linear mode, the grid voltage was typically set at 95% and the extraction delay time at 150 ns. The data shown are based on three accumulated acquisitions. Some spectra were obtained using 80 laser shots/spectrum, and spectra acquired in the linear mode from eight different positions were averaged. The analyte was prepared on target in the dried droplet mode using α-CHCA as matrix. ZipTipμ-C18 pipette tips were eluted in matrix containing 0.1% TFA onto the target. For peptide fragmentation, a 4700 Proteomics Analyzer was used and operated in the MS and MS/MS mode; typical laser power for MS was ∼3700; for MS/MS, ∼4500. Usually, 1000–2000 shots were acquired for MS; 2000–20,000 for MS/MS.
RESULTS AND DISCUSSION
Solid-Phase Chemical Strategy for Phosphopeptide Identification
This study represents the continuation of our effort to develop a simple and effective tool for comprehensive and rapid screening by MS for phosphopeptides in protein digests. The sample processing scheme is shown in Fig. 1A. The analytes are immobilized on ZipTipC18 or ZipTipμ-C18 pipette tips, followed by a desalting step. The tips are subsequently loaded with the β-elimination base. After completion of the reaction, an aliquot of the alkaline Michael addition mixture is aspirated onto the solid phase. The addition reaction is terminated by a solvent wash, followed by product elution for subsequent mass spectrometric analysis. During the incubations, the reversed-phase supports were left immersed in the reagents. For concurrent β-elimination with Michael addition, the desalted ZipTips are loaded with the single reactant mixture.17 In the expanded application of the solid-phase approach, the alkali-induced nucleophilic addition, is preceded by acetylation or guanidination. The use of these combined solid-phase reactions is discussed later in the text.
Figure 1.
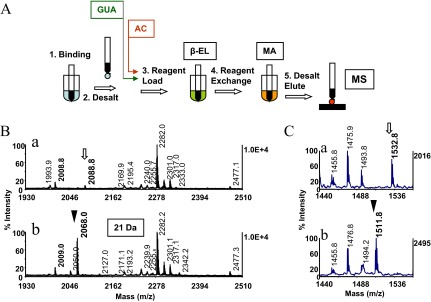
Solid-phase strategy for phosphopeptide identification in protein digests. (A) Schematic representation of the sample-handling steps used in the consecutive reaction scheme. Peptides are bound to reversed-phase pipette tips (1), followed by a solvent wash (2). The tips are then loaded with the β-elimination base (3), which after completion of the reaction [β-elimination (EL)], are exchanged directly on the solid phase for the alkaline Michael addition medium (4). The Michael addition reaction [(MA)] is terminated by a solvent wash (5), followed by product elution. Note this sample-handling format precludes analyte transfer between the reaction steps. The in situ guanidination (GUA) and acetylation (AC), depicted as preceding the β-elimination/Michael addition, are highlighted with boxes. Data obtained from the application of these solid-phase reactions to model peptides and protein digest are illustrated separately (see Figs. 2 and 8). (B) Application of the method to tryptic digests of gel-separated OVA. Conditions were as described (see Fig. 3F), except that each of the reactions was allowed to proceed for 1 h at 37°C. (B, a and b) Expanded sections of the MALDI spectra, acquired before and after derivatization, respectively. Open arrow and arrowhead denote the phosphopeptide at m/z 2088 (341EVVGpSAEAGVDAASVSEEFR360) and its thiol adduct at m/z 2068.0, respectively, with a derivatization mass signature of 21 Da, highlighted with a box. Asterisks denote the peptide's nonphosphorylated counterpart. Note prominent signal enhancement imparted to native species by the nucleophilic substitution. (C, a and b) Expanded sections of MALDI spectra of a tryptic digest of Maf1 before and after derivatization, respectively. Note comparable ionization efficiency of the highly basic, native phosphopeptide at m/z 1532.8 (88RDpSNSFWEQKR98; open arrow) and its tagged counterpart at m/z 1511.8 (arrowhead). One-tenth of the eluates corresponding to ∼2 pmol of the digests was applied to the target.
Representative data from an application of the solid-phase approach to a tryptic digest of OVA, a protein substoichiometrically phosphorylated at serine, are shown in Fig. 1B. The derivatization resulted in the formation of the prominent ion at m/z 2068.0 (arrowhead), representing the S-2-aminoethylcysteine analog of the native peptide recognized by the characteristic mass shift of −21 Da/phosphate as the low-intensity ion at m/z 2088.8 (open arrow). This mass shift arises from the loss of phosphoric acid (98 Da) and the addition of the nucleophile (Mr 77 Da). Notably, the derivatized peptide ionized with 11-fold-higher signal intensity than its native counterpart, enabling facile identification of the phosphopeptide in the unfractionated digest.17 The tagged peptide's strong MALDI response is attributable to replacement of the negatively charged phosphate by the S-2-aminoethyl group, which incorporates a protonatable hydrophobic moiety into the peptide. This is consistent with the hypothesis that basic and hydrophobic residues in peptides tend to enhance the desorption/ionization process.34
With prior knowledge of the protein sequence, phosphopeptides, identified in this manner, can be mass-matched to in silico candidate peptides. By this means, the tryptic peptide of OVA was aligned with 341EVVGSAEAGVDAASVSEEFR360, containing three potential phosphorylation sites. MALDI-TOF-MS/MS analysis of the peptide's tagged counterpart produced a nearly uninterrupted y ion series that confirmed the peptide sequence (data not shown). The substituted serine residue was readily identified by the unique mass difference of 146 Da between product ion y16 and y15 and hence, serine in position 345 as the site of phosphorylation, whereas the y16 counterpart from the native peptide was undetectable in its MS/MS spectrum, precluding phosphorylation-site determination. In the case of a phosphothreonyl peptide, the derivatized residue and hence, the phosphorylation site were recognized in the MS/MS spectrum by its unique difference mass of 160 Da.12
In contrast to the above OVA phosphopeptide, which contains multiple acidic amino acid residues, and the comparably highly acidic fragments recovered previously from α-S1 casein and α-catenin tryptic digests, phosphopeptides bearing multiple basic residues, such as those derived from the tryptic digest of in vitro-phosphorylated Maf1, were enhanced only moderately in MALDI-MS by the nucleophilic substitution.17 The incorporation of the protonatable moiety had only a minor impact on the ionization of these highly basic peptides, which is intrinsically favorable. As exemplified by the tryptic peptide of Maf1 (RDpSNSFWEQKR, residues 88–98, m/z 1532.8), the native peptide and its thiol adduct (at m/z 1511.8) appeared in the spectra at nearly equal abundance (see Fig. 1C in ref. 17). Accordingly, this highly basic peptide was readily identified by the characteristic derivatization mass shift of −21 Da, providing a viable means to recognize phosphopeptides in digest derived from basophilic kinase substrates. Additionally, this mass shift is a signature for the extent of phosphorylation.
We recognize that complex protein digests can be difficult to analyze by the method, as the species of interest may, owing to signal suppression, become undetectable in the abundant background of the nonphosphorylated species. Fractionation of the digest, before and after derivatization by nano-flow reversed-phase chromatography, coupled with on-target fraction collection using an automated eluent/matrix-dispensing device (LC-MALDI), may offer a means to alleviate the signal-suppression problem. In preliminary experiments, we have begun to explore the potential of this technique in model systems and found that low-level quantities of synthetic phosphopeptides spiked into digests of protein mixtures could be detected in the MALDI-MS spectra of the collected fractions. However, in cases where the stoichiometry of phosphorylation is very low (<10%), phosphopeptide enrichment becomes a prerequisite means to reduce the sample complexity. We have, therefore, been exploring the use of a solid-phase derivatization platform to render phosphopeptides amenable to purification by covalent chromatography. In this strategy, all amino groups in the digest are blocked by acetylation prior to β-elimination/Michael addition, making the amino group of S-2-aminoethylcysteine/β-methyl-S-2-aminoethylcysteine the sole target for subsequent thiolation. The thiol-functionalized analyte was then reversibly enriched on activated thiol sepharose. The isolates, by virtue of the affinity tag, proved highly suitable for phosphorylation-site localization by CID.
