

Abstract
Toxic superoxide radicals, generated via adventitious reduction of dioxygen, have been implicated in a number of disease states. The cysteinate-ligated non-heme iron enzyme superoxide reductase (SOR) degrades superoxide via reduction. Biomimetic analogues which provide insight into why nature utilizes a trans-thiolate to promote SOR function are described. Spectroscopic and/or structural characterization of the first examples of thiolate-ligated FeIII–peroxo complexes provides important benchmark parameters for the identification of biological intermediates. Oxidative addition of superoxide is favored by low redox potentials. The trans influence of the thiolate appears to significantly weaken the Fe–O peroxo bond, favoring proton-induced release of H2O2 from a high-spin Fe(III)–OOH complex.
Introduction
The bioinorganic chemistry of iron is rich and diverse,1–6 catalyzing reactions that range from the biosynthesis of neurotransmitters7 to the transport of O2.8 A number of iron-promoted biosynthetic pathways involve dioxygen as the oxidant and/or oxygen atom source.1,5–7 Although O2 oxidations are thermodynamically favored, they are kinetically slow because they are spin-forbidden, unless promoted by a transition metal, such as Fe2+.2,4 The intermediates formed in these reactions include iron–peroxo and oxo species,1,2,4,6 potent oxidants capable of functionalizing alkanes.6 Adventitious reduction of dioxygen can, on the other hand, result in the formation of toxic radicals, including superoxide (O2–•) which has been implicated in a number of disease states, including Alzheimer's, Parkinson's, and cancer.9 Due to its toxicity, organisms have evolved elaborate means for the degradation of O2–. In aerobic organisms, manganese-, nickel-, iron-, or copper- and zinc-containing enzymes known as superoxide dismutases (SODs) function to disproportionate (eq 2) adventitiously formed superoxide.10,11 In
| (1) |
| (2) |
anaerobic organisms, an iron-containing enzyme superoxide reductase (SOR) reduces (eq 1) superoxide;9,12–15 however, this requires an outside source of electrons. A cysteinate sulfur bound to the iron site, as well as the positioning of the metal ion on the surface (vs the interior) of the protein,12 alters the function of Fe-SOR relative to Fe-SOD.11
Superoxide Reductase Enzyme Active Site Structure and Mechanism
Superoxide reductases (SORs) are intense blue, cysteinate-ligated non-heme iron enzymes found in anaerobic microbes.3,12–14 The FeII active site is redox active, high-spin (S = 2), and ligated by four equatorial histidines and one apical cysteinate trans to an open site.12 The oxidized FeIII resting state is high-spin () and contains a glutamate (14Glu) coordinated to the sixth (axial) site.12 This conserved 14Glu, as well as a highly conserved lysine (47Lys), is proposed to be involved in the catalytic mechanism, possibly acting as a proton donor, and/or by attracting the anionic superoxide ion to the active site.12,16 Activity drops significantly in mutants lacking 47Lys.16 The SOR-catalyzed conversion of superoxide (O2–) to hydrogen peroxide (H2O2) requires one electron and two protons. Protons and solvent both play an important role in the SOR mechanism.14 Superoxide reduction is favored in the presence of a proton source [E1/2(O2–/H2O2) = 0.83 V at pH 7.5 vs –0.041 V at pH 14 (vs SCE)], whereas oxidation (to afford O2) is favored in the absence of a proton source [E1/2(O2–/O2) = –0.80 V at pH 14 vs –0.13 V at pH 0 (vs SCE)]. Our working hypothesis is that the unusual positioning of the metal ion on the surface of SOR provides the active site with a readily available source of protons which makes O2 formation disfavored in anaerobic bacteria.
The mechanism by which SOR is proposed to reduce superoxide has been somewhat controversial, particularly with regard to the number of intermediates involved.14,15,17 The first step is generally agreed to involve the oxidative addition of O2– to the open coordination site. Two transient intermediates (T1 and T2 in Figure 1) are observed in the reaction between SOR and O2–,15 and exogenous ligands (NO, N3–, and CN–) have been shown to bind to the SOR iron site,18,19 consistent with an inner-sphere mechanism. The first intermediate, T1, forms at nearly diffusion controlled rates15,17 and is proposed to be an FeIII–peroxo species.17 The second intermediate, T2, forms more slowly and displays a pH-dependent νFe–O stretch that shifts with 18OH2 and D2O,20 consistent with its assignment as an FeIII–OH species.15 This FeIII–OH species presumably forms via H2O-induced protonation of the proximal oxygen of the FeIII–peroxo species, followed by H2O2 release and OH– binding.15,20 Why protonation preferentially occurs at the proximal, as opposed to distal, oxygen is unclear at this point. Reduction of the FeIII–OH intermediate in the presence of a proton regenerates the catalytically active five-coordinate Fe(II) form. The glutamate-bound resting state (R) shown in Figure 1 eventually forms in the absence of additional substrate (O2–) or electrons. Although there is currently no evidence to suggest that this last step involves a proton, the difficulty with which Fe–OH bonds cleave relative to Fe–OH2 bonds would imply that protonation occurs prior to ligand dissociation. The exogenous electron donor is currently unknown. Given that the addition of a proton and electron would be equivalent to the addition of a hydrogen atom, this introduces the interesting (albeit remote) possibility that the last step of the SOR mechanism may, perhaps, resemble the Fe(III)–OH intermediate-induced H-atom abstraction step of lipoxygenases.21–23
FIGURE 1.

