

Summary
Intracerebral or parenchymal arterioles play an important role in the regulation of both global and regional blood flow within the brain. Brain cortex lacks significant collateral sources of blood and are thus at risk if blood flow through parenchymal arterioles is restricted. Increasingly, evidence is accumulating that abnormal parenchymal arteriolar constriction contributes to the development of neurological deficits caused by subarachnoid hemorrhage (SAH). For example, parenchymal arterioles isolated from SAH model rats exhibit enhanced constriction in response to increased intravascular pressure. This increased pressure-dependent constriction or myogenic tone would result in a shift in the cerebral autoregulatory response and decreased cerebral perfusion. Here, we summarize our current knowledge regarding cellular mechanisms contributing to enhanced contractility of parenchymal arteriolar myocytes following SAH. Our studies demonstrate SAH-induced membrane potential depolarization involving altered K+ homeostasis leads to enhanced voltage-dependent Ca2+ channel activity, increased smooth muscle cytosolic Ca2+ and parenchymal arteriolar constriction. In summary, emerging evidence demonstrates that SAH can profoundly affect parenchymal arteriolar tone promoting decreased cortical blood flow and compromised neuronal viability.
Keywords: Ca2+ channels, K+ channels, microcirculation, vascular smooth muscle, vasospasm
Role of parenchymal arterioles in the regulation of cerebral blood flow
Parenchymal arterioles are small diameter blood vessels containing a single layer of smooth muscle that are located in the brain parenchyma downstream of the Virchow-Robin space. Parenchymal arterioles can be distinguished from pial arteries, surface arterioles, and arterioles located within the transitional Virchow-Robin space by their lack of extrinsic innervation and encasement by astrocytic endfeet [2]. Considering Poiseuille’s law, which states that flow through a cylinder is proportional to the 4th power of the radius, it is not surprising that small changes in the diameter of these blood vessels will have a profound impact on the delivery of oxygen and nutrients to cells within the brain. Further, because collateral blood supply to the brain cortex is meager, hyper-constriction or occlusion of a parenchymal arteriole will severely limit tissue perfusion and nutrient supply to a given cortical region [12]. Another unique feature of parenchymal arterioles is their role in functional hyperemia, whereby focal increases in neuronal activity are coupled to parenchymal arteriolar dilation. This matching of local blood flow to regional brain function involves the coordinated activity of neurons, astrocytes and parenchymal arterioles (i.e. the neurovascular unit) [1]. Thus, parenchymal arterioles play an important role in local, as well as global, cerebral blood flow.
Enhanced pressure-induced constriction and elevated arteriolar wall Ca2+ of parenchymal arterioles from SAH model rats
Pial or brain surface arteries from SAH model animals exhibit enhanced constriction reflecting a combination of enhanced expression of voltage-dependent Ca2+ channels (VDCCs) and increased activity of these channels due to membrane potential depolarization [4, 5, 21]. Although available information is equivocal, a number of in vivo studies suggest that SAH can also negatively affect the microcirculation within the brain parenchyma [7, 15, 16]. However, because the small size of parenchymal arterioles poses a significant technical challenge, few in vitro studies have directly examined the impact of SAH on the parenchymal vasculature [19]. Most histological studies suggest that parenchymal arterioles from SAH model animals are more constricted [14]. Further, mechanistic information regarding the impact of SAH on the function of parenchymal arteriolar myocytes is limited. Our recent and ongoing work has begun to address these knowledge gaps.
Using the rat “double injection” SAH model, we have examined the impact of subarachnoid blood on parenchymal arteriolar function and Ca2+ signaling [13]. In this study, SAH rats received two intracisternal injections of autologous arterial blood via the cisterna magna at 24 hour intervals. This model recapitulates key pathologies observed in human SAH patients, including vasospasm, behavioral deficits and decreased cortical blood flow [10, 18, 20]. Importantly, we observed extravascular red blood cells along parenchymal arterioles for distances greater than 500 μm into the cerebral cortex, demonstrating that subarachnoid blood can pass beyond the Virchow-Robin space and directly interact with parenchymal arterioles within the brain cortex. These observations are consistent with previous reports of labeled (biotinylated) oxyhemoglobin penetrating a depth of greater than 1 mm into the cerebral cortex in a similar rat SAH model [17].