Sample Handling
Peptide chemical modification in contemporary proteomic studies relies almost exclusively on derivatization in solution. However, the use of the miniaturized reversed-phase support as a venue for chemical reactions offers a number of compelling advantages over the classical in-solution derivatization format. Solution-phase derivatization protocols often use an initial sample purification step to remove contaminants from the starting solution that may interfere with the chemical reactions. To this purpose, solid-phase extraction is commonly used, followed by SpeedVac dry-down of the desalted fractions. As noted for low-level samples (<2 pmol), sample concentration by vacuum drying, in general, can cause substantial adsorptive peptide loss ranging up to 50% of the starting solution.31 In the solid-phase strategy, in contrast, the immobilized analyte is purified on the same phase with no sample transfer prior to derivatization (see Fig. 1A). Importantly, the analyte during adsorption is concentrated on the support, thereby generating an environment that is essentially free of reagent dilution. As a result, reactions may proceed on the solid phase at higher efficiency and with faster kinetics than in solution.17,23 Peptides in dilute solutions are preferentially enriched on ZipTipμ-C18 pipette tips containing 200 nl resin beds. As the products can be transferred to a MALDI target from these reaction supports, nearly undiluted, femtomole-level mass detection can be readily attained.17 For optimal use of low-level samples, the starting material can be recovered after the initial mass analysis from the MALDI-TOF sample spot on the reversed-phase support for subsequent derivatization (see Materials and Methods). Finally, the solid-phase procedure should be readily amenable to automation on ZipTipC18 pipette and tip/ZipTipμ-C18 pipette tip-compatible liquid-handling stations. It is noteworthy that the benefits of solid-phase derivatization were recognized already a decade ago and exploited to improve significantly the accuracy, sensitivity, and throughput of the quantitative analysis of bioorganic compounds in complex matrixes. This reaction format and has since evolved as the predominant sample-preparation technique for trace detection of these compounds in toxicology, environmental, and pharmaceutical studies.23
As demonstrated in the current report and in an earlier communication,35 the solid-phase format proved particularly advantageous for combining chemistries into serial reaction schemes. On the solid-phase platform, reagents are exchanged directly on the stationary phase. At the conclusion of the reactions, the immobilized products are desalted by a brief solvent wash (see Fig. 1A). Owing to this limited sample handling, the analyte can be processed with minimal sample loss. As practiced in contemporary proteomics studies, classical desalting methods, such as microdialysis or reversed-phase HPLC, are commonly used to isolate the intermediate and final reaction products from excess reagent.14,15 These time-consuming sample-handling steps may, especially at low analyte level, incur substantial adsorptive sample loss, limiting the use of this serial peptide-modification technique.
Test peptide and proteolytic digest solutions used in our study were supplemented with trace amounts of OGS to minimize peptide adsorption to plastic surfaces during sample immobilization.17 This MALDI “friendly” nonionic detergent has been shown compatible with LC-MS. owing to its strong retention on reversed-phase columns.36
On-Resin Peptide Guanidination
In preparation for the experiments described below, we noticed that the GlcNAc-modified peptides were prone to metal-ion cluster formation. This observation was in contrast to an earlier report with synthetic O-glycopeptides used to assess, by MALDI-MS, the efficiency of β-elimination with concurrent nucleophilic addition of DTT.30 We found that guanidination eliminated this unwanted signal-dilution effect. In the adaptation of the solution-phase protocol,32 the bound peptides were incubated in an aliquot of the alkaline O-methylisourea hemisulfate solution (pH 10.6) for 10 min at 65°C and then desalted. Under these conditions, the conversion of lysyl residues to homoarginine was complete. The derivatives were then used as starting material for the experiments described below. Likewise, the model peptide P3 (SHNSALYpSQVQK, m/z 1441.2) reacted to completion. The derivative ionized with an estimated 40%-higher efficiency than the native counterpart (data not shown). These data suggest that the solid-phase guanidination protocol should be of general use for improving the mass detection of lysine-containing peptides in protein digests32 and can also be exploited for in situ N-terminal fragmentation-directed chemical modification.20 In addition, the method can be used in tandem with the β-elimination/Michael addition to prepare substrates for phosphorylation-site determination by chemical-targeted proteolysis, as discussed later in the text.14
Concurrent Reaction: Derivatization of O-GlcNAcylated Peptides
The structural characterization O-GlcNAcylated peptides is of considerable interest to gain insight in the dynamic roles that O-glycosylation and O-phosphorylation play in many cellular responses.30 Therefore, we sought to evaluate the use of a solid-phase derivatization format to prepared thiol adducts from this class of peptides to explore the potential for their detection in MALDI mass maps of unfractionated protein digests. Representative MALDI-TOF data from the derivatization experiments are shown in Fig. 2. In the experiments, the O-GlcNAc peptide modified at serine SVES(O-GlcNAc)GSADAK at m/z 1152.2 was guanidinated, as described above, and subsequently subjected to concurrent β-elimination with Michael addition for 1 h at 37°C, conditions that we previously found to afford complete phosphoseryl peptide conversion.16,17 For side-to-side comparison, the phosphoseryl peptide UOM9 at m/z 1422.8 (KRPpSQRHGSKY amide) was derivatized in parallel. As shown in Fig. 2A and B, the O-GlcNAc peptide modified at serine reacted to completion. Under these relatively mild reaction conditions, the bulk (>70%) of the guanidinated O-GlcNAc peptide, modified at threonine SVET(O-GlcNAc)GSADAHar, remained resistant to derivatization, consistent with the common perception that the residue's methyl group protects the β-carbon from nucleophilic attack (data not shown). Conversion yields improved when the peptide was incubated at increased temperatures. As illustrated in Fig. 2C and D, the thiol adduct of the peptide was fully formed after 1 h incubation at 55°C, conditions that were, as reported previously, sufficiently stringent to drive the conversion of the phosphothreonyl peptide UOM11 (KRpTIRR) to completion (Fig 2C and D, insets.17 The data show that the O-GlcNAcylated peptides reacted to completion with essentially the same kinetics as their phosphorylated counterparts. The result is in agreement with an earlier finding.15 Ions diagnostic for dehydroderivatives of serine or for base-induced hydrolysis products were not observed in the spectra, indicating that the integrity of the samples was preserved during derivatization.
Figure 2.
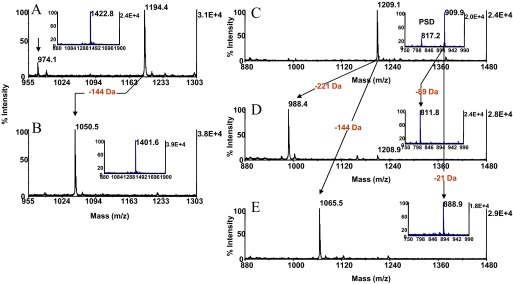
Solid-phase β-elimination with concurrent Michael addition. The O-GlcNAc peptide modified at serine SVES(O-GlcNAc)GSADAK, m/z 1152.4, was reacted for 10 min at 65°C in O-methylisourea hemisulfate solution (pH 10.6) and then with 66 mM barium hydroxide/33 mM 2-aminoethanethiol (pH 12.6) for 1 h at 37°C. The phosphoseryl peptide UOM9 (KRPpSQRHGSKY amide, m/z 1422.8) was reacted under the same conditions. MALDI-TOF spectra (A) after guanidination and (B) after β-elimination with concurrent Michael addition. Arrows across the spectra indicate the characteristic mass shift highlighted in red. (A and B, insets) Spectrum of the phosphoseryl peptide before and after β-elimination with concurrent Michael addition, respectively. Arrow in A denotes β-elimination product of the guanidinated species. One-tenth of the eluates corresponding to ∼2 pmol peptide was applied to the target. The O-GlcNAc peptide modified at threonine SVET(O-GlcNAc)GSADAK, m/z 1166.2, was guanidinated and subsequently β-eliminated for 30 min at 55°C in 50 mM barium hydroxide or subjected at the same temperature for 1 h to the concurrent β-elimination with Michael addition. The phosphothreonyl peptide UOM11 (KRpTIRR, m/z 909.9) was reacted under the same conditions MALDI-TOF spectra (C) after guanidination, (D) after β-elimination, and (E) after concurrent β-elimination with Michael addition. (C and D, inset) The spectrum of the phosphothreonyl peptide, the spectrum of its β-elimination product, and the spectrum of its thiol adduct, respectively. Arrows across the spectra indicate the characteristic mass shifts highlighted in red. PSD, Postsource decay product. One-tenth of the eluates corresponding to ∼2 pmol peptide was applied to the target.
