

Abstract
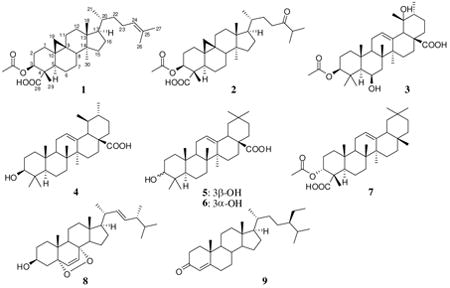
Three new triterpenoids, bonianic acids A (1), and B (2) and 3-O-acetyluncaric acid (3) were isolated from the leaves and twigs of Radermachera boniana (Bignoniaceae), together with six known compounds ursolic acid (4), oleanolic acid (5), 3-epi-oleanolic acid (6), 3α-O-acetyl-α-boswellic acid (7), ergosterol peroxide (8), and β-sitostenone (9). Ergosterol peroxide (8), bonianic acids A (1) and B (2) exhibited significant activity against Mycobacterium tuberculosis H37Rv strain.
Tuberculosis (TB) is a disease caused by Mycobacterium tuberculosis that most often affects the lungs.1 According to a 2009 estimate by the World Health Organization, 1.7 million deaths resulted from tuberculosis.2 The number of new cases recorded each year continued to rise globally, especially in Africa, the Eastern Mediterranean region, and South-East Asia. As part of our study in the search for new bioactive compounds from plants of Vietnam and Laos under the International Cooperative Biodiversity Groups (ICBG) Program,3 a plant extract (SV2933, Radermachera boniana Dop, Bignoniaceae) collected from the Cuc Phuong National Park was found to inhibit the growth of M. tuberculosis H37Rv with an MIC value of 78 μg/mL. Since a literature review showed that no chemical study of this plant had previously been reported, we selected this species for further studies. In this paper, we report the isolation and structural elucidation of three new triterpenoids (1-3), along with six known compounds, ursolic acid (4),4 oleanolic acid (5),5 3-epioleanolic acid (6),6 3α-O-acetyl-α-boswellic acid (7),7 ergosterol peroxide (8),8 and β-sitostenone (9).9 Ergosterol peroxide (8) was the most active compound against the M. tuberculosis H37Rv strain, followed by bonianic acids B (2) and A (1).
The dried and milled collected sample of the leaves and twigs of R. boniana (5.0 kg) was extracted with EtOAc at room temperature. The EtOAc soluble was purified by repeated open column chromatographies over silica gel to give compounds 1-9.
Compound 1 was obtained as a microcrystalline (mp 202-203 °C) material and was optically active [α]D25 +118 (c 0.2, CHCl3). In its positive HRESI mass spectrum, the pseudo-molecular ion was observed at m/z 521.3615 [M + Na]+, suggesting a molecular formula of C32H50O4. The 1D NMR spectra (1H and 13C) of 1 indicated the presence of an acetyl, six methyl (five singlets and one doublet), eleven methylenes, six methines (five sp3 and one sp2), one carboxylic, as well as six quaternary carbons (five sp3 and one sp2). The chemical shifts of CH2-19 (δC 29.6, δH 0.39 and 0.64, each dd, J = 4.5 Hz) were characteristic of a methylene function in a cyclopropane ring (Table 1).10a,b This observation suggested that 1 was a cycloartane triterpenoid. Analysis of the DEPT spectrum with the aid of 2D NMR determined the planar structure of 1 (Figure 1A), in which the methyl carbon C-28 was oxidized into a carboxylic group, which was established in turn by the presence of the HMBC correlation of the carboxylic carbonyl carbon at δC 180.5 (C-28) with H-3 at δH 5.23. The double bond was located between C-24 and C-25 as determined by the presence of the HMBC correlations of the proton at δH 5.10 (H-24) with two methyl carbons at δC 17.6 (C-26) and 25.7 (C-27). The cyclopropane ring formation involving C-9, C-10, and C-19 was determined by the presence of the HMBC cross-peaks of the protons at δH 0.39 and 0.64 (CH2-19) to the carbons at δC 44.3 (C-5) and 47.5 (C-8). The acetoxy group was assigned at C-3 due to the presence of the HMBC correlations between H-3 and the carbonyl carbon of the acetyl group at δC 170.2. This was also supported by the downfield chemical shift of H-3 at δH 5.23.