To examine the relationship between smooth muscle cytosolic Ca2+ and pressure-induced myogenic tone, simultaneous measurements of Ca2+ and diameter were obtained from isolated parenchymal arterioles using the ratiometric Ca2+ indicator fura-2 [13]. Within the physiological range of intravascular pressures (40–60 mmHg), parenchymal arterioles isolated from day 4 SAH rats exhibited significantly elevated arterial wall Ca2+ and enhance vasoconstriction (Figure 1A–E). Interestingly, the relationship between arteriolar Ca2+ and constriction (i.e. Ca2+ sensitivity) was similar between groups (Figure 1F). Further, selective L-type VDCC antagonists (e.g. nimodipine) caused a near maximum decrease in arteriolar Ca2+ and vasodilation (figure 1A–E). In the presence of L-type VDCC inhibitors, the R-type VDCC antagonist, SNX-482, and the purported T-type VDCC antagonist, mibefradil, did not alter cytosolic Ca2+ or diameter of parenchymal arterioles isolated from control or SAH model animals. These data demonstrate that elevated [Ca2+]i due to enhanced L-type VDCC activity underlies SAH- enhanced parenchymal arteriolar constriction.
Figure 1. Elevated cytosolic Ca2+ and enhanced myogenic tone in parenchymal arterioles from SAH animals.
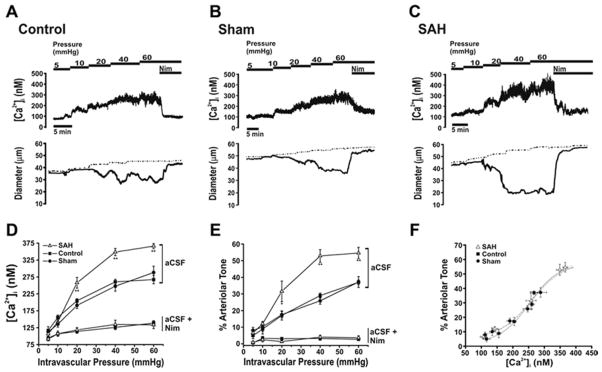
A–C: Representative simultaneous [Ca2+]i and diameter measurements obtained from intact arterioles isolated from unoperated (control; A), sham-operated (sham; B) and SAH (C) animals. Recordings were obtained during step-wise increases in intravascular pressure and subsequent nimodipine application (300 nM) at 60 mmHg. Dashed traces represent diameters in Ca2+-free aCSF containing nimodipine (300 nM). D, E. Summary of [Ca2+]i (D) and constriction (E) obtained in the absence and presence of 300 nM nimodipine. *P<0.05, **P<0.01 vs. control unoperated and sham-operated. F. Relationship between [Ca2+]i and constriction for arterioles isolated from control, sham-operated and SAH animals derived from summary data depicted in panel D and E. Reproduced from Nystoriak et al., American Journal of Physiology, 2011 [13].
SAH-enhanced VDCC activity in parenchymal arteriolar myocytes is due to suppression of voltage-dependent K+ channels and smooth muscle membrane potential depolarization
Enhanced L-type VDCC activity in parenchymal arteriolar myocytes from SAH animals could result from enhanced L-type VDCC expression, or membrane potential depolarization leading to increased activity of existing channels. Our data is consistent with the latter of these two possibilities. First, quantitative real-time PCR determined that expression of CaV1.2 mRNA, encoding the predominantly expressed L-type VDCC pore-forming α1 subunit in vascular smooth muscle, was similar in parenchymal arterioles isolated from control and SAH animals. Secondly, CaV1.2 protein levels were not different between parenchymal arteriolar homogenates obtained from control and SAH animals. Third, in vitro measurements using intracellular microelectrodes revealed a smooth muscle membrane potential depolarization of approximately 7 mV in pressurized parenchymal arterioles obtained from SAH animals, relative to control animals. As the open-state probability of L-type VDCCs is steeply voltage dependent in the physiological range of membrane potentials [11], a membrane potential depolarization of this magnitude (7 mV) would be expected to cause a substantial increase in Ca2+ channel activity [13]. These findings demonstrate that smooth muscle membrane potential depolarization, not increased L-type VDCC expression, is responsible for enhanced parenchymal arteriole constriction after SAH.