Proposed mechanism for SOR-catalyzed reduction of superoxide via hydroperoxo (T1) and solvent-bound (T2) intermediates.
Vibrational data to support the assignment of T1 as a peroxo species have yet to be reported. However, when H2O2 is added to a mutant form of SOR,13 isotopically sensitive νO–O (850 cm–1) and νFe–O (438 cm–1) stretches are observed by resonance Raman, consistent with the formation of a metal–peroxo species. This mutant peroxo species was originally proposed to be η2-O22– side-on bound,13 on the basis of data reported for N-ligated synthetic non-heme iron–peroxo complexes,24 but later was shown by X-ray crystallography to be an end-on Fe(III)–OOH species,25 revealing a need for more thiolate-bound iron–peroxo complexes to provide benchmark vibrational parameters for interpreting biophysical data.
Relationship between SOR and P450 Active Sites and Mechanism
The primary coordination sphere of the iron active site of SOR is structurally12 related to that of the heme enzyme cytochrome P450.26,27 Both enzymes have an apical cysteinate trans to an open coordination site (Figure 2), and both react with oxygen-derived substrates to afford an Fe(III)–OOH intermediate. The mechanistic pathways taken following formation of the Fe(III)–peroxo intermediate are very different for these two enzymes, however. With P450, the hydroperoxo O–O bond is cleaved to afford a high-valent iron–oxo intermediate,6,27,28 whereas with SOR, the Fe–O(peroxo) bond is cleaved, releasing H2O2. The cysteinate has been shown to play an important role in promoting P450-induced O–O bond cleavage,29 yet O–O bond cleavage has not been observed with SOR. It is not clear why these two structurally similar systems follow divergent reaction pathways. Although a porphyrin was originally thought to be necessary for the stabilization of a high-valent iron–oxo complex, this was later shown not to be the case upon structural characterization of the first non-heme Fe(IV)=O complex.30 Although theoretical calculations have shown that the site of protonation can influence the reaction pathway,27 controlled, site-specific protonation with the metal ion sitting on the surface of the protein seems unlikely. Prior to the development of spectroscopic methods for probing non-heme iron,31 less was known about non-heme iron, relative to heme iron, mechanisms. Solomon has shown that Fe–peroxo spin states can influence the energetically preferred O–O versus Fe–O bond cleaving pathways.2,31 We have shown, in collaboration with Solomon, that the intense S → Fe(III) charge transfer bands characteristic of Fe(III)–SR compounds make it convenient to detect Cys-ligated non-heme iron sites and probe their reactivity.32,33
FIGURE 2.

Comparison of the thiolate-ligated non-heme, and heme, iron active sites of SOR (left) and P450 (right).
Roles of Cysteinate Residues in Promoting Metalloenzyme Function
A comprehensive understanding of the influence of thiolate ligands on the properties of first-row transition metal ions is essential if we are to fully understand why nature utilizes cysteinate residues to promote specific biological metalloenzyme functions.3,34 Cysteinate-ligated metalloenzymes promote a number of critical biological processes, including electron transfer,35,36 and strong bond activation.6 Cysteinates form highly covalent bonds to transition metals, and this helps to facilitate redox changes.37 Our work has shown that thiolate ligands significantly lower redox potentials,38 make low-spin iron accessible in a non-heme environment,39–41 stabilize iron in the +3 oxidation state,39–41 and labilize sites trans to the thiolate, thereby promoting product release,38 even with the typically inert low-spin Co(III) ion.42,43 Others have shown that the trans coordinated cysteinate of P450 promotes O–O bond cleavage,6,29 and the subsequent Fe(IV)=O-promoted H-atom abstraction.44
Biomimetic Models
A precise description of the correlation among the structure, key properties, and function of metalloenzyme active sites can be most readily obtained by building small molecular analogues.34 Synthetic model complexes provide key parameters needed to fit spectroscopic data (e.g., EXAFS) and protein crystal structures.45 Multidentate ligands are generally required for maintenance of a relatively rigid, well-defined synthetic active site model. The incorporation of thiolate ligands into synthetic models can, however, be complicated by their propensity to oligomerize, as well as autoreduce and form disulfides,46 especially when there is an open coordination site. Peroxides tend to react with thiolates to form sulfoxides and sulfones47,48 and with iron to form rust. Despite these synthetic challenges, much progress has been made in the biomimetic modeling of cysteinate-ligated non-heme iron active sites in biology.32,38,40–42,48–57
Reactive Five-Coordinate, Thiolate-Ligated Iron Complexes
A biomimetic system capable of reproducing the SOR reaction (Figure 1) would have two requirements. First, it would require an open coordination site for superoxide to bind to the metal. Second, it would require that the +3 oxidation state be reversibly accessible. Given the challenges associated with the synthesis of mononuclear thiolate-ligated transition metal complexes, especially those with higher oxidation states, we initially had to demonstrate that molecules meeting the criteria listed above could be synthesized. By incorporating gem-dimethyls adjacent to the thiolate, we were able to isolate a number of coordinatively unsaturated, mononuclear five-coordinate thiolate iron complexes, including [FeIII(S2Me2N3(Pr,Pr))]+ (1) (Figure 3),41 [FeIII(S2Me2N3-(Et,Pr))]+,58 and [FeII(SMe2N4(tren))]+ (2) (Figure 4).59 We found that we could isolate an FeIII–SR complex such as 1 via the in situ oxidation of an FeII–SR precursor at low temperatures.41,58 Once oxidized, the FeIII thiolates were found to display rich spectroscopic features, including intense πS-to-metal charge transfer bands in the visible region,32 and low-spin EPR signals.33,40 The energy of these bands was found to be highly dependent on the local coordination environment and could therefore be used to monitor reactions.38,41,43,50,52–54 The FeIII–SR bonds were found to be highly covalent and favor a low-spin state, an observation that was surprising given the π-donor properties of thiolates.32
FIGURE 3.

ORTEP diagram of thiolate-ligated, five-coordinate [FeIII(S2Me2N3(Pr,Pr))]+ (1) synthesized by Kovacs group postdocs Steve Shoner and Jeff Ellison.
FIGURE 4.

