

Abstract
It is widely accepted that changes underlying depression and antidepressant-like effects involve not only alterations in the levels of neurotransmitters as monoamines and their receptors in the brain, but also structural and functional changes far beyond. During the last two decades, emerging theories are providing new explanations about the neurobiology of depression and the mechanism of action of antidepressant strategies based on cellular changes at the CNS level. The neurotrophic/plasticity hypothesis of depression, proposed more than a decade ago, is now supported by multiple basic and clinical studies focused on the role of intracellular-signalling cascades that govern neural proliferation and plasticity. Herein, we review the state-of-the-art of the changes in these signalling pathways which appear to underlie both depressive disorders and antidepressant actions. We will especially focus on the hippocampal cellularity and plasticity modulation by serotonin, trophic factors as brain-derived neurotrophic factor (BDNF), and vascular endothelial growth factor (VEGF) through intracellular signalling pathways—cAMP, Wnt/β-catenin, and mTOR. Connecting the classic monoaminergic hypothesis with proliferation/neuroplasticity-related evidence is an appealing and comprehensive attempt for improving our knowledge about the neurobiological events leading to depression and associated to antidepressant therapies.
1. Introduction
Major depressive disorder (MDD) constitutes the first leading cause of years lived with disability [1], and its incidence is on the rise globally. Yet, until recently, little was known about its pathogenesis, as these conditions are not associated with relevant brain alterations or clear animal models for spontaneous recurrent mood episodes.
The clinical phenomenology of major depression implicates brain neurotransmitter systems involved in the regulation of mood, anxiety, fear, reward processing, attention, motivation, stress responses, social interaction, and neurovegetative function [2]. MDD is associated with blunted reactivity to both positively and negatively stimuli [3]; thus, the decline in hedonic responses may be related to generalized affective insensitivity, instead of deficits in the capacity to feel pleasure at the level of basic sensory experience [4]. From the middle of the last century, a great effort has been made to elucidate the brain areas involved in emotion control and in the pathophysiology of mood disorders. Animal and human studies have indicated the involvement of the limbic system—including the hippocampal formation, cingulate gyrus, and anterior thalamus—the amygdala and different cortical structures as well as the hypothalamus in these processes [5, 6]. These structures are connected in two main networks: the “orbital” and the “medial prefrontal networks.” The orbital network appears to function both as a system for integration of multimodal stimuli and as a system for assessment of the value of those stimuli, and, probably, the support of abstract assessment of reward. The medial network is probably more significant for mood disorders [7].
In depressed subjects, the structures of the medial prefrontal network have been shown to contain alterations in gray matter volume, cellular elements, neurophysiological activity, receptor pharmacology, and gene expression. Dysfunction within this system underlies the disturbances in emotional behavior and other cognitive aspects of the major depressive disorder. Treatments for depression, involving pharmacological, neurosurgical, and deep brain stimulation methods, appear to suppress pathological activity within the components of medial prefrontal network such as the subgenual anterior cingulate cortex, ventromedial frontal cortex, striatum, and amygdala [6].
Although the causes of MDD are not yet completely known, genetic factors appear to play an important role although other factors deal with acute or chronic stress, childhood trauma, viral infections, and others [12, 13]. Regarding genetic causes, certain polymorphisms in genes related to the serotonergic system as the serotonin transporter, the brain-derived neurotrophic factor, the monoamine oxidase A, or the tryptophan hydroxylase 1, may increase the risk for depression or the vulnerability to stress [14]. Not all the studies published to date have found gene-environment interactions; however, the combination of both factors seems to predict more accurately a person's risk to suffer from major depressive disorder better than genes or environment alone.
The discovery that some drugs as iproniazid and imipramine exert an antidepressant effect dates back to the 1950s [15]. In 1965, it was shown that these drugs act through the monoaminergic system by increasing the brain levels of those monoamines [16]. These observations led to the development of the classical “monoaminergic hypothesis of depression,” which proposes that low monoamine brain levels in depressed individuals are responsible for this pathology. The classic antidepressants that increase monoamine neurotransmitters in the synaptic cleft are generally used for first-line treatment. However, the clinical benefit of these treatments is not immediate, taking 3-4 weeks to obtain a full response. Other therapeutic problems of currently used antidepressant drugs include relapses, drug side effects, incomplete resolution, residual symptoms, and drug resistance.
Traditionally, research in the neurobiology of major depressive disorder has been focused on monoamines. However, several lines of evidence [17] have led to the conclusion that the abnormalities associated to depression go beyond monoaminergic neurotransmission: thus, the development of better antidepressants will surely depend on the discovery and understanding of new cellular targets. In this regard, in the late 90's a new hypothesis has tried to explain major depression based on molecular mechanisms of neuroplasticity [18].
The “neuroplasticity hypothesis” was postulated based on several findings: first, stress decrease hippocampal neurogenesis and synaptic plasticity in prefrontal cortex (PFCx) [19–22]. Moreover, most known antidepressant therapies stimulate the proliferation of hippocampal progenitor cells, which constitutes the first stage of adult hippocampal neurogenesis [23]. However, the contribution of hippocampal neurogenesis to the pathogenesis of depression is far from being fully understood. Second, hippocampal morphologic analyses in depressed patients reveal volume loss and gray matter alterations. While some studies suggest that decreased adult neurogenesis could be responsible for such fluctuating changes, others show that the hippocampal volumetric reductions could be due to changes in neuropil, glial number, and/or dendritic complexity, and not necessarily to a cell proliferation decrease [24]. Third, different neuroplasticity- and proliferation-related intracellular pathways appear to be involved in the antidepressants' action as brain-derived neurotrophic factor (BDNF) [25], β-catenin [10, 26], or the mammalian target of rapamycin (mTOR) [27].
2. Cell Proliferation and Plasticity Role in Mood Disorders and Antidepressant Treatments
Dentate gyrus proliferation is decreased by stress [19–22], and in several animal models of depression as unpredictable stress, chronic administration of corticosterone, olfactory bulbectomy, or maternal deprivation [22, 28–31]. This loss in cell proliferation is correlated with a decreased hippocampal volume [32–34]. Hippocampal proliferation decrease is also observed in other disease models such as diabetic mice, which present a high incidence of depression reported in individuals with that primary illness [35]. In these animals, the reduced hippocampal proliferation is reversed with chronic antidepressant treatments [35]. In animals subjected to acute or chronic stress, a period of at least 24 h or 3 weeks, respectively, is required to get a recovery of the cellular proliferation [29]. However, although all these changes have been extensively studied, major depression is not generally considered as “hippocampal disorder.”
It is unlikely that impaired adult hippocampal neurogenesis alone may fully explain the neuropathology of major depression. In this sense, other studies have addressed cellular proliferation in anatomical structures quite relevant to depressive disorders, such as prefrontal cortex and amygdala, by using animal models of depression. Thus, medial frontal cortex presents a reduction in cell proliferation [36], downregulation of genes implicated in cell proliferation [37, 38], decreased cell growth and survival, and apoptosis inhibition [38]. Structures such as amygdala present an opposite pattern, with an increase in neuronal dendrite length in stress models [39]. Chronic administration of antidepressants leads to an increased proliferation in prefrontal cortex [36, 40] although the fate of the new generated cells goes toward the formation of glia rather than neurons, in contrast to hippocampus [36]. No data are available regarding antidepressant effects on amygdalar cell proliferation; however, this structure has been involved in the negative control of the hippocampal cell survival induced by antidepressant treatments, based on the increased cell survival observed in hippocampus, after the basolateral complex of the amygdale (BLA) lesion [41]. It is interesting to note that amygdala—implicated in fear-related learning that impairs the memory processing of the Hp-PFCx memory—shows an enhancement of LTP under stress situations, which is not reverted by antidepressants. Thus, antidepressants as tianeptine are able to restore the normal functionality of Hp and PFCx under stress situations, while the amygdala retains the ability to increase its activity in the same stress conditions [42].
The disturbed adult hippocampal neurogenesis cannot fully explain major depression. It could only be the most conspicuous feature of a more fundamental type of cellular plasticity, which could also govern the prefrontal cortex and other regions. It has also been proposed that, in addition to neural proliferation, changes in synaptic plasticity would also be involved in the biological basis of depression [19], being modulated by antidepressant treatments [43, 44]. PFCx is also a region sensitive to stress-induced effects, with a reduction in the number and length of spines [45] in apical dendrites of the pyramidal cells in the medial prefrontal cortex area [46, 47], as well as changes in the number, morphology, metabolism, and function of glial cells, that produce changes in the glutamatergic transmission, resulting in memory impairments [48–51], and reduced synaptic plasticity in the Hp-PFCx neuronal pathway [52]. The increase in extracellular glutamate could be one of the reasons underlying the molecular changes associated to stress [51]. However, while frontal cortex and hippocampus are reduced and hypofunctional in major depression, structures such as amygdala present hypertrophy and hyperactivity. Changes in synaptic plasticity will reflect a vulnerability to suffer a depression episode [39].
An increased apoptosis has also been related to a higher risk of suffering major depression since increased cell death in areas as dentate gyrus (DG), CA1, and CA4 areas of the hippocampus, entorhinal cortex, and subiculum are reported in studies using human postmortem brain samples though this phenomenon does not seem to account for the hippocampal volume reduction [53]. Animal studies also report that acute stress increase hippocampal apoptosis [29], while chronic stress induces no changes [29], increased apoptosis in cortex [54, 55], or hippocampus [55]. Antidepressant treatment decreases cell death by different mechanisms, as the activation of the expression of trophic factors (BDNF and its receptor TrkB) which results in increased cell survival [56, 57] or directly reducing cellular apoptosis in animal stress models as reported for fluoxetine [28, 58].
It has been suggested that an increase in serotonin levels mediates the raise in cell proliferation, while the depletion of this neurotransmitter does not lead to an immediate effect over the hippocampal cell division [59]. In line with that, the direct or indirect modulation leads to an increase in proliferation. Treatments exerting a direct action over the serotonergic system include chronic but not acute administration with drugs such as tricyclic antidepressants, monoamine oxidase inhibitors (MAOI), serotonin-selective reuptake inhibitors (SSRI), serotonin and noradrenaline reuptake inhibitors (SNRI), and 5HT4 agonists [8, 19, 20, 60–65]. A nonpharmacological intervention such as the silencing of the serotonin transporter (SERT) by RNAi in dorsal raphe serotonergic neurons also leads to increased hippocampal proliferation [66]. The administration of other drugs, such as lithium in combination with antidepressants as desipramine, produces an increase in hippocampal proliferation [67] and a decrease in apoptosis of hippocampal progenitor cells in irradiated animals [68]. Treatments with antidepressants that increase serotonin levels in brain act by targeting different progenitor cell populations. Thus, chronic administration of the SSRI fluoxetine [69] or subchronic treatment with a 5-HT4 agonist [8] increases cell proliferation and neurogenesis-targeting amplifying neural progenitors (ANPs) (Figure 1) while chronic electroconvulsive seizure (ECS) produces a fast-acting effect targeting both quiescent neural progenitors (QNPs) and ANPs [70]. An increased hippocampal proliferation as a consequence of chronic antidepressant treatment has been proven necessary for some [71–73], but not all the antidepressant-like effects in animals. The antidepressant-like effects have been related to the increased hippocampal proliferation [8, 71, 74], dendritic arborization, maturation, and functional integration of newborn neurons [75]. However, other drugs with potential antidepressant action do not mediate their effect through the activation of progenitor cells division, since the complete elimination of hippocampal proliferation by direct irradiation of this structure does not block the antidepressant response promoted by the blockade of drugs acting on other neurotransmitter systems as the corticotrophin-releasing factor receptor (CRF) or arginine vasopressin 1b (V1b) receptors [76].
Figure 1.
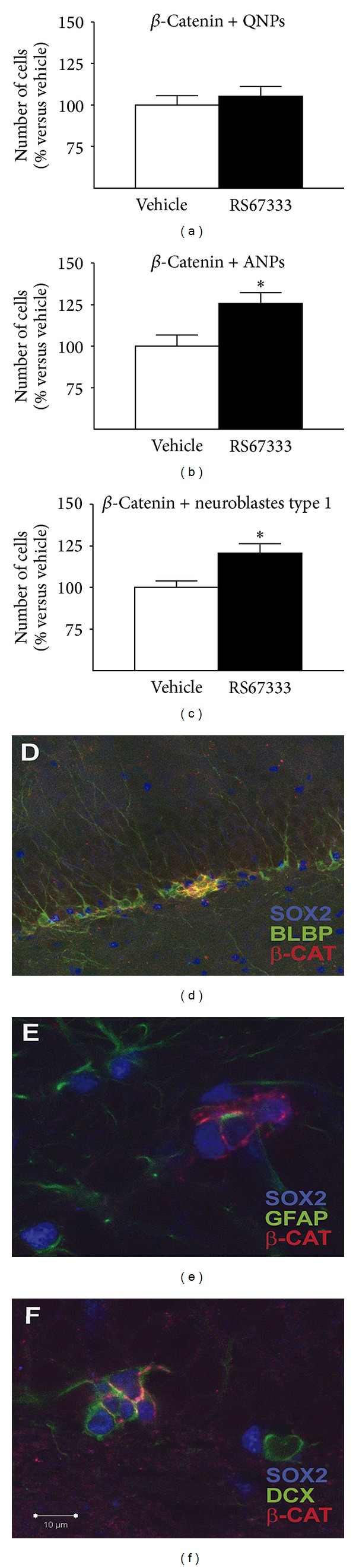
RS67333 increases the number of ANPs (b) and neuroblasts type 1 (c) that express β-catenin, but not total number of QNPs cells (a). Photomicrographs illustrating β-catenin expression in neural progenitors (d), ANPs (e), and neuroblasts (f). The results are the Mean ± S.E.M. *P < 0.05 versus vehicle. Bar: 10 μM, modified from Pascual-Brazo et al., 2012 [8].
3. Pathways Leading to Proliferation and Neural Plasticity Changes That Exert Antidepressant-Like Effect
Classically, the modulation of different neurotransmitter systems has been implicated in the mediation of the antidepressant effects, and, for some of them, a link with proliferative or plastic changes has been reported. The traditionally involved neurotransmitter systems include the serotonergic, adrenergic, and dopaminergic ones, while others, such as the glutamatergic and cannabinoid systems and the corticotropin-releasing factor (CRF) system implicated in the secretion of ACTH are acquiring increasing importance in the last years. Here we will focus on the serotonergic receptors most relevant to modulating neural proliferation and synaptic plasticity processes.
3.1. Serotonergic Receptors
Serotonin has a positive role in the regulation of hippocampal neurogenesis. The partial lesion of dorsal and medial raphe nuclei, which results in a decrease of serotonergic neurons that innervate the dentate gyrus of the hippocampus and other projection areas as cortex and amygdala, decreases the proliferation in the subgranular zone of the dentate gyrus [132]. Several serotonin receptors have been involved in the antidepressant-induced increase of cell proliferation in the hippocampus, together with neurite outgrowth and cell survival in cells expressing these receptors [80]. However, other authors report a lack of changes in proliferation and/or neurotrophic factors expression after chronic treatment with the antidepressant fluoxetine, questioning the importance of the serotonergic system in hippocampal proliferation [133–135].
3.1.1. 5-HT1A Receptors
The importance of this serotonergic subtype in the effects of antidepressants has been shown in studies in vivo using 3 day treatment with the 5-HT1A agonist 8-OH-DPAT [77], and chronic administration of this drug [74, 79, 80] that produce an increase in proliferation in the subgranular zone (SGZ) of the hippocampus that depends on the 5-HT1A postsynaptic receptors. In studies using hippocampal neural progenitors, the serotonin-mediated increase in proliferation is blocked by the 5-HT1A antagonist WAY100,635 [78]. On the contrary, the acute administration of 5-HT1A antagonists produces a decrease of hippocampal proliferation [136], or no changes after 14 days [79]. Knock out animals for the 5-HT1A receptor subtype present no changes in basal proliferation compared to wild type animals [9, 71], but present a decreased hippocampal cell survival [9]. The 5-HT1A receptor subtype has been proven necessary for the hippocampal proliferative effect of some antidepressants as fluoxetine [71], although other drugs as imipramine, acting on other neurotransmitter systems, increases hippocampal proliferation in a 5-HT1A-independent manner [71] (Table 1).