Table 1. NMR Data for Compounds 1-3 (1H: 500 MHz, 13C: 125 MHz).
| 1 | 2 | 3 | ||||
|---|---|---|---|---|---|---|
| position | δC | δH m (J, Hz) | δC | δH m (J, Hz) | δC | δH m (J, Hz) |
| 1 | 31.1, CH2 | 1.30, m | 31.0, CH2 | 1.30, m | 40.3, CH2 | 1.08, m |
| 1.74, ddd (3.5, 13.5, 13.5) | 1.76, m | 1.60, m | ||||
| 2 | 26.0, CH2 | 1.61, m | 26.0, CH2 | 1.60, m | 23.8, CH2 | 1.65, m |
| 1.95, m | 1.95, m | 1.74, m | ||||
| 3 | 77.3, CH | 5.23, dd (5.0, 12.0) | 76.9, CH | 5.22, d (4.5, 12.0) | 80.9, CH | 4.45, dd (4.0, 11.5) |
| 4 | 52.9, C | - | 52.9, C | - | 38.6, C | - |
| 5 | 44.3, CH | 2.10, dd (4.0, 12.5) | 44.2, CH | 2.12, dd (4.0, 12.5) | 55.7, CH | 0.87, m |
| 6 | 22.7, CH2 | 0.98, m | 22.7, CH2 | 0.97, m | 68.6, CH | 4.54, br.s |
| 1.26, m | 1.24, m | |||||
| 7 | 25.3, CH2 | 1.17, m | 25.3 | 1.17, m | 40.7, CH2 | 1.49, br. d (14.5) |
| 1.32, m | 1.32, m | |||||
| 8 | 47.5, CH | 1.58, m | 47.4, CH | 1.58, m | 39.1, C | - |
| 9 | 20.3, C | - | 20.2, C | - | 47.4, CH | 1.69, m |
| 10 | 24.9, C | - | 25.0, C | - | 36.4, C | - |
| 11 | 26.5, CH2 | 1.17, m | 26.4, CH2 | 1.17, m | 23.6, CH2 | 2.05, m |
| 1.99, m | 1.99, m | |||||
| 12 | 32.8, CH2 | 1.64, m | 32.7, CH2 | 1.62, m | 129.5, CH | 5.38, t (3.5) |
| 13 | 45.3, C | - | 45.3, C | - | 137.1, C | - |
| 14 | 48.8, C | - | 48.8, C | - | 41.8, C | - |
| 15 | 35.4, CH2 | 1.28, m | 35.3, CH2 | 1.29, m | 28.1, CH2 | 1.04, m |
| 1.74, m | ||||||
| 16 | 28.1, CH2 | 1.29, m | 28.0, CH2 | 1.31, m | 25.5, CH2 | 1.60, m |
| 1.90, m | 1.91, m | 2.51, ddd (4.5, 13.5, 13.5) | ||||
| 17 | 52.2, CH | 1.59, m | 52.2, CH | 1.59, m | 47.7, C | - |
| 18 | 17.9, CH3 | 0.95, s | 17.9, CH3 | 0.94, s | 52.9, CH | 2.54, s |
| 19 | 29.6, CH2 | 0.39, d (4.5) | 29.6, CH2 | 0.42, d (4.5) | 73.2, C | - |
| 0.64, d (4.5) | 0.67, d (4.5) | |||||
| 20 | 35.9, CH | 1.39, m | 35.7, CH | 1.38, m | 41.1, CH | 1.39, m |
| 21 | 18.3, CH3 | 0.88, d (6.5) | 18.1, CH3 | 0.88, d (6.5) | 26.0, CH2 | 1.31, m |
| 1.71, m | ||||||
| 22 | 36.3, CH2 | 1.04, m | 30.1, CH2 | 1.25, m | 37.3, CH2 | 1.65, m |
| 1.44, m | 1.77, m | 1.81, m | ||||
| 23 | 25.0, CH2 | 1.88, m | 37.5, CH2 | 2.40, ddd (6.0, 9.5, 27.9, CH3 16.5) | 0.96, s | |
| 2.06, m | 2.51, ddd (5.0, 10.0, 16.5) | |||||
| 24 | 125.2, CH | 5.10, dd (7.0, 7.0) | 215.5, C | - | 18.3, CH3 | 1.26, s |
| 25 | 130.9, C | - | 40.8, CH | 2.64, sept. (7.0) | 17.0, CH3 | 1.33, s |
| 26 | 17.6, CH3 | 1.60, s | 18.3, CH3 | 1.12, d (7.0) | 17.9, CH3 | 1.05, s |
| 27 | 25.7, CH3 | 1.68, s | 18.4, CH3 | 1.12, d (7.0) | 24.4, CH3 | 1.22, s |
| 28 | 180.5, C | - | 181.0, C | - | 183.8, C | - |
| 29 | 10.1, CH3 | 1.22, s | 10.1, CH3 | 1.21, s | 27.4, CH3 | 1.22, s |
| 30 | 19.2, CH3 | 0.90, s | 19.2, CH3 | 0.88, s | 16.1, CH3 | 0.94, d (6.5) |
| 31 | 170.2, C | - | 170.2, C | - | 171.0, C | - |
| 32 | 21.1, CH3 | 1.99, s | 21.1, CH3 | 1.99, s | 21.3, CH3 | 2.06, s |
Figure 1. Key HMBC (A) and NOE (B) cross-peaks for 1.

