

Abstract
Background
The endoribonuclease RNase-L is a type-I interferon (IFN)-regulatedcomponent of the innate immune response that functions in antiviral, antibacterial and antiproliferative activities. RNase-L produces RNA agonists of RIG-I-like receptors (RLRs), sensors of cytosolic pathogen-associated RNAs that induce cytokines including IFNβ. IFNβ and RLR signaling mediate protective responses against experimental colitis and colitis-associated cancer (CAC) and contribute to gastrointestinal (GI) homeostasis. Therefore, we investigated a role for RNase-L in murine colitis and CAC and its association with RLR signaling in response to bacterial RNA.
Methods
Colitis was induced in wild type (WT) and RNase-L-deficient mice (RNase-L−/−) by administration of dextran sulphate sodium (DSS). CAC was induced by DSS and azoxymethane (AOM). Histological analysis and immunohistochemistry were performed on colon tissue to analyze immune cell infiltration and tissue damage following induction of colitis. Expression of cytokines was measured by qRT-PCR and ELISA.
Results
DSS-treated RNase-L−/− mice exhibited a significantly higher clinical score, delayed leukocyte infiltration, reduced expression of IFNβ, TNFα, IL-1β and IL-18at early times post-DSS exposure and increased mortalityas compared to WT mice. DSS/AOM-treated RNase-L−/−mice displayed an increased tumor burden. Bacterial RNA triggeredIFNβproductionin an RNase-L-dependent manner and provided a potential mechanism by whichRNase-L contributes to the GI immune response to microbiota and protects against experimental colitis and CAC.
Conclusions
RNase-L promotes the innate immune response to intestinal damage and ameliorates murine colitis and CAC. The RNase-L-dependent production of IFNβ stimulated by bacterial RNA may be a mechanism to protectagainst GI inflammatory disease.
Keywords: Animal Models of IBD, Innate Immune System in IBD, Cytokines
INTRODUCTION
Inflammatory bowel disease (IBD), comprised of ulcerative colitis (UC) and Crohn’s disease (CD), is a major health concern that is diagnosed annually in approximately 1.4 million people in the United Statesand 2.2 million in Europe1. UCis characterized by chronic inflammation of the distal colon resulting from compromised GI barrier function which interferes with intestinal homeostasis2. In healthy individuals, pathogen-associated molecular patterns (PAMPs) from commensal microbes are sensed by pattern recognition receptors (PRRs) in host immune and epithelial cells which, in turn, activate signaling pathways to induce proinflammatory cytokines, chemokines and antibacterial effectors. This regulated response leads to bacterial clearance, repair of damaged intestinal epithelial cells (IECs) and resolution of inflammation. However an impaired capacity to detect pathogens or initiate an innate immune response to gut microflora results in excessive inflammation that is the hallmark of IBD and an important risk factor for the development of colon cancer3. Consistent with a critical role for innate immune components in intestinal homeostasis, mutation or aberrant expression of the cytosolic PRRs, NOD24,5, and NLRP36, the proinflammatory cytokine IL-187, and multiple TLRs that sense PAMPS on the cell surface and in endosomes8 are associated with CD in humans. Similarly, genetic disruption of PRRs9–12, their signaling components13, and effector cytokines14,15 in mice dramatically impacts susceptibility to DSS-induced GI epithelial injury in an experimental model of UC and demonstrated the functional roles of innate immune mediators in GI homeostasis. The dysregulated inflammatory response to DSS-induced epithelial damage in mice lacking innate immune components confers increased sensitivity to carcinogen-induced colorectal cancer and recapitulates the key role of inflammation in human malignancies9,12,13,16. Thus innate immune sensors and effectors are essential for intestinal homeostasis and their inactivation can predispose to chronic inflammation and malignant transformation1,17.
A complex array of gene products contributes to intestinal homeostasis and, if mutated or deleted, may result in pathologic outcomes. However, the full spectrum of host components that function to maintain a balanced interaction between host immune cells, IECs and intestinal microbiota remains to be determined. The endoribonucleaseRNase-L is an established component of the innate immune response that mediates Type-I interferon (IFN)-induced antiviral, antibacterial and antiproliferative activities. RNase-L is constitutively expressedas a latent monomer that upon binding to its allosteric activator 2-5A (pppA(2′p5′A)n,n ≥ 2), undergoes a conformational change resulting in dimerization and enzymatic activation. 2-5A is produced by a family of IFN-regulated 2′,5′-oligoadenylate synthetase (OAS) enzymes which are activated by double-stranded RNA (dsRNA). Active RNase-L cleaves host and viral RNAswith a preference forUU and UA dinucleotidesin single stranded regions; however the mechanisms by which endonucleolytic cleavage contribute to its biologic activities are not resolved. For example, while RNase-L may exert its antiviral effects through the direct cleavage of viral RNAs18, the products generated from RNase-L cleavage of host and viral RNAs can induce IFNβ and indirectly amplify antiviral activity19,20. RNase-L-mediated induction ofIFNβ is dependent on RLRs, cytosolic PRRs that bind viral RNA PAMPs to activate the mitochondrial antiviral signaling/IFNβ-promoter stimulator-1 (MAVS/IPS-1) adaptor protein. MAVS/IPS-1 signalosome-associated kinases then activate the IFN regulatory factors- (IRF) 3 and -7 and NFκB, resulting in their nuclear translocation and transcriptional inductionof proinflammatory cytokines21. Thus RNase-L-generated RNAs activate RLR signaling to induce proinflammatory cytokines. In addition to their roles in antiviral activity, RNase-L, RIG-I and MAVS have recently been reported to function in antibacterial defense. RNase-L-deficient (RNase-L−/−) mice exhibited increased susceptibility to gram-positive and -negative bacteria which corresponded with a diminished induction of proinflammatory cytokines including IFNβ, IL1β and TNFα22. LPS-induced bacterial phagocytosis was impaired in RIG-I-deficient macrophages and RIG-I knockout mice were more susceptible to Escherichia coli (E. coli) infection23. Consistent with this antibacterial activity, RIG-I and MAVS knockout mice displayed enhanced sensitivity to DSS-induced intestinal injury in experimental colitis24,25. Importantly, this study identified bacteria-derived RNA as a novel RLR PAMP providing a potential mechanism by which RLRs/MAVS monitorcommensal bacteria and regulate proinflammatory cytokines in the maintenance of GI homeostasis25.
In light of the antibacterial activities of RNase-L and RLRs/MAVS, their complementary roles in the production of RLR agonists and signal transduction induced by these ligands respectively, and the novel role of MAVS in monitoring commensal bacteria through sensing bacterial RNA, we hypothesized that RNase-L maycontribute to intestinal homeostasis and protection from pathological consequences of chronic GI damage. Consistent with this idea, a deficiency in the type I IFN receptor, which regulates 2-5A/RNase-L pathway activity, confers sensitivity to DSS-induced colitis26. Type I IFN promotes intestinal homeostasis in animal models and has shown efficacy in the treatment of UC27. Furthermore, OAS2, that functions upstream of RNase-L, interacts with the CD susceptibility gene NOD228.
To investigate a role for RNase-L in GI homeostasis we examined the response of RNase-L−/− mice to DSS-induced colitis. RNase-L−/−mice demonstrated delayed leukocyte infiltration anddiminished expression of IFNβ, TNFα, IL-1β and IL-18 at early times post DSS treatment. The impaired immune response corresponded with increased tissue damage and inflammation-associated pathology at later times post-DSS treatment. The increased sensitivity to DSS resulted in an increase in carcinogen-induced tumor burden and mortality. A potential mechanism by which RNase-L functions in the response to commensal bacteria was revealed by the finding that RNase-L promotes IFNβ production and suggests a model in which RNase-L cleaves bacterial RNA to stimulate cytokine induction via the RLR/MAVS pathway. Together these results identify anovel role for RNase-L in the homeostatic response to bacteria in the GI tract.
