

Abstract
Oculocutaneous albinism type III (OCA3), caused by mutations of TYRP1 gene, is an autosomal recessive disorder characterized by reduced biosynthesis of melanin pigment in the hair, skin, and eyes. The TYRP1 gene encodes a protein called tyrosinase-related protein-1 (Tyrp1). Tyrp1 is involved in maintaining the stability of tyrosinase protein and modulating its catalytic activity in eumelanin synthesis. Tyrp1 is also involved in maintenance of melanosome structure and affects melanocyte proliferation and cell death. In this work we implemented computational analysis to filter the most probable mutation that might be associated with OCA3. We found R326H and R356Q as most deleterious and disease associated by using PolyPhen 2.0, SIFT, PANTHER, I-mutant 3.0, PhD-SNP, SNP&GO, Pmut, and Mutpred tools. To understand the atomic arrangement in 3D space, the native and mutant (R326H and R356Q) structures were modelled. Finally the structural analyses of native and mutant Tyrp1 proteins were investigated using molecular dynamics simulation (MDS) approach. MDS results showed more flexibility in native Tyrp1 structure. Due to mutation in Tyrp1 protein, it became more rigid and might disturb the structural conformation and catalytic function of the structure and might also play a significant role in inducing OCA3. The results obtained from this study would facilitate wet-lab researches to develop a potent drug therapies against OCA3.
1. Introduction
Oculocutaneous albinism type 3 (OCA3) is an autosomal recessive disorder characterized by reduced biosynthesis of melanin pigment in the hair, skin, and eyes [MIM 203290]. This disorder is mostly caused by the genetic mutation in TYRP1 gene. OCA3 is also known as Rufous oculocutaneous albinism. The human TYRP1 gene consists of 8 exons and 7 introns, spanning almost 15–18 kb of genomic DNA in the region of 9p23 [1–4]. This gene encodes a protein called Tyrosinase-related protein 1 (Tyrp1), has a molecular weight of ~75 kDa, and appears to be the most abundant melanosomal protein of the melanocyte [5, 6]. Tyrp1 is comprising of 537 amino acid residues and shares 40–52% of amino acid homology to tyrosinase. The tyrosinase-related family includes tyrosinase, tyrosinase-related protein 1 (Tyrp1), and tyrosinase-related protein 2 (Tyrp2) involved in this enzymatic process that converts tyrosine to melanin pigments. Certainly, two types of melanin are produced by melanocytes, which are pheomelanins (red or yellow) and eumelanins (brown or black) [7]. The first two steps of both eumelanin and pheomelanin production involve tyrosinase catalysing the conversion of tyrosine to 3,4-dihydroxy-L-phenylalanine (DOPA) and of DOPA to DOPA quinone [8, 9]. Then pheomelanogenesis seems to be the default pathway in the absence of MC1R signalling, with a low tyrosinase activity and a high concentration of thiolic compounds, such as cysteine. In another way, eumelanin synthesis requires α-MSH binding to MC1R [10, 11], which transcriptionally activates tyrosinase and upregulates Tyrp1 and Tyrp2 [12–14]. In addition to their roles in pigmentation, tyrosinase family proteins also influence the biology of melanocyte and melanoma. There is evidence that Tyrp1 is involved in the maintenance of melanosome structure and affects melanocyte proliferation and cell death [15–18].
Based on its homology to tyrosinase, Tyrp1 has also been speculated to be another tyrosinase or reveal the tyrosinase-like activity. Tyrp1 shows tyrosine hydroxylase activity, albeit under low substrate (L-tyrosine) concentration, but no DOPA oxidase activity [19, 20]. Based on that human Tyrp1 is involved in conversion of L-tyrosine to DOPA with low turnover rates, sufficient to prime the system by the generation of low amounts of DOPA, a necessary co-factor for tyrosinase activity [21]. Tyrp1 has also been attributed with various other catalytic functions including dopachrome tautomerase (Dct), dihydroxyindole (DHI) oxidase [22] and 5,6-dihydroxyindole-2-carboxylic acid (DHICA) [23]. Mutagenesis studies have recently confirmed that Tyrp1 is actively involved in inactivation of the catalytic activity of tyrosinase [24]. Observing the more number of pathological genetic variants and their structural and functional aspects of OCA3 will aid in development of personalized medicine.
