

The N-methyl-D-aspartate (NMDA) receptor (NMDAR) is the predominant molecular device for controlling synaptic plasticity and memory function.1 Thus, an understanding of the control and action of the NMDAR at central synapses may provide clues to therapeutic strategies for treating memory disorders. Episodic and spatial memories, which are often compromised in persons with Alzheimer’s disease, critically depend on the hippocampus — in particular, the CA1 area — in both humans and laboratory animals. Blocking the NMDAR in the mouse brain impairs synaptic plasticity and compromises learning and memory. Conversely, genetic enhancement of NMDAR function improves memory in adult mice.1,2
Activation of the NMDAR, a major excitatory ligand-gated ion channel in the central nervous system, depends on a couple of coincidental events: the binding of its natural ligand (glutamate) and depolarization — which effects the removal of magnesium ions that otherwise block the ion-channel pore. The NMDAR constitutes the principal cellular machinery responsible for initiating many forms of synaptic plasticity in different areas of the brain. It is a heterotetramer consisting of two obligatory NR1 subunits and two of four possible NR2 subunits: NR2A, NR2B, NR2C, and NR2D. The NR2 subunits in the adult hippocampus and cortex are usually NR2A and NR2B, and the ratio of NR2B to NR2A decreases with age in diverse animal species (including humans), starting from or before the onset of sexual maturity. NR2 composition governs the properties of NMDAR channels and the extent of synaptic plasticity; a relative abundance of NR2B in the juvenile brain confers on it a greater plasticity than the adult brain. The core tetramer associates with a multiprotein complex that includes more than 70 associated proteins, many of which influence the transport, stability, subunit composition, or function of NMDARs.
In a recent study, Ng et al.3 showed that Neto1, a synaptic transmembrane protein known to be associated with the NMDAR, interacts with the receptor bivalently.3 The intracellular domain of Neto1 binds a synaptic protein (called PSD-95) that binds the receptor, and the extracellular domain of Neto1 interacts with NR2A and NR2B (Fig. 1).
Figure 1. Neuron Showing Glutamate Receptors and Synaptic Plasticity.
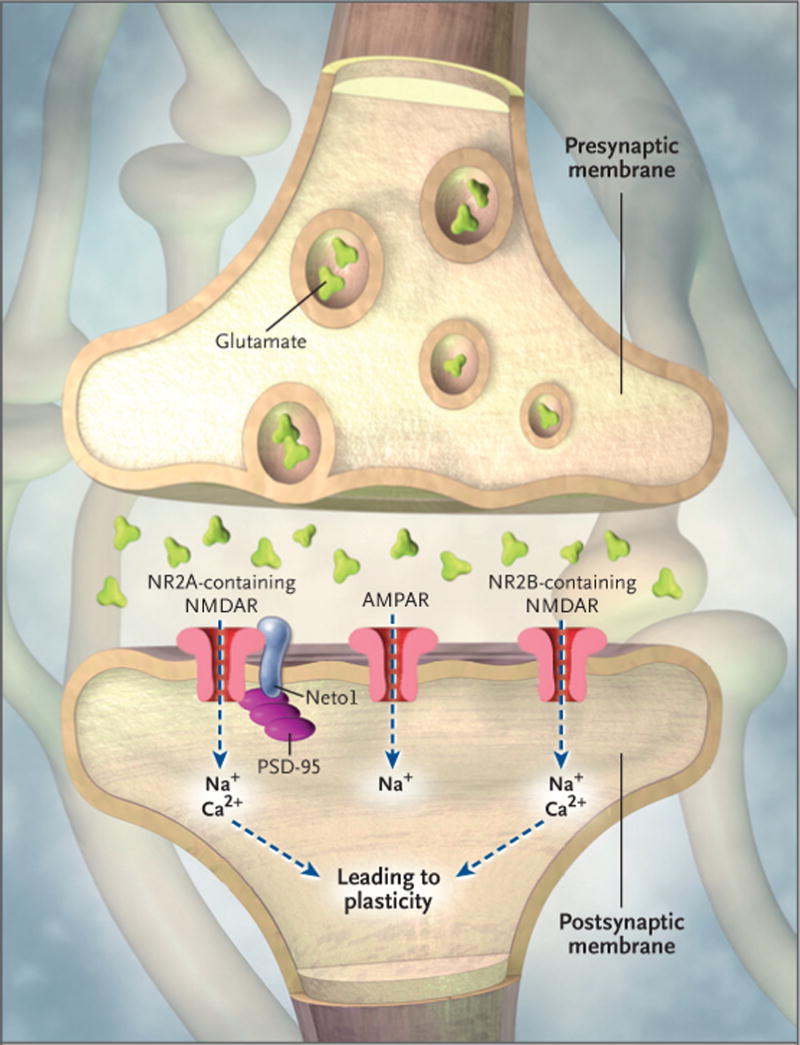
Various experiments suggest that memory formation involves two types of glutamate receptors: the N-methyl-D-aspartate receptor (NMDAR) and the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (AMPAR). These receptors sit on the surface of postsynaptic neurons. AMPARs allow sodium to flow into the postsynaptic cell, resulting in depolarization. NMDARs are permeable to both ionic sodium and calcium. The subsequent influx of ionic calcium into the postsynaptic terminals through the NMDAR activates biochemical cascades that trigger the up-regulation of AMPARs to the membrane while increasing the AMPAR’s sensitivity to glutamate and thus strengthen the synapses. A recent study by Ng et al.3 showed that the deletion of Neto1, a postsynaptic protein associated with the NMDAR complex, leads to deficits in both synaptic plasticity and cognition. Neto1 binds to PSD-95, a protein that is almost exclusively located in the postsynaptic density of neurons, and is important in anchoring synaptic proteins. Neto1 also binds to NR2A and NR2B, two isoforms of NR2 subunits that are essential for NMDAR functions. The composition of NR2A and NR2B in the NMDAR substantially modulates its channel properties. Stimulation of the AMPAR can partially compensate for deficits caused by Neto1 deletion.