As illustrated in Fig. 2, derivatization of O-glycosylated peptides imparts a mass shift of −144 Da to the native species, dictated by the nominal mass of the β-eliminated glycan (221.2 Da), in contrast to phosphopeptides, which display, in MALDI-MS, a mass shift of −21 Da. This unique mass signature may be exploited to facilitate identification and differentiation of O-GlcNAc-modified peptides from phosphopeptides in mass maps prepared from O-glycophosphoprotein digests. Experiments to test the validity of this approach using well-characterized model systems are currently being pursued in our laboratory. As the nucleophilic substitution precludes neutral loss of the glycan as the preferred pathway of peptide backbone fragmentation, the tagged glycopeptides are expected to provide more informative fragmentation upon CID than their unmodified counterparts. By analogy, DTT adducts of O-GlcNAc peptides derived from enriched postsynaptic density samples upon CID furnished easily interpretable sequence data, enabling unambiguous glycosylation-site determination.37
Chemistry Optimization–Consecutive Reactions
We next probed the CaMK2 substrate phosphopeptide (KKALRRQEpTVDAL, m/z 1608.5) with the concurrent β-elimination/Michael addition protocol. In contrast to the highly reactive UOM11 phosphopeptide (KRpTIRR), an estimated 75% of this peptide resisted conversion (data not shown). Upon incubation for 1 additional hour, a significant portion (>50%) of the peptide still remained in its dehydro-2-aminobutyric acid form, indicating that the concurrent β-elimination proceeded to completion, whereas the addition reaction was notably retarded (Fig. 3B). The data suggest that the chemistry may be subject to modulation by amino acids flanking the phosphorylation site. In particular, proline positioned immediately C-terminal to phosphoserine and threonine is well-recognized to protect the residues' β-carbon against nucleophilic attack. Prolonged exposure of the CaMK2 substrate phosphopeptide to the concurrent reaction mixture might be expected to increase the conversion yield but raise concerns about peptide stability under elevated temperatures. We therefore sought to explore alternative, less-stringent experimental conditions by decoupling the β-elimination from the Michael addition, which is advantageous, as the reactions can be examined separately. In the experiments, the CaMK2 substrate phosphopeptide was incubated for 10, 20, and 30 min in 50 mM barium hydroxide. MALDI-MS analysis of the samples showed that the peptides' β-elimination product at m/z 1510.6 was fully formed after 30 min incubation (Fig. 3C). We note that the UOM11 phosphopeptide was eliminated under these conditions as quickly as the CaMK2 substrate phosphopeptide, as was the O-GlcNAc peptide, modified at threonine. This serial reaction mode was then exploited to optimize the Michael addition chemistry, the time-limiting step of the overall reaction. In the experiments, the CaMK2 phosphopeptide was exposed to 50 mM barium hydroxide at 55°C for 30 min. The Michael addition reaction's efficiency was then semiquantitatively assessed by MALDI-MS over a range of nucleophile concentrations. The eliminated peptide was probed with reagent mixtures for a total of 2 h, in which the 2-aminoethanethiol concentration was adjusted to 25, 50, and 100 mM, whereas the barium hydroxide concentration was held constant at 75 mM. The pH measured for these solutions was 12.2, 11.8, and 10.6, respectively. As illustrated in Fig. 3D, at the lowest nucleophile concentration in the mixture, an estimated 75% of β-elimination product at m/z 1510.4 remained unconverted. The Michael adduct formation noticeably improved when the nucleophile was added to a final concentration of 50 mM (Fig. 3E) and reached near completion when the nucleophile was adjusted to the final concentration of 100 mM (Fig. 3F). When the protocol was applied to the UOM11 phosphopeptide and to the O-GlcNAc peptide, modified at threonine, the duration of the addition phase could be shortened by 30 min without affecting the product yield. This observation suggests that the structure-reactivity modulation effect still persists, albeit noticeably tempered. However, as discussed below, an alternative explanation can be proposed to account for the CaMK2 substrate phosphopeptide's distinct response to the two modes of derivatization.
Figure 3.
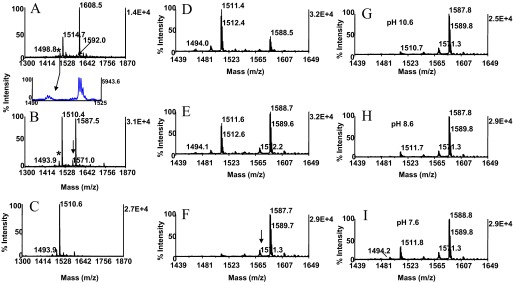
Chemistry optimization–consecutive reactions. Kinetics of Michael addition. The phosphothreonyl peptide CaMK2 (KKALRRQEpTVDAL, m/z 1608.5) was subjected to the concurrent conditions, as described in Fig. 2D, except that the reaction was performed for 2 h. MALDI-TOF spectra (A) of unmodified peptide and (B) after concurrent β-elimination/Michael addition. Note incompleteness of the addition reaction. Arrow and asterisk in A denote trace-level peptide synthesis byproduct at m/z 1592.0, separated from parent by 17 Da and its postsource decay product at m/z 1498.8, respectively. Cross-arrow indicates the expanded region containing the postsource decay product. Asterisk and arrow in B designate the byproduct's β-elimination product at m/z 1493.9 and its thiol adduct at m/z 1571.0, respectively. In the consecutive derivatization mode, the CaMK2 phosphopeptide was β-eliminated in 50 mM barium hydroxide for 30 min at 55°C (C). The efficiency of the addition reaction was probed at the same temperature for 2 h with reactants in which the 2-aminoethanethiol hydrochloride concentration was varied, whereas the base concentration was held constant. MALDI-TOF spectra (D) after incubation in 25 mM nucleophile/75 mM barium hydroxide, pH 12.2; (E) after incubation in 50 mM nucleophile/75 mM barium hydroxide, pH 11.8; and (F) after incubation in 100 mM nucleophile/75 mM barium hydroxide, pH 10.6. Arrow in F denotes thiol adduct of synthesis byproduct. The efficiency of the Michael addition was evaluated further with reactants in which the nucleophile concentration was kept constant, whereas the base concentration was varied. MALDI-TOF spectra (G) after incubation in 100 mM nucleophile/75 mM barium hydroxide, pH 10.6, used as reference; (H) after incubation in 100 mM nucleophile/37.5 mM barium hydroxide, pH 8.6; and (I) after incubation in 100 mM nucleophile/18.75 barium hydroxide, pH 7.6. Note reduction in efficiency of addition reaction at pH 7.6 attributable to diminished ionization of the nucleophile. One-tenth of the eluates corresponding to ∼2 pmol peptide was applied to the target.
As illustrated in Fig. 3B, the thiol adduct and the β-elimination product were accompanied by low-intensity ions, separated from the base peaks by 17 Da. Inspection of the spectrum, produced from the starting material, revealed an ion at m/z 1592.0, which was clearly identifiable as a phosphopeptide in the expanded region of the spectrum by its postsource decay product at m/z 1498.8 (Fig. 3A an B, cross-arrows). The β-elimination product and thiol adduct of this minor species became clearly discernible as the low-abundance ions at m/z 1493.9 and m/z 1571.0, respectively, in the spectrum of the derivatized sample (Fig. 3B). Evidently, the ion at m/z 1592.0 represents a peptide synthesis byproduct generated by loss of ammonia.
Thiols are, in general, considered to be the most potent nucleophiles in their ionized, thiolate form. To examine this pH-dependent phenomenon's potential effect on the Michael addition, the 2-aminoethanethiol solution (pKa 8.2) was adjusted with barium hydroxide to pH 8.6 and to pH 7.6, whereas the nucleophile concentration was kept constant at 100 mM. The CaMK2 substrate phosphopeptide was then probed with these additional media reactants, along with the pH 10.6 reagent mixture use as control (Fig. 3G). The spectra revealed that the reactivity was essentially unaffected when the pH was lowered to 8.6 (Fig. 3H). However, an ∼35% decline in thiol adduct formation was observed when the reaction was performed at pH 7.6 (Fig. 3I). The data suggest that the nucleophile exists near neutrality, partially in its protonated, less-reactive form.
As noted above, the β-elimination phase of the CaMK2 phosphopeptide during the single-mixture reaction proceeded to completion, whereas the addition reaction slowed considerably. A similar diminished efficiency of the Michael addition was observed during concurrent in-solution derivatization of the phosphothreonyl peptide EAIpTAAPFAK amide using 2-phenylethanethiol as nucleophile.38 Consistent with our results, the addition reaction to the β-eliminated product was markedly promoted when the peptide was subjected to β-elimination with consecutive Michael addition. This trend was also observed when ethanethiol was used as a nucleophile. In view of these data, we suspect that the concurrent reaction might proceed by a different mechanism than the stepwise β-elimination-Michael addition that would account for the inefficiency of the addition reaction in the concurrent reaction procedure. One could speculate that the concurrent reaction proceeds via a direct displacement of the phosphate as an alternate to the assumed elimination addition. However, more experimentation is needed to elucidate, in detail, the factors involved in this effect.
Additional phosphothreonyl peptides examined with the optimized protocol included the PKC substrate-5 phosphopeptide (RRGRpTGRGRRGIFR), the phosphorylated calcitonin fragment (1DFNKFHpTFPQTAIGV),20 and the O-GlcNAc peptide, modified at threonine [SVET(O-GlcNAc)GSADAHar]. The peptides, albeit differing in their amino acid sequence, reacted to completion (results not illustrated). We subsequently probed the model peptides (P1-RGApSPVE, m/z 795.5; and P2-RRApSPVA, m/z 835.5) with this protocol, in which phosphoserine is adjoined by proline. Both peptides converted to near completion (data not shown).
We next sought to evaluate the applicability of the serial reaction protocol with the EGFR phosphopeptide (KRELVEPLpTPSGEAPNQALLR amide, m/z 2398.7), in which phosphothreonine is followed in sequence by proline. Efficient conversion of phosphothreonyl peptides containing this sequence feature has thus far been met with limited success.29 Over the time course of the reactions, the β-methyl-S-2-aminoethylcysteine derivative was incompletely formed after 1 h incubation (Fig. 4B, inset), whereas an estimated 75% conversion was observed after 2 h incubation (Fig. 4B), and 1 additional hour incubation was required to drive the peptide's addition reaction to completion (Fig. 4C). The need for the more stringent reaction conditions to achieve almost quantitative product formation is consistent with the notion that the proximal proline imparts additional protection against nucleophilic attack on the β-carbon of the threonine residue. The derivatization the MBP phosphopeptide (APRpTPGGRR, m/z 1048.1) was diminished under these conditions, leaving an estimated 35% of the peptide in its β-eliminated form (results not shown). We note here that such moderately, reduced conversion of phosphoamino acids, in general, should not detract from the use of the method to scan for phosphopeptides in unfractionated protein digests. In fact, residual β-elimination products are recognizable in the spectra by the characteristic mass signature of −98 Da, providing complementary evidence for the presence of a phosphopeptide in a mixture.
Figure 4.