Several computational algorithms used for the accurate prediction of OCA3 uncharacterized alleles for their disease related property. Mutations involved in OCA3 disorder are hard to scrutinize using in vivo examinations. Hence, an efficient experimental design specific to these diseases are mandatory to observe the disease associated mutation of respective SNPs. Several research articles have stated is effectiveness in identifying the deleterious and disease-associated mutations, thus predicting the pathogenic nsSNPs in correlation to their functional and structural damaging properties [25–28]. Computational studies have previously provided an efficient platform for evaluation and analysis of genetic mutations for their pathological consequences and in determining their underlying molecular mechanism [27–33]. Moreover the conformational changes in the 3D structure of the protein account for the changes in its time dependent physiological affinities and various biochemical pathway alterations [34–37]. Here we used set of computational platforms that utilizes sequence-based conservation profile, homology-based structure profile information, and support vector algorithm used to examine the disease associated nsSNPs. In this study we have applied a set of tools like PolyPhen 2.0 [38], SIFT [39], I-mutant 3.0 [40], PANTHER [41], PhD-SNP [42], SNP&GO [43], Pmut [44], and MutPred [45] to show greater accuracy for the prediction of most disease-associated mutations in OCA3 gene and their structural consequence. Further, we carried out molecular dynamic simulations (MDS) to analyse the molecular and structural basis of predicted disease associated nsSNPs. MDS were applied to observe the motion trajectory and atomic interaction of native and mutant (R326H and R356Q) Tyrp1 protein. The overall strategy implemented in this work is shown in Figure 1.
Figure 1.
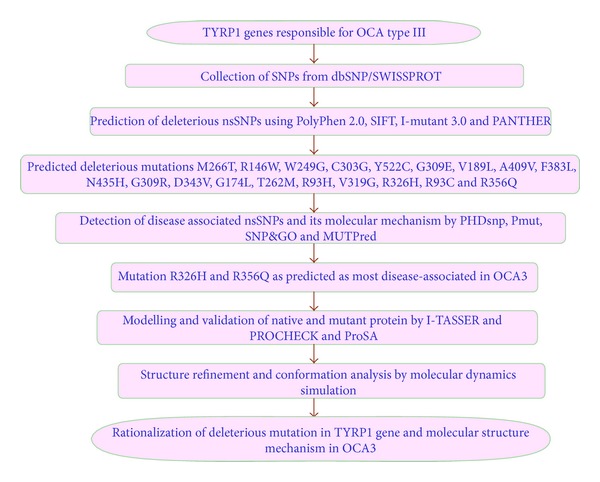
Flow chart of mutational analysis of OCA3.
2. Materials and Methods
2.1. Dataset
The data on human TYRP1 genes were collected from OMIM [46] and Entrez gene on National Center for Biotechnology Information (NCBI) Website. The SNP information of TYRP1 gene was obtained from dbSNP (http://www.ncbi.nlm.nih.gov/snp/) [47] and Swissprot databases [48–50]. The amino acid sequence of Tyrp1 protein was retrieved from the Uniprot database (Uniprot ID: P17643). In order to build the mutant structures, we induced the point mutations in the position of 326 and 356 of Tyrp1 protein using SPDB viewer package [51]. These structures were energetically optimized by applying the all atom OPLS force field available in GROMACS package 4.5.3 [52].