MATERIALS AND METHODS
Animals
WT and RNase-L−/− C57BL6 were housed in the UMB animal facility and were used in accordance with animal facilities at the University of Maryland School of Medicine and IACUC-approved protocols. Wild-type C57BL6 mice were purchased from the NCI and Jackson Laboratory or were derived from RNase-L/ C57BL6 backcrosses (Fig. A, B, Supplemental Digital Content 3, http://links.lww.com/IBD/A127). Helicobacter testing was performed on fecal samples by Idexx Radil.
Induction of DSS Colitis and DSS/AOM CAC
To induce colitis, sex-matched 8-10-week-old WT and RNase-L−/− mice were treated with 2.5% DSS (MP Biomedicals, 36,000-50,000 Da) in the drinking water for 9 days. For the DSS recovery assay, mice received DSS in the drinking water for 7 days followed by regular water for 7 days. Control mice received regular water only. To induce tumorigenesis, mice were administered AOM (10mg/kg) via IP injection at weeks 0 and 1, followed by 3 cycles of 2% DSS treatment beginning at weeks 2, 5 and 8 which lasted for 7, 4 and 4 days respectively. DSS/AOM control mice received PBS injections and regular water for the duration of the experiment. To minimize any influence of the microbiome that could obscure interpretation of the RNase-L−/− phenotype, purchased WT mice were acclimated for 12 days prior to beginning experimentation using an established flora transfer regimen adapted from that described in the Jackson Laboratory DSS-protocol29. During this acclimation period, imported WT mice were housed with bedding previously used for RNase-L−/− mice that were bred at the UMB animal facility. This bedding, including fecal material to promote transfer of GI flora and reduce the variability in microbiota between mice from different facilities, was changed at 12 and 7 days prior to DSS treatment.
Tissue Processing
Tissues were collected from mice at the time of euthanization and frozen at −70°C or fixed in 10% formalin overnight then embedded in paraffin. Colon tissue was rinsed 2x in PBS before processing. Whole blood was added to heparin then centrifuged at 1300 rpm. Serum was separated from erythrocytes and leukocytes and frozen until use.
Measuring Clinical Parameters
Clinical score is the sum of factors including weight loss, stool score and occult blood9. Weight loss is assigned a value of 0-4 for 0-20% loss. Stool is assigned a value of 0-4 for normal to diarrhea. Occult blood is assigned a score of 0 for no fecal blood, 2 for fecal blood (Hema Screen STAT FOBT) and 4 for rectal bleeding. Hemoglobin level was measured using 15 uL of whole blood (HgB Pro, ITC).
Histopathology: Paraffin-embedded colon tissue was cut into 5 μm sections and stained with H&E. Slides were coded then analyzed by two independent pathologists and scored according to previous methods30.
Myeloperoxidase Activity
Colon tissues were weighed then homogenized using an electric homogenizer in 0.5% hexa-decyl-trimethyl-ammonium bromide in 50 mmol/L potassium phosphate buffer (pH6) and centrifuged at 12,000 rpm for 10 min. Aliquots of the supernatant were mixed with a chromogenic peroxidase substrate (BM blue, Roche) and allowed to react for 20 min. Absorbance was measured by spectrophotometry at 450 nm. Human purified myeloperoxidase enzyme was used to create standard curves. Myeloperoxidase activity was determined by calculating OD vs. mass of tissue sample.
Multiplex ELISA
Multiplex ELISA of mouse serum for various cytokines was performed by the University of Maryland Cytokine Core Laboratory using the Luminex 100 System.
Quantitative Real-Time Polymerase Chain Reaction
Total RNA was isolated using TRIzol reagent (Invitrogen) following manufacturers protocol. Specific RNA sequences were quantified using the iScript One-Step RT-PCR Kit with SYBR Green reagents (Bio-Rad) and the C1000 Touch Thermal Cycler (Bio-Rad). Primers used were as follows: TNFα forward (ACGGCATGGATCTCAAAGAC) TNFα reverse (CGGACTCCGCAAAGTCTA AG), IL-6 forward (TGTCTATACCACTTCACAAGTCGGAG) IL-6 reverse (GCACAACTCTTTTCTCATTTCCAC), IL-1β forward (CAGGCAGGC AGTATCACTCA) IL-1β reverse (AGGCCACAGGTA TTTTGTCG), IFNβ forward (ATAAGCAGCTCCAGCTCCAA) IFNβ reverse (GCAACCACCACTCATTCTGA), IL-18 forward (GACAACTTTGGCCGACTTCACTGT) IL-18 reverse (CAGTCATATCCTCGAACACAGGCT). Expression levels of cytokines were normalized to GAPDH, primer sequence: GAPDH forward (CCTCGTCCCGTAGACAAAATC) GAPDH reverse (TGAAGGGGTCGTTGATGGC). For all primer sets, annealing temperature optimization and melting curve analysis were performed. Gene expression analysis was performed using CFX Bio-Rad software.
Immunohistochemistry
Colon tissue was frozen and cut into 6 μm sections. Sections were fixed in acetone, blocked, stained with primary antibody to F4/80 (Biolegend), 33D1 DC marker (eBioscience), Gr-1 (eBioscience), and CD3 (LifeSpan Biosciences) followed by biotinylated secondary antibody if necessary. Tissues were then incubated with Avidin (Vectastain ABC kit, Vector Laboratories), followed by incubation with peroxidase substrate (StableDAB, KPL). Tissues were counterstained with hematoxylin or Dapi (Invitrogen), dehydrated and mounted. Control sections were incubated with IgG from the host species.
Isolation of bone marrow macrophages and bacterial RNA
Bone marrows were flushed out from freshly-dissected mouse femurs and macrophages were cultured in DMEM containing 10% FBS, 15% L929 cell conditioned media (Bone marrow culture media). Cells were used for experiments after 7 d of culturing. Total bacterial RNA was isolated using Ambion® mirVana™ miRNA Isolation Kit (Life Technologies, Grand Island, NY).
Single ELISA assay for IFNβ
Transfections ofbone marrow macrophages with bacterial RNA were performed using Lipofectamine 2000 (Life Technologies). Following transfections, cell supernatants were collected and used to measure IFNβ with VeriKineTM Mouse Interferon-Beta ELISA KIT (PBL InterferonSource, Piscataway, NJ).E. coli strains TOP10 (Life Technologies) or LF82 were grown to exponential phase in L broth and used at multiplicities of infections of 5 and 20 to infect bone marrow macrophages in bone marrow cultured media (in the absence of antibiotics) for 1.5 hr after which gentamycin (Life Technologies) was added to the media at a concentration of 100 μg/ml. Cell supernatants were used for IFNβ determination by ELISA.
RESULTS
RNase-L has a protective role in the development of experimental colitis and promotes recovery following DSS treatment
To investigate a role for RNase-L in the development of experimental colitis, DSS (2.5% w/v in drinking water) or water vehicle was administered to RNase-L−/− and RNase-L+/+ (wild type, WT) mice for 9 days and clinical parameters were measured. Colitis-associated symptoms include weight loss, reduction in stool consistency, and increase in fecal occult blood, which can be combined to provide an overall clinical score. For each of these indicators, by 7–9 days of exposure DSS-treated RNase-L−/− mice exhibited more severe symptomsthan their WT counterparts (Fig. 1A–D). Decreased colon length is an additional macroscopic indicator of intestinal damage from colitis. Colon length in DSS-treated RNase-L−/− mice showed a significantly greater decrease than was observed in WT mice (Fig. 1E and S1 A). Blood loss due to ulceration from DSS-induced intestinal injury is reflected in a decrease in hemoglobin levels and provides a further measure of GI damage. Hemoglobin levels were lower in RNase-L−/− compared to WT mice at days 7 and 9 (Fig. B, Supplemental Digital Content 1, http://links.lww.com/IBD/A125) indicating more extensive colon damage. Thus RNase-L−/− mice displayed increased sensitivity to DSS treatment suggesting a novel protective function for RNase-L in response to GI damage. To determine if this protective role promoted recovery following DSS treatment, RNase-L−/− and WT mice were treated with DSS for seven days followed by a seven-day recovery period. The mortality rate was higher in RNase-L−/− compared to WT mice (Fig. 1F). Clinical parameters of colitis were monitored daily (as in Fig. 1D) and demonstrated the prompt recovery of WT mice, but not of RNase-L−/− mice (Fig., Supplemental Digital Content 2, http://links.lww.com/IBD/A126). These results suggest that RNase-L functions to protect from colitis-associated pathologies (Figs. 1 and 2).