ORTEP diagram of thiolate-ligated, biomimetic superoxide reducing catalyst [FeII(SMe2N4(tren))]+ (2) synthesized by Jason Shearer, a graduate student in the Kovacs lab.
A Functional SOR Model with a cis-Thiolate: [FeII(SMe2N4(tren))]+
Using a tripodal amine ligand as a scaffold, we were able assemble a functional SOR model, [FeII(SMe2N4(tren))]+ (2),59 that mimics each step of the proposed SOR mechanism (Figure 1). Like the SOR enzyme active site, complex 2 is high-spin (S = 2) and has a similar N4S1– ligand motif, but with the open site cis to the thiolate, rather than trans as in the enzyme. When superoxide is added to 2 in the presence of a proton donor, biomimetic activity is observed, resulting in the formation of H2O2.50,53 At low temperatures (less than or equal to –78 °C), a transient hydroperoxide intermediate, [FeIII(SMe2N4(tren))(OOH)]+ (3), is observed.53 In contrast to 2 which is colorless, intermediate 3 is tangerine orange and displays an intense charge transfer band at 452(2900) nm (Figure 5). When the solution is warmed, its color changes from orange to burgundy, and the band at 452 nm cleanly converts to a band at 511(1770) nm (Figure 5). The burgundy species was shown, via its independent synthesis, to be the solvent-derived methoxide-bound species [FeIII(SMe2N4(tren))(OMe)]+ (4). When the reaction between 2 and superoxide is monitored by stopped-flow at ambient temperature (in MeOH), intermediate 3 grows in at diffusion-controlled rates (too fast to measure) and then cleanly converts to 4 (releasing H2O2) at a rate [65(1) s–1]53 similar to that of the enzyme (50 s–1).60,61 The tangerine orange species displays a low-spin (S = ½) EPR signal (Figure 6), which converts to an intermediate-spin () signal as the solution develops a burgundy color. These changes to the electronic absorption and EPR spectra indicated that a transient intermediate forms (at low temperatures) during the reduction of superoxide but did not reveal its identity. Vibrational data, obtained at –78 °C, unambiguously identified this intermediate as a hydroperoxo species. A Fermi doublet (at 786 and 784 cm–1) is observed in the infrared spectrum of [FeIII(SMe2N4(tren))(OOH)]+ (3) in the νO–O stretching region (Figure 7). This doublet collapses to a sharpened singlet at 784 cm–1 upon addition of D2O, indicating that a proton is associated with the moiety responsible for the vibration.53 Addition of 18O-labeled (23%) superoxide to 2 results in a new shifted νO–O stretch at 753 cm–1, close to that predicted on the basis of Hooke's law for a diatomic oxygen species.
FIGURE 5.

Low-temperature detection of the hydroperoxo intermediate [FeIII(SMe2N4(tren))(OOH)]+ (3) using electronic absorption spectroscopy, and its conversion to solvent-bound [FeIII(SMe2N4(tren))(OMe)]+ (4) upon warming.
FIGURE 6.

X-Band EPR spectrum of tangerine orange [FeIII(SMe2N4(tren))(OOH)]+ (3) at 7 K in an MeOH/EtOH (5:2) glass.
FIGURE 7.

Low-temperature IR spectrum of 16O-labeled (· · ·) and 18O-labeled (23%; —) [FeIII(SMe2N4(tren))(OOH)]+ (3) showing the peroxo νO–O Fermi doublet that shifts upon incorporation of 18O.
Although we have yet to crystallize our hydroperoxo intermediate, we have structurally characterized [FeIII(SMe2N4(tren))(OOH)]+ (3) using X-ray absorption spectroscopy, in collaboration with Rob Scarrow. Fits to the EXAFS data for 3 require a new short Fe–O bond 1.86(3) Å in length, which is not present in either the FeII precursor (2) or the methoxide-bound product 4.53 The XANES spectrum of 3 is consistent with an oxidized six-coordinate FeIII intermediate. Addition of an outer-sphere oxygen at 2.79(6) Å improves the EXAFS fits slightly.53 Together, these data are consistent with the oxidative addition of superoxide to [FeII(SMe2N4(tren))]+ via an inner-sphere mechanism to form an end-on FeIII(η1-OOH) intermediate. Hydroperoxo [FeIII(SMe2N4(tren))(OOH)]+ (3) represented the first example of a thiolate-ligated peroxo species, and the first structurally characterized FeIII–OOH species in any ligand environment.53 The coexistence of a thiolate (a reductant) and a peroxide (an oxidant) in the same molecule is quite remarkable. Evidence that the peroxide binds cis with respect to the thiolate comes from the isolation and characterization of a number of more stable derivatives, including [FeIII(SMe2N4(tren))(MeCN)]2+ (Figure 8), [FeIII(SMe2N4(tren))(CN)]+, [FeIII(SMe2N4(tren))(N3)]+ (Figure 9), and [FeIII(SMe2N4(tren))(OAc)]+ (5; Figure 10),54 where it was shown that all ligands bind in this mode.
FIGURE 8.

ORTEP diagram of solvent-bound [FeIII(SMe2N4(tren))(MeCN)]2+ showing that MeCN binds trans to the imine nitrogen and cis to the thiolate sulfur.
FIGURE 9.

ORTEP diagram of thiolate-ligated [FeIII(SMe2N4(tren))(N3)]+.
FIGURE 10.

ORTEP diagram of acetate-bound [FeIII(SMe2N4(tren))(OAc)]+ (5), a mimic for the glutamate-bound SOR resting state.
Formation of hydroperoxo [FeIII(SMe2N4(tren))(OOH)]+ (3) is proton-dependent. No reaction occurs between [FeII(SMe2N4(tren))]+ (2) and O2– in rigorously dried THF until an external proton donor is added. This rules out a mechanism involving abstraction of H+ or a H-atom from the ligand. Addition of a variety of proton donors, including NH4+, MeOH, and PhOH, rapidly induces the formation of [FeIII(SMe2N4(tren))(OOH)]+ (3). The ammonium ion (NH4+) mimics the lysine residue proposed to be involved in the SOR mechanism.50 The rate of formation of 3 is highly dependent on both the HA concentration and pKa,62 indicating that a proton is transferred in the steps prior to or during the rate-determining step. The proton dependence of this reaction is consistent with three possible mechanisms involving initial protonation of O2–, the thiolate sulfur, or an Fe(II)–superoxo intermediate. These should be distinguishable on the basis of kinetics. Kinetic studies are currently underway in our laboratory, in an attempt to establish the most probable mechanism.62
The Fe–O(peroxo) bond of [FeIII(SMe2N4(tren))(OOH)]+ (3) can be cleaved via the addition of a second, more acidic, proton donor.50 This results in the release of H2O2, via what appears to be a proton-dependent dissociative mechanism. Nucleophiles, such as OAc–, do not react with 3, and the rate of H2O2 release is dependent on the pKa of the proton donor.62 The addition of HOAc releases H2O2 from 3 six orders of magnitude faster than NH4+ does. Noncoordinating acids (HBF4 and HClO4) cleanly afford a common eggplant purple intermediate, [FeIII(SMe2N4(tren)(MeOH))]2+ (6; λmax = 565 nm). Acetic acid also reacts with 3 to form solvent-bound 6 (Figure 11), which then converts to acetate-bound [FeIII(SMe2N4(tren)(OAc))]+ (5; Figure 10), a model for the Glu-bound oxidized SOR resting state, upon warming. A solvent-bound [FeII-I–OH(H)] intermediate has also recently been identified in the mechanism of SOR,15,20 as a species distinct from its FeIII–Glu resting state. Following the release of H2O2 from our hydroperoxo intermediate 3, we can regenerate the active catalyst, 2, by adding an external reductant such as cobaltacene (Figure 12). Subsequent addition of superoxide results in regeneration of the hydroperoxo intermediate. Eight turnovers have been achieved in this stepwise manner.50 The catalytic activity is most likely limited due to decomposition of the catalyst by H2O2-promoted oxidation of the thiolate.
FIGURE 11.