2.2. Disease Related SNP Prediction
The single nucleotide polymorphism occurring in the protein coding region may lead to the deleterious consequences and might affect its 3D structure. Here we applied PolyPhen 2.0 [38], SIFT [39], I-Mutant 3.0 [40], PANTHER [41], PhD-SNP [42], SNP&GO [43], Pmut [44], and MutPred [45] tools in order to examine the disease-associated nsSNP occurring in the Tyrp1 protein coding region. PolyPhen 2.0 is based on combination of sequence and structure based attributes and uses naive Bayesian classifier for the identification of an amino acid substitution and the impact of mutation. The output levels of probably damaging and possibly damaging were classified as functionally significant (≤0.5) and the benign level being classified as tolerated (≥0.51) [38]. SIFT prediction is based on the sequence homology and the physicochemical properties of amino acids which are dictated by the substituted amino acid. SIFT score ≥0.05 indicates the amino acid substitution is intolerant or deleterious, whereas the score ≤0.05 predicted it as tolerant [39]. I-Mutant 3.0 is a support vector machine (SVM) based tool. We used the sequence based version of I-Mutant 3.0 that classifies the prediction into three classes: neutral mutation (−0.5 ≤ DDG ≥ 0.5 kcal/moL), large decrease (<−0.5 kcal/moL), and a large increase (>0.5 kcal/moL). The free energy change (DDG) predicted by I-Mutant 3.0 is based on the difference between unfolding Gibbs free energy change of mutant and native protein (kcal/moL) [40]. PANTHER program is a protein family and subfamily database which predicts the frequency of occurrence of amino acid at a particular position in evolutionary related protein sequences. The threshold subPSEC score of −3 has been assigned below which the predictions are considered as deleterious [41]. We filtered the nsSNPs that were combinedly predicted to be deleterious and damaging from these four servers. Further we used PhD-SNP, SNP&GO, Pmut, and MutPred tools to examine the disease-associated nsSNPs. PhD-SNP is SVM based classifier, trained over the million amino acid polymorphism datasets using supervised training algorithm [42]. It predicts whether the given amino acid substitution leads to disease associated or neutral along with the reliability index score [42]. SNP&GO retrieves data from protein sequence, evolutionary information, and functions as encoded in the gene ontology terms [43]. Pmut is a neural network based program which is trained on large database of neutral and pathological mutations [44]. Pmut uses 3 parameters including mutation descriptors, solvent accessibility, and residue and sequence properties to calculate the pathogenicity indexes of given input mutation data ranging from 0 to 1. The mutations with index score greater than 0.5 are predicted to be pathologically significant [44]. MutPred is a web based tool, used to predict the molecular changes associated with amino acid variants [45]. It uses SIFT, PSI-BLAST, and Pfam profiles along with some structural disorder prediction algorithms, including TMHMM, MARCOIL, I-Mutant 2.0, B-factor prediction, and DisProt [45]. Functional analysis includes the prediction of DNA-binding site, catalytic domains, calmodulin-binding targets, and posttranslational modification sites [45]. Combining the scores of all four servers, the accuracy of prediction rises to a greater extent and finally we filtered the most disease-associated mutation.
2.3. Modelling of Native and Mutant TYRP1 Proteins
According to the annotated information available in UNIPROT entry-P17643, the predicted deleterious mutation sites of Tyrp1 protein were observed in the topological domain which comprised between the regions 190–385. Hence, we modeled Tyrp1 protein segment which consists of 196 amino acid residues by I-TASSER server [53]. This program works by combining the folds and secondary structure by profile-profile alignment threading techniques for non-aligned regions. For the submitted sequences, five 3D models were obtained and the best model was selected based on the lowest energy. Further the native structure was mutated with the most deleterious substitution predicted in this study. In order to build the mutant structures, we made a point mutation in native Tyrp1 protein at R326H (arginine to histidine) and R356Q (arginine to glutamine) using SPDB viewer [51]. The native and mutant structures were energetically optimized by applying the all atom OPLS force field available under the GROMACS 4.5.3 package [52]. The quality of model structures was verified using the PROCHECK [54] and PROSA [55] programs.