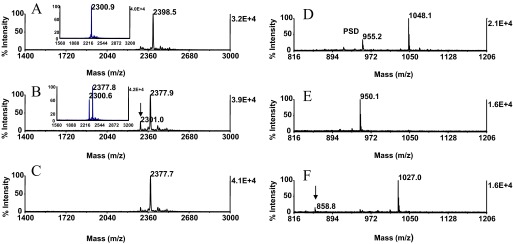
Solid-phase β-elimination with consecutive Michael addition. The EGFR phosphopeptide (KRELVEPLpTPSGEAPNQALLR amide, m/z 2398.5) was derivatized under the conditions, as described in Fig. 3F, except that the addition phase was allowed to proceed for 1 h and for 3 h. The MBP phosphopeptide (APRpTPGGRR) at m/z 1048.1 was reacted under the conditions, as described in Fig. 3F, except that the Michael addition was allowed to proceed overnight for 14 h at 37°C. MALDI-TOF spectra (A) of unmodified EGFR phosphopeptide; (B) after 2 h consecutive reaction; and (C) after 3 h consecutive reaction. (A, inset) The spectrum of the β-eliminated peptide at m/z 2300.9. (B, inset) The spectrum obtained after 1 h Michael addition. Note the relatively high efficiency of the overall reaction observed after 2.5 h of incubation. (B, arrow) Residual β-elimination product. Minor ions in A–C reflect sample heterogeneity. MALDI-TOF spectra (D) of unmodified MBP phosphopeptide; (E) after β-elimination; and (F) after 14 h Michael addition at 37°C. (F, arrow) Minor base-induced hydrolysis product. One-tenth of the eluates corresponding to ∼2 pmol peptide was applied to the target.
In a separate experiment, the MBP phosphopeptide was β-eliminated for 30 min at 55°C or for 2 h at 37°C and then converted at 37°C overnight for 14 h. As shown in Fig. 4D–F, the peptide reacted to completion and was efficiently recovered from the solid phase, suggesting that prolonged alkali exposure of the silica-based resin had little impact on the use of support (Fig. 5) This peptide contains the glycine-glycine sequence feature that had been identified in a study by Noll et al.39 as susceptible to base-induced hydrolysis. However, an ion corresponding in mass to the expected cleavage product was not observed in the spectrum (Fig 4F). Unexpectedly, cleavage occurred, albeit to a minor extent, C-terminal to the proline residue, giving rise to the low-intensity ion at m/z 858.8 (Fig 4F, arrow). The modified protocol offers the option to increase the sample throughput when needed.
Figure 5.

ZipTipC18 pipette tip alkali-compatibility evaluation. Angiotensin I [DRVYIHPFHL at m/z 1296.5 (50 pmol)] was immobilized on ZipTipC18 pipette tips that were washed briefly to remove unbound material. The tips were then exposed to the consecutive β-elimination/Michael conditions, as described in Fig 3E, or left untreated. Peptides with and without incubation were desorbed, methyl-d3-esterified, and mixed in equimolar amounts with the methyl-d0-esterified reference peptide. Experiments were in triplicate. MALDI-TOF spectra were acquired using 400 laser shots/spectrum, accumulated from five different spot positions. MALDI-TOF spectra of (A) methyl-d3-esterified peptide from untreated sample mixed with methyl-d0-esterfied internal reference peptide and (B) methyl-d3-esterified peptide from alkali-exposed sample mixed with methyl-d0-esterified internal reference peptide. Labels in A and B designate the relative peptide-abundance ratios. The alkali-induced minor sample loss is highlighted with a box. Approximately 0.5 pmol peptides was applied to the target.
We recognize the need for expanded data to fully establish the general use of the method. However, this is, thus far, the first demonstration of a method enabling efficient conjugation of a nucleophile to phosphothreonyl residues adjoined by proline.29 Notably, the phosphothreonyl peptides were derivatized faster and more efficiently with this protocol than with the protocols reported in the literature.14,15,28 Accordingly, we have adopted this protocol as standard procedure in our ongoing work to map phosphorylation sites at serine and threonine in experimental digest. The more stringent reaction conditions, as described in Fig. 4C, were optionally used in separate experiments to afford enhanced detection of phosphopeptide candidates derived from proline-directed kinase substrates. Alternatively, the reaction conditions described in Fig. 4D–F may be used to drive the derivatization of phosphothreonine-proline sequences to completion. In the practical implementation of this dual-reaction scheme, when aimed at the conservation of low-level samples, the digest (<5 pmol) was split in two equal portions and immobilized on ZipTipμ-C18 pipette tips. One ZipTip was then exposed to the reaction condition described in Fig. 4B, whereas its untreated counterpart was set aside as a control and then deposited along with the derivatized sample onto the MALDI target. After mass analysis, the MALDI spot of the control digest was solubilized directly on the target (see Methods and Materials). The material was then adsorbed from the target on a ZipTipμ-C18 pipette tip and submitted in a second pass manner to the more stringent chemical treatment. In this manner, only two samples were consumed to furnish the desired MS data.
As mentioned previously, the use of ZipTipμ-C18 pipette tips as reaction beds enables the entire sample to be deposited onto the MALDI-plate. By this means, 200 fmol CaMK2 substrate phosphopeptide spiked into 2 pmol of a OVA/α-S1 casein digest was readily identified in the background of the nonphosphorylated species, suggesting that this sample handling format may provide for a tool to characterize substoichiometric phosphorylation events (<20%). In other experiments, the immobilized peptide mixture was subjected to concurrent β-elimination with Michael addition using the protocol described in Fig. 2B and analyzed by MALDI-MS. The phosphoseryl peptide components of the mixture were recognized in the mass maps by the resultant signal enhancement, consistent with earlier data,17 whereas the phosphothreonyl peptide remained under these mild reaction conditions, essentially underivatized, and hence, was excluded from identification. Consequently, this reaction condition modulation may be exploited as a tool to differentiate between protein serine and protein threonine phosphorylation.
The use of the protocol is expected to be readily expanded to screen digest for the presence of O-glycosylated or O-sulfonated peptides, as O-GlcNAc and O-sulfonyl groups have been shown to undergo β-elimination with essentially the same kinetics as their phosphorylated counterparts15,40 (see Fig. 2C–E). It has been reported that the nearly complete gas-phase elimination of the sulfono moiety (80 Da) upon CID results in fragment ion spectra, which are identical to those of the unmodified peptides. Likewise, the sulfono group is poorly retained under the conditions of ETD and hence, precluded in most cases; sulfonation-site determination highlights the advantage of the chemical proteomics approach to characterize this post-translational modification.40 Furthermore, the protocol is expected to be applicable to peptide conjugation with alternative water-soluble nucleophiles, such as mercapto-functionalized mono-N-(2-mercaptoethyl)amide of 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA).41 Unexpectedly, this bulky reagent proved to be as reactive toward dehydroalanine as the 2-aminoethanethiol nucleophile.41 Because of the chelating property of the DOTA moiety, the peptide conjugates are expected to reversibly bind to metal-ion solid supports, which may provide a means for their enrichment from peptide mixtures. Additionally, these DOTA-peptide conjugates may find application as element-coded tags in MS.42
Chemistry Side-Reactions
Alkali-catalyzed peptide-bond hydrolysis is of concern, as it could introduce unwanted complexity into the spectra, confounding data interpretation. Amide bonds in model dipeptides most prone to hydrolysis at pH 12.6 were identified as those linking glycine to glycine and glycine to C-terminal serine.39 The MALDI-MS spectra, prepared from the UOM9 phosphopeptide (KRPpSQRHGSKY amide) and the MBP phosphopeptide (APRpTPGGRR) under the alkaline conditions (pH 10.6) described in Fig. 3F, were devoid of ions that correspond to the predicted cleavage products (data not shown). A tryptic digest prepared from an equimolar mixture of HSA, bovine carbonic anhydrase, bovine lactoglobulin, and bovine myoglobin was then examined under these conditions or left untreated as a control. A total of 640 laser shots sampled from eight different spot positions were summed for each MALDI-TOF spectrum. Eighty laser shots/spot were typically required to obtain stable signals. Comparison of the complex tryptic mass maps revealed only minor ion-intensity changes in the spectra (<15%), indicating that the peptides were alkali-stable (results not illustrated). Furthermore, these spectra were devoid of mass signals corresponding to dehydroderivatives of serine and threonine, as were the spectra produced from the EGFR phosphopeptide (KRELVEPLpTPSGEAPNQALLR amide; Fig. 4B and C), the calcitonin fragment (DFNKFHpTFPQTAIGV), the cholecystokinin fragment (IKNLQpSLDPSH), and the UOM9 phosphopeptide (KRPpSQRHGSKY amide; data not shown). This side-reaction is promoted at an elevated temperature (50°C) by considerably more alkaline-reaction conditions than used in our protocols (i.e., 0.52 N sodium hydroxide).43 Ions corresponding to water addition across the double-bond of the β-eliminated residues, which would regenerate the hydroxyl amino acid residues, were not observed in any of the spectra. This side-reaction is known to proceed at a markedly slow reaction rate, requiring 24 h to yield detectable product levels (<20%).12 The data show that the reaction conditions preserved the integrity of the samples.