The relative configuration of 1 was defined on the basis of analysis of 1H-1H vicinal coupling constants and NOE interactions. H-3 displayed a gauche (5.0 Hz) and an anti (12.0 Hz) coupling constant, indicating its axial disposition on the A-ring. In addition, H-5 appeared as a doublet of doublets in the 1H NMR spectrum with small (4.0 Hz) and large (12.5 Hz) coupling constants, suggesting its axial orientation. In the NOESY spectrum, the proton Hβ of CH2-19 of the cyclopropane ring at δH 0.64 correlated with the protons at δH 1.22 (CH3-29) and 1.58 (H-8). The latter proton (H-8) showed a further cross-peak with the protons at δH 0.95 (CH3-18). This observation indicated that H-8, CH2-19, CH3-18 and CH3-29 were cofacial. In addition, CH3-30 at δH 0.90 exhibited a cross-peak with Hα of CH2-11 at δH 1.99 that suggested a boat conformation for the C-ring. The structure of 1 was finally established as drawn in Figure 1B. This new cycloartane was identified as 3β-O-acetylcycloart-24-en-28-oic acid and named bonianic acid A. Cycloartane triterpenoids bearing carboxylic functions are rare in nature.11a,b
Compound 2 was obtained in micro-crystalline form and was optically active, [α]D25 +68 (c 0.4, CHCl3). The negative HRESI mass spectrum exhibited the base peak at m/z 513.3595 [M-H]-, suggesting a molecular formula of C32H50O5. The 1H and 13C NMR spectroscopic data of 2 were similar to those of 1, except for the presence of a carbonyl group at δC 215.5 and a methine function at δC 40.8 and δH 2.64 (CH-25) instead of the olefinic signals in 1. 2D NMR analysis allowed the determination of a planar structure for 2. The presence of a C-24 keto group, was shown by the presence of the HMBC cross-peaks of the carbonyl at δC 215.5 (C-24) with two methyl groups CH3-26 and CH3-27 at δH 1.12, as well as with the protons at δH 2.40 and 2.51 (CH2-23). The C-28 carboxylic carbon was confirmed by the 3J-HMBC correlation with the proton at δH 5.22 (H-3). H-3 was further correlated to the carbon at δC 10.1 (C-29), the carbonyl of the acetyl group at δC 170.2 and the carbon at δC 44.2 (C-5), depicting the linkage of the acetate group to C-3.
Analyses of 1H-1H vicinal coupling constants and NOE interactions showed that this compound had the same relative configuration as 1: H-3 had a gauche (J = 4.5 Hz) and a trans-diaxial (J = 12.5 Hz) coupling constant, indicating its axial disposition on the A-ring. Similarly, H-5 was a doublet of doublets (J = 4.0 and 12.5 Hz) in the 1H NMR spectrum and had NOE interaction with H-3. An axial orientation was thus assigned for H-5. Similar to 1, a boat conformation of the C-ring was also observed for 2 which was determined from the presence of the NOE interaction of the protons at δH 0.88 (CH3-30) with the proton at δH 1.99 (Hα of CH2-11). Compound 2 was thus 3β-O-acetylcycloart-24-one-28-oic acid and named bonianic acid B.