Voltage-dependent delayed rectifier K+ (KV) channels are expressed in the cerebral vasculature and are key regulators of smooth muscle membrane potential and arterial diameter [11]. Decreased KV channel activity would cause membrane potential depolarization, increased Ca2+ influx via VDCCs and vasoconstriction [6]. Thus, membrane potential depolarization and enhanced constriction of parenchymal arterioles from SAH animals could reflect a reduction in KV channel activity. We have previously demonstrated in pial cerebral areteries that the blood component, oxyhemoglobin, suppresses KV currents in cerebral artery myocytes by 30 %–40 % and causes vasoconstriction within minutes of application [3, 9]. This acute oxyhemoglobin-induced KV channel suppression is mediated via a cell signaling pathway involving activation of matrix metalloproteases (MMPs) leading to shedding of heparin-binding EGF-like growth factor (HB-EGF), activation of the tyrosine kinase EGF receptor, and KV channel internalization [3, 9] (figure 2). Currently, it is not known if this novel mechanism of KV channel suppression contributes to the sustained membrane potential depolarization and constriction that we have observed in parenchymal arterioles obtained from 4 day SAH model rats. Consistent with this possibility, using conventional whole-cell patch clamp electrophysiology, we have preliminary evidence indicating that outward voltage-dependent K+ currents are indeed suppressed in parenchymal arteriolar myocytes freshly isolated from day 4 SAH model rats (figure 3). Consistent with our previous studies using myocytes isolated from pial arteries [3, 8, 9], we have found that 4-AP-sensitive KV current density is dramatically decreased in freshly isolated parenchymal arteriolar myocytes from SAH animals. We have also observed that suppression of KV currents by HB-EGF was reduced in parenchymal arteriolar myocytes from SAH animals. Further, we have found that MMP-2 activity, but not expression, is enhanced in homogenates of cerebral arteries obtained from SAH animals. These data are consistent with SAH-induced suppression of KV currents in parenchymal arteriolar myocytes through a mechanism involving MMP and EGF receptor activation.
Figure 2. Proposed signaling pathway of OxyHb-induced KV current suppression involving HB-EGF and EGFR activation.
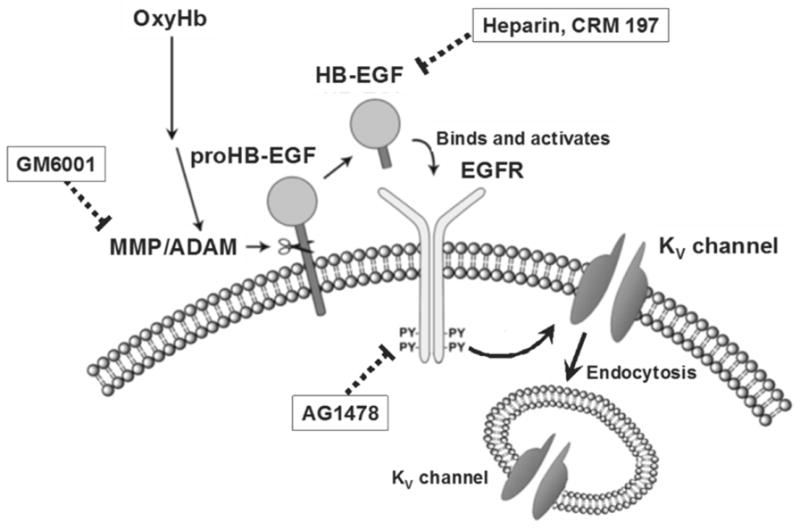
Schematic diagram illustrates OxyHb-induced KV current suppression via enhanced MMP activation and HB-EGF shedding. Abbreviations: OxyHb: oxyhemoglobin, MMP: matrix metalloprotease, ADAM: a disintegrin and metalloprotease, HB-EGF: heparin binding epidermal growth factor like growth factor, EGFR: epidermal growth factor receptor, PY: phosphorylated tyrosine residue, KV channel: voltage-dependent potassium channel. Reproduced from Koide et al., American Journal of Physiolgy, 2007 [9].
Figure 3. Voltage-dependent K+ channel currents are decreased in parenchymal arteriolar myocytes following SAH.