Proton-induced conversion of [FeIII(SMe2N4(tren))(OOH)]+ (3) to solvent-bound [FeIII(SMe2N4(tren)(MeOH))]2+ (6) as monitored by electronic absorption spectroscopy at low temperatures.
FIGURE 12.

Catalytic cycle involving [FeII(SMe2N4(tren))]+ (2)-promoted reduction of superoxide to afford H2O2, via the sequential protonation and Cp2Co-promoted reduction of hydroperoxo- and solvent-bound intermediates.
Influence of the trans-Thiolate Ligand in Promoting SOR Chemistry
A few examples of structural SOR models containing a trans-thiolate have been reported,28,55 and the thiolate ligand has been shown to lower Fe3+/2+ redox potentials.55 Although none of these structural models has been reported to react with superoxide, in one case, H2O2 addition was shown to afford a high-valent FeIV=O species,28 presumably via the cleavage of a transient FeIII–peroxo O–O bond. A macrocyclic thiolate-ligated ferrous complex was recently reported,49 which reacts with ROOH to afford the first example of a thiolate-ligated alkyl peroxo complex [([15]aneN4)FeIII(SPh)(OOR)]+ (7), with the thiolate presumably trans to the peroxo. Although 7 is a low-spin complex (g = 2.20 and 1.97), its νFe–O stretch (612 cm–1) is significantly lower than those of most low-spin FeIII–OOR complexes.24 Most likely, this is due to the trans influence of the thiolate sulfur, although one cannot conclusively say that ROO– binds in this position. Macrocyclic cyclam ligands have been shown to fold to afford cis-ligated six-coordinate geometries.63 Recently, we reported a rare example of a functional metalloenzyme active site model, [FeII(cyclam-PrS)](BPh4) (8; Figure 13), that like SOR reduces O2–, presumably via a trans-thiolate-ligated FeIII–peroxo intermediate.38 The thiolate ligand of 8 was shown to lower the redox potential by 565 mV, alter the spin state of the peroxo intermediate, and dramatically weaken the Fe–O(peroxo) bond, favoring O2– reduction and H2O2 release.38 Consistent with previous observations regarding amine substituents,56,64 the secondary amines of 8 also appear to play an important role in the observed superoxide reduction chemistry. Upon addition of O2– (18-crown-6-K+ salt) and a proton donor (i.e., MeOH) to 8 at –78 °C in CH2Cl2, a metastable, high-spin (g = 7.72, 5.40, and 4.15) burgundy [λmax = 530(1350) nm] intermediate (9) is observed (Figure 14). No reaction occurs in the absence of a proton donor, and O2– does not convert to H2O2 under the same conditions in the absence of 8.38 An νO–O stretch (Fermi doublet) is observed at 891 cm–1 in the vibrational (resonance Raman) spectrum of 9, which shifts to 856 cm–1 in the 18O-labeled (50% label) spectrum. Addition of D+ (i.e., MeOD) causes the Fermi doublet to collapse. An intense νFe–S stretch is also observed at 352 cm–1, indicating that the thiolate remains coordinated to the metal, and an 18O-sensitive νFe–O stretch is observed at 419 cm–1, which shifts to 400 cm–1.38 Altogether, these data are consistent with the proton-dependent oxidative addition of superoxide to 8 which forms a thiolate-ligated hydroperoxo intermediate [FeIII(cyclam-PrS)(OOH)]+ (9). The remarkably low νFe–O stretch, and the high-spin state of this intermediate, strongly suggest that the hydroper-oxide ligand binds trans to the thiolate, although one cannot conclusively assign its structure in the absence of crystallographic evidence.
FIGURE 13.
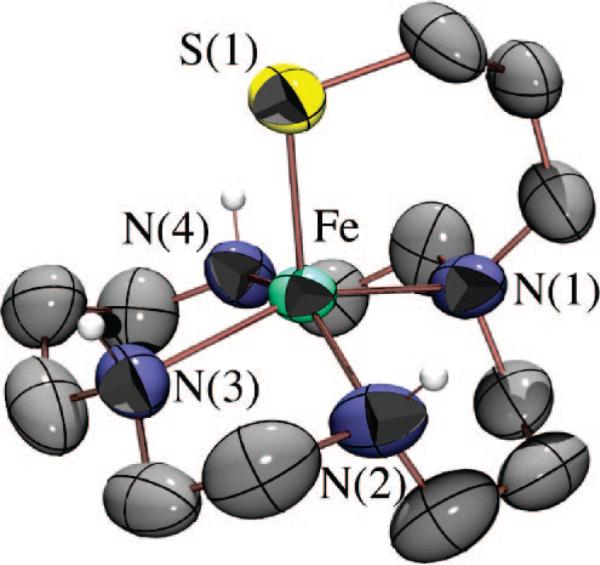
ORTEP diagram of trans-thiolate-ligated, biomimetic superoxide reducing catalyst [FeII(cyclam-PrS)]+ (8) synthesized by Terutaka Kitagawa, a graduate student in the Kovacs lab.
FIGURE 14.