ZipTipC18 Pipette Tips Alkali-Compatibility Evaluation
Alkali-induced silica resin decomposition has been reported to impact to a minor extent (∼2%) the retention of the silane-bonded stationary phase upon long-term C18 reversed-phase column operation (>1 week) with mobile phases at pH 9–12.3.44 To examine whether this effect could potentially reduce analyte recovery from the silica-based ZipTip support, we assessed the relative recovery of the model peptide angiotensin I (DRVYIHPFHL, m/z 1296.6) from ZipTips before and after the alkali treatment using a modified version of an earlier-reported stable isotope-dilution technique.17 To assess the relative peptide recovery from untreated solid-phase support, 50 pmol peptide was immobilized in replicates, washed to remove unbound material, eluted, methyl-d3-esterified, and mixed with the methyl-d3-esterified internal reference peptide. As estimated from the spectral-abundance ratio calculated for the replicates, an average of 85.3% of peptide was reproducibly recovered in the eluted fractions (Fig. 5A). The reproducibility and high efficiency of peptide recovery from untreated resin noted here agree with an earlier evaluation of ZipTipc18 pipette tips using 14C-accelerator MS for peptide quantitation.45
To assess the relative peptide recovery after alkali exposure, ZipTipC18 pipette tips were loaded with 50 pmol peptide and washed briefly. Samples were then carried through the consecutive β-elimination/Michael addition reaction scheme, as described in Fig. 3F, desalted, and eluted. The eluates were methyl-d3-esterified, reconstituted, and mixed with the methyl-d0-esterified peptide internal reference. As estimated from the spectral-abundance ratio calculated for the replicates, an average of 72.4% peptide could be reproducibly recovered from the chemically treated supports (Fig. 5B). The comparison data show that a small fraction of the sample (average 13.9%) could not be retrieved from the support after the alkali exposure. Taken together, the overall benefits gained from this sample preparation format more than compensate for this minor sample loss.
The potential impact of the alkali treatment on peptide retention during guanidination and acetylation was assessed in a semiquantitative mode by MALDI-MS analysis. Fibrinopeptide B (pyrGVNDNEEGFFSAR), N-terminally blocked by pyroglutamate and devoid of lysine, was selected as a model peptide. The peptide was loaded onto ZipTipC18 pipette tips and incubated in the alkaline O-methylisourea hemisulfate solution for 10 min at 65°C (pH 10.6) or with sulfo-NHS acetate for 20 min at 55°C (pH 8.0). Untreated samples were left aside as controls. A total of 640 laser shots sampled from eight different spot positions was averaged for each spectrum. The MALDI-TOF spectra showed that the peptides were recovered at comparable signal strength from the alkali-exposed and the untreated samples, suggesting that the peptide retention on the reversed-phase support was unaffected by the chemical treatment (results not shown).
Phosphorylation Site Mapping by MALDI Tandem TOF (TOF/TOF)-MS/MS Analysis
Fragmentation of phosphopeptides by CID using MALDI-TOF or electrospray MS/MS commonly results in preferential fragmentation of the phosphate ester bond. As exemplified by the MALDI-TOF/TOF product spectrum of the model peptide P4 (GIKSHNpSALYSQVQK, m/z 1739.3), the ensuing neutral loss event of phosphoric acid precluded phosphorylation-site determination for lack of informative fragmentation, the main obstacle to phosphorylation-site analysis by CID (Fig. 6A). In contrast, the product ion spectrum of the peptides' thiol adduct at m/z 1719.4 displayed a nearly uninterrupted b and y ion series produced in high abundance, including y9 and b7, representing the first y and b ions that contain the modification, as well as y8 and b6 that are contiguous to the modification (Fig. 6B). The location of the modification could be readily identified by its unique residue mass (146 Da) and hence, serine in Position 7 as the site of phosphorylation. Ions corresponding to dehydroproducts of serine or a diagnostic for deamidation of asparagine and glutamine were not observed in the spectrum. Importantly, the tag remained stable under the conditions of low-energy, collisionally induced fragmentation. The data underscore the use of the chemical proteomics approach as a viable means to render protein digests amenable to high-fidelity phosphorylation-site determination.
Figure 6.
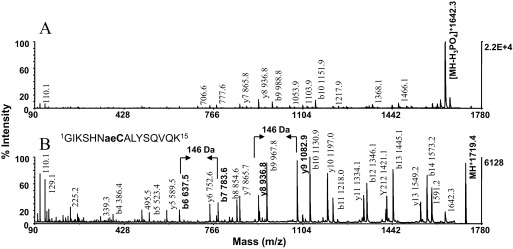
Phosphorylation-site mapping by MALDI-MS/MS. The model peptide P4 (GIKSHNpSALYSQVQK, m/z 1739.3) was subjected to β-elimination with concurrent Michael addition using 2-aminoethanethiol as nucleophile. MALDI-TOF/TOF spectra of (A) unmodified peptide and (B) S-2-aminoethylcysteine derivative. Note that the neutral loss of phosphate from the parent as the major fragmentation pathway is precluded by the incorporation of the nucleophile. As a result, the nucleophilic substitution markedly increased peptide-backbone fragmentation, yielding a nearly uninterrupted b- and y-type ion series in high abundance, and discriminates the site of phosphorylation as the unique residue mass of 146 Da. This mass signature is contained in the product ions y9 and b7, indicated in bold in the spectra, as are y8 and b6, which are contiguous to the modification. The presence of the ion pair b6, b7 and the ion pair y8, y9 in the spectra afforded unambiguous assignment of serine in Position 7 as the site of phosphorylation. Cae, S-2-aminoethylcysteine. Note stability of the label. Approximately 1 pmol native peptide and derivative was applied to the target.
Solid-Phase Dephosphorylation
AP treatment of phosphoprotein digests has been an early method to detect phosphopeptides in MALDI-MS by the resultant, characteristic mass shift of 80 Da/released phosphate.5 In our adaptation of this procedure, the substrate was immobilized on ZipTipC18 pipette tips or ZipTipμ-C18 pipette tips abrogating the need for detrimental sample dry-down, as practiced in current in-solution digestion protocols. The ZipTips were then briefly rinsed to remove salts and contaminants that might interfere with the enzymatic reaction. Aliquots of the enzyme solutions were then loaded onto the supports. After incubation, the digestion was terminated by a solvent wash, and eluates were analyzed by MALDI-TOF. An alternative procedure, which we examined initially, on MALDI target enzymatic dephosphorylation,33 resulted in very limited or no digestion when applied to peptides bearing basic amino acids in close proximity to the phosphorylated residues (e.g., PKC substrate-3 phosphopeptide: KRPpSQRHGSKY amide). This finding was in agreement with an earlier report.46
Representative data from the experiments are shown in Fig. 7. Mixtures of the β-casein fragment T6 (FQpSEEQQQTEDELQDK) at m/z 2061.5 and the CaMK2 substrate phosphopeptide (KKALRRQEpTVDAL) at m/z 1608.3, adsorbed in equimolar amounts to ZipTipC18 pipette tips, were incubated for 15 min at 37°C in a 2-U/μl or in a 4-U/μl AP solution. As shown in Fig. 7A–C, complete removal of phosphate from the phosphothreonyl peptide occurred at the higher enzyme concentration, which is in accord with an earlier observation that the phosphate group is released more readily from phosphoseryl peptides than from the phosphothreonyl counterparts.47 In fact, T6 was already completely dephosphorylated at the enzyme concentration of 0.8 U/μl after 15 min incubation at 37°C, whereas ∼50% of the CaMK2 substrate phosphopeptide remained intact under these conditions (results not shown). Complete removal of phosphate from the model peptide P2 (RRQpSPVA) at m/z 836.8, in which an arginine residue is positioned in close proximity to the phosphorylated residue, required 2 h incubation at 37°C in a 4-U/μl enzyme solution (Fig. 7A and C, insets). Over the same time period, 8 U/μl was needed to complete the digestion of the UOM9, PKC substrate phosphopeptide (KRPpSQRHGSKY amide), and at the same enzyme concentration, the incubation had to be extended for an additional 2 h to afford efficient dephosphorylation of the UOM11, PKC substrate phosphopeptide (KRpTIRR) and of the PKC substrate-5 phosphopeptide (RRGRpTGRGRRGIFR; data not shown). Although AP is known to exhibit broad substrate specificity, the data show that peptides with basic amino acid residues flanking the site of phosphorylation required progressively more stringent digestion conditions to drive the enzymatic reaction to completion.
Figure 7.