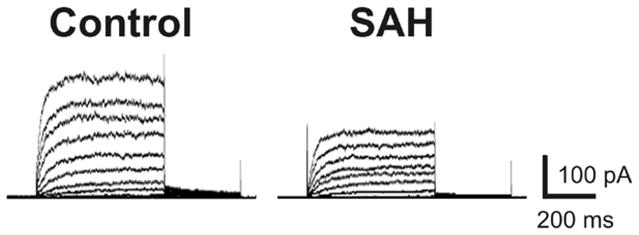
Representative traces of voltage-dependent K+ channel currents recorded using the conventional whole-cell patch clamp technique from control (cell capacitance: 8.3 pF) and SAH animals (cell capacitance: 8.2 pF).
Conclusions
Parenchymal arterioles play a critical role and represent a potential bottleneck in the delivery of blood to brain cortex. Subarachnoid blood causes enhanced parenchymal arteriole constriction at physiological intravascular pressures. This enhanced vasoconstriction is due to KV channel suppression leading to membrane potential depolarization and increased Ca2+ influx due to enhanced VDCC activity. SAH-induced parenchymal arteriolar constriction may contribute to decreased cerebral blood and the development of ischemic neuronal damage commonly observed in patients following cerebral aneurysm rupture.
Acknowledgments
This work was supported by the Totman Medical Research Trust Fund, the Peter Martin Brain Aneurysm Endowment, the NIH NIH (P01 HL095488, R01 HL078983, R01 HL078983-05S1 and R01 HL044455). The authors would also like to acknowledge the University of Vermont Neuroscience COBRE molecular biology and imaging core facilities.
Footnotes
Conflict of interest statement: None.
Reference List
- 1.Filosa JA, Bonev AD, Nelson MT. Calcium dynamics in cortical astrocytes and arterioles during neurovascular coupling. Circ Res. 2004;95:e73–e81. doi: 10.1161/01.RES.0000148636.60732.2e. [DOI] [PubMed] [Google Scholar]
- 2.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 3.Ishiguro M, Morielli AD, Zvarova K, Tranmer BI, Penar PL, Wellman GC. Oxyhemoglobin-induced suppression of voltage-dependent K+ channels in cerebral arteries by enhanced tyrosine kinase activity. Circ Res. 2006;99:1252–1260. doi: 10.1161/01.RES.0000250821.32324.e1. [DOI] [PubMed] [Google Scholar]
- 4.Ishiguro M, Puryear CB, Bisson E, Saundry CM, Nathan DJ, Russell SR, Tranmer BI, Wellman GC. Enhanced myogenic tone in cerebral arteries from a rabbit model of subarachnoid hemorrhage. Am J Physiol Heart Circ Physiol. 2002;283:H2217–H2225. doi: 10.1152/ajpheart.00629.2002. [DOI] [PubMed] [Google Scholar]
- 5.Ishiguro M, Wellman TL, Honda A, Russell SR, Tranmer BI, Wellman GC. Emergence of a R-type Ca2+ channel (CaV 2.3) contributes to cerebral artery constriction after subarachnoid hemorrhage. Circ Res. 2005;96:419–426. doi: 10.1161/01.RES.0000157670.49936.da. [DOI] [PubMed] [Google Scholar]
- 6.Knot HJ, Nelson MT. Regulation of membrane potential and diameter by voltage-dependent K+ channels in rabbit myogenic cerebral arteries. Am J Physiol. 1995;269:H348–H355. doi: 10.1152/ajpheart.1995.269.1.H348. [DOI] [PubMed] [Google Scholar]
- 7.Knuckey NW, Fox RA, Surveyor I, Stokes BA. Early cerebral blood flow and computerized tomography in predicting ischemia after cerebral aneurysm rupture. J Neurosurg. 1985;62:850–855. doi: 10.3171/jns.1985.62.6.0850. [DOI] [PubMed] [Google Scholar]
- 8.Koide M, Nystoriak MA, Krishnamoorthy G, O’Connor KP, Bonev AD, Nelson MT, Wellman GC. Reduced Ca2+ spark activity after subarachnoid hemorrhage disables BK channel control of cerebral artery tone. J Cereb Blood Flow Metab. 2011;31:3–16. doi: 10.1038/jcbfm.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koide M, Penar PL, Tranmer BI, Wellman GC. Heparin-binding EGF-like growth factor mediates oxyhemoglobin-induced suppression of voltage-dependent potassium channels in rabbit cerebral artery myocytes. Am J Physiol Heart Circ Physiol. 2007;293:H1750–H1759. doi: 10.1152/ajpheart.00443.2007. [DOI] [PubMed] [Google Scholar]