Table 1.
Role of the activation and blockade of the different serotonin receptor subtypes in neural proliferation and synaptic plasticity.
| Serotonin receptor subtypes | Effect of pharmacological manipulation on hippocampal proliferation | ko | ||||
|---|---|---|---|---|---|---|
| Agonist | Antagonist | |||||
| Change | Reference | Change | Reference | Change | Reference | |
| 5-HT1A | ↑ proliferation (subchronic) | [77] | ↓ proliferation (acute) | [78] | = proliferation | [9, 71] |
| = proliferation (chronic) | [79] | |||||
| ↑ proliferation (chronic) | [74, 79, 80] | blocks 5-HT-induced proliferation | [78] | ↓ cell survival | [9] | |
|
| ||||||
| 5-HT2A/2C | = proliferation (+SSRI) | [74] | ↑ proliferation (chronic) | [81, 82] | No data | |
| = proliferation (acute) | [74, 81] | |||||
| ↑ plasticity markers and BDNF (+SSRI, subchronic) | [83] | |||||
|
| ||||||
| 5-HT4 | ↑ proliferation (subchronic) | [8, 64] | Blocks 5-HT-induced proliferation | [78] | No data | |
| ↑ plasticity markers and BDNF (subchronic) | [8, 63–65] | |||||
|
| ||||||
| 5-HT6 | No data | = proliferation | [84] | No data | ||
| ↑ plasticity markers (PSA-NCAM) | [84] | |||||
|
| ||||||
| 5-HT7 | No data | ↑ proliferation (subchronic) | [85] | = proliferation | [86] | |
↑: increase; ↓: decrease; ko: knock-out mice.
3.1.2. 5-HT2A/C Receptors
The role of the 5-HT2A/C receptors in the regulation of neurogenesis is less clear. The chronic administration of 5-HT2A antagonists as ketanserin [81], and 5-HT2C antagonists as SB243,213 and S32006 [82], produce the increase in hippocampal proliferation, while the acute treatment with 5-HT2A/C agonists or antagonists produce no changes or a decrease in proliferation, respectively [74, 81]. Interestingly, the subchronic treatment with ketanserine in combination with the SSRI fluoxetine increases a series of synaptic plasticity markers as β-catenin and N-cadherin present in the membrane fraction, together with BDNF gene expression, however, hippocampal proliferation is not significantly modified [83]. The increased proliferation or synaptic plasticity parallel the antidepressant-like effect observed for the treatments with antagonists [83, 137, 138], while the administration of 5-HT2A agonist counteracts the effect of SSRIs [137]. The blockade of 5-HT2A receptor subtype located in GABAergic interneurons produces the activation of hippocampal pyramidal neurons [139] that modulates dendritic activation and synaptic plasticity [140] (Table 1).
3.1.3. 5-HT4 Receptors
In the last years, the 5-HT4 receptor subtype has been proven to have an outstanding role on the depressive pathology. This receptor subtype density and signaling cascade through cAMP are up-regulated in the frontal cortex and caudate-putamen of depressed humans [141]. Chronic treatments with classical antidepressants produce a desensitization of this subtype in structures as hippocampus [97, 98]. In the last years, it has been described a short-term antidepressant-like response mediated by 5-HT4 partial agonists [8, 64, 65], or when coadministered with classical antidepressants [63]. The antidepressant effect of 5-HT4 agonist is mediated by an increase in hippocampal proliferation in vivo [8, 64], together with other proliferative and plasticity markers as β-catenin, Akt [8], BDNF [8, 65], phosphorylated cAMP response element binding (CREB) protein [8, 63–65]. The 5-HT4 implication in serotonin-induced hippocampal proliferation has been observed blocking this receptor with the 5-HT4 antagonist DAU 6285 in primary hippocampal progenitor cell cultures [78] (Table 1).
3.1.4. 5-HT6 Receptors
The role of the 5-HT6 receptor subtype in depression is not clear, but tricyclic antidepressants as amitriptyline and atypical antidepressants as mianserin have high affinity for this serotonin receptor subtype, acting as antagonists [142]. Moreover, the expression of the 5-HT6 receptors is regulated by glucocorticoid levels [142]. This receptor subtype is present postsynaptically in brain areas as cortex and hippocampus and is implicated in learning and memory [142]. The action over this receptor to date is contradictory since it has been published that both antagonists and agonists exert antidepressant and anxiolytic effects alone [142, 143] or enhance the beneficial effect when combined with antidepressant drugs [144]. When locally administered on hippocampus, 5-HT6 antagonists produce antidepressant-like effect [145]. However, this effect is not mediated by increased neurogenesis but for an increase in neural cell adhesion molecule polysialylation (PSA-NCAM) that may mediate memory consolidation [84] through long-term changes in synaptic plasticity [146] (Table 1).
3.1.5. 5-HT7 Receptors
The 5-HT7 receptor subtype is also involved in the antidepressant effect. Recent studies have shown that the blockade of the 5-HT7 receptor subtype produces antidepressant-like behaviour [147, 148]. This is supported by studies in animal depression models as the olfactory bulbectomy [85], the antidepressant-like behaviour of knock-out mice for the 5-HT7 receptor subtype [149], and clinical data using the antagonist Lu AA21004 [150]. Moreover, a 7-day treatment with the 5-HT7 antagonist SB-269970 produces an increase in proliferation in the subgranular zone of the hippocampus [85] although changes in the number of dividing cells do not appear in 5-HT7 knock-out animals [86] (Table 1).
3.2. Neurotrophic Factors
In an attempt to explain those brain changes implicated on depression and/or antidepressant effect that could not be included in the initial monoaminergic hypothesis of depression, it was postulated the so-called “Neurotrophic hypothesis of Depression” that later was revised to a “new” “hypothesis of neuroplasticity” [18]. This hypothesis links the changes in depression models to a decrease of brain-derived neurotrophic factor (BDNF) and the antidepressant effect to an increase in BDNF in hippocampus [18, 19, 151, 152]. Moreover, the decreased BDNF observed in heterozygous knock-out mice (BDNF+/−) is related to a depression-like phenotype [18]. These changes in brain BDNF expression are paralleled by serum levels, so that it has been proposed as a biomarker for depression disease, positive or negative response of the individuals to the antidepressant treatment [153–156], and even a marker of suicidal depression [157]. However, the role of BDNF is still not clear in the depressive pathology since some authors describe a lack of changes on the BDNF levels associated to stress animal models [57, 158–160].
The infusion of BDNF in brain [161, 162] or more specifically in hippocampus [163, 164] produces antidepressant-like effects. Moreover, within the hippocampus, the infusion of BDNF in the DG but not in the CA1 region produces an antidepressant-like effect [163], which is supported by the lack of antidepressant action in mice selectively knocked out for the BDNF gene in the DG and not in the CA1 [160]. Even peripherally administered BDNF is able to display antidepressant-like actions [165], resembling the increased serum BDNF observed after antidepressant treatments [166].
Chronic administration of antidepressants produces an increase in hippocampal BDNF mRNA expression and BDNF protein levels (Figure 2) [8, 65, 167]. The blockade of 5-HT2A receptor reverses the effect of stress-induced downregulation of BDNF mRNA expression in hippocampus [168]. Also, the subchronic treatment with SSRI and 5-HT2A antagonists is able to increase BDNF expression in the dentate gyrus of the hippocampus; however, the protein level is not yet modified in subchronic treatments (Figure 2) [83].
Figure 2.
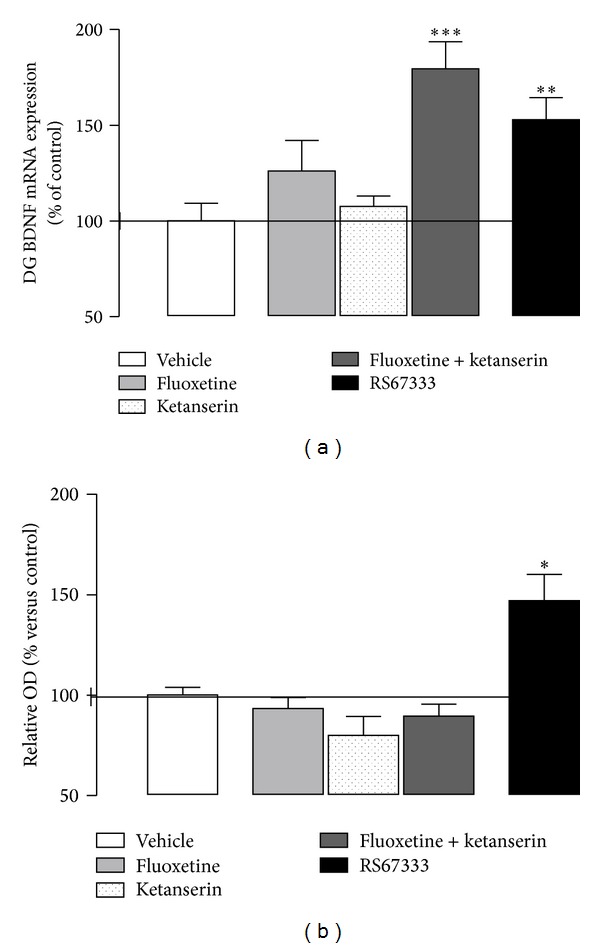
BDNF mRNA expression (a) and protein level (b) in the dentate gyrus of the hippocampus (DG) or total hippocampus, respectively, after 7-day treatment with the 5-HT4 partial agonist RS67333 (1.5 mg/kg/day) (modified from [8]) and 7-day coadministration of the SSRI fluoxetine (5 mg/kg/day) and the 5-HT2A antagonist ketanserin (0.1 mg/kg/day). *P < 0.05 versus vehicle. Modified from Pascual-Brazo et al., 2012 [8], and Pilar-Cuéllar et al., 2012 [83].
The main role of BDNF regarding adult neurogenesis is not linked to proliferation, but to the increase in cell survival, as described using BDNF and its receptor TrkB knock-out animals which present a reduced BDNF expression [61, 169]. BDNF is implicated in synaptic plasticity, and proteins as neuritin that are induced by BDNF are decreased in stress-induced animal models of depression [170] and increased after chronic antidepressant treatment, contributing to the BDNF antidepressant effect [170, 171].
The existence of a single-nucleotide polymorphism (SNP) in the human BDNF gene, BDNF (Val66Met) is associated to reduced BDNF secretion [172], and to an increased incidence of neuropsychiatric disorders [173, 174]. In animals BDNF (Val66Met) predisposes to a depression-like behaviour after stress situations that recover normal values after the administration of antidepressants [175]. This polymorphism is also associated to nonresponders after antidepressant treatment [176].
Other important trophic factor is the vascular endothelial growth factor (VEGF) implicated in the “vascular niche hypothesis of adult neurogenesis.” This theory proposes the need of vascular recruitment associated to active sites of neurogenesis formed by proliferative cells that present an endothelial phenotype in 37% of the cases [177]. VEGF expression is reduced in hippocampal dentate gyrus after irradiation [178], and in stress models [179] although other authors do not show changes associated to stressed animal models [180]. From studies using irradiated rats, it was proposed that the decrease of progenitor cells responsible for the expression of VEGF would underlie the decrease of this factor [178].
Some antidepressant treatments, as the electroconvulsive therapy (ECS) [178, 181, 182], approache with antidepressant-like effect as exercise [180], or mood stabilizers as lamotrigine [183], result in the upregulation of VEGF expression. Moreover, the local administration of this trophic factor produces an increase in hippocampal proliferation [178]. In addition, the silencing of hippocampal VEGF [184] or the use of antagonists for its receptor Flk-1 [180] blocks its antidepressant-like effect and decreases markers of newborn neurons as doublecortin (DCX).
Even though these data indicate the importance of VEGF brain levels in the depressive disorder, preliminary reports do not show a clear correlation between peripheral VEGF and depressive disorders, not allowing for the use of this molecule as a marker of depression and/or antidepressant response [185, 186].
The activation of receptor tyrosine kinases by neurotrophic factors promotes the activation of the PI3K/Akt pathway that is linked to the Wnt/β-catenin through the inhibition of GSK-3β and to the mTOR pathway through the phosphorylation of mTOR protein [187] that are discussed below. The PI3K/Akt pathway per se has an outstanding role in promoting adult hippocampal proliferation and the inhibition of cell differentiation [188]. Antidepressant treatments also produce increases in Akt levels in structures as hippocampus [8, 10] and frontal cortex [27].
3.3. Intracellular Pathways
3.3.1. Cyclic Adenosine Monophosphate (cAMP) Cascade
The upstream and downstream components of the cAMP signaling pathway have been extensively involved in the pathophysiology of mood disorders as well as in the actions of antidepressant drugs. Alterations in several elements of this pathway, such as G proteins (Gs or Gi), adenylate cyclase (AC), cAMP levels, cAMP-dependent protein kinase (PKA), and the cAMP response element-binding protein (CREB) transcription factor, have been described in peripheral cells and the postmortem brain of patients with affective disorders, both untreated or after antidepressant therapy [11, 100, 189, 190]. Various elements along this pathway have been identified as potential targets for antidepressant drugs (Table 2).
Table 2.
Involvement of cAMP, Wnt/β-catenin, and mTOR-signaling pathways in major depression (MDD) and antidepressant treatment.
| Signaling pathways | Changes related to disease or treatment | Direct effect | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Nontreated MDD (versus control) | Treated MDD (versus untreated) | ||||||||
| Change | Region | Reference | Change | Region | Reference | Change | Reference | ||
| cAMP/PKA | cAMP | ↓ AC (induced by FK, β-AR, α2-AR) | [87–94] | ↑ cAMP | [95, 96] | ||||
| ↓ 5-HT1A-induced AC inhibition/↓ 5-HT4-induced AC stimulation | Human PFCx | [8, 97, 98] | cAMP (administration/degradation inhibition) → AD-like effect | [99] | |||||
| ↑ AC CB1-induced inhibition | [100] | ||||||||
| PKA | No data | ↑ PKA activity | [101] | No data | |||||
| CREB | ↓ CREB | [102–104] | ↑ CREB (protein, expression, activity) | Hp, Cx | [11, 103–110] | CREB viral expression in Hp → AD-like effect | [44] | ||
| CREB viral expression in Acb → depression-like effect | [111] | ||||||||
| Epac | ↑ Epac-2 | Hp, PFCx | [112] | No data | No data | ||||
|
| |||||||||
| Wnt/β-catenin | GSK-3β | ↓ GSK-3β | Hp | [113] | ↑ GSK-3β | Hp | [8, 10, 113–117] | GSK-3β inhibition → AD-like and anxiolytic effect | [117–120] |
| GSK-3β knock in → stress-induced depression | [121] | ||||||||
| GSK-3β SNP (−50 T/C; rs334558) → later onset of bipolar depression | [122] | ||||||||
| GSK-3β SNP (−50 T/C; rs334558) → ↑ antidepressant response |
[123] | ||||||||
| β-catenin | ↓β-catenin | PFCx | [124] | ↑ β-catenin | Hp | [8, 10, 26, 83, 113–117] | β-catenin ko mice → depression-like effect | [125] | |
| β-catenin OE mice → AD-like effect | [126] | ||||||||
|
| |||||||||
| mTOR | mTOR | ↓ mTOR, p70S6K, eIF4B, p-eIF4B | [127–129] | ↑ p-mTOR/mTOR | PFCx | [27, 130] | Activation (NMDAr antagonists) → AD-like effect | [27, 131] | |
↑: increase; ↓: decrease; AC: adenylyl cyclase activity; FK: forskolin; Hp: hippocampus; PFCx: prefrontal cortex; Acb: accumbens nucleus; OE: overexpressing.