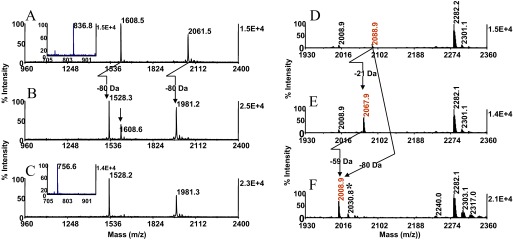
Solid-phase dephosphorylation. Peptides were adsorbed on ZipTipC18 pipette tips, washed briefly, and loaded with AP solutions. After digestion, the samples were desalted. MALDI-TOF spectra (A) of a mixture of 8 pmol β-casein tryptic peptide T6 (FQpSEEQQQTEDELQDK) and 10 pmol CaMK2 substrate phosphopeptide (KKALRRQEpTVDAL); (B) after incubation for 15 min at 37°C in 2 U/μl; and (C) after incubation for 15 min at 37°C in 4 U/μl. (A and C, insets) The spectrum of 20 pmol P2 (RRQpSPVA) before and after incubation for 2 h at 37°C in 4 U/μl, respectively. (B, arrow) Residual, undigested material. Cross-arrows indicate the 80-Da mass shift/phosphate. Note comparable ionization efficiency of P2 before and after digestion. One-tenth of the eluates from the mixture, and 1/20 of the eluates from the single species were applied to the target. A tryptic in-gel digest of OVA (10 pmol), immobilized onto a ZipTipC18 pipette tip, was and subjected to β-elimination with concurrent Michael addition, as described in Fig. 2A and B. An equimolar amount of digest was immobilized and exposed to 2 U/μl AP for 15 min at 37°C. MALDI-TOF spectra (D) of untreated digest; (E) after derivatization; and (F) after AP treatment. Masses highlighted in red designate the phosphopeptide at theoretical m/z 2088. 9, its thiol adduct at m/z 2067.8, and its dephosphorylation product at m/z 2008.8. Cross-arrows indicate the mass shifts that could not be unambiguously ascertained. (E and F, arrows) Mass signature of 59 Da diagnostic for the presence of the phosphopeptide in the digest. Asterisk denotes sodium adduct. One-tenth of the eluates corresponding to ∼1 pmol of the digest was applied to the target.
We next evaluated this protocol with an in-gel OVA digest, of which ∼20 pmol was immobilized on the ZipTipC18 pipette tip and treated with 2 U/μl AP for 15 min at 37°C. MALDI-MS analysis of this sample revealed a prominent signal at m/z 2008.9, identified by the mass shift of 80 Da as the dephosphorylated species. Comparable results were obtained with digests of α-S1 casein, an additional acidophilic kinase substrate (results not shown). Complete removal of phosphate from the tryptic fragments recovered from Maf1, a basophilic kinase substrate, required more stringent conditions (i.e., 4 U/μl/2 h/37°C). Dephosphorylation of these fragments (e.g., RDpSNSFWEQKR) had only a minor impact on the ionization of these highly basic peptides (unpublished results). Taken together, the solid-phase procedure combines analyte concentration, enzymatic digestion, and sample clean-up into a simple and highly efficient operation, eliminating the limitations associated with the on-target dephosphorylation protocol.
As discussed previously, phosphopeptide identification relies on inspecting the mass maps prepared before and after derivatization for ions that differ in mass by −21 Da, indicative of thiol-adduct formation. At low sample levels (10 pmol or less of protein applied to the gel), the native monophosphorylated peptide in an in-gel digest of OVA could not be unambiguously discerned from the background noise (Fig. 7D) but remained clearly detectable as its tagged counterpart (Fig. 7E) and ionized at comparable high abundance in its dephosphorylated form (Fig. 7F). Owing to the specificity of the enzyme, the mass difference of 59 Da between this ion pair is a signature diagnostic for the identity of the phosphopeptide's thiol adduct and therefore, for the presence of the phosphopeptide in the digest. We have adopted the use of on-resin dephosphorylation as an orthogonal method to confirm the identity of the species of interest in our ongoing effort to map phosphorylation sites in proteins of limited structural information or whose sequences are not found in current databases. In the practical implementation of this strategy, the starting material may be retrieved from the mass-analyzed MALDI spot on a ZipTipμ-C18 pipette tip and subjected to the enzymatic treatment (see Methods and Materials). In this manner, only two samples need to be committed for analysis. In an expanded application of this protocol, the OVA digest was treated with AP and subsequently submitted to concurrent β-elimination with Michael addition. MALDI-MS of this sample revealed that the digestion product was unaffected by the derivatization (data not illustrated). Accordingly, only glycosylated or sulfonated peptides in a mixture with a phosphopeptide would be labeled by the nucleophile. We have been exploring the use of this sequential reaction scheme as a tool to render glycosylated or sulfonated peptides in mixtures with phosphopeptides amenable to selective affinity purification.
Sample Preparation for Chemoenzymatic O-Modification Site Determination
In this strategy, the substituted phospho/O-GlcNAc/O-sulfono moiety, since mimicking a lysine residue, can be cleaved on the C-terminal side of the site by Lys-C, enabling O-modification-site localization by mass-matching the cleavage products to the known protein sequence. To confine the proteolysis to the lysine analog, internal lysine residues in tryptic miscleavage products and in nontryptic proteolytic fragments need to be blocked efficiently. To this purpose, guanidination or acetylation preceding β-elimination with Michael addition was commonly used.14,15 Intermediates and final reaction products were in this step-wise, in solution procedures purified by microdialysis14 or reversed-phase HPLC,15 followed by SpeedVac concentration of the desalted fractions, manipulations that may incur substantial adsorptive sample loss.31 In the solid-phase adaptation of this serial reaction scheme, traditional sample purification was strictly avoided, as reagents were exchanged directly on the support and removed at the conclusion of a nucleophilic conversion step by a brief solvent wash (see Fig. 1A). Consequently, the analyte could be carried through the consecutive reactions with minimal adsorptive sample loss, as demonstrated by the experiment described below.
To explore the efficiency of the solid-phase sample preparation method, a tryptic digest of α-S1 casein (2 pmol), bound to a ZipTipμ-C18 pipette tip, was exposed to a phosphate- buffered 20 mM sulfo-NHS acetate solution (pH 8.0) for 20 min at 55°C. The reagent was then replaced on the support with 66 mM barium hydroxide/33 mM 2-aminoethanethiol. After 1 h incubation at 37°C, the sample was desalted and deposited in matrix onto the MALDI plate. Comparison of the mass spectra acquired before and after acetylation, coupled to β-elimination with concurrent Michael addition, showed that the acetylation reaction resulted in the expected mass additions of 42 Da/amine. Ions corresponding to underivatized material were not observed in the spectrum, indicating that the reactions proceeded to completion (Fig. 8A and B). Accordingly, the phosphopeptides recognized in the initial spectrum at m/z 1660.8 (121VPQLEIVPNpSAEER134) and its miscleavage product at m/z 1951.9 (19YKVPQLEIVPNpSAEER134), carrying an internal lysine, were recovered at m/z 1682.2 and m/z 2014.8 as fully monoacetylated and diacetylated 2-aminoethyllcysteine analogs, respectively. Consistent with an earlier report,35 guanidination of the immobilized protein digest proved as effective as acetylation to block the miscleavage product's internal lysine.
Figure 8.
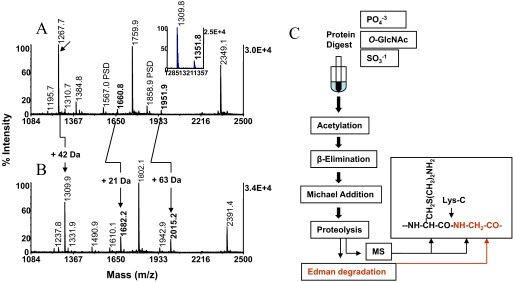
Preparation of substrates for phosphorylation-site determination by chemical targeted proteolysis and chemoenzymatic solid-phase workflow. ZipTipμ-C18 pipette tip-bound α-S1 protein digests (2 pmol) were subjected to on-column acetylation for 20 min at 55°C and subsequently to concurrent β-elimination with Michael addition for 1 h at 37°C. The ZipTips were eluted in matrix onto the MALDI plate. MALDI-TOF spectra of (A) unmodified digest and (B) digest after acetylation and subsequent β-elimination with concurrent Michael addition. Cross-arrows designate the native phosphoseryl peptide at m/z 1660.8 and its miscleavage product at m/z 1951.9, respectively, and their acetylated thiol adducts at m/z 1682.2 and at m/z 2015.2, respectively. Note the characteristic net mass addition of 21 Da and of 63 Da imparted to the native peptides and its miscleavage product, respectively, and completeness of serial derivatization. (A, arrow) Tryptic peptide at m/z 1267.7, corresponding to YLGYLEQLLR, residues 106–115, susceptible to low-level O-acylation. (A, inset) Expanded section containing the peptide's N-acetylated derivative and its O-acylation product at m/z 1351.8. Note reversal of this side-reaction upon subsequent β-elimination with Michael addition by base-induced ester-bond hydrolysis. (C) Schematic representation of the chemoenzymatic solid-phase workflow and postreaction phosphorylation/O-glycosylation/O-sulfonation-site determination by MS and/or Edman degradation. (C, inset) The structure of 2-aminoethylcysteine, adjoined by glycine with a Lys-C-sensitive cleavage site indicated by an arrow. Note that amine-blocking by acetylation affords selective Edman degradation of the cleavage product C-terminal to the site of the chemically modified residue. MS, MALDI-MS or optional LC-MS/MS.
The tryptic peptide at m/z 1267.7 corresponding to 106YLGYLEQLLR115 proved susceptible to O-acylation, a well-documented NHS-ester side-reaction targeting hydroxyl amino acid residues (Fig. 8A, inset.48 The resultant minor product at m/z 1351.8 was not observed after β-elimination with Michael addition, as a result of base-induced ester-bond hydrolysis of the O-linked derivative. If N-acetylation were to be used as a general amine-labeling method, the side-reaction can be readily reversed in situ via ester-bond hydrolysis by a brief hydroxylamine treatment.48 In the solid-phase reaction format, the acetylation reagent was replaced on the support by alkaline 2% hydroxyl amine. The ZipTip was then incubated for 15 min at 37°C, while immersed in the reagent solution. As an application, the method provides for a viable tool to make lysine-rich proteolytic fragments amenable to sequence determination by CID and to differentiate in CID spectra this residue from isobaric glutamine.49 Taken together, the results validate the use of the on-resin sequential derivatization to prepare phosphoprotein digests for chemoenzymatic phosphorylation-site determination. By analogy, the scope of this sample-preparation method may be extended to digest prepared from O-GlcNAcylated and O-sulfonated proteins.