Low-temperature electronic absorption spectrum and DFT-calculated structure of the hydroperoxo intermediate [FeIII(cyclam-PrS)(OOH)]+ (9) formed upon addition of superoxide to reduced [FeII(cyclam-PrS](BPh4) (8) in the presence of a proton source.
Although clearly more examples are needed if one hopes to correlate spectroscopic parameters to peroxo and thiolate binding modes and ultimately SOR function, with three examples (3, 7, and 9) now in hand, we38,50,53 and others25,28,49 are closer to this goal. In comparison to other reported synthetic iron peroxides,24 the νFe–O stretch of 9 is significantly weakened (419 cm–1 for 9 vs reported range, 450–639 cm–1),24 but it compares well with the only reported SOR peroxo stretch (438 cm–1),13 which was recently shown by X-ray crystallography to contain a trans-thiolate and end-on hydroperoxo.25 The νO–O stretch of 9 is unusually high (reported range, 820–860 cm–1). The DFT-optimized structure of 9 (Figure 14) contains an Fe–O distance of 1.95 Å and a force constant (kFe–O = 1.20 mdyn/cm) which is significantly longer (reported range, 1.76–1.86 Å) and weaker (reported force constant range, 2.2–2.1 mdyn/cm) than those of all other reported Fe–(η1-OOH) species.24 These data strongly support a trans positioning of the thiolate in 9 and indicate that its influence is to significantly weaken the Fe–O(peroxo) bond, favoring Fe–O, as opposed to O–O, bond cleavage.
The Fe–O(peroxo) bond of [FeIII(cyclam-PrS)(OOH)]+ (9) can be cleaved via the addition of proton donors, resulting in the release of H2O2. A blue acetate-bound derivative, [FeIII(cyclam-PrS)(OAc)]+ (10), modeling the glutamate-bound SOR resting state, forms following the low-temperature addition of acetic acid.38 Once H2O2 is released, the active catalyst 8 can be regenerated via the addition of a sacrificial reductant (Cp2Co). Subsequent addition of superoxide results in regeneration of hydro-peroxo intermediate 9. Five turnovers have been achieved in this stepwise manner. In comparison to our cis-thiolate-ligated hydroperoxo complex 3 which releases H2O2 extremely slowly in MeOH at –78 °C (t1/2 = 63.9 h), hydroperoxo complex 9 rapidly releases H2O2 under the same conditions (within seconds). The macrocyclic amine ligand in combination with the presumed trans positioning of the thiolate converts the spin state from an S = ½ state in cis-ligated 353 to an state, promoting faster Fe–O bond cleavage under similar conditions. If the thiolate is indeed trans, it would also increase the basicity of the proximal peroxo oxygen relative to that of the cis-thiolate-ligated system, and this would be expected to influence product release rates if Fe–OOH bond cleavage is proton-induced. The relative rates of proximal oxygen Fe–OOH protonation (to induce H2O2 release) versus O–O(peroxide) bond cleavage must play a critical role in governing whether a SOR or P450 reaction pathway is followed.
Concluding Remarks
The biological chemistry of iron is exquisitely controlled by its ligand environment. Supporting ligands play an important role in determining function by controlling metal ion redox potential and spin state, as well as the basicity of bound substrates. Spin states control reaction pathways by fine-tuning relative bond strengths. Although there are many parallels between heme and non-heme iron systems, porphyrin ligands alter reaction pathways by facilitating oxidation via the delocalization of charge onto the ligand. Thiolate ligands also have the potential of facilitating the delocalization of charge.65 As described in this Account, we are beginning to understand the functional role of the SOR thiolate ligand in promoting superoxide reduction. The intense π-thiolate sulfur-to-FeIII charge transfer band, and its sensitivity to local coordination environment, provide a convenient method for monitoring reactions involving thiolate-ligated non-heme iron complexes.32 By successfully mimicking the individual proton and electron transfer steps of the SOR catalytic cycle,38,50 we have begun investigating the molecular level details of the SOR mechanism and its dependence on the positioning of the thiolate ligand relative to the substrate binding site. The thiolate ligand favors superoxide reduction, regardless of its position in the coordination sphere, by lowering the redox potential of the metal ion.56 In a protein environment, the thiolate provides an efficient electron transfer pathway.35,37 Spectroscopic characterization of the first reported examples of SOR peroxo intermediate analogues has allowed us,38,53 and others,49,66 to determine how the thiolate influences key properties affecting reaction pathways. Despite the structural similarities between the heme iron enzyme P450 and non-heme iron enzyme SOR peroxo intermediates, the two systems follow very different reaction pathways.4 The spin state favored by the supporting amine ligands influences the relative strength of the Fe–O(peroxo) versus O–O bond, and presumably the basicity of the proximal peroxo oxygen. The combination of a high-spin state and trans-thiolate affords a labile FeIII–OOH species susceptible to protonation by protic solvents (e.g., MeOH or H2O), resulting in facile H2O2 release. The biomimetic analogues described herein have allowed us to establish many of the key properties that are critical for the identification and function of cysteinate-ligated metalloenzymes.
FIGURE 15.