In peripheral cells and postmortem brains of patient with mayor depression, there is a reduction of the adenylyl cyclase (AC) activity in response to forskolin [87], β 2-adrenergic agonists [88–93], and α2-adrenoceptor agonists [94]. Chronic treatment with antidepressant drugs produces the increase in cAMP levels in rat hippocampus, cortex, and striatum, as well as in postmortem human frontal cortex samples from depressed patients (Figure 3(a); personal observation). This effect has been attributed to both enhanced coupling of Gs proteins to adenylyl cyclase and increased adenylyl cyclase activity [95, 96]. The direct injection of cAMP or inhibition of cAMP degradation by rolipram produces antidepressant-like effect in animals [99]. Chronic antidepressant treatment in rat desensitizes cAMP response to serotonergic receptor as 5-HT1A receptor (Figure 3(b)) and 5-HT4 receptor [8, 97, 98] and increases the CB1-mediated inhibition of adenylyl cyclase (AC) in prefrontal cortex, an effect that is modulated by 5-HT1A receptors [100].
Figure 3.
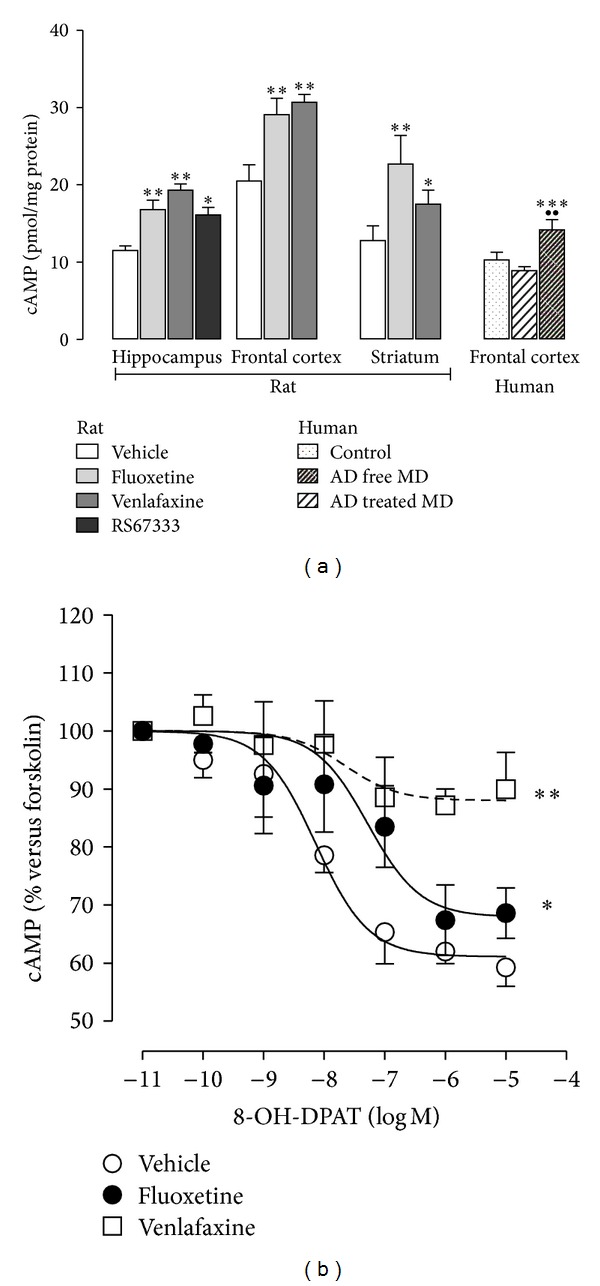
Antidepressant treatment increases basal cAMP in both rat and human. (a) Increase of basal cAMP levels in rat hippocampus, frontal cortex and striatum after chronic (14 days) antidepressant treatment with fluoxetine (10 mg/kg/day) and venlafaxine (40 mg/kg/day), and subchronic treatment (3 days) with the 5HT4 agonist RS67333 (1.5 mg/kg/day) and in postmortem frontal cortex samples from control, antidepressant free-depressed subjects (AD-free MD) and antidepressant-treated depressed subjects (AD-treated MD). cAMP is expressed in pmoles/mg protein. *P < 0.05; **P < 0.01 and ***P < 0.001 versus vehicle or control subjects; ●● P < 0.01 versus antidepressant-free depressed subjects. (b) Modulation of 5-HT1A receptor subtype-mediated inhibition of cAMP accumulation by antidepressant drugs. Chronic antidepressant treatment with fluoxetine (10 mg/kg/day) and venlafaxine (40 mg/kg/day) for 14 days downregulates 8-OH-DPAT inhibition of forskolin-induced cAMP accumulation. (a) Modified from Mostany et al., 2008 [10], Pascual-Brazo et al., 2012 [8], and unpublished results and (b) unpublished results.
The next step in this signaling pathway is the activation of cAMP-dependent protein kinase (PKA) by cAMP, so that PKA activity is increased after chronic antidepressant administration [101].Active PKA phosphorylates proteins as CREB, a transcription factor that regulates the expression of several genes involved in neuroplasticity, cell survival, and cognition [191–195].
CREB has been widely involved in the pathophysiology of depression and both behavioural and cellular responses to antidepressant treatments [11, 190]. Hippocampal expression of CREB is reduced in response to stress exposure [102, 103]. In contrast, chronic but not acute antidepressant therapy and electroconvulsive shock (ECS) increase levels of CREB mRNA, CREB protein (Figure 4), and CREB activity—promoting the phosphorylation of this protein—effects that seems to be area and drug dependent [11, 103, 105–108]. Thus, increased phosphorylated CREB levels in hippocampus are linked to antidepressant-like behaviour [44], as observed after viral-mediated overexpression of CREB in hippocampus in behavioural models of depression [44]. Contrary to what could be expected, CREB overexpression in the nucleus accumbens produces prodepressive effects [111], and lowered CREB in the nucleus accumbens in mice exhibits an antidepressant-like response [196]. A different pattern appears also for amygdala, in which high CREB levels produce opposite effects depending on the timing. Thus, when CREB overexpression is induced before learned helplessness training, there is a prodepressant effect, while the increase of CREB after the training is antidepressant [109]. Studies in postmortem human brain indicate lower levels of CREB protein in depressed antidepressant-free subjects, in contrast to the increased CREB level in patients taking an antidepressant at the time of death [104]. These results are parallel to studies in human fibroblasts of patients with major depression [110], which is consistent with animal studies. Among the several target genes regulated by CREB, two of the more relevant, are the brain-derived neurotrophic factor (BDNF) and the vascular endothelium growth factor (VEGF) [19, 60, 197, 198].
Figure 4.
Western blot analyses of hippocampal CREB (a) and p-CREB (b) levels and ratio p-CREB/CREB (c) after chronic antidepressant treatment (14 days) with fluoxetine (10 mg/kg/day) and venlafaxine (40 mg/kg/day). Note that p-CREB levels and ratio p-CREB/CREB are increased after fluoxetine treatment, but not total CREB, or after venlafaxine treatment in total cell lysate from hippocampus of rats. Values are means ± S.E.M. Corresponding to densitometry levels of the proteins expressed as the percentage of the same proteins in vehicle-treated animals. *P < 0.05 versus vehicle. Modified from Mostany et al., 2008 [10], and Mato et al., 2010 [100].
A growing body of data shows that other signalling cascades can modulate CREB activity through phosphorylation, such as the calcium/calmodulin-dependent kinase (CaMKII) and the mitogen-activated protein (MAP) kinase cascades, and may also be implicated in the mechanism of action of antidepressants [11, 199].
Initially, all effects of cAMP increase were attributed to the activation of PKA/CREB, but two novel targets as the cAMP-regulated ion channels and Epac (exchange protein directly activated by cAMP) are now known to be involved in mediating cAMP responses. An increase in Epac-2 levels, but not Epac-1, has been found in postmortem samples of prefrontal cortex and hippocampus of depressed subjects [112].
3.3.2. Wnt/β-Catenin Pathway
The Wingless-type (Wnt) family of proteins has key roles in many fundamental processes during neurodevelopment [200]. The role of this pathway in neural development, through the modulation of neural stem cells' (NSC) proliferation and differentiation, has been clearly demonstrated [201]. Some of the processes regulated by Wnt/β-catenin pathway activity are neural differentiation [202], hippocampal formation [203, 204], dendritic morphogenesis [205, 206], axon guidance [207, 208], and synapse formation [209]. Moreover, it also plays an important role in spatial learning [210] and memory, including long-term potentiation (LTP) phenomena [159].
In the absence of Wnt signaling, β-catenin function is blocked by a destruction complex consisting of Axin, APC, and GSK-3β and CK1a kinases [211], which phosphorylates β-catenin for destruction in the proteasome [212, 213]. Wnts act through both canonical and noncanonical signal transduction pathways. Canonical Wnt signaling results in the inhibition of GSK-3β which is constitutively active, and the non-phosphorylated β-catenin is stabilized in the cytoplasm and translocated to the nucleus, which is essential for canonical Wnt signaling [214]. Once in the nucleus, β-catenin forms a complex with the T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors, to activate the expression of Wnt target genes. TCF/LEF transcription factors are bound to Groucho, a protein producing repressive effects [214]. Nuclear β-catenin promotes the displacement of Groucho and the binding of the histone acetylase cyclic AMP response element-binding protein (CREB), activating the transcription machinery [214, 215]. The noncanonical pathway or β-catenin independent is mediated through Rac/Rho (Wnt/PCP) or through calcium (Wnt/Ca2+) [201].
In the last years several evidences have implicated Wnt-signaling pathway in the pathophysiology and treatment of mood disorders and other cognitive pathologies. GSK-3β and β-catenin are regulated either directly or indirectly by lithium, valproate, antidepressants, and antipsychotics [8, 10, 113–117], while GSK-3β has also been identified as a target for the treatment of Alzheimer's disease [216] (Table 2).
Postmortem human brain samples from depressed subjects and teenage suicide victims present a dysregulation of Wnt/GSK-3β signaling with a decrease in β-catenin expression in prefrontal cortex [124]. β-catenin knock-out mice with 50–70% decrease of β-catenin expression in forebrain regions present an increased immobility time in the tail suspension test indicating a depression-like state, but not in other anxiety tests [125].
The inhibition of GSK-3β activity, either pharmacologically [117–119], or through deletion in mouse forebrain, results in an increase in brain β-catenin levels, as well as in antidepressant-like effects or decreased anxiety [120], as observed by the direct overexpression of β-catenin in mouse brain [126]. GSK-3β inhibition by lithium is an important regulator of cell survival related to mood stabilizers [215] and displays antidepressant efficacy [115, 118, 215, 217, 218]. In contrast, GSK-3β knockin mice displayed increased susceptibility to stress-induced depressive-like behaviour [121], presenting decreased cell proliferation in the subgranular zone of the dentate gyrus, accompanied by a reduction in VEGF, but not BDNF, and blunted neurogenesis in response to antidepressant treatments [219]. These data support the importance of the Wnt pathway activation and β-catenin levels associated to mood disorders and their treatment. In addition, SNP variation in the promoter region of GSK-3β plays a protective role in the onset of bipolar illness [122] and increased antidepressant response [123].
Recent studies have identified the Wnt/GSK-3β/β-catenin-signaling pathway as a key regulator of adult neurogenesis in hippocampus [220, 221] or subventricular zone [222], highlighting the role of GSK-3β on neural progenitor homeostasis [200]. Wnt proteins are signaling molecules that are released from hippocampal neural stem cells (NSC) and astrocytes, acting autocrinally to regulate proliferation via Wnt canonical pathway [220, 221].
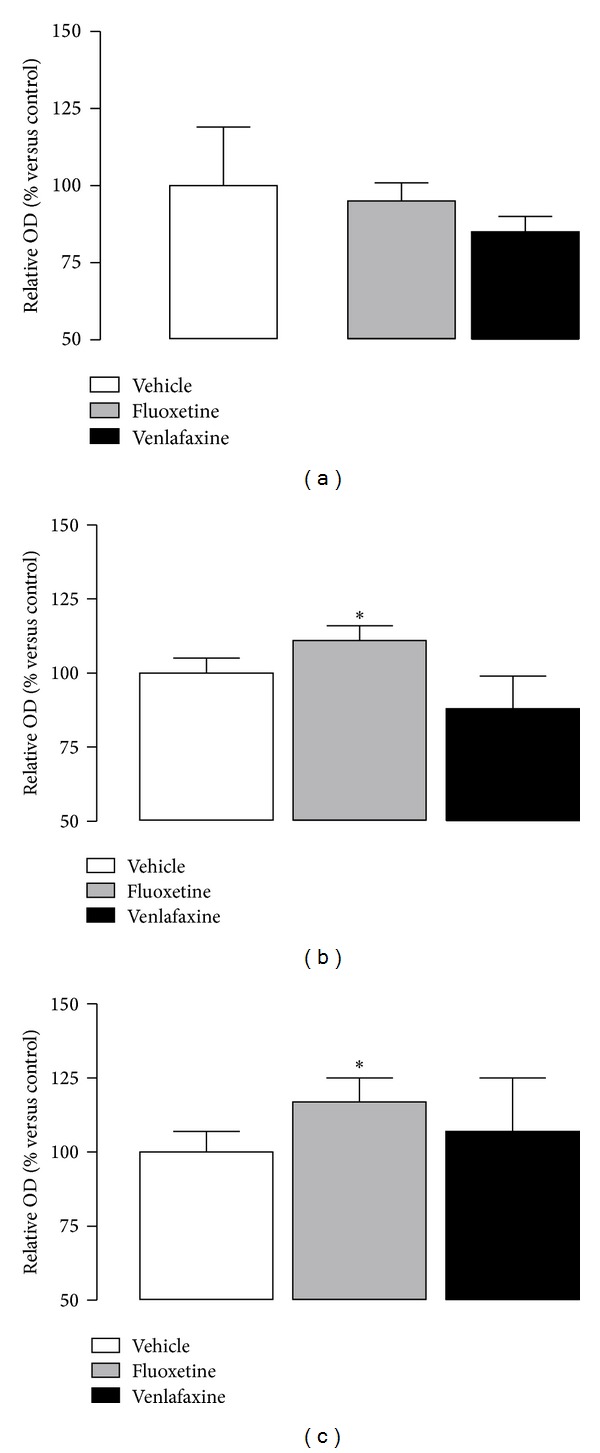
Wnt/β-catenin pathway is activated by antidepressant treatments as electroconvulsive therapy [26], chronic treatments with classical antidepressants as the dual serotonin-noradrenaline reuptake inhibitor (SNRI) venlafaxine (Figure 5(a)) [10], and 5-HT4 partial agonists [8]. The antidepressant-induced β-catenin increase is observed in the subgranular zone (SGZ) of the dentate gyrus (DG) of the hippocampus, in membrane and nuclear fractions [10, 26]. The increased proliferation observed in SGZ after chronic antidepressant treatments is localized in cell clusters that also show a positive β-catenin staining [8, 10].
Figure 5.
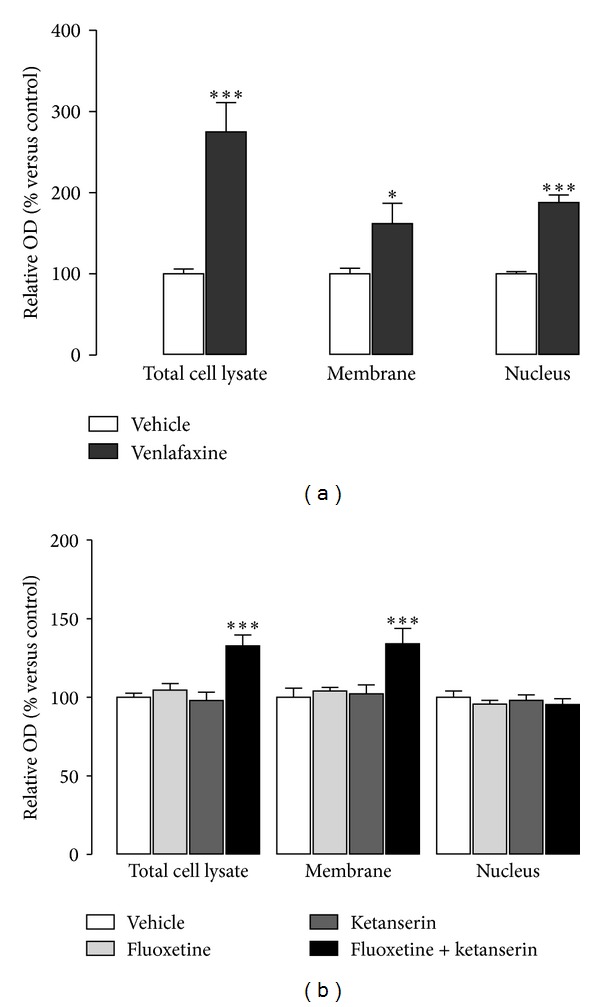
Implication on β-catenin subcellular distribution of the chronic (14 days) antidepressant treatment with the SNRI venlafaxine (40 mg/kg/day) (a), and 7-day treatment with the SSRI fluoxetine (5 mg/kg/day), the 5-HT2A antagonist ketanserin (0.1 mg/kg/day), and the combination of both (b). Note that chronic treatment with venlafaxine produces an increase in both membrane-associated and nuclear β-catenin, while the subchronic treatment with fluoxetine + ketanserin, only increases β-catenin in the membrane but not in the nuclear fraction. *P < 0.05 and ***P < 0.001 versus vehicle. (a) Modified from Mostany et al., 2008 [10], and (b) modified from Pilar-Cuellar et al., 2012 [83].