Compound 3, a micro-crystalline solid, was optically active, [α]D30 +34 (c 0.5, CHCl3). The base peak was observed at m/z 531.3693 for [M + H]+ in its positive mass spectrum, suggesting a molecular formula of C32H50O6. The 1H NMR spectrum exhibited signals of eight methyl groups (seven singlets and one doublet). The 13C NMR and DEPT spectra showed the presence of 32 carbons, including the presence of an acetyl group. The NMR signals of 3 resembled those of uncaric acid,12 except for the additional signal of an acetyl group and the signal of H-3 shifted downfield at δH 4.45. Analyses of the 2D NMR allowed to assign the structure of 3 as shown. The acetyl group at C-3 was established from the presence of the HMBC correlation of the proton at δH 4.45 (H-3) with the carbonyl carbon at δC 171.0 of the acetyl group. The β-configuration of the C-3 substituent was determined from vicinal coupling constants of H-3 which exhibited a gauche (J = 4.0 Hz) and an anti (J = 11.5 Hz) coupling constant. Similarly, H-6 appeared as a broad singlet in the 1H NMR spectrum, indicating its equatorial orientation. Comparison of the chemical shifts of C-19 of 3 (δC 73.2 in CDCl3) and uncaric acid (δC 72.2 in CDCl3+pyridine-d5),12 suggested the α-orientation of the C-19 OH group for 3. The compound was determined as 3β-O-acetyl-6β,19α-diol-12-ursen-28-oic acid and was named 3-O-acetyluncaric acid.
The known compounds, ursolic acid (4),4 oleanolic acid (5),5 3-epioleanolic acid (6),6 3α-O-acetyl-α-boswellic acid (7),7 ergosterol peroxide (8),8 and/β-sitostenone (9)9 were also isolated and characterized. Their NMR data were compared with reported data.
The fractions obtained from the first chromatography column were evaluated for their activity against M. tuberculosis H37Rv. Subsequent separation of the active fractions led to the isolation of the pure compounds, ergosterol peroxide (8) with an MIC value of 3.5 μM, followed by the new triterpene, bonianic acid B (2) (MIC value: 9.9 μM) (Table 2). It is important to note that ergosterol peroxide (8) had no toxicity against Vero cells at 200 μM, while bonianic acid B (2) exhibited weak cytotoxicity with an IC50 value of 74.2 μM. Furthermore, the new compound bonianic acid A (1) demonstrated a moderate anti-TB activity with an MIC value of 34.8 μM. The other compounds showed no or weak anti-TB activity. The activity and selectivity of ergosterol peroxide (8) is consistent with previous reports.13a,b
Table 2. Anti-TB Activities of Compounds 1-9.