Catalytic cycle involving [FeII(cyclam-PrS](BPh4) (8)-promoted reduction of superoxide to afford H2O2, via the sequential protonation and Cp2Co-promoted reduction of hydroperoxo- and acetate-bound intermediates.
Acknowledgments
J.A.K. is grateful to have had the privilege to work with a number of very talented graduate students and postdocs who have contributed in one way or another to the work described herein. The National Institutes of Health is also gratefully acknowledged for its support of the work carried out in the Kovacs laboratory (Grant GM 45881-16).
Biography
Julie Kovacs was born in Lansing, MI, on March 15, 1959, and received her B.S. degree from Michigan State University (where her father was on the faculty as a theoretical physicist) in 1981. She received her Ph.D. from Harvard University in 1986, where she synthesized the first analogue of the VFeS cluster site of nitrogenase with Richard Holm. She did her postdoctoral work at the University of California, Berkeley, with Bob Bergman and has been on the faculty at the University of Washington since 1988, where she is currently a full Professor. She currently serves as Chair-elect of the bioinorganic ACS subdivision and will Chair the 2008 Metals in Biology Gordon conference. Her research focuses on understanding how sulfur ligands influence function in metalloenzymes.
Lisa M. Brines was born in Spokane, WA, in 1980. She received a Bachelor of Science in Chemistry from Seattle University in 2002 and is currently a graduate student at the University of Washington. Her research in Julie Kovacs’ lab involves studying the active sites of non-heme metalloenzymes using synthetic modeling techniques.
References
- 1.Costas M, Mehn MP, Jensen MP, Que LJ. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 2.Solomon EI, Decker A, Lehnert N. Non-heme iron enzymes: Contrasts to heme catalysis. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3589–3594. doi: 10.1073/pnas.0336792100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kovacs JA. Synthetic Analogues of Cysteinate-Ligated Non-Heme Iron, and Non-Corrinoid Cobalt Enzymes. Chem. Rev. 2004;104:825–848. doi: 10.1021/cr020619e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kovacs JA. How Iron Activates O2. Science. 2003;299:1024–1025. doi: 10.1126/science.1081792. [DOI] [PubMed] [Google Scholar]
- 5.Hausinger RP. Fe(II) α-Ketoglutarate-Dependent Hydroxylases and Related Enzymes. Crit. Rev. Biochem. Mol. Biol. 2004;39:1–47. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 6.Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and Chemistry of Cytochrome P450. Chem. Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 7.Kappock TJ, Caradonna JP. Pterin-Dependent Amino Acid Hydroxylases. Chem. Rev. 1996;96:2659–2756. doi: 10.1021/cr9402034. [DOI] [PubMed] [Google Scholar]
- 8.Momenteau M, Reed CA, Guiset F. Synthetic Heme Dioxygen Complexes. Chem. Rev. 1994;94:659–698. [Google Scholar]
- 9.Kurtz DM., Jr. Microbial Detoxification of Superoxide: The Non-Heme Iron Reductive Paradigm for Combating Oxidative Stress. Acc. Chem. Res. 2004;37:902–908. doi: 10.1021/ar0200091. [DOI] [PubMed] [Google Scholar]
- 10.Wuerges J, Lee J-W, Yim Y-I, Yim H-S, Kang S-O, Carugo KD. Crystal structure of nickel-containing superoxide dismutase reveals another type of active site. Proc. Natl. Acad. Sci. U.S.A. 2004;101:8569–8574. doi: 10.1073/pnas.0308514101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jackson TA, Brunold TC. Combined Spectroscopic/Computational Studies on Fe and Mn-Dependent Superoxide Dismutases: Insights into Second Sphere Tuning of Active Site Properties. Acc. Chem. Res. 2004;37:461–470. doi: 10.1021/ar030272h. [DOI] [PubMed] [Google Scholar]
- 12.Yeh AP, Hu Y, Jenney FE, Jr., Adams MWW, Rees DC. Structures of the superoxide reductase from Pyrococcus furiosus in the oxidized and reduced states. Biochemistry. 2000;39:2499–2508. doi: 10.1021/bi992428k. [DOI] [PubMed] [Google Scholar]
- 13.Mathe C, Mattioli TA, Horner O, Lombard M, Latour J-M, Fontecave M, Niviere V. Identification of Iron(III) Peroxo Species in the Active Site of the Superoxide Reductase SOR from Desulfoarculus baarsii. J. Am. Chem. Soc. 2002;124:4966–4967. doi: 10.1021/ja025707v. [DOI] [PubMed] [Google Scholar]
- 14.Niviere V, Asso M, Weill CO, Lombard M, Guigliarelli B, Favaudon V, Houe'e-Levin C. Superoxide Reductase from Desulfoarculus baarsii: Identification of Protonation Steps in the Enzymatic Mechanism. Biochemistry. 2004;43:808–818. doi: 10.1021/bi035698i. [DOI] [PubMed] [Google Scholar]
- 15.Rodrigues JV, Abreu IA, Cabelli D, Teixeira M. Superoxide Reduction Mechanism of Archaeoglobus fulgidus One-Iron Su-peroxide Reductase. Biochemistry. 2006;45:9266–9278. doi: 10.1021/bi052489k. [DOI] [PubMed] [Google Scholar]
- 16.Lombard M, Houee-Levin C, Touati D, Fontecave M, Niviere V. Superoxide Reductase from Desulfoarculus baarsii: Reaction Mechanism and Role of Glutamate 47 and Lysine 48 in Catalysis. Biochemistry. 2001;40:5032–5040. doi: 10.1021/bi0023908. [DOI] [PubMed] [Google Scholar]
- 17.Kurtz DM., Jr. Avoiding high-valent iron intermediates: Super-oxide reductase and rubrerythrin. J. Inorg. Biochem. 2006;100:679–693. doi: 10.1016/j.jinorgbio.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 18.Clay MD, Jenney FE, Jr., Hagedoorn PL, George GN, Adams MWW, Johnson MK. Spectroscopic Studies of Pyrococcus furiosus Superoxide Reductase: Implications for Active-Site Stuctures and the Catalytic Mechanism. J. Am. Chem. Soc. 2002;124:788–805. doi: 10.1021/ja016889g. [DOI] [PubMed] [Google Scholar]