Other treatments with antidepressant-like efficacy, such as the subchronic administration of SSRI fluoxetine together with the 5-HT2A antagonist ketanserin, also produce a β-catenin increase in the membrane fraction but not in the nuclear one, which corresponds with a lack of changes in hippocampal proliferation (Figure 5(b)) [83]. The increase in membrane-associated β-catenin is parallel to an elevation of N-cadherin protein [83], both members of the β-catenin/N-cadherin complex present in pre- and postsynaptic terminals [223, 224], where β-catenin recruits scaffolding proteins [225], conforming cell-cell adhesion complexes [226], recruiting synaptic vesicles [209, 227], and acting on the development of new synapses [225]. This suggests a preference of modifications in synaptic plasticity instead of proliferation, as previously reported for other antidepressant treatments [60].
In addition, Frizzled receptors and GPCRs can interact through several pathways [228, 229]. Some GPCRs act through Gq and/or Gi proteins activating PKB (protein kinase B)/Akt which inhibits GSK-3β via phosphorylation. These receptors can also activate Gs proteins that activate prostaglandin E2 (PGE2), phosphoinositide 3-kinase (PI3K), and PKB/Akt, leading to the inhibition of GSK-3β. Other receptors act on Gq or G12/13 proteins, activating the phospholipase Cβ (PLCβ) and protein kinase C (PKCs) and inhibiting GSK-3β [228]. Taken together, these data support the possible existence of interactions between the GSK-3β/β-catenin pathway and other neurotransmitter systems involved in depression, including serotonin. The pharmacological modulation of the different elements of the Wnt/β-catenin pathway with antidepressant purposes has to be clarified in the near future, probably modulating at the level of Wnts or β-catenin activity. Interestingly, a number of patents regarding GSK-3β inhibition as the therapeutic mechanism for treatment of neuropsychiatric disorders are being launched, including treatment of depression.
3.3.3. mTOR Pathway
Target of rapamycin (TOR) genes, members of the phosphoinositol kinase-related kinase (PIKK) family of kinases [230], was first described in yeast as the pharmacological targets of the microbicide rapamycin [231]. TORs were subsequently described in other invertebrate and vertebrate organisms. mTOR, the mammalian form of this protein, exists in two different functional multiprotein complexes within the cells, mTORC1 and mTORC2, which are evolutionarily conserved from yeast to mammals [232, 233]. mTORC2 is involved in cytoskeletal remodeling [234] and in the regulation of cell survival and cell cycle progression. mTORC1, the primary target of rapamycin, is involved in cell proliferation, cell growth and survival by protein translation, energy regulation, and autophagy in response to growth factors, mitogens, nutrients, and stress [235–237].
In neurons, mTORC1 activity is regulated by phosphorylation in response to growth factors, as BDNF, mitogens, hormones, and neurotransmitters through the activation of G protein-coupled receptors (GPCRS) or ionotropic receptors. The mTORC1 phosphorylation is mediated by ERK/MAPK, PI3K, PKA, and Epac. The activation of mTORC1 results in the phosphorylation and activation of several downstream targets as the eukaryotic initiation factor 4E-binding protein 1 (4E-BP1), p70 ribosomal S6 kinase (p70S6K), RNA helicase cofactor eIF4A, extracellular signal-regulated kinase (ERK, including both ERK1 and ERK2), or PKB/Akt; and the inhibition of the eukaryotic elongation factor 2 kinase (eEF2) [238, 239].
mTOR has been extensively studied related to cancer, development, metabolism, and more recently to the central nervous system (CNS) physiology and diseases [238, 240, 241]. mTOR-signaling pathway is involved in synaptic plasticity, memory retention, neuroendocrine regulation associated with food intake and puberty, and modulation of neuronal repair following injury. The target proteins of mTOR, 4E-BP1, and eukaryotic initiation factor-4E (eIF4E) have been detected in cell bodies and dendrites in cultured hippocampal neurons and their distribution completely overlaps with the postsynaptic density protein-95 (PSD-95) at synaptic sites, suggesting the postsynaptic localization of these proteins [242]. The activation of mTOR has been functionally linked with local protein synthesis localized presynaptically as synapsin I, or postsynaptically as PSD-95 and GluR1, and cytoskeletal proteins as the activity-regulated cytoskeletal-associated protein (Arc) [27, 241, 243].
mTOR-signaling pathway has been also related to a number of neurological diseases, such as Alzheimer's disease, Parkinson's disease, and Huntington's disease, tuberous sclerosis, neurofibromatosis, fragile X syndrome, epilepsy, brain injury, and ischemic stroke [244]. Dysfunction of mTORC1 is associated with the pathogenic mechanisms of Alzheimer's disease, and the activation of p70S6K, downstream of mTORC1, has been identified as a contributor to hyperphosphorylated tau accumulation in neurons with neurofibrillary tangles [245].
Recent studies have also associated mTOR signaling in affective disorders since the administration of ketamine produces a fast-acting antidepressant-like effect in animals [27] and human [131]. In stressed rats, a reduction in PI3K-Akt-mTOR-signaling pathway has been reported in PFCx [127, 128] or amygdala [129]. The inhibition in mPFCx of calcineurin, a serine/threonine protein phosphatase that participates in the regulation of neurotransmission, neuronal structure and plasticity, and neuronal excitability, induces a depression-like behaviour [246], accompanied by a decrease in mTOR activity [130]. This effect can be reverted by the activation of mTOR by NMDA or the chronic administration of the antidepressant venlafaxine, promoting an antidepressant-like effect [130]. In human postmortem samples of prefrontal cortex of depressed subjects, there is a decrease in the expression of mTOR, as well as some of the downstream targets of this pathway, as p70S6 kinase (p70S6K), eIF4B, and its phosphorylated form, which suggests the impairment of the mTOR pathway in major depressive disorder (MDD) that would lead to a reduction in protein translation [247] (Table 2).
The subchronic, but not acute, administration of rapamycin in rodents has an antidepressant-like effect shown in two behavioural tests as forced swimming and tail suspension tests [248]. Acute administration of different NMDA receptor antagonists as the ketamine [27], Ro 25-6981 [27], and MK-801 [249] or antagonists of the group II of the metabotropic glutamate receptors (mGlu2/3), as MGS0039 and LY341495, produce a fast antidepressant effect [250, 251] mediated by mTOR-signaling pathway activation. Ketamine rapidly activates the mammalian target of rapamycin (mTOR) pathway, increases synaptogenesis, including increased density and function of spine synapses, in the prefrontal cortex of rats [27, 243], and increases hippocampal BDNF expression [252], that results in a rapid antidepressant-like effect in rats [27, 243] and humans [253]. Moreover, blockade of mTOR signalling by the specific antagonist rapamycin completely blocks the ketamine induction of synaptogenesis and behavioural responses in models of depression [27]. Other antidepressant strategies as the electroconvulsive treatment (ECT) also activate the mTOR pathway, leading to an increase in VEGF [254]. Therefore, modulation of mTOR could be a novel approach to develop strategies for the treatment of affective disorders [255].
4. A Further Step: Neuroplasticity versus Proliferation
The neurogenesis hypothesis of depression was based upon the demonstration that stress decreased adult neurogenesis in the hippocampus. This reduction in the production of newborn granule cells in the hippocampal dentate gyrus is related to the pathophysiology of depression. Since then, several studies have established that newborn neurons in the dentate gyrus are required for mediating some of the beneficial effects of antidepressant treatments since the increase in cell proliferation after antidepressant treatment is only observed in the SGZ and not in SVZ, suggesting a specificity of the antidepressants to regulate hippocampal neurogenesis. Moreover, psychotropic drugs without antidepressant activity do not increase neurogenesis [135, 256]. The disruption of hippocampal proliferation by irradiation is not sufficient to drive a depression-like phenotype. Both X-irradiation and genetic manipulation approaches demonstrated a requirement of hippocampal neurogenesis in mediating some of the antidepressant treatment effects [71, 76, 257], while mice exposed to X-irradiation of the SVZ or cerebellum responded normally to the antidepressants. However, some drugs with potential antidepressant action do not mediate their effect through the increase in hippocampal proliferation, as drugs acting on corticotrophin releasing factor receptor (CRF) or arginine vasopressin 1b (V1b) receptors [76], as indicated previously.
The appearance of the antidepressant-like effect in behavioural tests after 2-3 weeks parallels the time needed for the growth of newborn cells in hippocampus [258]. However, this time course does not always take so long. For classic antidepressants as the serotonin transporter inhibitors, a chronic regime is needed to observe that increased proliferation rate [10, 60], while, for others as ECS and 5-HT4 agonists [8, 64], an acute or subacute treatment, respectively, is enough to increase proliferation.
The putative role of changes in synaptic plasticity and/or neural proliferation in the depressive pathology is proposed some time ago [19]. Synaptic plasticity, as indicated for proliferation, is also modulated by antidepressant treatments [43, 44]. The neural plasticity is not only functional but structural and is impaired in animal models [259]. For example, there is a decrease in spine number in hippocampal CA1 and CA3 areas in bulbectomized animals that are reverted with antidepressant treatment [259–261]. This structural plasticity is more striking when new neurons are born [53], or there is an increase in neuron survival as a consequence of antidepressant treatment or ECS [262]. The new dendritic spines formed are associated to smaller postsynaptic densities (PSDs) and a higher frequency of miniexcitatory postsynaptic currents (mEPSCs), suggesting an increased number of new and active glutamatergic synapses [263].
The rapid antidepressant response to drugs as ketamine acting through the blockade of NMDA receptors appears as a new target for having fast-acting effects on the treatment of mood disorders compared to the weeks or months required for standard medications. Ketamine and other glutamate antagonists through the increase of the number and function of new spine synapses in rat prefrontal cortex by the activation of mTOR [27] do not modify hippocampal cell proliferation [264].
It would also be critical for future work to validate the relative importance of antidepressant-induced neurogenesis and synaptic plasticity in the antidepressant effects. However, evidence is strong that neurogenesis is required for at least some of the beneficial effects of antidepressant treatment. The exact role of neuroplastic/neuroproliferative changes in other brain structures as mPFCx and amygdala should be elucidated.
5. Conclusion
As indicated in this review, the importance of either proliferation or plasticity, or both, is still a matter of debate. As the involvement of proliferation and plasticity has been mainly studied in hippocampus, we might be underestimating its role in the antidepressant effect. In this sense, as the hippocampus is responsible for the learning and cognition part of the depressive disorder, the fact that the impairment of hippocampal proliferation would not block the antidepressant effect of some drugs does not necessarily conclude that the proliferation is only dependent on hippocampus. In the last years, prefrontal cortex, a structure with a great importance in mood control and working memory, is gaining increasing relevance in the plastic changes linked to antidepressant effects promoted by drugs as ketamine. In this sense, hippocampal proliferation would be only a small part of the plastic changes that are taking place within the hippocampus, and other brain areas. Thus, we must not underestimate the implication of synaptic plasticity in those antidepressant treatments that are not accompanied with increased proliferation.
Acknowledgments
The authors wish to thank Rebeca Madureira, Alicia Martín, Beatriz Romero, Isabel Ruiz, María Josefa Castillo, and Lourdes Lanza for their technical assistance. The scientific work of former coworkers Olga Guitérrez, Susana Mato, Elena del Olmo, Antonio Rodríguez-Gaztelumendi, and María Luisa Rojo is kindly acknowledged. This research was supported by Ministry of Science, SAF04-00941, SAF07-61862, Fundación Alicia Koplowitz, Fundación de Investigación Médica Mutua Madrileña, Instituto de Salud Carlos III, and University of Cantabria-FAES research contract. Rebecca Vidal has been the recipient of a fellowship from University of Cantabria-FAES, and a CIBERSAM contract.
References
- 1.WHO. The world health report. 2001.