Figure 1.
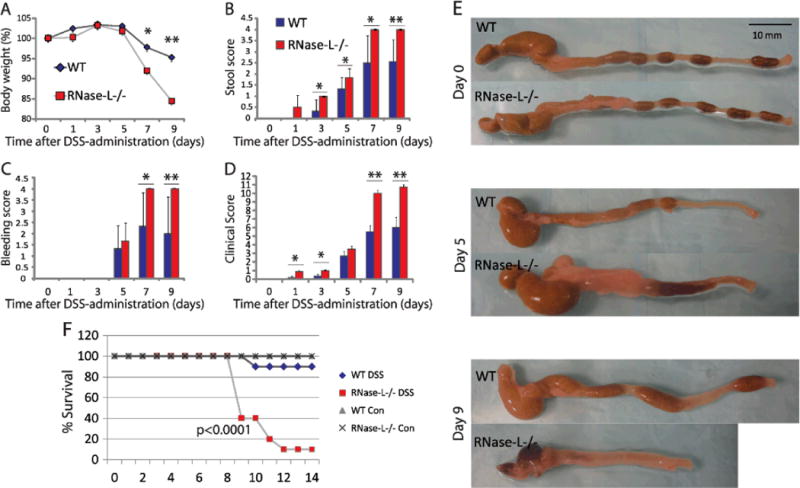
RNase-L is protective against DSS-induced colitis and promotes recovery following GI injury. RNase-L−/− and WT mice were treated with 2.5% DSS w/v drinking water or regular water control for 9 days. Mice were euthanized at days 0, 1, 3, 5, 7 (n = 6) and 9 (n = 7). The clinical score (D) is representative of colitis severity and is comprised of: (A) % weight loss (B) stool score- a measure of stool consistency and (C) bleeding score- a measure of occult blood. (E) Colon tissue was measured and photographed before processing. RNase-L−/− and WT mice were treated with 2.5% DSS w/v drinking water for 7 days followed by 7 days of regular water (con = untreated) to examine recovery. Survival (F) is expressed as a percent of the surviving mice from the total number treated (n = 15 for days1-7, n = 10 post day 7) and untreated (n = 3). Significance was determined by Kaplan-Meier analysis. Data represent means ± SE. *p < 0.05, **p < 0.005.
Figure 2.
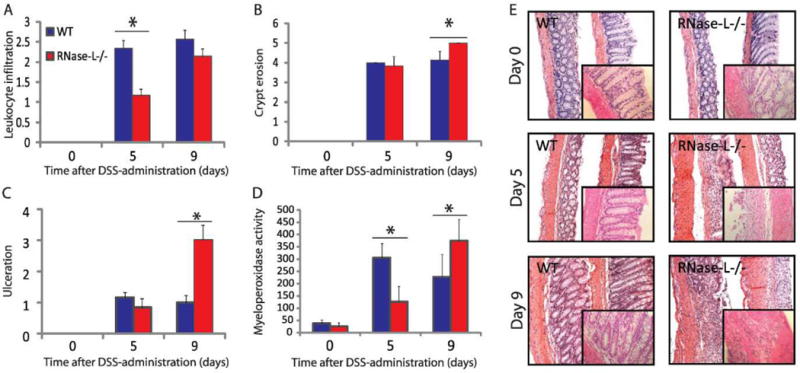
RNase-L promotes an early inflammatory response resulting in attenuated tissue damage in WT mice. H&E-stained colon sections were prepared from DSS-treated mice for histological analysis. Samples from days 0, 5 (n = 6) and 9 (n = 7) were analyzed. (A) Leukocyte infiltration, (B) crypt damage and (C) ulceration were measured by two independent pathologists. (D) Neutrophil activity was determined by myeloperoxidase assay using RNase-L−/− and WT mouse distal colon tissue (n = 6). (E) Representative images are shown (100x) with enlargements in insets (400x). Data represent means ± SE. *p < 0.05, **p < 0.005.
RNase-L promotes immune activity at early times of DSS treatment and reduces tissue injury at later times of exposure
To analyze the RNase-L DSS response phenotype at the cellular and molecular level, markers of inflammation and tissue damage were assayed in colons from DSS-treated RNase-L−/− and WT mice. H&E stained colon sections were analyzed by two pathologists who were masked to sample genotype and treatment. Histological analysis determined that leukocyte infiltration was greater in WT compared toRNase-L−/− mice at day 5, with no significant difference at day 9 (Fig. 2A), whereas overall tissue injury(crypt erosion and ulceration) was not significantly different at day 5 but was greater in RNase-L−/− mice at day 9 (Fig. 2B, C and E). The histology data reflects examinations of the entire colon, whereas examination of the distal colon only shows an increase in crypt erosion in RNase-L−/− compared to WT mice at days 5 and 9 (Fig. 2E). These results validate data from an initial study which demonstrated increased inflammation in RNase-L−/− as compared to backcrossed WT mice (Fig. A, B, Supplemental Digital Content 3, http://links.lww.com/IBD/A127). To determine differences in leukocyte activity, we measured myeloperoxidase (MPO) activity of colon tissue, an indicator of neutrophil activation. WT mice displayed higher MPO activity at day 5, whereas RNase-L−/− mice had significantly higher activity at day 9 (Fig. 2D). The diminished MPO activity at day 9 in WT mice is indicative of decreased tissue damage as increased neutrophil activity is correlated with colitis severity31. Thus, leukocyte infiltration and activity are lower in RNase-L−/− compared to WT mice at day 5 which may prevent a robust antibacterial response and contribute to the increased tissue damage observed in RNase-L−/− mice at day 9.
To investigate the influence of RNase-L on the infiltration of specific immune cells following DSS administration, we performed immunohistochemistry on distal colon tissue from RNase-L−/− and WT mice following treatment with DSS for 7 days. Neutrophil and monocyte infiltration are early events in the development of DSS-colitis and stimulate an immediate innate immune response followed by an adaptive response at later time points32. Both inefficient or unresolved innate immune activity can promote tissue injury2; similarly, adaptive immunity can be beneficial or detrimental in colitis33. We analyzed colon tissue for markers of macrophages, dendritic cells (DCs), neutrophils and T-cells. At day 7 of DSS treatment, dendritic cell, neutrophil and, to a lesser degree, macrophage infiltration was greater in RNase-L−/− compared to WT colon (Fig. 3B, D and A respectively) and corresponded with the elevated MPO activity observed at day 9. In contrast, CD3+ T-cell infiltration was reduced in RNase-L−/− mice (Fig. 3C); consistently, low CD3 expression within tumor tissue is correlated with decreased survival in CRC patients34. Interestingly, an attenuated T-cell response was previously observed following skin allografts in RNase-L−/− compared to WT mice35. The diminished CD3+ T-cell infiltrate may reflect the reduced early immune response observed in RNase-L−/− mice. Furthermore, the lack of an early response may inhibit the resolution of inflammation in mice lacking RNase-L.
Figure 3.