- 19.Clay M, Cosper CA, Jenney FE, Jr., Adams MWW, Johnson MK. Nitric oxide binding at the mononuclear active site of reduced Pyrococcus furiosus superoxide reductase. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3796–3801. doi: 10.1073/pnas.0636858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mathé C, Nivière V, Mattioli TA. Fe3+-Hydroxide Ligation in the Superoxide Reductase from Desulfoaculus baarsii Is Associated with pH Dependent Spectral Changes. J. Am. Chem. Soc. 2005;127:16436–16441. doi: 10.1021/ja053808y. [DOI] [PubMed] [Google Scholar]
- 21.Lehnert NISE. Density-functional investigation on the mechanism of H-atom abstraction by lipoxygenase. J. Biol. Inorg. Chem. 2003;8:294–305. doi: 10.1007/s00775-002-0415-6. [DOI] [PubMed] [Google Scholar]
- 22.Goldsmith CR, Stack TDP. Hydrogen Atom Abstraction by a Mononuclear Ferric Hydroxide Complex: Insight into the Reactivity of Lipoxygenase. Inorg. Chem. 2006;45:6048–6055. doi: 10.1021/ic060621e. [DOI] [PubMed] [Google Scholar]
- 23.Roth JP, Mayer JM. Hydrogen Transfer Reactivity of a Ferric Bi-imidazoline Complex That Models the Activity of Lipoxygenase Enzymes. Inorg. Chem. 1999;38:2760–2761. doi: 10.1021/ic990251c. [DOI] [PubMed] [Google Scholar]
- 24.Roelfes G, Vrajmasu V, Chen K, Ho RYN, Rohde J-U, Zondervan C, Crois RM, Schudde EP, Lutz M, Spek AL, Hage R, Feringa BL, Munck E, Que L., Jr. End-on and Side-on Peroxo Derivatives of Non-Heme Iron Complexes with Pentadentated Ligands: Models for Putative Intermedates in Biological Iron/Dioxygen Chemistry. Inorg. Chem. 2003;42:2639–2653. doi: 10.1021/ic034065p. [DOI] [PubMed] [Google Scholar]
- 25.Katona G, Capentier P, Niviere V, Amara P, Adam V, Ohana J, Tsanov N, Bourgeois D. Raman-Assisted Crystallography Reveals End-On Peroxide Intermediates in a Nonheme Iron Enzyme. Science. 2007;316:449–453. doi: 10.1126/science.1138885. [DOI] [PubMed] [Google Scholar]
- 26.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. The Catalytic Pathway of Cytochrome P450cam at Atomic Resolution. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 27.Harris DL, Loew GH. Theoretical Investigation of the Proton Assisted Pathway to Formation of Cytochrome P450 Compound I. J. Am. Chem. Soc. 1998;120:8941–8948. [Google Scholar]
- 28.Bukowski MR, Koehntop KD, Stubna A, Bominaar EL, Halfen JA, Münck E, Nam W, Que L., Jr. A Thiolate-Ligated Nonheme Oxoiron(IV) Complex Relevant to Cytochrome P450. Science. 2005;310:1000–1002. doi: 10.1126/science.1119092. [DOI] [PubMed] [Google Scholar]
- 29.Auclair K, Moenne-Loccoz P, Ortiz de Montellano PR. Roles of the Proximal Heme Thiolate Ligand in Cytochrome P450cam. J. Am. Chem. Soc. 2001;123:4877–4885. doi: 10.1021/ja0040262. [DOI] [PubMed] [Google Scholar]
- 30.Rohde J-U, In JH, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Munck E, Nam W, Que L., Jr. Crystallographic and Spectroscopic Characterization of a Nonheme Fe(IV)=O complex. Science. 2003;299:1037–1039. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]
- 31.Solomon EI. Geometric and Electronic Structure Contributions to Function in Bioinorganic Chemistry: Active Sites in Non-Heme Iron Enzymes. Inorg. Chem. 2001;40:3656–3669. doi: 10.1021/ic010348a. [DOI] [PubMed] [Google Scholar]
- 32.Kennepohl P, Neese F, Schweitzer D, Jackson HL, Kovacs JA, Solomon EI. Spectroscopy of Non-Heme Iron Thiolate Complexes: Insight into the Electronic Structure of the Low-Spin Active Site of Nitrile Hydratase. Inorg. Chem. 2005;44:1826–1836. doi: 10.1021/ic0487068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lugo-Mas P, Dey A, Xu L, Davin SD, Benedict J, Kaminsky W, Hodgson KO, Hedman B, Solomon EI, Kovacs JA. How Does Single Oxygen Atom Addition Affect the Properties of an Fe-Nitrile Hydratase Analogue? The Compensatory Role of the Unmodified Thiolate. J. Am. Chem. Soc. 2006;128:11211–11221. doi: 10.1021/ja062706k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holm RH, Kennepohl P, Solomon EI. Structural and Functional Aspects of Metal Sites in Biology. Chem. Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 35.Gray HB, Malström B, Williams RJP. Copper coordination in blue proteins. J. Biol. Inorg. Chem. 2000;5:551–559. doi: 10.1007/s007750000146. [DOI] [PubMed] [Google Scholar]
- 36.Rao PV, Holm RH. Synthetic Analogues of the Active Sites of Iron-Sulfur Proteins. Chem. Rev. 2004;104:527–560. doi: 10.1021/cr020615+. [DOI] [PubMed] [Google Scholar]
- 37.Glaser T, Hedman B, Hodgson KO, Solomon EI. Ligand K-Edge X-ray Absorption Spectroscopy: A Direct Probe of Ligand-Metal Covalency. Acc. Chem. Res. 2000;33:859–868. doi: 10.1021/ar990125c. [DOI] [PubMed] [Google Scholar]
- 38.Kitagawa T, Dey A, Lugo-Mas P, Benedict J, Kaminski W, Solomon E, Kovacs JA. A Functional Model for the Metalloenzyme Superoxide Reductase. J. Am. Chem. Soc. 2006;128:14448–14449. doi: 10.1021/ja064870d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shoner S, Barnhart D, Kovacs JA. A Model for the Low-Spin, Non-Heme, Thiolate-Ligated Iron Site of Nitrile Hydratase. Inorg. Chem. 1995;34:4517–4518. [Google Scholar]
- 40.Jackson HL, Shoner SC, Rittenberg D, Cowen JA, Lovell S, Barnhart D, Kovacs JA. Probing the Influence of Local Coordination Environment on the Properties of Fe-Type Nitrile Hydratase Model Complexes. Inorg. Chem. 2001;40:1646–1653. doi: 10.1021/ic001271d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ellison JJ, Nienstedt A, Shoner SC, Barnhart D, Cowen JA, Kovacs JA. Reactivity of Five-Coordinate Models for the Thiolate-Ligated Fe Site of Nitrile Hydratase. J. Am. Chem. Soc. 1998;120:5691–5700. [Google Scholar]