As acetylation, in contrast to guanidination, blocks α- and ε-amino groups, the Lys-C cleavage product, immediately C-terminal to the substituted phosphate, becomes the sole peptide in the digest, bearing a free amino group (see Fig. 8C, inset). Consequently, chemical sequence analysis offers a means for phosphorylation/O-glycosylation/O-sulfonation-site determination in complex peptide mixtures.15 In this strategy, the O-modification sites are ascertained by alignment of the Edman sequence tag with the known protein sequence. Sorting of Edman sequence readouts derived from peptide mixtures has been demonstrated using algorithms developed for this task, thus facilitating the localization of multiple phosphorylation sites distributed along the polypeptide chain.50 Although the chemoenzymatic strategy combined with chemical sequencing extends the use of the method to complex protein digests, at least 1 pmol peptide is typically needed to obtain readily interpretable midfemto-mol-level sequence data, confining the use of the method to analyze substoichiometric phosphorylation events (<50%) to relatively high sample levels (>10 pmol). Accordingly, the benefits of this approach are best exploited for the analysis of low-level amounts of in vitro phosphorylated proteins.
Current and Future Developments
As noted in the course of method optimization, the nearly complete conversion of the CaMK2 substrate phosphopeptide to its thiol adduct remained unaffected when the pH of the addition medium was lowered from 10.6 to 8.6. In pilot experiments, we exploited this finding to explore other gentler reaction conditions using Tris (hydroxymethyl) aminomethane as the base component in the Michael addition medium. This reagent is comparable in its moderate alkalinity with Tris-HCl buffer systems (i.e., 0.2 M Tris-HCl, pH 7.8–8.6) normally used for tryptic protein digestion. Notably, elevated temperature protein digestion (i.e., 50–58°C) in such buffer systems has been exploited to afford increased throughput of analysis without compromising the integrity of the samples.51 In our modified protocol, the CaMK2 substrate phosphopeptide was first exposed to 50 mM barium hydroxide for 2 h at 37°C conditions that proved effective to afford complete β-elimination of all peptides listed in Materials and Methods. The addition reaction was then allowed to proceed for 2 h at 55°C. The phosphopeptide as well as the phosphoseryl/threonyl peptides listed in Materials and Methods reacted to completion, and hydrolytic degradation products were not observed in the spectra. Under the same conditions, EGFR phosphopeptide, containing the phosphothreonyl-proline sequence, formed a single species corresponding to the β-methyl-S-2-aminoethylcysteine analog, indicating that the addition phase proceeded to near completion. We are currently evaluating this protocol with a panel of additional phosphopeptides containing the phosphothreonine-proline sequence feature with a future aim to exploit these mild, overall reaction conditions for the analysis of digests from experimental samples.
Conclusions
We earlier adapted the barium hydroxide catalyzed β-elimination/Michael addition reaction to solid-phase derivatization on ZipTipC18 pipette tips using 2-aminoethanethiol as nucleophile and demonstrated its feasibility of this concurrent reaction method to identify selectively, by MS phosphoseryl peptides in unfractionated protein digests, and to exploit the derivatives for facile phosphorylation-site determination. Here, we have expanded the evaluation of the solid-phase chemical strategy to phosphothreonyl peptides, to phosphopeptides derived from substrates targeted by proline-directed kinases, and to O-GlcNAc peptides modified at serine and threonine. An experimental parameter explored during chemistry optimization included nucleophile concentration, reactant basicity, temperature, and duration of reaction. Sample processing on the solid phase in the consecutive reaction mode was adopted to optimize the Michael addition reaction for throughput and completeness of derivatization. Notably, this is thus far the first demonstration of a method enabling efficient conjugation of a nucleophile to phosphothreonyl peptides adjoined by proline. The analyte remained intact during derivatization and was recovered efficiently from the silica-based, reversed-phase support with minimal sample loss. The solid-phase format readily accommodated the use of AP and was used as an orthogonal method to confirm the identity of phosphopeptides in dilute protein digests. The use of the solid-phase approach to prepare substrates for phosphorylation-site determination by chemical-targeted proteolysis was demonstrated with low-level amounts of protein digests. The solid-phase method provides for a simple, rugged, and efficient tool to prepare samples for comprehensive phosphopeptide screening by differential MALDI-MS mass mapping using standard equipment available in most biological laboratories. The solid-phase analytical platform is expected to accommodate readily digests prepared from O-glyco- and O-sulfonoproteins for MS-based modification-site determination and additional chemistries to expand the scope of solid-phase derivatization for peptide structural characterization.
ACKNOWLEDGMENTS
This work has been funded by the U.S. National Institutes of Health grants R33CA101150 and P20-DA026149 to R.H.A. The authors thank the Albert Einstein College of Medicine for generous support, Dr. Richard Stanley for helpful discussions, and Ms. Junko Hihara for editorial assistance in preparing the manuscript.
DISCLOSURE
The authors declare no conflict of interest associated with financial support.
REFERENCES
- 1. Helmbrecht K, Zeise F, Rensing L. Chaperones in cell cycle regulation and mitotic signal transduction: a review. Cell Proliferation 2000;33:341–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Annan RS, Carr SA. Phosphopeptide analysis by matrix-assisted laser desorption time-of-flight mass spectrometry. Anal Chem 1996;68:3413–3421 [DOI] [PubMed] [Google Scholar]
- 3. Annan RS, Huddleston MJ, Verma R, Deshaies RJ, Carr SA. A multidimensional electrospray MS-based approach to phosphopeptide mapping. Anal Chem 2001;73:393–404 [DOI] [PubMed] [Google Scholar]
- 4. Tholey A, Reed J, Lehmann W. Electrospray tandem mass spectrometric studies of phosphopeptides and phosphopeptide analogues. J Mass Spectrom 1999;34:117–123 [DOI] [PubMed] [Google Scholar]
- 5. Liao PC, Leykam J, Andrews PC, Gage DA, Allison J. An approach to locate phosphorylation sites in a phosphoprotein: mass mapping by combining specific enzymatic degradation with matrix-assisted laser desorption/ionization mass spectrometry. Anal Biochem 1994;219:9–20 [DOI] [PubMed] [Google Scholar]
- 6. Larsen MR, Thingholm TE, Jense ON, Roepstorff P, Jorgensen JD. Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide micro columns. Mol Cell Proteomics 2005;4:873–886 [DOI] [PubMed] [Google Scholar]
- 7. Beausoleil SA, Jedrychowki M., Schwartz D. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci USA 2004;101:12130–12135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Molina H, Horn DM, Tang N, Mathivanan S, Pandey A. Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc Natl Acad Sci USA 2007;104:2199–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jedrychowski MP, Huttlin EL, Haas W, Sowa ME, Rad R, Gygi SP. Evaluation of HCD- and CID-type fragmentation within their respective detection platforms for murine phosphoproteomics. Mol Cell Proteomics 2011;10:M111.009910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jaffe H, Veeranna, Harish CP. Characterization of serine and threonine phosphorylation in β-elimination/ethanethiol addition-modified proteins by electrospray tandem mass spectrometry and database searching. Biochemistry 1998;37:16211–16224 [DOI] [PubMed] [Google Scholar]
- 11. Shen J, Smith RA, Stoll VS. Characterization of protein kinase A phosphorylation: multi-technique approach to phosphate mapping. Anal Biochem 2004;324:204–218 [DOI] [PubMed] [Google Scholar]
- 12. Thompson AJ, Hart SR, Franz C, Barnouin K, Ridley A, Cramer R. Characterization of protein phosphorylation by mass spectrometry using immobilized metal ion affinity chromatography with on-resin β-elimination and Michael addition. Anal Chem 2003;75:3232–3243 [DOI] [PubMed] [Google Scholar]
- 13. Reinders J, Meyer HE, Sickman A. Applications of highly sensitive phosphopeptide derivatization methods without the need of organic solvents. Proteomics 2006;6:2647–2649 [DOI] [PubMed] [Google Scholar]