- 2.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) APA Press; 1994. [Google Scholar]
- 3.Bylsma LM, Morris BH, Rottenberg J. A meta-analysis of emotional reactivity in major depressive disorder. Clinical Psychology Review. 2008;28(4):676–691. doi: 10.1016/j.cpr.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Treadway MT, Zald DH. Reconsidering anhedonia in depression: lessons from translational neuroscience. Neuroscience and Biobehavioral Reviews. 2011;35(3):537–555. doi: 10.1016/j.neubiorev.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koenigs M, Grafman J. The functional neuroanatomy of depression: distinct roles for ventromedial and dorsolateral prefrontal cortex. Behavioural Brain Research. 2009;201(2):239–243. doi: 10.1016/j.bbr.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Price JL, Drevets WC. Neurocircuitry of mood disorders. Neuropsychopharmacology. 2010;35(1):192–216. doi: 10.1038/npp.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Price JL, Drevets WC. Neural circuits underlying the pathophysiology of mood disorders. Trends in Cognitive Sciences. 2012;16:61–71. doi: 10.1016/j.tics.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 8.Pascual-Brazo J, Castro E, Diaz A, et al. Modulation of neuroplasticity pathways and antidepressant-like behavioural responses following the short-term (3 and 7 days) administration of the 5-HT(4) receptor agonist RS67333. International Journal of Neuropsychopharmacology. 2012;15(5):631–643. doi: 10.1017/S1461145711000782. [DOI] [PubMed] [Google Scholar]
- 9.Xia L, Delomenie C, David I, et al. Ventral hippocampal molecular pathways and impaired neurogenesis associated with 5-HT(1)A and 5-HT(1)B receptors disruption in mice. Neuroscience Letters. 2012;521(1):20–25. doi: 10.1016/j.neulet.2012.05.046. [DOI] [PubMed] [Google Scholar]
- 10.Mostany R, Valdizán EM, Pazos A. A role for nuclear β-catenin in SNRI antidepressant-induced hippocampal cell proliferation. Neuropharmacology. 2008;55(1):18–26. doi: 10.1016/j.neuropharm.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 11.Tardito D, Perez J, Tiraboschi E, Musazzi L, Racagni G, Popoli M. Signaling pathways regulating gene expression, neuroplasticity, and neurotrophic mechanisms in the action of antidepressants: a critical overview. Pharmacological Reviews. 2006;58(1):115–134. doi: 10.1124/pr.58.1.7. [DOI] [PubMed] [Google Scholar]
- 12.Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. 2002;34(1):13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- 13.Berton O, Nestler EJ. New approaches to antidepressant drug discovery: beyond monoamines. Nature Reviews Neuroscience. 2006;7(2):137–151. doi: 10.1038/nrn1846. [DOI] [PubMed] [Google Scholar]
- 14.Caspi A, Sugden K, Moffitt TE, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301(5631):386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- 15.Ban TA. Pharmacotherapy of depression: a historical analysis. Journal of Neural Transmission. 2001;108(6):707–716. doi: 10.1007/s007020170047. [DOI] [PubMed] [Google Scholar]
- 16.Schildkraut JJ. The catecholamine hypothesis of affective disorders: a review of supporting evidence. The American Journal of Psychiatry. 1965;122(5):509–522. doi: 10.1176/ajp.122.5.509. [DOI] [PubMed] [Google Scholar]
- 17.Ruhé HG, Mason NS, Schene AH. Mood is indirectly related to serotonin, norepinephrine and dopamine levels in humans: a meta-analysis of monoamine depletion studies. Molecular Psychiatry. 2007;12(4):331–359. doi: 10.1038/sj.mp.4001949. [DOI] [PubMed] [Google Scholar]
- 18.Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Archives of General Psychiatry. 1997;54(7):597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- 19.Duman RS, Malberg J, Thome J. Neural plasticity to stress and antidepressant treatment. Biological Psychiatry. 1999;46(9):1181–1191. doi: 10.1016/s0006-3223(99)00177-8. [DOI] [PubMed] [Google Scholar]
- 20.Czéh B, Michaelis T, Watanabe T, et al. Stress-induced changes in cerebral metabolites, hippocampal volume, and cell proliferation are prevented by antidepressant treatment with tianeptine. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(22):12796–12801. doi: 10.1073/pnas.211427898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malberg JE, Duman RS. Cell proliferation in adult hippocampus is decreased by inescapable stress: reversal by fluoxetine treatment. Neuropsychopharmacology. 2003;28(9):1562–1571. doi: 10.1038/sj.npp.1300234. [DOI] [PubMed] [Google Scholar]
- 22.Warner-Schmidt JL, Duman RS. Hippocampal neurogenesis: opposing effects of stress and antidepressant treatment. Hippocampus. 2006;16(3):239–249. doi: 10.1002/hipo.20156. [DOI] [PubMed] [Google Scholar]
- 23.Sahay A, Hen R. Adult hippocampal neurogenesis in depression. Nature Neuroscience. 2007;10(9):1110–1115. doi: 10.1038/nn1969. [DOI] [PubMed] [Google Scholar]
- 24.Sahay A, Drew MR, Hen R. Dentate gyrus neurogenesis and depression. Progress in Brain Research. 2007;163:697–722. doi: 10.1016/S0079-6123(07)63038-6. [DOI] [PubMed] [Google Scholar]
- 25.Duman CH, Schlesinger L, Russell DS, Duman RS. Voluntary exercise produces antidepressant and anxiolytic behavioral effects in mice. Brain Research C. 2008;1199:148–158. doi: 10.1016/j.brainres.2007.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madsen TM, Newton SS, Eaton ME, Russell DS, Duman RS. Chronic electroconvulsive seizure up-regulates β-catenin expression in rat hippocampus: role in adult neurogenesis. Biological Psychiatry. 2003;54(10):1006–1014. doi: 10.1016/s0006-3223(03)00700-5. [DOI] [PubMed] [Google Scholar]
- 27.Li N, Lee B, Liu RJ, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329(5994):959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee HJ, Kim JW, Yim SV, et al. Fluoxetine enhances cell proliferation and prevents apoptosis in dentate gyrus of maternally separated rats. Molecular Psychiatry. 2001;6(6):725–728. doi: 10.1038/sj.mp.4000954. [DOI] [PubMed] [Google Scholar]
- 29.Heine VM, Maslam S, Zareno J, Joëls M, Lucassen PJ. Suppressed proliferation and apoptotic changes in the rat dentate gyrus after acute and chronic stress are reversible. European Journal of Neuroscience. 2004;19(1):131–144. doi: 10.1046/j.1460-9568.2003.03100.x. [DOI] [PubMed] [Google Scholar]
- 30.Jaako-Movits K, Zharkovsky T, Pedersen M, Zharkovsky A. Decreased hippocampal neurogenesis following olfactory bulbectomy is reversed by repeated citalopram administration. Cellular and Molecular Neurobiology. 2006;26(7-8):1559–1570. doi: 10.1007/s10571-006-9090-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu Y, Barish PA, Pan J, Ogle WO, O'Donnell JM. Animal models of depression and neuroplasticity: assessing drug action in relation to behavior and neurogenesis. Methods in Molecular Biology. 2012;829:103–124. doi: 10.1007/978-1-61779-458-2_6. [DOI] [PubMed] [Google Scholar]
- 32.Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW. Hippocampal atrophy in recurrent major depression. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(9):3908–3913. doi: 10.1073/pnas.93.9.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bremner JD, Narayan M, Anderson ER, Staib LH, Miller HL, Charney DS. Hippocampal volume reduction in major depression. American Journal of Psychiatry. 2000;157(1):115–117. doi: 10.1176/ajp.157.1.115. [DOI] [PubMed] [Google Scholar]
- 34.Stockmeier CA, Mahajan GJ, Konick LC, et al. Cellular changes in the postmortem hippocampus in major depression. Biological Psychiatry. 2004;56(9):640–650. doi: 10.1016/j.biopsych.2004.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beauquis J, Roig P, Homo-Delarche F, De Nicola A, Saravia F. Reduced hippocampal neurogenesis and number of hilar neurones in streptozotocin-induced diabetic mice: reversion by antidepressant treatment. European Journal of Neuroscience. 2006;23(6):1539–1546. doi: 10.1111/j.1460-9568.2006.04691.x. [DOI] [PubMed] [Google Scholar]
- 36.Czéh B, Müller-Keuker JIH, Rygula R, et al. Chronic social stress inhibits cell proliferation in the adult medial prefrontal cortex: hemispheric asymmetry and reversal by fluoxetine treatment. Neuropsychopharmacology. 2007;32(7):1490–1503. doi: 10.1038/sj.npp.1301275. [DOI] [PubMed] [Google Scholar]
- 37.Tochigi M, Iwamoto K, Bundo M, Sasaki T, Kato N, Kato T. Gene expression profiling of major depression and suicide in the prefrontal cortex of postmortem brains. Neuroscience Research. 2008;60(2):184–191. doi: 10.1016/j.neures.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 38.Tordera RM, Garcia-García AL, Elizalde N, et al. Chronic stress and impaired glutamate function elicit a depressive-like phenotype and common changes in gene expression in the mouse frontal cortex. European Neuropsychopharmacology. 2011;21(1):23–32. doi: 10.1016/j.euroneuro.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 39.Blugeot A, Rivat C, Bouvier E, et al. Vulnerability to depression: from brain neuroplasticity to identification of biomarkers. Journal of Neuroscience. 2011;31(36):12889–12899. doi: 10.1523/JNEUROSCI.1309-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kodama M, Fujioka T, Duman RS. Chronic olanzapine or fluoxetine administration increases cell proliferation in hippocampus and prefrontal cortex of adult rat. Biological Psychiatry. 2004;56(8):570–580. doi: 10.1016/j.biopsych.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 41.Castro JE, Varea E, Márquez C, Cordero MI, Poirier G, Sandi C. Role of the amygdala in antidepressant effects on hippocampal cell proliferation and survival and on depression-like behavior in the rat. PLoS ONE. 2010;5(1) doi: 10.1371/journal.pone.0008618.e8618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diamond DM, Campbell A, Park CR, Vouimba RM. Preclinical research on stress, memory, and the brain in the development of pharmacotherapy for depression. European Neuropsychopharmacology. 2004;14(supplement 5):S491–S495. doi: 10.1016/j.euroneuro.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 43.Thome J, Sakai N, Shin KH, et al. cAMP response element-mediated gene transcription is upregulated by chronic antidepressant treatment. Journal of Neuroscience. 2000;20(11):4030–4036. doi: 10.1523/JNEUROSCI.20-11-04030.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen ACH, Shirayama Y, Shin KH, Neve RL, Duman RS. Expression of the cAMP response element binding protein (CREB) in hippocampus produces an antidepressant effect. Biological Psychiatry. 2001;49(9):753–762. doi: 10.1016/s0006-3223(00)01114-8. [DOI] [PubMed] [Google Scholar]
- 45.Penzes P, Cahill ME, Jones KA, Vanleeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nature Neuroscience. 2011;14(3):285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cook SC, Wellman CL. Chronic stress alters dendritic morphology in rat medial prefrontal cortex. Journal of Neurobiology. 2004;60(2):236–248. doi: 10.1002/neu.20025. [DOI] [PubMed] [Google Scholar]
- 47.Radley JJ, Sisti HM, Hao J, et al. Chronic behavioral stress induces apical dendritic reorganization in pyramidal neurons of the medial prefrontal cortex. Neuroscience. 2004;125(1):1–6. doi: 10.1016/j.neuroscience.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 48.Öngür D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(22):13290–13295. doi: 10.1073/pnas.95.22.13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bowley MP, Drevets WC, Öngür D, Price JL. Low glial numbers in the amygdala in major depressive disorder. Biological Psychiatry. 2002;52(5):404–412. doi: 10.1016/s0006-3223(02)01404-x. [DOI] [PubMed] [Google Scholar]
- 50.Rajkowska G, Miguel-Hidalgo JJ, Wei J, et al. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biological Psychiatry. 1999;45(9):1085–1098. doi: 10.1016/s0006-3223(99)00041-4. [DOI] [PubMed] [Google Scholar]
- 51.Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nature Reviews Neuroscience. 2012;13:22–37. doi: 10.1038/nrn3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cerqueira JJ, Mailliet F, Almeida OFX, Jay TM, Sousa N. The prefrontal cortex as a key target of the maladaptive response to stress. Journal of Neuroscience. 2007;27(11):2781–2787. doi: 10.1523/JNEUROSCI.4372-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lucassen PJ, Müller MB, Holsboer F, et al. Hippocampal apoptosis in major depression is a minor event and absent from subareas at risk for glucocorticoid overexposure. American Journal of Pathology. 2001;158(2):453–468. doi: 10.1016/S0002-9440(10)63988-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bachis A, Cruz MI, Nosheny RL, Mocchetti I. Chronic unpredictable stress promotes neuronal apoptosis in the cerebral cortex. Neuroscience Letters. 2008;442(2):104–108. doi: 10.1016/j.neulet.2008.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kosten TA, Galloway MP, Duman RS, Russell DS, D'Sa C. Repeated unpredictable stress and antidepressants differentially regulate expression of the Bcl-2 family of apoptotic genes in rat cortical, hippocampal, and limbic brain structures. Neuropsychopharmacology. 2008;33(7):1545–1558. doi: 10.1038/sj.npp.1301527. [DOI] [PubMed] [Google Scholar]
- 56.Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. Journal of Neuroscience. 1995;15(11):7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saarelainen T, Hendolin P, Lucas G, et al. Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. Journal of Neuroscience. 2003;23(1):349–357. doi: 10.1523/JNEUROSCI.23-01-00349.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lucassen PJ, Fuchs E, Czéh B. Antidepressant treatment with tianeptine reduces apoptosis in the hippocampal dentate gyrus and temporal cortex. Biological Psychiatry. 2004;55(8):789–796. doi: 10.1016/j.biopsych.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 59.Ueda S, Sakakibara S, Yoshimoto K. Effect of long-lasting serotonin depletion on environmental enrichment-induced neurogenesis in adult rat hippocampus and spatial learning. Neuroscience. 2005;135(2):395–402. doi: 10.1016/j.neuroscience.2005.06.065. [DOI] [PubMed] [Google Scholar]
- 60.Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. Journal of Neuroscience. 2000;20(24):9104–9110. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sairanen M, Lucas G, Ernfors P, Castrén M, Castrén E. Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. Journal of Neuroscience. 2005;25(5):1089–1094. doi: 10.1523/JNEUROSCI.3741-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alonso R, Griebel G, Pavone G, Stemmelin J, Le Fur G, Soubrié P. Blockade of CRF1 or V1b receptors reverses stress-induced suppression of neurogenesis in a mouse model of depression. Molecular Psychiatry. 2004;9(3):278–286. doi: 10.1038/sj.mp.4001464. [DOI] [PubMed] [Google Scholar]
- 63.Lucas G, Du J, Romeas T, et al. Selective serotonin reuptake inhibitors potentiate the rapid antidepressant-like effects of serotonin4 receptor agonists in the rat. PLoS ONE. 2010;5(2) doi: 10.1371/journal.pone.0009253.e9253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lucas G, Rymar VV, Du J, et al. Serotonin4 (5-HT4) receptor agonists are putative antidepressants with a rapid onset of action. Neuron. 2007;55(5):712–725. doi: 10.1016/j.neuron.2007.07.041. [DOI] [PubMed] [Google Scholar]
- 65.Tamburella A, Micale V, Navarria A, Drago F. Antidepressant properties of the 5-HT4 receptor partial agonist, SL65.0155: behavioral and neurochemical studies in rats. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2009;33(7):1205–1210. doi: 10.1016/j.pnpbp.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 66.Ferres-Coy A, Pilar-Cuellar F, Vidal R, et al. RNAi-mediated serotonin transporter suppression rapidly increases serotonergic neurotransmission and hippocampal neurogenesis. Translational Psychiatry. 2013;3(article e211) doi: 10.1038/tp.2012.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O'Leary OF, Zandy S, Dinan TG, Cryan JF. Lithium augmentation of the effects of desipramine in a mouse model of treatment-resistant depression: a role for hippocampal cell proliferation. Neuroscience. 2013;228:36–46. doi: 10.1016/j.neuroscience.2012.09.072. [DOI] [PubMed] [Google Scholar]