- 42.Shearer J, Kung IY, Lovell S, Kaminsky W, Kovacs JA. Why Is There an “Inert” Metal Center in the Active Site of Nitrile Hydratase? Reactivity and Ligand Dissociation from a Five-Coordinate Co(III) Nitrile Hydratase Model. J. Am. Chem. Soc. 2001;123:463–468. doi: 10.1021/ja002642s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shearer J, Jackson HL, Schweitzer D, Rittenberg DK, Leavy TM, Kaminsky W, Scarrow RC, Kovacs JA. The first example of a nitrile hydratase model complex that reversibly binds nitriles. J. Am. Chem. Soc. 2002;124:11417–11428. doi: 10.1021/ja012555f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Green MT, Dawson JH, Gray HB. Oxoiron(IV) in Chloroperoxidase Compound II Is Basic: Implications for P450 Chemistry. Science. 2004;304:1653–1656. doi: 10.1126/science.1096897. [DOI] [PubMed] [Google Scholar]
- 45.Sommerhalter M, Lieberman RL, Rosenzweig AC. X-ray Crystallography and Biological Metal Centers: Is Seeing Believing. Inorg. Chem. 2005;44:770–778. doi: 10.1021/ic0485256. [DOI] [PubMed] [Google Scholar]
- 46.Corwin DT, Jr., Gruff ES, Koch S. Zinc, cobalt, and cadmium thiolate complexes. J. Chem. Soc., Chem. Commun. 1987;13:966–967. [Google Scholar]
- 47.Grapperhaus CA, Darensbourg MY. Oxygen Capture by Sulfur in Nickel Thiolates. Acc. Chem. Res. 1998;31:451–459. [Google Scholar]
- 48.Kung IY, Schweitzer D, Shearer J, Taylor WD, Jackson HL, Lovell S, Kovacs JA. How Do Oxidized Thiolate Ligands Affect the Electronic and Reactivity Properties of a Nitrile Hydratase Model Compound. J. Am. Chem. Soc. 2000;122:8299–8300. [Google Scholar]
- 49.Krishnamurthy D, Kasper GD, Namuswe F, Kerber WD, Sarjeant AAN, Moënne-Loccoz P, Goldberg DP. Fe(III)-OOR SOR model. J. Am. Chem. Soc. 2006;128:14222–14223. doi: 10.1021/ja064525o. [DOI] [PubMed] [Google Scholar]
- 50.Theisen RM, Kovacs JA. The Role of Protons in Superoxide Reduction by a Superoxide Reductase Analogue. Inorg. Chem. 2005;44:1169–1171. doi: 10.1021/ic048818z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tyler LA, Noveron JC, Olmstead MM, Mascharak PK. Modulation of the pKa of Metal-Bound Water via Oxidation of Thiolato Sulfur in Model Complexes of Co(III) Containing Nitrile Hydratase. Inorg. Chem. 2003;42:5751–5761. doi: 10.1021/ic030088s. [DOI] [PubMed] [Google Scholar]
- 52.Schweitzer D, Ellison JJ, Shoner SC, Lovell S, Kovacs JA. A Synthetic Model for the NO-Inactivated Form of Nitrile Hydratase. J. Am. Chem. Soc. 1998;120:10996–10997. [Google Scholar]
- 53.Shearer J, Scarrow RC, Kovacs JA. Synthetic models for the cysteinate-ligated Non-Heme Iron Enzyme Superoxide Reductase: Observation and Structural Characterization by XAS of an FeIII–OOH Intermediate. J. Am. Chem. Soc. 2002;124:11709–11717. doi: 10.1021/ja012722b. [DOI] [PubMed] [Google Scholar]
- 54.Shearer J, Fitch SB, Kaminsky W, Benedict J, Scarrow RC, Kovacs JA. How does cyanide inhibit superoxide reductase? Insight from synthetic FeIIIN4S model complexes. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3671–3676. doi: 10.1073/pnas.0637029100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fiedler AT, Halfen HL, Halfen JA, Brunold TC. Synthesis, Structure Determination, and Spectroscopic/Computational Characterization of a Series of Fe(II)-Thiolate Model Complexes: Implications for Fe-S Bonding in Superoxide Reductases. J. Am. Chem. Soc. 2005;127:1675–1689. doi: 10.1021/ja046939s. [DOI] [PubMed] [Google Scholar]
- 56.Theisen RM, Shearer JWK, Kovacs JA. Steric and Electronic Control Over the Reactivity of a Thiolate-Ligated Fe(II) Complex with Dioxygen and Superoxide. Reversible mu-oxo Dimer Formation. Inorg. Chem. 2004;43:7682–7690. doi: 10.1021/ic0491884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Noveron JC, Olmstead MM, Mascharak PK. A Synthetic Analogue of the Active Site of Fe-Containing Nitrile Hydratase with Carboxamido N and Thiolato S as Donors: Synthesis, Structure, and Reactivities. J. Am. Chem. Soc. 2001;123:3247–3259. doi: 10.1021/ja001253v. [DOI] [PubMed] [Google Scholar]
- 58.Schweitzer D, Shearer J, Rittenberg DK, Shoner SC, Ellison JJ, Loloee R, Lovell SC, Barnhart D, Kovacs JA. Enhancing Reactivity via Ligand Constraints. Inorg. Chem. 2002;41:3128–3136. doi: 10.1021/ic0109187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shearer J, Nehring J, Kaminsky W, Kovacs JA. Modeling the Reactivity of Superoxide Reducing Metalloenzymes with a Nitrogen and Sulfur Coordinated Iron Complex. Inorg. Chem. 2001;40:5483–5484. doi: 10.1021/ic010221l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kurtz DM, Coulter ED. The mechanism(s) of superoxide reduction by superoxide reductases in vitro and in vivo. J. Biol. Inorg. Chem. 2002;7:653–658. doi: 10.1007/s00775-002-0360-4. [DOI] [PubMed] [Google Scholar]
- 61.Emerson JP, Coulter ED, Cabelli DE, Phillips RS, Kurtz DM., Jr. Kinetics and Mechanism of Superoxide Reduction by Two-Iron Superoxide Reductase from Desulfovibrio vulgaris. Biochemistry. 2002;41:4348–4357. doi: 10.1021/bi0119159. [DOI] [PubMed] [Google Scholar]
- 62.Nam E, Alokolaro P, Kovacs JA. Kinetics paper, 2007, manuscript in preparation.
- 63.Lemma K, Ellern A, Bakac A. 1H NMR Studies of Ligand and H/D Exchange Reactions of cis- and trans-([14]aneN4)(H2O)RhH2+ in Aqueous Solutions. Inorg. Chem. 2003;42:3662–3669. doi: 10.1021/ic034130v. [DOI] [PubMed] [Google Scholar]
- 64.Berry JF, Bill E, Garcia-Serres R, Neese F, Weyhermuller T, Wieghardt K. Effect of N-Methylation of Macrocyclic Amine Ligands on the Spin State of Fe(III): A Tale of Two Fluoro Complexes. Inorg. Chem. 2006;45:2027–2037. doi: 10.1021/ic051823y. [DOI] [PubMed] [Google Scholar]
- 65.Green MT. Evidence for Sulfur-Based Radicals in Thiolate Compound I Intermediates. J. Am. Chem. Soc. 1999;121:7939–7940. [Google Scholar]
- 66.Bukowski MR, Halfen HL, van der Berg TA, Halfen JA, Que L., Jr. Spin-State Rationale for the Peroxo-Stabilizing Role of the Thiolate Ligand in Superoxide Reductase. Angew. Chem., Int. Ed. 2005;44:584–587. doi: 10.1002/anie.200461527. [DOI] [PubMed] [Google Scholar]