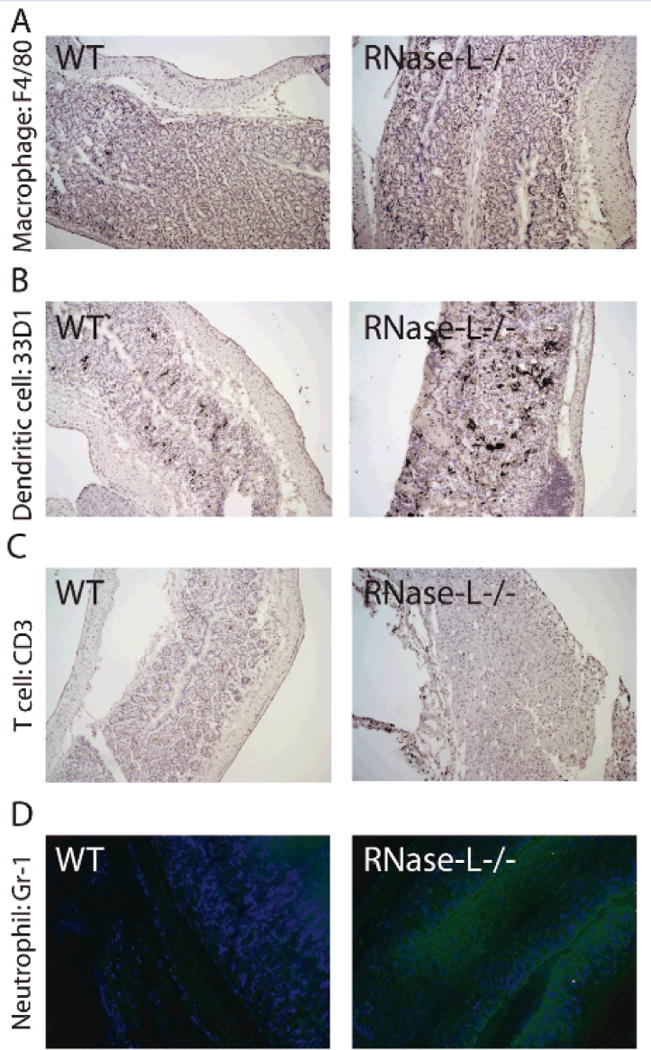
RNase-L influences immune cell infiltration during experimental colitis. Colon from DSS-treated mice was utilized for immunohistochemistry for immune cell markers. Day 7 frozen colon tissue was stained with antibody to (A) F4/80, (B) 33D1, (C) CD3 and (D) Gr-1 to determine the presence of macrophage, dendritic cells, CD3+ T cells and neutrophils respectively.
DSS-induced GI damage results in a compromised barrier function, release of bacteria from the gut lumen, activation of an innate immune response and induction of proinflammatory cytokines. We previously reported that RNase-L exerts antibacterial activity, in part, by stimulating the induction of proinflammatory cytokines22, therefore we hypothesized that RNase-L may also impact cytokine induction in response to DSS. Consistent with this prediction, qPCR analysis of cytokine mRNAs from colon tissue at day 5 of DSS treatment revealed significantly lower amounts of IFNβ, IL-1β, TNFα and IL-18 transcripts in RNase-L−/−compared to WT mice (Fig. 4A–D). Notably, critical roles in intestinal homeostasis and response to DSS have been reported for each of these cytokines9,26,27,31,32. IL-6 expression did not significantly differ in RNase-L−/− and WT mice (Fig. 4E). At day 9 of DSS treatment RNase-L−/− mice continued to exhibit diminished levels of IFNβ and IL-18 mRNAs whereas, the amount of IL1-β mRNA in RNase-L−/− mice was increased (Fig. 4A, D and B). In addition, serum levels of the key inflammatory cytokines IL-1β, TNFα, KC and MCP-1 were elevated at day 9 in RNase-L−/− mice and corresponded with an increase in inflammation-associated tissue damage at that time point (Fig. A–D, Supplemental Digital Content 4, http://links.lww.com/IBD/A128). Thus, the reduced expression of several proinflammatory cytokines in RNase-L−/− mice at early time-points post-DSS treatment correlated with diminished leukocyte recruitment and neutrophil activation. The reduced leukocyte recruitment may reflect an altered expression of cytokine-regulated chemokines in RNase-L−/− mice1,27,31,32.
Figure 4.
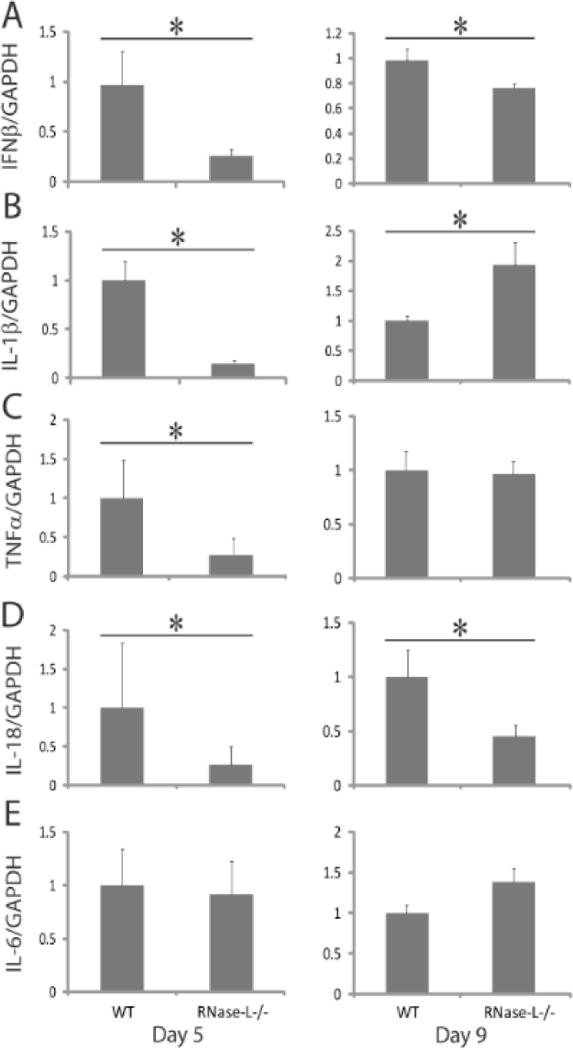
RNase-L is required for optimal induction of cytokine expression following DSS-induced damage. Colon tissue collected from DSS-treated mice at days 5 and 9 (n = 6) was analyzed by qRT-PCR for the expression of (A) IFNβ, (B) IL-1β, (C) TNFα, (D) IL-18 and (E) IL-6 mRNA. Cytokine expression was normalized to GAPDH. Data represent means ± SE. *p < 0.05.
RNase-L deficiency increases tumor burdenin inflammation-induced colon cancer
DSS-induced inflammation sensitizes mice to tumorigenesis induced by the carcinogen azoxymethane (AOM) and is a well-established model of inflammation-associated colon cancer9,36. RNase-L is implicated in tumor suppressor functionsdue to its proapoptotic and antiproliferative activities and the correlation of mutations in the RNASEL gene with multiple human malignancies37–40. In light of our data indicating a protective role for RNase-L in the development of DSS-induced experimental colitis (Fig. 1 and 2), we hypothesized that RNase-L will functionto suppress inflammation-associated tumorigenesis in the DSS/AOM model. Accordingly, to induce colon tumorigenesisRNase-L−/− and WT mice were injected with AOM followed by 3 cycles of DSS (Fig. 5A). RNase-L−/− mice displayed a significantly greater tumor burden compared to WT mice as measured by composite tumor volume at week 14 of the DSS/AOM regimen (Fig. 5B and Fig., Supplemental Digital Content 5, http://links.lww.com/IBD/A129), demonstrating its protective, antitumor function in this model. Furthermore, although the difference was not significant, RNase-L−/− mice tended to develop an increased number of tumors than WT mice (Fig. 5C). Moreover, mapping of macroscopic tumors to specific areas of the colon revealed differences in the location of tumorigenesis; specifically, tumors in RNase-L−/− mice were distributed in the cecum, proximal and distal colon whereas tumors in WT mice were limited to the distal colon (Fig., Supplemental Digital Content 6, http://links.lww.com/IBD/A130). The increased weight loss and mortality following DSS/AOM treatment of RNase-L−/− mice (Fig. 5D and Fig., Supplemental Digital Content 7, http://links.lww.com/IBD/A131) was similar to that observed following treatment with DSS alone (Fig. 1E) suggesting that the enhanced response to DSS was primarily responsible for the increased sensitivity to the DSS/AOM regimen. Therefore, we performed histological analysis of DSS/AOM-treated colon tissue to assess the presence of carcinoma pathology distinct from colitis-associated hyperplasia. This analysis revealed the presence of well-differentiated adenocarcinoma within areas of adenoma in both WT and RNase-L−/− mice (Fig. 5E) demonstrating an impact of RNase-L on inflammation-associated oncogenesis.