- 68.Huo K, Sun Y, Li H, et al. Lithium reduced neural progenitor apoptosis in the hippocampus and ameliorated functional deficits after irradiation to the immature mouse brain. Molecular and Cellular Neuroscience. 2012;51:32–42. doi: 10.1016/j.mcn.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 69.Encinas JM, Vaahtokari A, Enikolopov G. Fluoxetine targets early progenitor cells in the adult brain. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(21):8233–8238. doi: 10.1073/pnas.0601992103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Segi-Nishida E, Warner-Schmidt JL, Duman RS. Electroconvulsive seizure and VEGF increase the proliferation of neural stem-like cells in rat hippocampus. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(32):11352–11357. doi: 10.1073/pnas.0710858105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Santarelli L, Saxe M, Gross C, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301(5634):805–809. doi: 10.1126/science.1083328. [DOI] [PubMed] [Google Scholar]
- 72.Malberg JE, Schecter LE. Increasing hippocampal neurogenesis: a novel mechanism for antidepressant drugs. Current Pharmaceutical Design. 2005;11(2):145–155. doi: 10.2174/1381612053382223. [DOI] [PubMed] [Google Scholar]
- 73.Dranovsky A, Hen R. Hippocampal eurogenesis: regulation by stress and antidepressants. Biological Psychiatry. 2006;59(12):1136–1143. doi: 10.1016/j.biopsych.2006.03.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Banasr M, Hery M, Printemps R, Daszuta A. Serotonin-induced increases in adult cell proliferation and neurogenesis are mediated through different and common 5-HT receptor subtypes in the dentate gyrus and the subventricular zone. Neuropsychopharmacology. 2004;29(3):450–460. doi: 10.1038/sj.npp.1300320. [DOI] [PubMed] [Google Scholar]
- 75.Wang JW, David DJ, Monckton JE, Battaglia F, Hen R. Chronic fluoxetine stimulates maturation and synaptic plasticity of adult-born hippocampal granule cells. Journal of Neuroscience. 2008;28(6):1374–1384. doi: 10.1523/JNEUROSCI.3632-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Surget A, Saxe M, Leman S, et al. Drug-dependent requirement of hippocampal neurogenesis in a model of depression and of antidepressant reversal. Biological Psychiatry. 2008;64(4):293–301. doi: 10.1016/j.biopsych.2008.02.022. [DOI] [PubMed] [Google Scholar]
- 77.Arnold SA, Hagg T. Serotonin 1A receptor agonist increases species- and region-selective adult CNS proliferation, but not through CNTF. Neuropharmacology. 2012;63:1238–1247. doi: 10.1016/j.neuropharm.2012.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pilar-Cuéllar F, Vidal R, Díaz A, et al. Pathways involved in antidepressant-induced proliferation and synaptic plasticity. doi: 10.2174/13816128113196660736. Current Pharmaceutical Design. In press. [DOI] [PubMed] [Google Scholar]
- 79.Huang GJ, Herbert J. The role of 5-HT1A receptors in the proliferation and survival of progenitor cells in the dentate gyrus of the adult hippocampus and their regulation by corticoids. Neuroscience. 2005;135(3):803–813. doi: 10.1016/j.neuroscience.2005.05.056. [DOI] [PubMed] [Google Scholar]
- 80.Fricker AD, Rios C, Devi LA, Gomes I. Serotonin receptor activation leads to neurite outgrowth and neuronal survival. Molecular Brain Research. 2005;138(2):228–235. doi: 10.1016/j.molbrainres.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 81.Jha S, Rajendran R, Fernandes KA, Vaidya VA. 5-HT2A/2C receptor blockade regulates progenitor cell proliferation in the adult rat hippocampus. Neuroscience Letters. 2008;441(2):210–214. doi: 10.1016/j.neulet.2008.06.028. [DOI] [PubMed] [Google Scholar]
- 82.Soumier A, Banasr M, Lortet S, et al. Mechanisms contributing to the phase-dependent regulation of neurogenesis by the novel antidepressant, agomelatine, in the adult rat hippocampus. Neuropsychopharmacology. 2009;34(11):2390–2403. doi: 10.1038/npp.2009.72. [DOI] [PubMed] [Google Scholar]
- 83.Pilar-Cuellar F, Vidal R, Pazos A. Subchronic treatment with fluoxetine and ketanserin increases hippocampal brain-derived neurotrophic factor, beta-catenin and antidepressant-like effects. British Journal of Pharmacology. 2012;165:1046–1057. doi: 10.1111/j.1476-5381.2011.01516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Foley AG, Hirst WD, Gallagher HC, et al. The selective 5-HT6 receptor antagonists SB-271046 and SB-399885 potentiate NCAM PSA immunolabeling of dentate granule cells, but not neurogenesis, in the hippocampal formation of mature Wistar rats. Neuropharmacology. 2008;54(8):1166–1174. doi: 10.1016/j.neuropharm.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 85.Mnie-Filali O, Faure C, Lambas-Senas L, et al. Pharmacological blockade of 5-HT7 receptors as a putative fast acting antidepressant strategy. Neuropsychopharmacology. 2011;36(6):1275–1288. doi: 10.1038/npp.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sarkisyan G, Hedlund PB. The 5-HT7 receptor is involved in allocentric spatial memory information processing. Behavioural Brain Research. 2009;202(1):26–31. doi: 10.1016/j.bbr.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Menninger JA, Tabakoff B. Forskolin-stimulated platelet adenylyl cyclase activity is lower in persons with major depression. Biological Psychiatry. 1997;42(1):30–38. doi: 10.1016/S0006-3223(96)00245-4. [DOI] [PubMed] [Google Scholar]
- 88.Valdizán EM, Gutierrez O, Pazos A. Adenylate cyclase activity in postmortem brain of suicide subjects: reduced response to β-adrenergic stimulation. Biological Psychiatry. 2003;54(12):1457–1464. doi: 10.1016/s0006-3223(03)00589-4. [DOI] [PubMed] [Google Scholar]
- 89.Extein I, Tallman J, Smith CC, Goodwin FK. Changes in lymphocyte beta-adrenergic receptors in depression and mania. Psychiatry Research. 1979;1(2):191–197. doi: 10.1016/0165-1781(79)90061-1. [DOI] [PubMed] [Google Scholar]
- 90.Halper JP, Brown RP, Sweeney JA, Kocsis JH, Peters A, Mann JJ. Blunted β-adrenergic responsivity of peripheral blood mononuclear cells in endogenous depression. Isoproterenol dose-response studies. Archives of General Psychiatry. 1988;45(3):241–244. doi: 10.1001/archpsyc.1988.01800270053006. [DOI] [PubMed] [Google Scholar]
- 91.Mann JJ, Halper JP, Wilner PJ, et al. Subsensitivity of adenylyl cyclase-coupled receptors on mononuclear leukocytes from drug-free inpatients with a major depressive episode. Biological Psychiatry. 1997;42(10):859–870. doi: 10.1016/S0006-3223(97)00154-6. [DOI] [PubMed] [Google Scholar]
- 92.Mazzola-Pomietto P, Azorin JM, Tramoni V, Jeanningros R. Relation between lymphocyte beta-adrenergic responsivity and the severity of depressive disorders. Biological Psychiatry. 1994;35:920–925. doi: 10.1016/0006-3223(94)91238-6. [DOI] [PubMed] [Google Scholar]
- 93.Pandey GN, Sudershan P, Davis JM. Beta adrenergic receptor function in depression and the effect of antidepressant drugs. Acta Pharmacologica et Toxicologica Supplementum. 1985;56(1):66–79. doi: 10.1111/j.1600-0773.1985.tb02500.x. [DOI] [PubMed] [Google Scholar]
- 94.Valdizn EM, Dez-Alarcia R, Gonzlez-Maeso J, et al. α2-adrenoceptor functionality in postmortem frontal cortex of depressed suicide victims. Biological Psychiatry. 2010;68(9):869–872. doi: 10.1016/j.biopsych.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ozawa H, Rasenick MM. Chronic electroconvulsive treatment augments coupling of the GTP-binding protein G(s) to the catalytic moiety of adenylyl cyclase in a manner similar to that seen with chronic antidepressant drugs. Journal of Neurochemistry. 1991;56(1):330–338. doi: 10.1111/j.1471-4159.1991.tb02599.x. [DOI] [PubMed] [Google Scholar]
- 96.Watts VJ, Neve KA. Sensitization of adenylate cyclase by Galpha i/o-coupled receptors. Pharmacology & Therapeutics. 2005;106:405–421. doi: 10.1016/j.pharmthera.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 97.Vidal R, Valdizan EM, Mostany R, Pazos A, Castro E. Long-term treatment with fluoxetine induces desensitization of 5-HT4 receptor-dependent signalling and functionality in rat brain. Journal of Neurochemistry. 2009;110(3):1120–1127. doi: 10.1111/j.1471-4159.2009.06210.x. [DOI] [PubMed] [Google Scholar]
- 98.Vidal R, Valdizan EM, Vilaro MT, Pazos A, Castro E. Reduced signal transduction by 5-HT4 receptors after long-term venlafaxine treatment in rats. British Journal of Pharmacology. 2010;161(3):695–706. doi: 10.1111/j.1476-5381.2010.00903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang HT. Cyclic AMP-specific phosphodiesterase-4 as a target for the development of antidepressant drugs. Current Pharmaceutical Design. 2009;15(14):1688–1698. doi: 10.2174/138161209788168092. [DOI] [PubMed] [Google Scholar]
- 100.Mato S, Vidal R, Castro E, Díaz A, Pazos A, Valdizán EM. Long-term fluoxetine treatment modulates cannabinoid type 1 receptor-mediated inhibition of adenylyl cyclase in the rat prefrontal cortex through 5-hydroxytryptamine 1A receptor-dependent mechanisms. Molecular Pharmacology. 2010;77(3):424–434. doi: 10.1124/mol.109.060079. [DOI] [PubMed] [Google Scholar]
- 101.Nestler EJ, Terwilliger RZ, Duman RS. Chronic antidepressant administration alters the subcellular distribution of cyclic AMP-dependent protein kinase in rat frontal cortex. Journal of Neurochemistry. 1989;53(5):1644–1647. doi: 10.1111/j.1471-4159.1989.tb08564.x. [DOI] [PubMed] [Google Scholar]
- 102.Alfonso J, Frick LR, Silberman DM, Palumbo ML, Genaro AM, Frasch AC. Regulation of hippocampal gene expression is conserved in two species subjected to different stressors and antidepressant treatments. Biological Psychiatry. 2006;59(3):244–251. doi: 10.1016/j.biopsych.2005.06.036. [DOI] [PubMed] [Google Scholar]
- 103.Morley-Fletcher S, Mairesse J, Soumier A, et al. Chronic agomelatine treatment corrects behavioral, cellular, and biochemical abnormalities induced by prenatal stress in rats. Psychopharmacology. 2011;217(3):301–313. doi: 10.1007/s00213-011-2280-x. [DOI] [PubMed] [Google Scholar]
- 104.Dowlatshahi D, MacQueen GM, Wang JF, Young LT. Increased temporal cortex CREB concentrations and antidepressant treatment in major depression. Lancet. 1998;352(9142):1754–1755. doi: 10.1016/S0140-6736(05)79827-5. [DOI] [PubMed] [Google Scholar]
- 105.Dowlatshahi D, MacQueen GM, Wang JF, Reiach JS, Young LT. G protein-coupled cyclic AMP signaling in postmortem brain of subjects with mood disorders: effects of diagnosis, suicide, and treatment at the time of death. Journal of Neurochemistry. 1999;73(3):1121–1126. doi: 10.1046/j.1471-4159.1999.0731121.x. [DOI] [PubMed] [Google Scholar]
- 106.Odagaki Y, García-Sevilla JA, Huguelet P, La Harpe R, Koyama T, Guimón J. Cyclic AMP-mediated signaling components are upregulated in the prefrontal cortex of depressed suicide victims. Brain Research. 2001;898(2):224–231. doi: 10.1016/s0006-8993(01)02188-6. [DOI] [PubMed] [Google Scholar]
- 107.Yamada S, Yamamoto M, Ozawa H, Riederer P, Saito T. Reduced phosphorylation of cyclic AMP-responsive element binding protein in the postmortem orbitofrontal cortex of patients with major depressive disorder. Journal of Neural Transmission. 2003;110(6):671–680. doi: 10.1007/s00702-002-0810-8. [DOI] [PubMed] [Google Scholar]
- 108.Laifenfeld D, Karry R, Grauer E, Klein E, Ben-Shachar D. Antidepressants and prolonged stress in rats modulate CAM-L1, laminin, and pCREB, implicated in neuronal plasticity. Neurobiology of Disease. 2005;20(2):432–441. doi: 10.1016/j.nbd.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 109.Wallace TL, Stellitano KE, Neve RL, Duman RS. Effects of cyclic adenosine monophosphate response element binding protein overexpression in the basolateral amygdala on behavioral models of depression and anxiety. Biological Psychiatry. 2004;56(3):151–160. doi: 10.1016/j.biopsych.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 110.Manier DH, Shelton RC, Sulser F. Noradrenergic antidepressants: does chronic treatment increase or decrease nuclear CREB-P? Journal of Neural Transmission. 2002;109(1):91–99. doi: 10.1007/s702-002-8239-6. [DOI] [PubMed] [Google Scholar]
- 111.Pliakas AM, Carlson RR, Neve RL, Konradi C, Nestler EJ, Carlezon WA. Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated cAMP response element-binding protein expression in nucleus accumbens. Journal of Neuroscience. 2001;21(18):7397–7403. doi: 10.1523/JNEUROSCI.21-18-07397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dwivedi Y, Mondal AC, Rizavi HS, et al. Differential and brain region-specific regulation of Rap-1 and Epac in depressed suicide victims. Archives of General Psychiatry. 2006;63(6):639–648. doi: 10.1001/archpsyc.63.6.639. [DOI] [PubMed] [Google Scholar]
- 113.Liu R, Dang W, Jianting M, et al. Citalopram alleviates chronic stress induced depression-like behaviors in rats by activating GSK3 beta signaling in dorsal hippocampus. Brain Research. 2012;1467:10–17. doi: 10.1016/j.brainres.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 114.Chen G, Huang LD, Jiang YM, Manji HK. The mood-stabilizing agent valproate inhibits the activity of glycogen synthase kinase-3. Journal of Neurochemistry. 1999;72(3):1327–1330. doi: 10.1046/j.1471-4159.2000.0721327.x. [DOI] [PubMed] [Google Scholar]
- 115.Gould TD, Chen G, Manji HK. In vivo evidence in the brain for lithium inhibition of glycogen synthase kinase-3. Neuropsychopharmacology. 2004;29(1):32–38. doi: 10.1038/sj.npp.1300283. [DOI] [PubMed] [Google Scholar]
- 116.Li X, Zhu W, Roh MS, Friedman AB, Rosborough K, Jope RS. In vivo regulation of glycogen synthase kinase-3β (GSK3β) by serotonergic activity in mouse brain. Neuropsychopharmacology. 2004;29(8):1426–1431. doi: 10.1038/sj.npp.1300439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Beaulieu JM, Zhang X, Rodriguiz RM, et al. Role of GSK3β in behavioral abnormalities induced by serotonin deficiency. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(4):1333–1338. doi: 10.1073/pnas.0711496105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kaidanovich-Beilin O, Milman A, Weizman A, Pick CG, Eldar-Finkelman H. Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on β-catenin in mouse hippocampus. Biological Psychiatry. 2004;55(8):781–784. doi: 10.1016/j.biopsych.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 119.O’Brien WT, Harper AD, Jové F, et al. Glycogen synthase kinase-3β haploinsufficiency mimics the behavioral and molecular effects of lithium. Journal of Neuroscience. 2004;24(30):6791–6798. doi: 10.1523/JNEUROSCI.4753-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Latapy C, Rioux V, Guitton MJ, Beaulieu JM. Selective deletion of forebrain glycogen synthase kinase 3beta reveals a central role in serotonin-sensitive anxiety and social behaviour. Philosophical Transactions of the Royal Society B. 2012;367:2460–2474. doi: 10.1098/rstb.2012.0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Polter A, Beurel E, Yang S, et al. Deficiency in the inhibitory serine-phosphorylation of glycogen synthase kinase-3 increases sensitivity to mood disturbances. Neuropsychopharmacology. 2010;35(8):1761–1774. doi: 10.1038/npp.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Benedetti F, Serretti A, Colombo C, Lorenzi C, Tubazio V, Smeraldi E. A glycogen synthase kinase 3-β promoter gene single nucleotide polymorphism is associated with age at onset and response to total sleep deprivation in bipolar depression. Neuroscience Letters. 2004;368(2):123–126. doi: 10.1016/j.neulet.2004.06.050. [DOI] [PubMed] [Google Scholar]
- 123.Benedetti F, Dallaspezia S, Lorenzi C, et al. Gene-gene interaction of glycogen synthase kinase 3-beta and serotonin transporter on human antidepressant response to sleep deprivation. Journal of Affective Disorders. 2012;136:514–519. doi: 10.1016/j.jad.2011.10.039. [DOI] [PubMed] [Google Scholar]
- 124.Karege F, Perroud N, Burkhardt S, et al. Protein levels of beta-catenin and activation state of glycogen synthase kinase-3beta in major depression. A study with postmortem prefrontal cortex. Journal of Affective Disorders. 2012;136:185–188. doi: 10.1016/j.jad.2011.09.024. [DOI] [PubMed] [Google Scholar]
- 125.Gould TD, O’Donnell KC, Picchini AM, Dow ER, Chen G, Manji HK. Generation and behavioral characterization of β-catenin forebrain-specific conditional knock-out mice. Behavioural Brain Research. 2008;189(1):117–125. doi: 10.1016/j.bbr.2007.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gould TD, Einat H, O’Donnell KC, Picchini AM, Schloesser RJ, Manji HK. Β-catenin overexpression in the mouse brain phenocopies lithium-sensitive behaviors. Neuropsychopharmacology. 2007;32(10):2173–2183. doi: 10.1038/sj.npp.1301338. [DOI] [PubMed] [Google Scholar]
- 127.Howell KR, Kutiyanawalla A, Pillai A. Long-term continuous corticosterone treatment decreases VEGF receptor-2 expression in frontal cortex. PLoS ONE. 2011;6(5) doi: 10.1371/journal.pone.0020198.e20198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sui L, Wang J, Li BM. Role of the phosphoinositide 3-kinase-Akt-mammalian target of the rapamycin signaling pathway in long-term potentiation and trace fear conditioning memory in rat medial prefrontal cortex. Learning and Memory. 2008;15(10):762–776. doi: 10.1101/lm.1067808. [DOI] [PubMed] [Google Scholar]