| Compd. | MIC (μM) | Compd. | MIC (μM) |
|---|---|---|---|
| 1 | 34.8 | 6 | > 200 |
| 2 | 9.9 | 7 | 94.8 |
| 3 | 75.5 | 8 | 3.5 |
| 4 | 94.8 | 9 | 39.5 |
| 5 | 96.5 | Rifampin | 0.14 |
Experimental section
General Experimental Procedures
Plant Material
Extraction and Isolation
The dried and ground mixture of the twigs and leaves (5.0 kg) of R. boniana was extracted with EtOAc three times at room temperature. The EtOAc extract was concentrated under reduced pressure and the residue (254 g) was purified by silica gel column chromatography (600 g), eluted with a solvent gradient of n-hexane/EtOAc (4 L) and then EtOAc/MeOH (3 L), to yield 15 fractions. Fraction 3 (16.12 g) was purified by column chromatography over silica gel (150 g), eluted with n-hexane/acetone (2 to 40% of acetone in n-hexane, 1.5 L) to afford 9 (14 mg). Fraction 4 (11.91 g) was separated on a silica gel column (150 g) eluting with a gradient of n-hexane/EtOAc (5 to 30% of EtOAc in n-hexane, 2.1 L) to give 5 subfractions. Subfraction 3 (0.7 g) was separated by silica gel column chromatography (20 g), eluted with a mixture of n-hexane/EtOAc (5 to 30% of EtOAc in n-hexane, 280 mL), followed by recrystallization from EtOAc to yield 1 (15 mg) and 7 (50 mg). Fraction 6 (12.36 g) was subjected to column chromatography on silica gel (150 g), eluted with mixture of CH2Cl2/acetone (5 to 30% of acetone in CH2Cl2, 2.4 L), then crystallized from MeOH to afford 2 (13 mg), 3 (7 mg), 5 (100 mg) and 6 (20 mg). Crystallization of fraction 7 (7.0 g) from EtOAc yielded 4 (1.5 g). Fractions 8 and 9 were recombined (13.17 g) and separated on a silica gel column (150 g), eluted with a mixture of n-hexane/EtOAc (5 to 70% of EtOAc in n-hexane, 310 mL), to yield 5 subfractions. Subfraction 3 (2.0 g) was purified by column chromatography on silica gel (10 to 100% of EtOAc in n-hexane, 460 mL), followed by crystallization from a mixture of n-hexane/EtOAc (8/2) to give 8 (10 mg).
Bonianic acid A (1)
Micro-crystals, mp 202-203 °C (EtOAc); [α]D25 +118 (c 0.2, CHCl3); IR νmax 3456, 2941, 2875, 1738, 1693, 1465, 1378, 1241, 1025, 998 cm−1; HRESIMS (positive mode) m/z 521.3615 [M + Na]+ (calcd for C32H50NaO4, 521.3607). NMR data see Table 1.
Bonianic acid B (2)
Micro-crystals, mp 193-194 °C (MeOH); [α]D25 +68 (c 0.4, CHCl3). IR νmax 3498, 2933, 2869, 1727, 1628, 1469, 1377, 1264, 1030, 997 cm−1; HRESIMS (negative mode) m/z 513.3595 [M - H]- (calcd for C32H49O5, 513.3580). NMR data see Table 1.
3-O-Acetyluncaric acid (3)
Micro-crystals, mp 203-204 °C (MeOH); [α]D30 +34 (c 0.5, CHCl3); IR νmax 3430, 2932, 1733, 1690, 1467, 1372, 1246, 1029, 893 cm−1; HRESIMS (positive mode) m/z 531.3693 [M + H]+ (calcd for C32H51O6, 531.3686). NMR data see Table 1.
Bioassays
The details for the anti-TB bioassays are described in the Supporting Information.14a,b
Supplementary Material
Figure 2. Selected HMBC correlations for 3.

Acknowledgments
The authors express their thanks to the Director of Cuc Phuong National Park for the permission and auspices, and to Mr. Mai Van Xinh, Cuc Phuong National Park, in the collection and recollection of samples of Radermachera boniana. This research was supported under the ICBG grants from the Fogarty International Center, NIH, 1-UO1-TW001015-01 (1998-2003), 2-UO1-TW001015-06 (2003-2008), 3U01TW001015-10S1 and 2U01TW001015-11A1, administered by the Fogarty International Center as part of an International Cooperative Biodiversity Groups (ICBG) program, through funds from NIH, NSF, and Foreign Agricultural Service of the USDA.
Footnotes
Supporting Information. 1D and 2D NMR spectra of 1-3. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Southwick F. Infectious Diseases: A Clinical Short Course. 2nd. Vol. 104 McGraw-Hill Medical Publishing Division; 2007. [Google Scholar]
- 2.Http://www.who.int/tb/publications/2010/factsheet_tb_2010.pdf.