Figure 5.
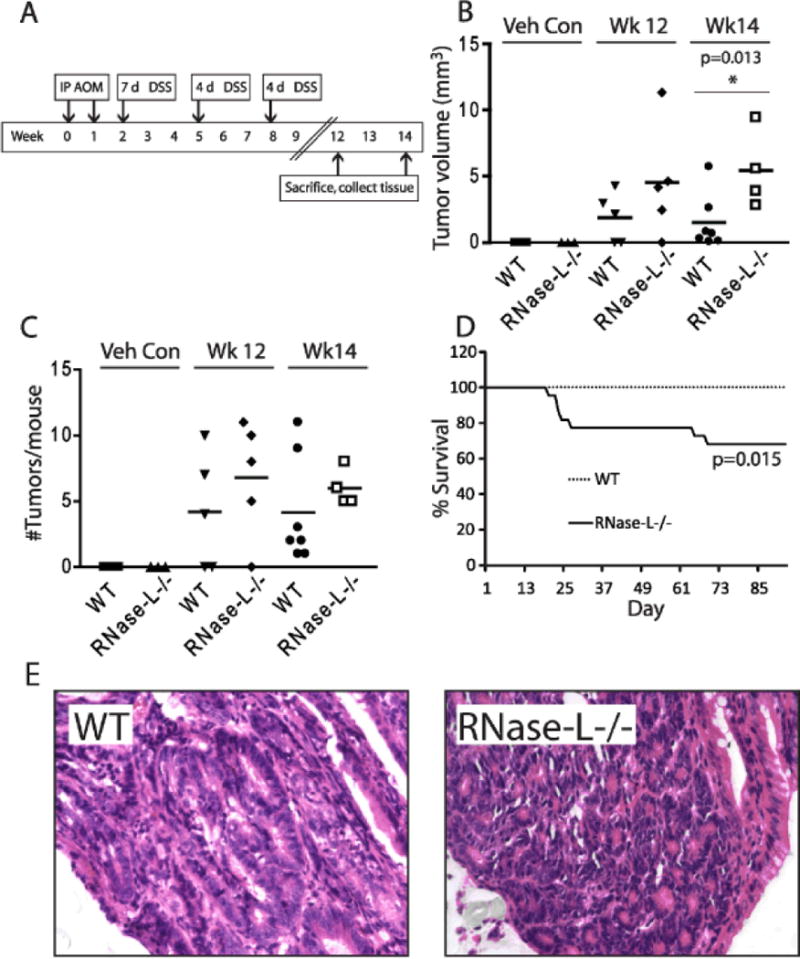
Tumor burden from colitis-associated cancer is increased in RNase-L−/− compared to WT mice. (A) In the treatment regimen, mice were administered 10 mg/kg azoxymethane (AOM) by intraperitoneal (IP) injection followed by three cycles of 2% DSS (WT: n = 21, RNase-L−/−: n = 22). Veh con = sham injection of PBS without AOM and regular water without DSS (WT: n = 8, RNase-L−/−: n = 8). (B) The total tumor burden per mouse was determined using the equation 1/2(Lx2W) as mm3. Significance was determined by Kaplan-Meier analysis. (C) Scatter plot of the number of tumors per mouse colon; bars indicate mean values. (D) Mortality due to DSS/AOM treatment is shown as % survival according to day. Significance was determined by Kaplan-Meier analysis. For tumor number (C) and volume (D), WT DSS/AOM: n = 12, RNase-L−/− DSS/AOM: n = 9, WT and RNase-L−/− sham: n = 3. Data represent means ± SE. *p < 0.05. (E) Histopathological analysis was performed on H&E-stained colon tissue sections from 14 weeks post DSS/AOM-treated mice. 400x images show well-differentiated adenocarcinoma in both WT and RNase-L−/− colon sections.
Bacteria-derived RNA is a trigger of RNase-L-dependent IFNβinduction
RNase-L-mediated regulation of cytokine induction is thought to contribute to its antibacterial activity and may be a mechanism by which it functions in DSS-induced inflammation. The RNase-L-dependent regulation of IFNβ and possibly other cytokines is proposed to occur by the cleavage of viral and host RNAs to generate RLR agonists leading to MAVS signaling and the induction of proinflammatory cytokines20. In this regard, MAVS−/− mice exhibited enhanced susceptibility to DSS-induced colitis25 and this phenotype corresponded with the MAVS-dependent sensing of RNA from commensal bacteria and resulting induction of IFNβ. In light of this finding and the role of RNase-L in the production of RLR agonists 41, we hypothesized that bacteria-derived RNA functions as a PAMP to trigger IFNβ induction in a RNase-L-dependent manner. To test this model, we first infected WT and RNase-L−/− bone marrow-derived macrophages (BMDMs) with a laboratory strain of E. coli (Top 10) or an adherent-invasive strain that is associated with Crohn’s disease (LF82) and IFNβ induction was measured by ELISA. Both E. coli strains induced 3-6-fold more IFNβ in WT as compared to RNase-L−/− BMDMs (Fig. 6A) consistent with a previous observation with E. coli Bort strain22. To determine if bacterial RNA is the trigger for RNase-L-dependent induction of IFNβ, RNase-L−/− and WT BMDMs were transfected with bacterial RNA and IFNβ induction was measured. Bacteria-derived RNA induced significantly more IFNβ production in WT compared to RNase-L−/− BMDMs (Fig. 6B). This diminished IFNβ induction in RNase-L−/− BMDMs mimicked that observed in MAVS−/− BMDMs suggesting that RNase-L functions upstream of RLR/MAVS to signal IFNβ induction by bacterial RNA25. In this manner, the RNase-L-RLR/MAVS axis may contribute to intestinal homeostasis by monitoring intestinal bacteria and regulating cytokine production.
Figure 6.
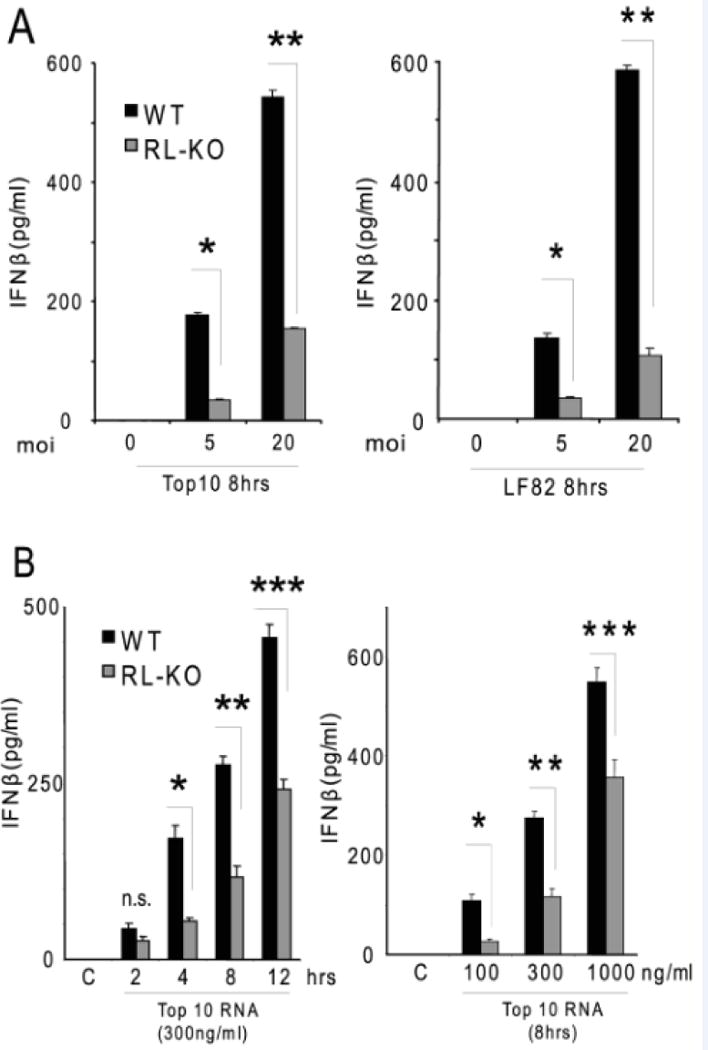
RNase-L mediates IFNβ production by BMDM in response to bacterial RNA. (A) BMDM were infected with Top10 or LF82 strains of E. coli at an MOI of 5 or 20 for 8 hours and IFNβ was measured by ELISA. (B) BMDMs were transfected with E. coli (Top10) RNA at the indicated times and doses, then IFN production was measured by ELISA. Data represent mean ± SE. *p < 0.05, **p < 0.01, ***p < 0.005.