- 129.Chandran A, Iyo AH, Jernigan CS, Legutko B, Austin MC, Karolewicz B. Reduced phosphorylation of the mTOR signaling pathway components in the amygdala of rats exposed to chronic stress. Progress in Neuro-Psychopharmacology & Biological Psychiatry. 2013;40:240–245. doi: 10.1016/j.pnpbp.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yu JJ, Zhang Y, Wang Y, et al. Inhibition of calcineurin in the prefrontal cortex induced depressive-like behavior through mTOR signaling pathway. Psychopharmacology. 2013;225:361–372. doi: 10.1007/s00213-012-2823-9. [DOI] [PubMed] [Google Scholar]
- 131.Mathew SJ, Keegan K, Smith L. Glutamate modulators as novel interventions for mood disorders. Revista Brasileira de Psiquiatria. 2005;27(3):243–248. doi: 10.1590/s1516-44462005000300016. [DOI] [PubMed] [Google Scholar]
- 132.Brezun JM, Daszuta A. Serotonergic reinnervation reverses lesion-induced decreases in PSA-NCAM labeling and proliferation of hippocampal cells in adult rats. Hippocampus. 2000;10:37–46. doi: 10.1002/(SICI)1098-1063(2000)10:1<37::AID-HIPO4>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 133.Cowen DS, Takase LF, Fornal CA, Jacobs BL. Age-dependent decline in hippocampal neurogenesis is not altered by chronic treatment with fluoxetine. Brain Research C. 2008;1228:14–19. doi: 10.1016/j.brainres.2008.06.059. [DOI] [PubMed] [Google Scholar]
- 134.Marlatt MW, Lucassen PJ, van Praag H. Comparison of neurogenic effects of fluoxetine, duloxetine and running in mice. Brain Research C. 2010;1341:93–99. doi: 10.1016/j.brainres.2010.03.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hanson ND, Nemeroff CB, Owens MJ. Lithium, but not fluoxetine or the corticotropin-releasing factor receptor 1 receptor antagonist R121919, increases cell proliferation in the adult dentate gyrus. Journal of Pharmacology and Experimental Therapeutics. 2011;337(1):180–186. doi: 10.1124/jpet.110.175372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Radley JJ, Jacobs BL. 5-HT1A receptor antagonist administration decreases cell proliferation in the dentate gyrus. Brain Research. 2002;955(1-2):264–267. doi: 10.1016/s0006-8993(02)03477-7. [DOI] [PubMed] [Google Scholar]
- 137.Marek GJ, Carpenter LL, McDougle CJ, Price LH. Synergistic action of 5-HT2A antagonists and selective serotonin reuptake inhibitors in neuropsychiatric disorders. Neuropsychopharmacology. 2003;28(2):402–412. doi: 10.1038/sj.npp.1300057. [DOI] [PubMed] [Google Scholar]
- 138.Pandey DK, Mahesh R, Kumar AA, Rao VS, Arjun M, Rajkumar R. A novel 5-HT(2A) receptor antagonist exhibits antidepressant-like effects in a battery of rodent behavioural assays: approaching early-onset antidepressants. Pharmacology Biochemistry and Behavior. 2010;94(3):363–373. doi: 10.1016/j.pbb.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 139.Vaidya VA, Terwilliger RMZ, Duman RS. Role of 5-HT(2a) receptors in the stress-induced down-regulation of brain-derived neurotrophic factor expression in rat hippocampus. Neuroscience Letters. 1999;262(1):1–4. doi: 10.1016/s0304-3940(99)00006-3. [DOI] [PubMed] [Google Scholar]
- 140.Gulyas AI, Acsady L, Freund TF. Structural basis of the cholinergic and serotonergic modulation of GABAergic neurons in the hippocampus. Neurochemistry International. 1999;34:359–372. doi: 10.1016/s0197-0186(99)00041-8. [DOI] [PubMed] [Google Scholar]
- 141.Rosel P, Arranz B, Urretavizcaya M, Oros M, San L, Navarro MA. Altered 5-HT2A and 5-HT4 postsynaptic receptors and their intracellular signalling systems IP3 and cAmP in brains from depressed violent suicide victims. Neuropsychobiology. 2004;49(4):189–195. doi: 10.1159/000077365. [DOI] [PubMed] [Google Scholar]
- 142.Wesołowska A. Potential role of the 5-HT6 receptor in depression and anxiety: an overview of preclinical data. Pharmacological Reports. 2010;62(4):564–577. doi: 10.1016/s1734-1140(10)70315-7. [DOI] [PubMed] [Google Scholar]
- 143.Wesołowska A, Nikiforuk A. Effects of the brain-penetrant and selective 5-HT6 receptor antagonist SB-399885 in animal models of anxiety and depression. Neuropharmacology. 2007;52(5):1274–1283. doi: 10.1016/j.neuropharm.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 144.Wesołowska A, Nikiforuk A. The selective 5-HT6 receptor antagonist SB-399885 enhances anti-immobility action of antidepressants in rats. European Journal of Pharmacology. 2008;582(1–3):88–93. doi: 10.1016/j.ejphar.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 145.Wesołowska A, Nikiforuk A, Stachowicz K. Anxiolytic-like and antidepressant-like effects produced by the selective 5-HT6 receptor antagonist SB-258585 after intrahippocampal administration to rats. Behavioural Pharmacology. 2007;18(5-6):439–446. doi: 10.1097/FBP.0b013e3282d28f9c. [DOI] [PubMed] [Google Scholar]
- 146.Callaghan CK, Hok V, Della-Chiesa A, Virley DJ, Upton N, O'Mara SM. Age-related declines in delayed non-match-to-sample performance (DNMS) are reversed by the novel 5HT6 receptor antagonist SB742457. Neuropharmacology. 2012;63:890–897. doi: 10.1016/j.neuropharm.2012.06.034. [DOI] [PubMed] [Google Scholar]
- 147.Mnie-Filali O, Lambás-Señas L, Zimmer L, Haddjeri N. 5-HT7 receptor antagonists as a new class of antidepressants. Drug News and Perspectives. 2007;20(10):613–618. doi: 10.1358/dnp.2007.20.10.1181354. [DOI] [PubMed] [Google Scholar]
- 148.Stahl SM. The serotonin-7 receptor as a novel therapeutic target. Journal of Clinical Psychiatry. 2010;71(11):1414–1415. doi: 10.4088/JCP.10bs06601gry. [DOI] [PubMed] [Google Scholar]
- 149.Hedlund PB, Huitron-Resendiz S, Henriksen SJ, Sutcliffe JG. 5-HT7 receptor inhibition and inactivation induce antidepressantlike behavior and sleep pattern. Biological Psychiatry. 2005;58(10):831–837. doi: 10.1016/j.biopsych.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 150.Alvarez E, Perez V, Dragheim M, Loft H, Artigas F. A double-blind, randomized, placebo-controlled, active reference study of Lu AA21004 in patients with major depressive disorder. International Journal of Neuropsychopharmacology. 2012;15:589–600. doi: 10.1017/S1461145711001027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Calabrese F, Molteni R, Racagni G, Riva MA. Neuronal plasticity: a link between stress and mood disorders. Psychoneuroendocrinology. 2009;34(supplement 1):S208–S216. doi: 10.1016/j.psyneuen.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 152.Taliaz D, Loya A, Gersner R, Haramati S, Chen A, Zangen A. Resilience to chronic stress is mediated by hippocampal brain-derived neurotrophic factor. Journal of Neuroscience. 2011;31(12):4475–4483. doi: 10.1523/JNEUROSCI.5725-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hashimoto K. Brain-derived neurotrophic factor as a biomarker for mood disorders: an historical overview and future directions. Psychiatry and Clinical Neurosciences. 2010;64(4):341–357. doi: 10.1111/j.1440-1819.2010.02113.x. [DOI] [PubMed] [Google Scholar]
- 154.Sen S, Duman R, Sanacora G. Serum brain-derived neurotrophic factor, depression, and antidepressant medications: meta-analyses and implications. Biological Psychiatry. 2008;64(6):527–532. doi: 10.1016/j.biopsych.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Umene-Nakano W, Yoshimura R, Ikenouchi-Sugita A, et al. Serum levels of brain-derived neurotrophic factor in comorbidity of depression and alcohol dependence. Human Psychopharmacology. 2009;24(5):409–413. doi: 10.1002/hup.1035. [DOI] [PubMed] [Google Scholar]
- 156.Gonul AS, Akdeniz F, Taneli F, Donat O, Eker C, Vahip S. Effect of treatment on serum brain-derived neurotrophic factor levels in depressed patients. European Archives of Psychiatry and Clinical Neuroscience. 2005;255(6):381–386. doi: 10.1007/s00406-005-0578-6. [DOI] [PubMed] [Google Scholar]
- 157.Kim YK, Lee HP, Won SD, et al. Low plasma BDNF is associated with suicidal behavior in major depression. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2007;31(1):78–85. doi: 10.1016/j.pnpbp.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 158.Monteggia LM, Barrot M, Powell CM, et al. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(29):10827–10832. doi: 10.1073/pnas.0402141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chen ZY, Jing D, Bath KG, et al. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314(5796):140–143. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Adachi M, Barrot M, Autry AE, Theobald D, Monteggia LM. Selective loss of brain-derived neurotrophic factor in the dentate gyrus attenuates antidepressant efficacy. Biological Psychiatry. 2008;63(7):642–649. doi: 10.1016/j.biopsych.2007.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Siuciak JA, Lewis DR, Wiegand SJ, Lindsay RM. Antidepressant-like effect of brain-derived neurotrophic factor (BDNF) Pharmacology Biochemistry and Behavior. 1997;56(1):131–137. doi: 10.1016/S0091-3057(96)00169-4. [DOI] [PubMed] [Google Scholar]
- 162.Hoshaw BA, Malberg JE, Lucki I. Central administration of IGF-I and BDNF leads to long-lasting antidepressant-like effects. Brain Research. 2005;1037(1-2):204–208. doi: 10.1016/j.brainres.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 163.Shirayama Y, Chen ACH, Nakagawa S, Russell DS, Duman RS. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. Journal of Neuroscience. 2002;22(8):3251–3261. doi: 10.1523/JNEUROSCI.22-08-03251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ye Y, Wang G, Wang H, Wang X. Brain-derived neurotrophic factor (BDNF) infusion restored astrocytic plasticity in the hippocampus of a rat model of depression. Neuroscience Letters. 2011;503:15–19. doi: 10.1016/j.neulet.2011.07.055. [DOI] [PubMed] [Google Scholar]
- 165.Schmidt HD, Duman RS. Peripheral BDNF produces antidepressant-like effects in cellular and behavioral models. Neuropsychopharmacology. 2010;35(12):2378–2391. doi: 10.1038/npp.2010.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Tadić A, Wagner S, Schlicht KF, et al. The early non-increase of serum BDNF predicts failure of antidepressant treatment in patients with major depression: a pilot study. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2011;35(2):415–420. doi: 10.1016/j.pnpbp.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 167.Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biological Psychiatry. 2006;59(12):1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 168.Vaidya VA, Marek GJ, Aghajanian GK, Duman RS. 5-HT(2A) receptor-mediated regulation of brain-derived neurotrophic factor mRNA in the hippocampus and the neocortex. Journal of Neuroscience. 1997;17(8):2785–2795. doi: 10.1523/JNEUROSCI.17-08-02785.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Choi SH, Li Y, Parada LF, Sisodia SS. Regulation of hippocampal progenitor cell survival, proliferation and dendritic development by BDNF. Molecular Neurodegeneration. 2009;4(1, article 52) doi: 10.1186/1750-1326-4-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Son H, Banasr M, Choi M, et al. Neuritin produces antidepressant actions and blocks the neuronal and behavioral deficits caused by chronic stress. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(28):11378–11383. doi: 10.1073/pnas.1201191109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Alme MN, Wibrand K, Dagestad G, Bramham CR. Chronic fluoxetine treatment induces brain region-specific upregulation of genes associated with BDNF-induced long-term potentiation. Neural Plasticity. 2007;2007:9 pages. doi: 10.1155/2007/26496.26496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Egan MF, Kojima M, Callicott JH, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112(2):257–269. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- 173.Sen S, Nesse RM, Stoltenberg SF, et al. A BDNF coding variant is associated with the NEO personality inventory domain neuroticism, a risk factor for depression. Neuropsychopharmacology. 2003;28(2):397–401. doi: 10.1038/sj.npp.1300053. [DOI] [PubMed] [Google Scholar]
- 174.Verhagen M, Van Der Meij A, Van Deurzen PM, et al. Meta-analysis of the BDNF Val66Met polymorphism in major depressive disorder: effects of gender and ethnicity. Molecular Psychiatry. 2010;15(3):260–271. doi: 10.1038/mp.2008.109. [DOI] [PubMed] [Google Scholar]
- 175.Yu H, Wang DD, Wang Y, Liu T, Lee FS, Chen ZY. Variant brain-derived neurotrophic factor Val66Met polymorphism alters vulnerability to stress and response to antidepressants. Journal of Neuroscience. 2012;32(12):4092–4101. doi: 10.1523/JNEUROSCI.5048-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kocabas NA, Antonijevic I, Faghel C, et al. Brain-derived neurotrophic factor gene polymorphisms: influence on treatment response phenotypes of major depressive disorder. International Clinical Psychopharmacology. 2011;26:1–10. doi: 10.1097/yic.0b013e32833d18f8. [DOI] [PubMed] [Google Scholar]
- 177.Palmer TD, Willhoite AR, Gage FH. Vascular niche for adult hippocampal neurogenesis. Journal of Comparative Neurology. 2000;425:479–494. doi: 10.1002/1096-9861(20001002)425:4<479::aid-cne2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 178.Warner-Schmidt JL, Madsen TM, Duman RS. Electroconvulsive seizure restores neurogenesis and hippocampus-dependent fear memory after disruption by irradiation. European Journal of Neuroscience. 2008;27(6):1485–1493. doi: 10.1111/j.1460-9568.2008.06118.x. [DOI] [PubMed] [Google Scholar]
- 179.Heine VM, Zareno J, Maslam S, Joëls M, Lucassen PJ. Chronic stress in the adult dentate gyrus reduces cell proliferation near the vasculature and VEGF and Flk-1 protein expression. European Journal of Neuroscience. 2005;21(5):1304–1314. doi: 10.1111/j.1460-9568.2005.03951.x. [DOI] [PubMed] [Google Scholar]
- 180.Kiuchi T, Lee H, Mikami T. Regular exercise cures depression-like behavior via VEGF-Flk-1 signaling in chronically stressed mice. Neuroscience. 2012;207:208–217. doi: 10.1016/j.neuroscience.2012.01.023. [DOI] [PubMed] [Google Scholar]
- 181.Newton SS, Collier EF, Hunsberger J, et al. Gene profile of electroconvulsive seizures: induction of neurotrophic and angiogenic factors. Journal of Neuroscience. 2003;23(34):10841–10851. doi: 10.1523/JNEUROSCI.23-34-10841.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Altar CA, Laeng P, Jurata LW, et al. Electroconvulsive seizures regulate gene expression of distinct neurotrophic signaling pathways. Journal of Neuroscience. 2004;24(11):2667–2677. doi: 10.1523/JNEUROSCI.5377-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sun R, Li N, Li T. VEGF regulates antidepressant effects of lamotrigine. European Neuropsychopharmacology. 2012;22:424–430. doi: 10.1016/j.euroneuro.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 184.Lee JS, Jang DJ, Lee N, et al. Induction of neuronal vascular endothelial growth factor expression by cAMP in the dentate gyrus of the hippocampus is required for antidepressant-like behaviors. Journal of Neuroscience. 2009;29(26):8493–8505. doi: 10.1523/JNEUROSCI.1321-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ventriglia M, Zanardini R, Pedrini L, et al. VEGF serum levels in depressed patients during SSRI antidepressant treatment. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2009;33(1):146–149. doi: 10.1016/j.pnpbp.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 186.Halmai Z, Dome P, Dobos J, et al. Peripheral vascular endothelial growth factor level is associated with antidepressant treatment response: results of a preliminary study. Journal of Affective Disorders. 2013;144:269–273. doi: 10.1016/j.jad.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 187.Cowen DS. Serotonin and neuronal growth factors—a convergence of signaling pathways. Journal of Neurochemistry. 2007;101(5):1161–1171. doi: 10.1111/j.1471-4159.2006.04420.x. [DOI] [PubMed] [Google Scholar]
- 188.Peltier J, O’Neill A, Schaffer DV. PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Developmental Neurobiology. 2007;67(10):1348–1361. doi: 10.1002/dneu.20506. [DOI] [PubMed] [Google Scholar]
- 189.Gould TD, Manji HK. Signaling networks in the pathophysiology and treatment of mood disorders. Journal of Psychosomatic Research. 2002;53:687–697. doi: 10.1016/s0022-3999(02)00426-9. [DOI] [PubMed] [Google Scholar]