- 3.Soejarto DD, Fong HHS, Tan GT, Zhang HJ, Ma CY, Franzblau SG, Gyllenhaal C, Riley MC, Kadushin MR, Pezzuto JM, Xuan LT, Hiep NT, Hung NV, Vu BM, Loc PK, Dac LX, Binh LT, Chien NQ, Hai NV, Bich TQ, Cuong NM, Southavong B, Sydara K, Bouamanivong S, Ly HM, Thuy TV, Rose WC, Dietzman GR. J Nat Prod. 2006;69:473–481. doi: 10.1021/np058107t. [DOI] [PubMed] [Google Scholar]
- 4.Poehland BL, Carté BK, Francis TA, Hyland LJ, Allaudeen HS, Troupe N. J Nat Prod. 1987;50:706–713. doi: 10.1021/np50052a022. [DOI] [PubMed] [Google Scholar]
- 5.(a) Maheshwari PJ. Nat Prod. 1989;52:623–628. [Google Scholar]; (b) Djarmati Z, Jankov RM, Djordjevic A, Ribar B, Lazar D, Engel P. Phytochemistry. 1992;31:1307–1309. [Google Scholar]; (c) Maillard M, Adewunmi CO, Hostettmann K. Phytochemistry. 1992;31:1321–1323. [Google Scholar]
- 6.(a) Srivastava OP, Khare A, Khare MPJ. Nat Prod. 1983;46:458–461. [Google Scholar]; (b) Huneck S. Tetrahedron. 1963;19:479–482. [Google Scholar]
- 7.Belsner K, Buchele B, Werz U, Syrovets T, Simmet T. Magn Reson Chem. 2003;41:115–122. [Google Scholar]
- 8.(a) Wojciechowski ZA, Goad LJ, Goodwin Trevor W. Phytochemistry. 1973;12:1433–1436. [Google Scholar]; (b) Gunatilaka AA, Gopichand Y, Schmitz FJ, Djerassi C. J Org Chem. 1981;46:3860–3866. [Google Scholar]; (c) González AG, León F, Rivera A, Muñoz CM, Bermejo J. J Nat Prod. 1999;62:1700–1701. [Google Scholar]; (d) Batrakov SG, Konova IV, Sheichenko VI, Esipov SE, Galanina LA, Istratova LN, Sergeeva YE. Phytochemistry. 2004;65:1239–1246. doi: 10.1016/j.phytochem.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 9.(a) Migliuolo A, Piccialli A, Sica D. J Nat Prod. 1990;52:1262, 1266. [Google Scholar]; (b) Gaspar EMM, das Neves HJC. Phytochemistry. 1993;34:523–527. [Google Scholar]
- 10.(a) Sun H, Qiu S, Lin L, Wan Z, Lin Z, Pengsuparp T, Pezzuto JM, Fong HHS, Cordell GA, Farnsworth NR. J Nat Prod. 1996;59:525–527. doi: 10.1021/np960149h. [DOI] [PubMed] [Google Scholar]; (b) Ciau Z, Brito-Leoza W, Quijino L. J Nat Prod. 2001;64:953–955. doi: 10.1021/np0100744. [DOI] [PubMed] [Google Scholar]
- 11.(a) Banskota AH, Tezukaa Y, Le KP, Tran KQ, Saikia I, Miwa Y, Taga T, Kadota S. Bioorg Med Chem Lett. 1998;8:3519–3524. doi: 10.1016/s0960-894x(98)00644-1. [DOI] [PubMed] [Google Scholar]; (b) Banskota AH, Tezuka K, Tran KQ, Tanaka K, Saiki I, Kadota S. Chem Pharm Bull. 2000;48:496–504. doi: 10.1248/cpb.48.496. [DOI] [PubMed] [Google Scholar]
- 12.Diyabalanage TKK, Wannigama GP, Weerasuriya A, Jayasinghe L, Simmonds P. Phytochemistry. 1995;40:1311–1312. [Google Scholar]
- 13.(a) Cantrell CL, Rajab MS, Franzblau SG, Fronczek FR, Fischer NH. Planta Med. 1999;65:732–734. doi: 10.1055/s-1999-14053. [DOI] [PubMed] [Google Scholar]; (b) Case RJ, Wang Y, Franzblau SG, Soejarto DD, Matainaho L, Piskaut P, Pauli GF. J Chromatogr A. 2007;1151:169–174. doi: 10.1016/j.chroma.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 14.(a) Collins LA, Franzblau SG. Antimicrob Agents Chemother. 1997;41:1004–1009. doi: 10.1128/aac.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Falzari K, Zhu Z, Pan D, Liu H, Hongmanee P, Franzblau SG. Antimicrob Agents Chemother. 2005;49:1447–1454. doi: 10.1128/AAC.49.4.1447-1454.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.