DISCUSSION
We previously demonstrated that RNase-L functions in antibacterial immunity and in the regulation of proinflammatory cytokines22, two important components of intestinal homeostasis2. Therefore, we examined a role for RNase-L in the response to the DSS-induced intestinal damage in amodel of UC. RNase-L−/− mice displayed a compromised innate immune response at early times post-DSS and increased intestinal damage at later times indicating that RNase-L functions to protect from colitis-associated pathologies (Figs. 1 and 2). The increased sensitivity to DSS-induced colitis observed in mice lacking mediators of GI homeostasis is associated with enhanced susceptibility to CAC induced by DSS/AOM9,13,16. Similarly, DSS/AOM treatment resulted inan increased tumor burden in RNase-L−/− as compared to WTmice (Fig. 5D). Furthermore, RNase-L−/− but not WT mice developed tumors that were distributed throughout the colon (Figs, Supplemental Digital Content 5 and 6, http://links.lww.com/IBD/A129 and http://links.lww.com/IBD/A130) which may be due to differences in the immune response to infiltrating bacteria. For example, tumors induced following injection of high titres of Helicobacter into immunocompromised mice develop in the proximal colon42 suggesting that low levels of Helicobacter, that are common in laboratorymice but do not induce tumors, may influence the location of tumors induced by GI damaging agents. However, our data demonstrating the enhanced DSS-induced inflammation and pathology in RNase-L−/− mice as compared to backcrossed WT mice indicates that RNase-L-deficiency impacts the response to GI damage beyond a background contribution of Helicobacter in our mice. Consistent with this association of RNase-L deficiency with enhanced inflammation and tumorigenesis in mice, altered expression or activity of RNase-L has been implicated in colorectal cancer in humans43,44 suggesting a link between its roles in GI homeostasis and cancer.
The induction of proinflammatory cytokines was altered in RNase-L−/− colons following DSS treatment suggesting a mechanism underlying the protective phenotype of RNase-L in colitis. Specifically, RNase-L contributes to the induction of IFNβ and potentially other proinflammatory cytokines via the production of RLR agonists to activate the RLR/MAVS signalosome20. Thus the diminished cytokine induction in RNase-L−/− mice may reflect decreased signaling through the RLR/MAVS axis. Consistent with a role for RNase-L upstream of MAVS in the response to GI damage, both MAVS−/− and RNase-L−/− mice exhibited an increased susceptibility to DSS-induced colitis which corresponded with a diminished induction of IFNβ and other antimicrobial effectors25. Transfection of bacterial RNA mimicked the MAVS-dependent induction of IFNβ seen in response to bacterial infection suggesting that bacterial RNA represents a novel RNase-L substrate. Importantly, bacterial RNA induced IFNβ in an RNase-L-dependent manner (Fig. 6) providing further evidence that RNase-L functions upstream of MAVS in the response to bacterial RNA. The induction of IFNβ by transfected bacterial RNA is consistent with its cytosolic processing into RLR agonists by RNase-L and subsequent activation of the RLR/MAVS signalosome. In the case of bacterial infection, cytosolic RNase-L may gain access to bacterial RNA that has escaped the endosomal compartment; however, we cannot rule out the possibility that bacterial infection and RNA transfection induce IFNβ by separate, yet RNase-L-dependent, pathways.
The nature of bacterial RNA PAMPs and their precise mechanism of action in the GI tract remain to be determined. One study reported that feces-derived RNA, as compared to that from laboratory strains of E. coli or host RNA, induced significantly more IFNβ following transfection of immortalized mouse macrophage (RAW264.7) or human embryonic kidney (293) cells suggesting a means to distinguish commensal bacteria25. In contrast, we observed comparable RNase-L-dependent IFNβ induction by laboratory and invasive strains of E. coli in BMDMs; this difference may reflect differential responsiveness to bacterial stimuli in primary BMDMs and immortalized cell lines. Bacterial RNAs exhibit many modifications that may permit discrimination from host RNAs and lead to the selective activation of innate immune pathways including RNase-L/RLR/MAVS45–47. In this regard, RNA modifications can alter both the capacity to activate OAS and to be cleaved by RNase-L suggesting that OAS may function in sensing bacterial RNA45. Relatedly, the IFN-induced RNA sensor PKR, was recently demonstrated to function in DSS-induced inflammation and may also contribute to the detection of bacterial RNA48.
In addition to the regulation of proinflammatory cytokines, RNase-L may function in the response to GI damage through the modulation of IEC repair. Indeed, the expression of innate immune effectors in IECs is critical for the regulated response to GI injury1,25. Moreover, RNase-L serves as an endogenous constraint on cell proliferation and can promote apoptosis38. Defects in apoptosis in RNase-L-deficient cells have been reported in primary murine lymphocytes and fibroblasts and multiple human cancer cell lines49–51. Interestingly, IFNβ can promote apoptosis in some contexts52,53 suggesting that RNase-L may function both to induce IFNβ expression and as an effector of its apoptotic activity; however evidence for this regulatory circuit requires experimental validation. A compromised apoptotic response following damage of RNase-L-deficient IECs may promote dysregulated proliferation and oncogenesis; our finding that RNase-L−/− mice exhibit increased tumor burden in the DSS/AOM model is consistent with this prediction and experiments to directly compare the apoptotic response in WT and RNase-L−/− IECs are the subject of ongoing investigations. The increased tumor burden in DSS/AOM treated RNase-L−/− mice reflected a larger tumor volume as tumor frequency did not markedly differ from that seen in WT mice (Fig. 5B and C). This observation suggests that RNase-L deficiency impacts tumor progression rather than initiation; it will be of interest to further dissect the role of RNase-L in distinct steps of oncogenesis. The increased mortality in RNase-L−/− mice following DSS treatment likely accounted for the similar increase observed with the DSS/AOM regimen. However, histological analysis of DSS/AOM-treated colon tissue confirmed the carcinoma pathology, as opposed to DSS/colitis-induced hyperplasia. These findings suggest that the tumor suppressor activity of RNase-L is mediated, in part, via its role in the inflammatory response.
Most studies, including the DSS/AOM model described here, have reported antiproliferative/ tumor suppressor functions for RNase-L40,54,55. However elevated expression of RNase-L was observed in colorectal adenocarcinomas and noncancerous polyps as compared to normal mucosa in familial adenomatous polyposis patients, thus representing a potential early event in colorectal tumorigenesis43 and enhanced enzyme activity was implicated as a feature of chronic myelogenous leukemia56. These findings suggest that RNase-L may mediate oncogenic activities in a subset of malignancies. This dual tumor suppressor/oncogene functionality is frequently observed for critical regulators of gene expression (e.g. c-myc, miR-29)57,58 and is thought to reflect the presence of distinct targets or cofactors that direct the expression of tumor suppressor or oncogenic gene products in specific tumors. Similarly, the context-specific functions of RNase-L may be mediated by the profile of RNA binding proteins and RNA targets that are expressed in different cell types or cancers that, in turn, determine if RNase-L functions as a tumor suppressor or oncogene. Futher analysis of RNase-L activity and regulation in distinct tumors is required to assess its role and potential as a therapeutic target in these settings.