- 190.Blendy JA. The role of CREB in depression and antidepressant treatment. Biological Psychiatry. 2006;59(12):1144–1150. doi: 10.1016/j.biopsych.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 191.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annual Review of Biochemistry. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 192.Viola H, Furman M, Izquierdo LA, et al. Phosphorylated cAMP response element-binding protein as a molecular marker of memory processing in rat hippocampus: effect of novelty. The Journal of Neuroscience. 2000;20(23, article RC112) doi: 10.1523/JNEUROSCI.20-23-j0002.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hardingham GE, Arnold FJL, Bading H. Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nature Neuroscience. 2001;4(3):261–267. doi: 10.1038/85109. [DOI] [PubMed] [Google Scholar]
- 194.Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35(4):605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 195.Carlezon WA, Jr., Duman RS, Nestler EJ. The many faces of CREB. Trends in Neurosciences. 2005;28(8):436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 196.Valverde O, Mantamadiotis T, Torrecilla M, et al. Modulation of anxiety-like behavior and morphine dependence in CREB-deficient mice. Neuropsychopharmacology. 2004;29(6):1122–1133. doi: 10.1038/sj.npp.1300416. [DOI] [PubMed] [Google Scholar]
- 197.Nibuya M, Nestler EJ, Duman RS. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. Journal of Neuroscience. 1996;16(7):2365–2372. doi: 10.1523/JNEUROSCI.16-07-02365.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Perera TD, Park S, Nemirovskaya Y. Cognitive role of neurogenesis in depression and antidepressant treatment. Neuroscientist. 2008;14(4):326–338. doi: 10.1177/1073858408317242. [DOI] [PubMed] [Google Scholar]
- 199.Johannessen M, Delghandi MP, Moens U. What turns CREB on? Cellular Signalling. 2004;16(11):1211–1227. doi: 10.1016/j.cellsig.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 200.Kim WY, Wang X, Wu Y, et al. GSK-3 is a master regulator of neural progenitor homeostasis. Nature Neuroscience. 2009;12:1390–1397. doi: 10.1038/nn.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Li F, Chong ZZ, Maiese K. Vital elements of the Wnt-frizzled signaling pathway in the nervous system. Current Neurovascular Research. 2005;2(4):331–340. doi: 10.2174/156720205774322557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ciani L, Salinas PC. WNTs in the vertebrate nervous system: from patterning to neuronal connectivity. Nature Reviews Neuroscience. 2005;6(5):351–362. doi: 10.1038/nrn1665. [DOI] [PubMed] [Google Scholar]
- 203.Galceran J, Miyashita-Lin EM, Devaney E, Rubenstein JLR, Grosschedl R. Hippocampus development and generation of dentate gyrus granule cells is regulated by LEF1. Development. 2000;127(3):469–482. doi: 10.1242/dev.127.3.469. [DOI] [PubMed] [Google Scholar]
- 204.Zhou CJ, Zhao C, Pleasure SJ. Wnt signaling mutants have decreased dentate granule cell production and radial glial scaffolding abnormalities. Journal of Neuroscience. 2004;24(1):121–126. doi: 10.1523/JNEUROSCI.4071-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Yu X, Malenka RC. Beta-catenin is critical for dendritic morphogenesis. Nature Neuroscience. 2003;6:1169–1177. doi: 10.1038/nn1132. [DOI] [PubMed] [Google Scholar]
- 206.Gao X, Arlotta P, Macklis JD, Chen J. Conditional knock-out of β-catenin in postnatal-born dentate gyrus granule neurons results in dendritic malformation. Journal of Neuroscience. 2007;27(52):14317–14325. doi: 10.1523/JNEUROSCI.3206-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zaghetto AA, Paina S, Mantero S, et al. Activation of the Wnt-βcatenin pathway in a cell population on the surface of the forebrain is essential for the establishment of olfactory axon connections. Journal of Neuroscience. 2007;27(36):9757–9768. doi: 10.1523/JNEUROSCI.0763-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Purro SA, Ciani L, Hoyos-Flight M, Stamatakou E, Siomou E, Salinas PC. Wnt regulates axon behavior through changes in microtubule growth directionality: a new role for adenomatous polyposis coli. Journal of Neuroscience. 2008;28(34):8644–8654. doi: 10.1523/JNEUROSCI.2320-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Bamji SX, Shimazu K, Kimes N, et al. Role of β-catenin in synaptic vesicle localization and presynaptic assembly. Neuron. 2003;40(4):719–731. doi: 10.1016/s0896-6273(03)00718-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Hernández F, Borrell J, Guaza C, Avila J, Lucas JJ. Spatial learning deficit in transgenic mice that conditionally over-express GSK-3β in the brain but do not form tau filaments. Journal of Neurochemistry. 2002;83(6):1529–1533. doi: 10.1046/j.1471-4159.2002.01269.x. [DOI] [PubMed] [Google Scholar]
- 211.Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of β-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes and Development. 1996;10(12):1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]
- 212.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. β-catenin is a target for the ubiquitin-proteasome pathway. EMBO Journal. 1997;16(13):3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Li L, Yuan H, Weaver CD, et al. Axin and Frat1 interact with Dvl and GSK, bridging Dvl to GSK in Wnt-mediated regulation of LEF-1. EMBO Journal. 1999;18(15):4233–4240. doi: 10.1093/emboj/18.15.4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annual Review of Cell and Developmental Biology. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 215.Jope RS, Bijur GN. Mood stabilizers, glycogen synthase kinase-3β and cell survival. Molecular Psychiatry. 2002;7(supplement 1):S35–S45. doi: 10.1038/sj.mp.4001017. [DOI] [PubMed] [Google Scholar]
- 216.Hernández F, Nido JD, Avila J, Villanueva N. GSK3 inhibitors and disease. Mini-Reviews in Medicinal Chemistry. 2009;9(9):1024–1029. doi: 10.2174/138955709788922647. [DOI] [PubMed] [Google Scholar]
- 217.Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Wada A, Yokoo H, Yanagita T, Kobayashi H. Lithium: potential therapeutics against acute brain injuries and chronic neurodegenerative diseases. Journal of Pharmacological Sciences. 2005;99(4):307–321. doi: 10.1254/jphs.crj05009x. [DOI] [PubMed] [Google Scholar]
- 219.Eom TY, Jope RS. Blocked inhibitory serine-phosphorylation of glycogen synthase kinase-3alpha/beta impairs in vivo neural precursor cell proliferation. Biological Psychiatry. 2009;66(5):494–502. doi: 10.1016/j.biopsych.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Lie DC, Colamarino SA, Song HJ, et al. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005;437(7063):1370–1375. doi: 10.1038/nature04108. [DOI] [PubMed] [Google Scholar]
- 221.Wexler EM, Andres P, Kornblum HI, Palmer TD, Geschwind DH. Endogenous Wnt signaling maintains neural progenitor cell potency. Stem Cells. 2009;27(5):1130–1141. doi: 10.1002/stem.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Adachi K, Mirzadeh Z, Sakaguchi M, et al. β-catenin signaling promotes proliferation of progenitor cells in the adult mouse subventricular zone. Stem Cells. 2007;25(11):2827–2836. doi: 10.1634/stemcells.2007-0177. [DOI] [PubMed] [Google Scholar]
- 223.Uchida N, Honjo Y, Johnson KR, Wheelock MJ, Takeichi M. The catenin/cadherin adhesion system is localized in synaptic junctions bordering transmitter release zones. Journal of Cell Biology. 1996;135(3):767–779. doi: 10.1083/jcb.135.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Miskevich F, Zhu Y, Ranscht B, Sanes JR. Expression of multiple cadherins and catenins in the chick optic tectum. Molecular and Cellular Neurosciences. 1998;12(4-5):240–255. doi: 10.1006/mcne.1998.0718. [DOI] [PubMed] [Google Scholar]
- 225.Yao I, Iida J, Tanaka N, Hata Y, Medical T, University D. Interaction of synaptic scaffolding molecule and β-catenin. Journal of Neuroscience. 2002;22(3):757–765. doi: 10.1523/JNEUROSCI.22-03-00757.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303(5663):1483–1487. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Togashi H, Abe K, Mizoguchi A, Takaoka K, Chisaka O, Takeichi M. Cadherin regulates dendritic spine morphogenesis. Neuron. 2002;35(1):77–89. doi: 10.1016/s0896-6273(02)00748-1. [DOI] [PubMed] [Google Scholar]
- 228.Shevtsov SP, Haq S, Force T. Activation of β-catenin signaling pathways by classical G-protein-coupled receptors: mechanisms and consequences in cycling and non-cycling cells. Cell Cycle. 2006;5(20):2295–2300. doi: 10.4161/cc.5.20.3357. [DOI] [PubMed] [Google Scholar]
- 229.Jin T, George Fantus I, Sun J. Wnt and beyond Wnt: multiple mechanisms control the transcriptional property of β-catenin. Cellular Signalling. 2008;20(10):1697–1704. doi: 10.1016/j.cellsig.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 230.Kunz J, Henriquez R, Schneider U, Deuter-Reinhard M, Movva NR, Hall MN. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell. 1993;73(3):585–596. doi: 10.1016/0092-8674(93)90144-f. [DOI] [PubMed] [Google Scholar]
- 231.Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253(5022):905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- 232.Helliwell SB, Wagner P, Kunz J, Deuter-Reinhard M, Henriquez R, Hall MN. TOR1 and TOR2 are structurally and functionally similar but not identical phosphatidylinositol kinase homologues in yeast. Molecular Biology of the Cell. 1994;5(1):105–118. doi: 10.1091/mbc.5.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Loewith R, Jacinto E, Wullschleger S, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Molecular Cell. 2002;10(3):457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 234.Jacinto E, Loewith R, Schmidt A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nature Cell Biology. 2004;6(11):1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 235.Takahara T, Hara K, Yonezawa K, Sorimachi H, Maeda T. Nutrient-dependent multimerization of the mammalian target of rapamycin through the N-terminal HEAT repeat region. Journal of Biological Chemistry. 2006;281(39):28605–28614. doi: 10.1074/jbc.M606087200. [DOI] [PubMed] [Google Scholar]
- 236.Harwood FC, Shu L, Houghton PJ. mTORC1 signaling can regulate growth factor activation of p44/42 mitogen-activated protein kinases through protein phosphatase 2A. Journal of Biological Chemistry. 2008;283(5):2575–2585. doi: 10.1074/jbc.M706173200. [DOI] [PubMed] [Google Scholar]
- 237.Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORCl association with the ULK1-Atg13-FIP200 complex required for autophagy. Molecular Biology of the Cell. 2009;20(7):1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends in Neurosciences. 2010;33(2):67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Livingstone M, Atas E, Meller A, Sonenberg N. Mechanisms governing the control of mRNA translation. Physical Biology. 2010;7(2) doi: 10.1088/1478-3975/7/2/021001.021001 [DOI] [PubMed] [Google Scholar]
- 240.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes and Development. 2004;18(16):1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 241.Gong R, Chang SP, Abbassi NR, Tang SJ. Roles of glutamate receptors and the mammalian target of rapamycin (mTOR) signaling pathway in activity-dependent dendritic protein synthesis in hippocampal neurons. Journal of Biological Chemistry. 2006;281(27):18802–18815. doi: 10.1074/jbc.M512524200. [DOI] [PubMed] [Google Scholar]
- 242.Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(1):467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Duman RS, Li N, Liu RJ, Duric V, Aghajanian G. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology. 2012;62:35–41. doi: 10.1016/j.neuropharm.2011.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Chong ZZ, Shang YC, Zhang L, Wang S, Maiese K. Mammalian target of rapamycin: hitting the bull’s-eye for neurological disorders. Oxidative Medicine and Cellular Longevity. 2010;3(6):374–391. doi: 10.4161/oxim.3.6.14787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.An WL, Cowburn RF, Li L, et al. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer's disease. American Journal of Pathology. 2003;163:591–607. doi: 10.1016/S0002-9440(10)63687-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Zhu WL, Shi HS, Wang SJ, Wu P, Ding ZB, Lu L. Hippocampal CA3 calcineurin activity participates in depressive-like behavior in rats. Journal of Neurochemistry. 2011;117(6):1075–1086. doi: 10.1111/j.1471-4159.2011.07285.x. [DOI] [PubMed] [Google Scholar]
- 247.Jernigan CS, Goswami DB, Austin MC, et al. The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2011;35(7):1774–1779. doi: 10.1016/j.pnpbp.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Cleary C, Linde JAS, Hiscock KM, et al. Antidepressive-like effects of rapamycin in animal models: implications for mTOR inhibition as a new target for treatment of affective disorders. Brain Research Bulletin. 2008;76(5):469–473. doi: 10.1016/j.brainresbull.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 249.Yoon SC, Seo MS, Kim SH, et al. The effect of MK-801 on mTOR/p70S6K and translation-related proteins in rat frontal cortex. Neuroscience Letters. 2008;434(1):23–28. doi: 10.1016/j.neulet.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 250.Maeng S, Zarate CA, Jr., Du J, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biological Psychiatry. 2008;63(4):349–352. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 251.Koike H, Iijima M, Chaki S. Involvement of the mammalian target of rapamycin signaling in the antidepressant-like effect of group II metabotropic glutamate receptor antagonists. Neuropharmacology. 2011;61:1419–1423. doi: 10.1016/j.neuropharm.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 252.Yang C, Hu YM, Zhou ZQ, Zhang GF, Yang JJ. Acute administration of ketamine in rats increases hippocampal BDNF and mTOR levels during forced swimming test. Upsala Journal of Medical Sciences. 2013;118(1):3–8. doi: 10.3109/03009734.2012.724118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Zarate CA, Jr., Singh JB, Carlson PJ, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Archives of General Psychiatry. 2006;63(8):856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 254.Elfving B, Wegener G. Electroconvulsive seizures stimulate the vegf pathway via mTORC1. Synapse. 2012;66:340–345. doi: 10.1002/syn.21518. [DOI] [PubMed] [Google Scholar]
- 255.Dwyer JM, Lepack AE, Duman RS. mTOR activation is required for the antidepressant effects of mGluR(2)/(3) blockade. International Journal of Neuropsychopharmacology. 2012;15:429–434. doi: 10.1017/S1461145711001702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Eisch AJ. Adult neurogenesis: implications for psychiatry. Progress in Brain Research. 2002;138:315–342. doi: 10.1016/S0079-6123(02)38085-3. [DOI] [PubMed] [Google Scholar]
- 257.Li Y, Luikart BW, Birnbaum S, et al. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron. 2008;59(3):399–412. doi: 10.1016/j.neuron.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Pittenger C, Duman RS. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology. 2008;33(1):88–109. doi: 10.1038/sj.npp.1301574. [DOI] [PubMed] [Google Scholar]
- 259.Fuchs E, Czéh B, Kole MHP, Michaelis T, Lucassen PJ. Alterations of neuroplasticity in depression: the hippocampus and beyond. European Neuropsychopharmacology. 2004;14(supplement 5):S481–S490. doi: 10.1016/j.euroneuro.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 260.Watanabe Y, Gould E, McEwen BS. Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Research. 1992;588(2):341–345. doi: 10.1016/0006-8993(92)91597-8. [DOI] [PubMed] [Google Scholar]
- 261.Norrholm SD, Ouimet CC. Altered dendritic spine density in animal models of depression and in response to antidepressant treatment. Synapse. 2001;42(3):151–163. doi: 10.1002/syn.10006. [DOI] [PubMed] [Google Scholar]
- 262.Nakagawa S, Kim JE, Lee R, et al. Regulation of neurogenesis in adult mouse hippocampus by cAMP and the cAMP response element-binding protein. Journal of Neuroscience. 2002;22(9):3673–3682. doi: 10.1523/JNEUROSCI.22-09-03673.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Christoffel DJ, Golden SA, Russo SJ. Structural and synaptic plasticity in stress-related disorders. Annual Review of Neuroscience. 2011;22:535–549. doi: 10.1515/RNS.2011.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Lamont SR, Stanwell BJ, Hill R, Reid IC, Stewart CA. Ketamine pre-treatment dissociates the effects of electroconvulsive stimulation on mossy fibre sprouting and cellular proliferation in the dentate gyrus. Brain Research. 2005;1053(1-2):27–32. doi: 10.1016/j.brainres.2005.06.041. [DOI] [PubMed] [Google Scholar]