- 10.Lee JY, Huang DL, Keep R, Sagher O. Characterization of an improved double hemorrhage rat model for the study of delayed cerebral vasospasm. J Neurosci Methods. 2008;168:358–366. doi: 10.1016/j.jneumeth.2007.10.029. [DOI] [PubMed] [Google Scholar]
- 11.Nelson MT, Patlak JB, Worley JF, Standen NB. Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol. 1990;259:C3–18. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- 12.Nishimura N, Schaffer CB, Friedman B, Lyden PD, Kleinfeld D. Penetrating arterioles are a bottleneck in the perfusion of neocortex. Proc Natl Acad Sci U S A. 2007;104:365–370. doi: 10.1073/pnas.0609551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nystoriak MA, O’Connor KP, Sonkusare SK, Brayden JE, Nelson MT, Wellman GC. Fundamental increase in pressure-dependent constriction of brain parenchymal arterioles from subarachnoid hemorrhage model rats due to membrane depolarization. Am J Physiol Heart Circ Physiol. 2011;300:H803–H812. doi: 10.1152/ajpheart.00760.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohkuma H, Itoh K, Shibata S, Suzuki S. Morphological changes of intraparenchymal arterioles after experimental subarachnoid hemorrhage in dogs. Neurosurgery. 1997;41:230–235. doi: 10.1097/00006123-199707000-00036. [DOI] [PubMed] [Google Scholar]
- 15.Ohkuma H, Manabe H, Tanaka M, Suzuki S. Impact of cerebral microcirculatory changes on cerebral blood flow during cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Stroke. 2000;31:1621–1627. doi: 10.1161/01.str.31.7.1621. [DOI] [PubMed] [Google Scholar]
- 16.Takeuchi H, Handa Y, Kobayashi H, Kawano H, Hayashi M. Impairment of cerebral autoregulation during the development of chronic cerebral vasospasm after subarachnoid hemorrhage in primates. Neurosurgery. 1991;28:41–48. doi: 10.1097/00006123-199101000-00007. [DOI] [PubMed] [Google Scholar]
- 17.Turner CP, Bergeron M, Matz P, Zegna A, Noble LJ, Panter SS, Sharp FR. Heme oxygenase-1 is induced in glia throughout brain by subarachnoid hemoglobin. J Cereb Blood Flow Metab. 1998;18:257–273. doi: 10.1097/00004647-199803000-00004. [DOI] [PubMed] [Google Scholar]
- 18.Vatter H, Weidauer S, Konczalla J, Dettmann E, Zimmermann M, Raabe A, Preibisch C, Zanella FE, Seifert V. Time course in the development of cerebral vasospasm after experimental subarachnoid hemorrhage: clinical and neuroradiological assessment of the rat double hemorrhage model. Neurosurgery. 2006;58:1190–1197. doi: 10.1227/01.NEU.0000199346.74649.66. [DOI] [PubMed] [Google Scholar]
- 19.Vollmer DG, Takayasu M, Dacey RG., Jr An in vitro comparative study of conducting vessels and penetrating arterioles after experimental subarachnoid hemorrhage in the rabbit. J Neurosurg. 1992;77:113–119. doi: 10.3171/jns.1992.77.1.0113. [DOI] [PubMed] [Google Scholar]
- 20.Weidauer S, Vatter H, Dettmann E, Seifert V, Zanella FE. Assessment of vasospasm in experimental subarachnoid hemorrhage in rats by selective biplane digital subtraction angiography. Neuroradiology. 2006;48:176–181. doi: 10.1007/s00234-005-0021-8. [DOI] [PubMed] [Google Scholar]
- 21.Wellman GC. Ion channels and calcium signaling in cerebral arteries following subarachnoid hemorrhage. Neurol Res. 2006;28:690–702. doi: 10.1179/016164106X151972. [DOI] [PubMed] [Google Scholar]