Our study identifies RNase-L as one of many genetic loci that contribute to intestinal homeostasis; accordingly, mutation or dysregulation of RNase-L may predispose individuals to develop IBD. Consistent with this role, RNase-L was identified as a gene that is altered in experimental colitis59. RNase-L is a tightly regulated effector that is amenable to pharmacologic activation51 or inhibition60, therefore further dissection of its role in GI homeostasis and pathogenesis may identify settings in which the therapeutic targeting of RNase-L is indicated.
Supplementary Material
Acknowledgments
The authors thank William Twaddell and Mike Lipsky, Department of Pathology, for histological analysis,. Rena Lapidus, Translational Services Shared Core, for mouse work with the DSS and DSS/AOM models and the Cytokine Core, for multiplex ELISA, Thomas Blanchard and Jessica Shiu for helpful discussion, School of Medicine (all from UMB). We alsothank Christine McDonald and Kourtney Nickerson for infecting macrophages withE.coli (LF82)andSean Kessler and Carol De La Motte for valuable discussions (all from the Department of Pathobiology, Cleveland Clinic).
Funding: These investigations were supported by NIH, NCI grant CA044059 (to R.H.S.), NCI grant CA120407 (to J-P.R.), NIAID grant AI077556 (to B.A.H.) and a Marlene and Stewart Greenebaum Cancer Center Pilot Grant (to B.A.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: RHS is a consultant and inventor on patents relating to RNase-L licensed to Alios BioPharma. BAH is an inventor on patents relating to RNase-L licensed to AliosBiopharma.
References
- 1.Saleh M, Trinchieri G. Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat Rev Immunol. 2011 Jan;11(1):9–20. doi: 10.1038/nri2891. [DOI] [PubMed] [Google Scholar]
- 2.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007 Jul 26;448(7152):427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 3.Danese S, Mantovani A. Inflammatory bowel disease and intestinal cancer: a paradigm of the Yin-Yang interplay between inflammation and cancer. Oncogene. 2010 Jun 10;29(23):3313–3323. doi: 10.1038/onc.2010.109. [DOI] [PubMed] [Google Scholar]
- 4.Strober W, Kitani A, Fuss I, Asano N, Watanabe T. The molecular basis of NOD2 susceptibility mutations in Crohn’s disease. Mucosal Immunol. 2008 Nov;1(Suppl 1):S5–9. doi: 10.1038/mi.2008.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001 May 31;411(6837):603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 6.Villani AC, Lemire M, Fortin G, et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat Genet. 2009 Jan;41(1):71–76. doi: 10.1038/ng285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pizarro TT, Michie MH, Bentz M, et al. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: expression and localization in intestinal mucosal cells. J Immunol. 1999 Jun 1;162(11):6829–6835. [PubMed] [Google Scholar]
- 8.Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol. 2010 Feb;10(2):131–144. doi: 10.1038/nri2707. [DOI] [PubMed] [Google Scholar]
- 9.Allen IC, TeKippe EM, Woodford RM, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med. 2010 May 10;207(5):1045–1056. doi: 10.1084/jem.20100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Natividad JM, Petit V, Huang X, et al. Commensal and probiotic bacteria influence intestinal barrier function and susceptibility to colitis in Nod1(−/−) ;Nod2(−/−) Mice. Inflamm Bowel Dis. 2011 Dec 11; doi: 10.1002/ibd.22848. [DOI] [PubMed] [Google Scholar]
- 11.Barreau F, Madre C, Meinzer U, et al. Nod2 regulates the host response towards microflora by modulating T cell function and epithelial permeability in mouse Peyer’s patches. Gut. 2010 Feb;59(2):207–217. doi: 10.1136/gut.2008.171546. [DOI] [PubMed] [Google Scholar]
- 12.Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010 Mar 26;32(3):379–391. doi: 10.1016/j.immuni.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salcedo R, Worschech A, Cardone M, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med. 2010 Aug 2;207(8):1625–1636. doi: 10.1084/jem.20100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McNamee EN, Masterson JC, Jedlicka P, et al. Interleukin 37 expression protects mice from colitis. Proc Natl Acad Sci U S A. 2011 Oct 4;108(40):16711–16716. doi: 10.1073/pnas.1111982108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elinav E, Strowig T, Kau AL, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011 May 27;145(5):745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dupaul-Chicoine J, Yeretssian G, Doiron K, et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity. 2010 Mar 26;32(3):367–378. doi: 10.1016/j.immuni.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 17.Grivennikov SI, Karin M. Inflammation and oncogenesis: a vicious connection. Curr Opin Genet Dev. 2010 Feb;20(1):65–71. doi: 10.1016/j.gde.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chakrabarti A, Jha BK, Silverman RH. New insights into the role of RNase L in innate immunity. J Interferon Cytokine Res. 2011 Jan;31(1):49–57. doi: 10.1089/jir.2010.0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luthra P, Sun D, Silverman RH, He B. Activation of IFN-{beta} expression by a viral mRNA through RNase L and MDA5. Proc Natl Acad Sci U S A. 2011 Feb 1;108(5):2118–2123. doi: 10.1073/pnas.1012409108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malathi K, Dong B, Gale M, Jr, Silverman RH. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature. 2007 Aug 16;448(7155):816–819. doi: 10.1038/nature06042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loo YM, Gale M., Jr Immune signaling by RIG-I-like receptors. Immunity. 2011 May 27;34(5):680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li XL, Ezelle HJ, Kang TJ, et al. An essential role for the antiviral endoribonuclease, RNase-L, in antibacterial immunity. Proc Natl Acad Sci U S A. 2008 Dec 30;105(52):20816–20821. doi: 10.1073/pnas.0807265105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kong L, Sun L, Zhang H, et al. An essential role for RIG-I in toll-like receptor-stimulated phagocytosis. Cell Host Microbe. 2009 Aug 20;6(2):150–161. doi: 10.1016/j.chom.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Zhang HX, Sun YP, et al. Rig-I-/- mice develop colitis associated with downregulation of G alpha i2. Cell Res. 2007 Oct;17(10):858–868. doi: 10.1038/cr.2007.81. [DOI] [PubMed] [Google Scholar]
- 25.Li XD, Chiu YH, Ismail AS, et al. Mitochondrial antiviral signaling protein (MAVS) monitors commensal bacteria and induces an immune response that prevents experimental colitis. Proc Natl Acad Sci U S A. 2011 Oct 18;108(42):17390–17395. doi: 10.1073/pnas.1107114108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McFarland AP, Savan R, Wagage S, et al. Localized delivery of interferon-beta by Lactobacillus exacerbates experimental colitis. PLoS One. 2011;6(2):e16967. doi: 10.1371/journal.pone.0016967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012 Jan 6; doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dugan JW, Albor A, David L, et al. Nucleotide oligomerization domain-2 interacts with 2′-5′-oligoadenylate synthetase type 2 and enhances RNase-L function in THP-1 cells. Mol Immunol. 2009 Dec;47(2–3):560–566. doi: 10.1016/j.molimm.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.JAX Notes No. 507. 2007. pp. 3–4. [Google Scholar]
- 30.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993 Aug;69(2):238–249. [PubMed] [Google Scholar]