To evaluate the physiological function of Neto1, Ng et al. bred mutant mice that were deficient in Neto1. A comparison of the mutant mice with their wild-type littermates showed that the total expression of NR1, NR2A, NR2B, and PSD-95 was the same in the hippocampus, but that synaptic NR2A (but not NR2B) was diminished in the mutant mice. This selective decrease in synaptic NR2A suggests that Neto1 may be required for achieving a normal abundance of NR2A-containing synaptic NMDARs, and it supports the hypothesis that different mechanisms are involved in the processing of NR2A and NR2B. The authors also observed a major defect in CA1 long-term potentiation in the mutant mice. Although the mutant mice performed similarly to wild-type mice in the hidden-platform water maze and recognition of new objects, they showed deficits in more complex tests of learning and memory.
Because NR2B subunits are present in the brains of mutant mice and are known to be the rate-limiting factor in controlling NMDAR-mediated synaptic plasticity and memory formation,1 the authors reasoned that the deficits in the Neto1-deficient mice might be compensated for by pharmacologically prolonging the excitatory pulses generated by the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor (the other main type of excitatory receptor at the postsynaptic membrane). Thus, they administered the ampakine CX546, which leads to a secondary increase of NMDA currents by relieving the magnesium blockade of the NR2B-containing NMDARs.
Indeed, the application of CX546 successfully compensated for both deficits in long-term potentiation and deficits in spatial learning in the mutant mice and had no discernible effect on wild-type mice. Ng et al. state that this was the first report of a pharmacologic solution to an NMDAR impairment. More importantly, this study suggests that the enhancement of NR2B-containing NMDAR function can overcome inherited defects in plasticity and restore memory functions. Other strategies to boost the NR2B-containing NMDA-receptor functions involve manipulating the NR2B transport pathway4 or modulating levels of magnesium in the cerebrospinal fluid through diet.
The study by Ng et al. may have relevance to therapeutic approaches to brain disorders with reduced NMDAR functions, such as schizophrenia and Alzheimer’s disease. For instance, one study suggested an association between a polymorphism in the promoter region of NR2B, reduced levels of NR2B expression, and an increased risk of Alzheimer’s disease.5 That said, memory processing is vastly more complicated in humans than in mice, and diseases affecting the brain often have diverse causes; positive results obtained with the use of a mutant mouse may not necessarily translate into similar results with the use of a similar approach to treat human disease.
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.Tsien JZ. Building a brainier mouse. Sci Am. 2000;282:62–8. doi: 10.1038/scientificamerican0400-62. [DOI] [PubMed] [Google Scholar]
- 2.Cao X, Cui Z, Feng R, et al. Maintenance of superior learning and memory function in NR2B transgenic mice during ageing. Eur J Neurosci. 2007;25:1815–22. doi: 10.1111/j.1460-9568.2007.05431.x. [DOI] [PubMed] [Google Scholar]
- 3.Ng D, Pitcher GM, Szilard RK, et al. Neto1 is a novel CUB-domain NMDA receptor-interacting protein required for synaptic plasticity and learning. PLoS Biol. 2009;7(2):e41. doi: 10.1371/journal.pbio.1000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang H, Jia J. Association between NR2B subunit gene (GRIN2B) promoter polymorphisms and sporadic Alzheimer’s disease in the North Chinese population. Neurosci Lett. 2009;450:356–60. doi: 10.1016/j.neulet.2008.10.075. [DOI] [PubMed] [Google Scholar]
- 5.Slutsky I, Sadeghpour S, Li B, Liu G. Enhancement of synaptic plasticity through chronically reduced Ca2+ flux during uncorrelated activity. Neuron. 2004;44:835–49. doi: 10.1016/j.neuron.2004.11.013. [DOI] [PubMed] [Google Scholar]