- 14. Knight ZA, Schilling B, Row RH, Kenski DM, Gibson BW, Shokat KM. Phosphospecific proteolysis for mapping sites of protein phosphorylation. Nat Biotech 2003;21:1047–1054 [DOI] [PubMed] [Google Scholar]
- 15. Rusnak F, Zhou J, Hathaway GM. Identification of phosphorylated and glycosylated sites in peptides by chemically targeted proteolysis. J Biomol Tech 2002;13:228–237 [PMC free article] [PubMed] [Google Scholar]
- 16. Nika H, Hawke DH, Kobayashi R. Derivatization on reversed-phase supports for enhanced detection of phosphorylated peptides. 52nd American Society for Mass Spectrometry Conference on Mass Spectrometry & Allied Topics, Nashville, TN, USA Abstract ThPN 292, 2004 [Google Scholar]
- 17. Nika H, Lee JH, Willis IM, Angeletti RH, Hawke DH. Phosphopeptide characterization by mass spectrometry using reversed-phase supports for solid-phase β-elimination/Michael addition. J Biomol Tech 2012;23:2012–2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pluskal MG. Microscale sample preparation. Nat Biotech 2000;18:104–105 [DOI] [PubMed] [Google Scholar]
- 19. Aitken A. Sample cleanup by solid phase extraction/pipet-tip chromatography. In Walker J. (ed): The Proteomics Protocols Handbook, 1st ed Totowa, NJ, USA: Humana, 2005;307–309 [Google Scholar]
- 20. Conrotto P, Hellman U. Sulfonation chemistry as a powerful tool for MALDI TOF/TOF de novo sequencing and posttranslational modification analysis. J Biomol Tech 2005;16:441–452 [PMC free article] [PubMed] [Google Scholar]
- 21. Cindric M, Cepo T, Skrlin A, Vuletic M, Bindila L. Accelerated on-column lysine derivatization and cysteine methylation by imidazole reaction in a deuterated environment for enhanced product ion analysis. Rapid Commun Mass Spectrom 2006;20:694–702 [DOI] [PubMed] [Google Scholar]
- 22. Farmar JG, Nika H, Che F-Y, Weiss L, Hogue Angeletti R. IVICAT for masses: an improved technique for permethylation of peptides. J Biomol Tech 2009;20:285–292 [PMC free article] [PubMed] [Google Scholar]
- 23. Johnson ME, Carpenter TS. The use of solid-phase supports for derivatization in chromatography and spectroscopy. Appl Spectrosc Rev 2005;40:391–412 [Google Scholar]
- 24. Jemere AB, Oleschuk RD, Ouchen P, Fajuyigbe F, Harrison DJ. An integrated solid-phase extraction system for sub-picomolar detection. Electrophoresis 2002;23:3537–2544 [DOI] [PubMed] [Google Scholar]
- 25. Ubersax JA, Ferrrell JE. Mechanism of specificity in protein phosphorylation. Nat Rev Mol Cell Biol 2007;8:530–541 [DOI] [PubMed] [Google Scholar]
- 26. Ryo A, Liou Y-C, Lu KP, Wulf G. Prolyl isomerase Pin1: a catalyst for oncogenesis and a potential therapeutic target in cancer. J Cell Sci 203;116:773–783 [DOI] [PubMed] [Google Scholar]
- 27. Hilden TJ, Valmu L, Karkainen S, Gahmberg CG. Threonine phosphorylation sites in the β2 and β7 leucocyte integrin polypeptide. J Immunol 2003;170:4170–4177 [DOI] [PubMed] [Google Scholar]
- 28. Molloy MP, Andrews PC. Phosphopeptide derivatization signatures to identify serine and threonine phosphorylated peptides by mass spectrometry. Anal Chem 2001;73:5387–5394 [DOI] [PubMed] [Google Scholar]
- 29. Thaler F, Valsasina B, Baldi R. A new approach to phosphoserine and phosphothreonine analysis in peptides and proteins: chemical modification, enrichment via solid-phase reversible binding, and analysis by mass spectrometry. Anal Bioanal Chem 2003;376:366–373 [DOI] [PubMed] [Google Scholar]
- 30. Wells L, Vosseller K, Cole RN, Cronshaw JM, Matunis MJ, Hart GW. Mapping sites of O-GlcNAc modification using affinity tags for serine and threonine post-translational modifications. Mol Cell Proteomics 2002;1:791–804 [DOI] [PubMed] [Google Scholar]
- 31. Speicher DT, Kolbas O, Harper D, Speicher DW. Systematic analysis of peptide recoveries from in-gel digestions for protein identifications in proteome studies. J Biomol Tech 2000;11:74–86 [PMC free article] [PubMed] [Google Scholar]
- 32. Beardsley RL, Reilly JP. Optimization of guanidination procedures for MALDI mass mapping. Anal Chem 2002;74:1884–1890 [DOI] [PubMed] [Google Scholar]
- 33. Larsen MR, Sorensen GL, Fey SJ, Larsen PM, Roepstorff P. Phospho-proteomics: evaluation of the use of enzymatic de-phosphorylation and differential mass spectrometric peptide mass mapping for site specific phosphorylation assignment in proteins separated by gel electrophoresis. Proteomics 2001;1:223–238 [DOI] [PubMed] [Google Scholar]
- 34. Baumgart S, Lindner Y, Kuhne R, Oberemm A, Wenschu H, Krause E. The contribution of specific amino acid side chains to signal intensities of peptides in matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun Mass Spectrom 2004;18:863–868 [DOI] [PubMed] [Google Scholar]
- 35. Nika H, Hawke DH, Kobayashi R. Derivatization on reversed phase supports for chemoenzymatic phosphorylation site determination. Association of Biomolecular Resource Facilities 2005, Biomolecular Technologies: Discovery to Hypothesis, Savannah, GA, USA: Abstr. P75-T, 2005 [Google Scholar]
- 36. Katayama H, Tabata T, Ishihama Y, Sato T, Oda Y, Nagasu T. Efficient in-gel digestion procedure using 5-cyclohexyl-1-pentyl-β-d-maltoside as an additive for gel-based membrane proteomics. Rapid Commun Mass Spectrom 2004;18:2388–2394 [DOI] [PubMed] [Google Scholar]
- 37. Vosseller K., Trinidad JC, Chalkley RJ. O-Linked N-acetylglucosamine proteomics of postsynaptic density preparations using lectin weak affinity chromatography and mass spectrometry. Mol Cell Proteomics 2006;5:923–934 [DOI] [PubMed] [Google Scholar]
- 38. Klemm C, Schroeder S, Gluckmann M, Beyermann M, Krause E. Derivatization of phosphorylated peptides with S- and N-nucleophiles for enhanced ionization efficiency in matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun Mass Spectrom 2004;18:2697–2705 [DOI] [PubMed] [Google Scholar]
- 39. Noll BW, Jarboe CJ, Hass LF. Kinetic studies on the alkali-catalyzed hydrolysis and epimerization of model alkyl and hydroxyalkyl di- and tripeptides. Biochemistry 1974;13:5164–5169 [DOI] [PubMed] [Google Scholar]
- 40. Medzihradszky KF, Darula Z, Perlson E. O-Sulfonation of serine and threonine: mass spectrometric detection and characterization of a new posttranslational modification in diverse proteins throughout the eukaryotes. Mol Cell Proteomics 2004;3:429–440 [DOI] [PubMed] [Google Scholar]
- 41. Mattila K, Siltainsuu J, Balaspiri L, Ora M, Lonnberg H.Derivatization of phosphopeptides with mercapto- and amino-functionalized conjugate groups by phosphate elimination and subsequent Michael addition. Org Biomol Chem 2005;3:3039–3044 [DOI] [PubMed] [Google Scholar]
- 42. Whetstone PA, Butlin NG, Corneillie TM, Meares CF. Element-coded affinity tags for peptides and proteins. Bioconjugate Chem 2004;15:3–6 [DOI] [PubMed] [Google Scholar]
- 43. Li W, Backlund PS, Boykins RA, Wang G, Chen K-C. Susceptibility of the hydroxyl groups in serine and threonine to β-elimination/Michael addition under commonly used moderately high-temperature conditions. Anal Biochem 2003;323:94–102 [DOI] [PubMed] [Google Scholar]
- 44. Kirkland JJ, van Straten MA, Claessens HA. High pH mobile phase effects on silica-based reversed-phase high-performance liquid chromatography. J Chromatogr A 1995;691:3–19 [DOI] [PubMed] [Google Scholar]
- 45. Palmblad M, Vogel JS. Quantitation of binding, recovery and desalting efficiency of peptides and proteins in solid phase extraction micropipette tips. J Chromatogr B 2005;814:309–313 [DOI] [PubMed] [Google Scholar]
- 46. Chen SL, Huddleston MJ, Shou W, Deshaies RJ, Annan RS, Carr SA. Mass spectrometry-based methods for phosphorylation site mapping of hyperphosphorylated proteins applied to Net1, a regulator of exit from mitosis on yeast. Mol Cell Proteomics 2002;1:186–196 [DOI] [PubMed] [Google Scholar]
- 47. Donella-Deana A, Meyer HE, Pinna IA. The use of phosphopeptides to distinguish between protein phosphatase and acid/alkaline phosphatase activities: opposite effect specificity toward phosphoseryl/phosphothreonyl substrates. Biochim Biophys Acta 1991;1094:130–133 [DOI] [PubMed] [Google Scholar]
- 48. Miller BT, Collins TJ, Rogers ME, Kurosky A. Peptide biotinylation with amine-reactive esters: differential side chain reactivity. Peptides 1997;18:1585–1595 [DOI] [PubMed] [Google Scholar]
- 49. Biemann K. Sequencing of peptides by tandem mass spectrometry and high-energy collision-induced dissociation. Methods Enzymol 1990;193:455–479 [DOI] [PubMed] [Google Scholar]
- 50. Henzel W, Tropea J, Dupont D. Protein identification using 20-minute Edman cycles and sequence mixture analysis. Anal Biochem 1999;267:148–160 [DOI] [PubMed] [Google Scholar]
- 51. Finehout EJ, Cantor JR, Lee KH. Kinetic characterization of sequencing grade modified trypsin. Proteomics 2005;5:2319–2321 [DOI] [PubMed] [Google Scholar]