- 31.Yan Y, Kolachala V, Dalmasso G, et al. Temporal and spatial analysis of clinical and molecular parameters in dextran sodium sulfate induced colitis. PLoS ONE. 2009;4(6):e6073. doi: 10.1371/journal.pone.0006073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Popivanova BK, Kitamura K, Wu Y, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. 2008 Feb;118(2):560–570. doi: 10.1172/JCI32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waldner MJ, Neurath MF. Colitis-associated cancer: the role of T cells in tumor development. Semin Immunopathol. 2009 Jul;31(2):249–256. doi: 10.1007/s00281-009-0161-8. [DOI] [PubMed] [Google Scholar]
- 34.Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006 Sep 29;313(5795):1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 35.Silverman RH, Zhou A, Auerbach MB, Kish D, Gorbachev A, Fairchild RL. Skin allograft rejection is suppressed in mice lacking the antiviral enzyme, 2′,5′-oligoadenylate-dependent RNase L. Viral immunology. 2002;15(1):77–83. doi: 10.1089/088282402317340242. [DOI] [PubMed] [Google Scholar]
- 36.Clapper ML, Cooper HS, Chang WC. Dextran sulfate sodium-induced colitis-associated neoplasia: a promising model for the development of chemopreventive interventions. Acta Pharmacol Sin. 2007 Sep;28(9):1450–1459. doi: 10.1111/j.1745-7254.2007.00695.x. [DOI] [PubMed] [Google Scholar]
- 37.Zhou A, Hassel BA, Silverman RH. Expression cloning of 2-5A-dependent RNAase: a uniquely regulated mediator of interferon action. Cell. 1993 Mar 12;72(5):753–765. doi: 10.1016/0092-8674(93)90403-d. [DOI] [PubMed] [Google Scholar]
- 38.Castelli JC, Hassel BA, Maran A, et al. The role of 2′-5′ oligoadenylate-activated ribonuclease L in apoptosis. Cell Death Differ. 1998 Apr;5(4):313–320. doi: 10.1038/sj.cdd.4400352. [DOI] [PubMed] [Google Scholar]
- 39.Madsen BE, Ramos EM, Boulard M, et al. Germline mutation in RNASEL predicts increased risk of head and neck, uterine cervix and breast cancer. PLoS One. 2008;3(6):e2492. doi: 10.1371/journal.pone.0002492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andersen JB, Li XL, Judge CS, et al. Role of 2-5A-dependent RNase-L in senescence and longevity. Oncogene. 2007 May 10;26(21):3081–3088. doi: 10.1038/sj.onc.1210111. [DOI] [PubMed] [Google Scholar]
- 41.Malathi K, Paranjape JM, Bulanova E, et al. A transcriptional signaling pathway in the IFN system mediated by 2′-5′-oligoadenylate activation of RNase L. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(41):14533–14538. doi: 10.1073/pnas.0507551102. %U http://www.ncbi.nlm.nih.gov/pubmed/16203993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mangerich A, Knutson CG, Parry NM, et al. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc Natl Acad Sci U S A. 2012 Jul 3;109(27):E1820–1829. doi: 10.1073/pnas.1207829109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang L, Zhou A, Vasavada S, et al. Elevated levels of 2′,5′-linked oligoadenylate-dependent ribonuclease L occur as an early event in colorectal tumorigenesis. Clin Cancer Res. 1995 Nov;1(11):1421–1428. [PubMed] [Google Scholar]
- 44.Kruger S, Silber AS, Engel C, et al. Arg462Gln sequence variation in the prostate-cancer-susceptibility gene RNASEL and age of onset of hereditary non-polyposis colorectal cancer: a case-control study. Lancet Oncol. 2005 Aug;6(8):566–572. doi: 10.1016/S1470-2045(05)70253-9. [DOI] [PubMed] [Google Scholar]
- 45.Anderson BR, Muramatsu H, Jha BK, Silverman RH, Weissman D, Kariko K. Nucleoside modifications in RNA limit activation of 2′-5′-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res. 2011 Nov;39(21):9329–9338. doi: 10.1093/nar/gkr586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wright JR, Keffer-Wilkes LC, Dobing SR, Kothe U. Pre-steady-state kinetic analysis of the three Escherichia coli pseudouridine synthases TruB, TruA, and RluA reveals uniformly slow catalysis. RNA. 2011 Dec;17(12):2074–2084. doi: 10.1261/rna.2905811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eberle F, Sirin M, Binder M, Dalpke AH. Bacterial RNA is recognized by different sets of immunoreceptors. European journal of immunology. 2009 Sep;39(9):2537–2547. doi: 10.1002/eji.200838978. [DOI] [PubMed] [Google Scholar]
- 48.Cao SS, Song B, Kaufman RJ. PKR protects colonic epithelium against colitis through the unfolded protein response and prosurvival signaling. Inflamm Bowel Dis. 2012 Jan 24; doi: 10.1002/ibd.22878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou A, Paranjape J, Brown TL, et al. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J. 1997 Nov 3;16(21):6355–6363. doi: 10.1093/emboj/16.21.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malathi K, Paranjape JM, Ganapathi R, Silverman RH. HPC1/RNASEL mediates apoptosis of prostate cancer cells treated with 2′,5′-oligoadenylates, topoisomerase I inhibitors, and tumor necrosis factor-related apoptosis-inducing ligand. Cancer Res. 2004 Dec 15;64(24):9144–9151. doi: 10.1158/0008-5472.CAN-04-2226. [DOI] [PubMed] [Google Scholar]
- 51.Xiang Y, Wang Z, Murakami J, et al. Effects of RNase L mutations associated with prostate cancer on apoptosis induced by 2′,5′-oligoadenylates. Cancer Res. 2003 Oct 15;63(20):6795–6801. [PubMed] [Google Scholar]
- 52.Zhang T, Sun HC, Zhou HY, et al. Interferon alpha inhibits hepatocellular carcinoma growth through inducing apoptosis and interfering with adhesion of tumor endothelial cells. Cancer Lett. 2010 Apr 28;290(2):204–210. doi: 10.1016/j.canlet.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 53.Qu J, Zhao M, Teng Y, et al. Interferon-alpha sensitizes human gastric cancer cells to TRAIL-induced apoptosis via activation of the c-CBL-dependent MAPK/ERK pathway. Cancer biology & therapy. 2011 Sep 15;12(6):494–502. doi: 10.4161/cbt.12.6.15973. [DOI] [PubMed] [Google Scholar]
- 54.Liu W, Liang SL, Liu H, Silverman R, Zhou A. Tumour suppressor function of RNase L in a mouse model. Eur J Cancer. 2007 Jan;43(1):202–209. doi: 10.1016/j.ejca.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 55.Andersen JB, Mazan-Mamczarz K, Zhan M, Gorospe M, Hassel BA. Ribosomal protein mRNAs are primary targets of regulation in RNase-L-induced senescence. RNA Biol. 2009 Jul;6(3):305–315. doi: 10.4161/rna.6.3.8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hubbell HR, Kariko K, Suhadolnik RJ, Brodsky I. RNase L and increased endoribonuclease activities in the mononuclear cells of patients with chronic myelogenous leukemia. Anticancer Res. 1994 Mar-Apr;14(2A):341–346. [PubMed] [Google Scholar]
- 57.Larsson LG, Henriksson MA. The Yin and Yang functions of the Myc oncoprotein in cancer development and as targets for therapy. Experimental cell research. 2010 May 1;316(8):1429–1437. doi: 10.1016/j.yexcr.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 58.Pekarsky Y, Croce CM. Is miR-29 an oncogene or tumor suppressor in CLL? Oncotarget. 2010 Jul;1(3):224–227. doi: 10.18632/oncotarget.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fang K, Bruce M, Pattillo CB, et al. Temporal genomewide expression profiling of DSS colitis reveals novel inflammatory and angiogenesis genes similar to ulcerative colitis. Physiol Genomics. 2011 Jan 7;43(1):43–56. doi: 10.1152/physiolgenomics.00138.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jha BK, Polyakova I, Kessler P, et al. Inhibition of RNase L and RNA-dependent protein kinase (PKR) by sunitinib impairs antiviral innate immunity. J Biol Chem. 2011 Jul 29;286(30):26319–26326. doi: 10.1074/jbc.M111.253443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.