2.4. Molecular Dynamics Simulation
Molecular dynamics simulation was performed by using gromacs 4.5.3 package [52] running on a single Intel Core2Duo machine with 3 GB RAM and running Ubuntu 11.10 Linux package. Structure of native and mutant Tyrp1 protein was used as starting point for MD simulations. Systems were solvated in a cubic box with simple point charge (SPC) water molecules at 10 Å marginal radius. At physiological pH the structures were found to be negatively charged; thus in order to make the simulation system electrically neutral, we added 10 sodium ions (Na+) to the simulation box using the “genion” tool that accompanies with gromacs package. Initially the solvent molecules were relaxed while all the solute atoms were harmonically restrained to their original positions with a force constant of 100 kcal/moL for 5000 steps. After this, whole molecular system was subjected to energy minimization for 5000 iterations by steepest descent algorithm implementing GROMOS96 43a1 force field. Berendsen temperature coupling method [56] was used to regulate the temperature inside the box. Electrostatic interactions were computed using the Particle Mesh Ewald method [57]. The ionization states of the residues were set appropriate to pH 7 with all histidines assumed neutral. The pressure was maintained at 1 atm with the allowed compressibility range of 4.5e − 5 atm. SHAKE algorithm was used to constrain bond lengths involving hydrogen, permitting a time step of 2 fs. Van der Waals and coulomb interactions were truncated at 1.0 nm. The nonbonded pair list was updated every 10 steps and conformations were stored every 0.5 ps. Position restraint simulation for 500 ps was implemented to allow solvent molecules to enter the cavity region of structure. Finally, systems were subjected to MD simulation for 20 ns. We then computed the comparative analysis of structural deviations in native and mutant structure. RMSD, RMSF, SASA, Rg, DSSP, and density plot analysis were carried out by using g_rms, g_rmsf, g_sas, g_gyrate, do_dssp, and g_density tool, respectively. Number of distinct hydrogen bonds formed by specific residues to other amino acids within the protein during the simulation (NH bond) was calculated using g_hbond. NH bond determined on the basis of donor-acceptor distance smaller than 0.35 nm and of donor-hydrogen-acceptor. All the graphs were plotted using XMGRACE [58] program.
2.5. Principal Component Analysis
The calculation of the eigenvectors and eigenvalues, and their projection along the first two principal components, was carried out using essential dynamics (ED) method according to protocol [59] within the GROMACS software package. The principle component analysis or ED is a technique that reduces the complexity of the data and extracts the concerted motion in simulations that are essentially correlated and presumably meaningful for biological function [59]. In the ED analysis, a variance/covariance matrix was constructed from the trajectories after removal of the rotational and translational movements. A set of eigenvectors and eigenvalues was identified by diagonalizing the matrix. The eigenvalues represents the amplitude of the eigenvector along the multidimensional space, and the displacement of atoms along each eigenvector shows the concerted motions of protein along each direction. The movements of structures in the essential subspace were identified by projecting the Cartesian trajectory coordinates along the most important eigenvectors from the analysis. Backbone C-alpha bonds trajectories were obtained using g_covar and g_anaeig of gromacs utilities.
3. Results and Discussion
To determine the deleterious nonsynonymous single nucleotide polymorphisms (nsSNPs), which might be involved in inducing disease associated phenomena, is now among the most important field of computational genomic research. The disease associated mutations can be identified with the help of genome sequencing and its analysis. The advanced method in computational biology has now enabled us to determine the deleterious nsSNPs in the target candidate genes. Computational methods were applied to study the protein structural and functional effect on point mutation at molecular level. In this investigation we implemented multiple computational methods to identify the most likely pathogenic mutations in TYRP1 gene. Our results also revealed that implementations of different algorithms often serve as powerful tools for prioritizing candidate functional nsSNPs. Here we used SIFT, PolyPhen, I-Mutant 3.0, PANTHER, PhD-SNP, SNP&GO, Pmut, and MutPred tools to examine the most deleterious and disease associated nsSNPs from the SNP dataset. The combination of methods based on evolutionary information and protein structure and/or functional parameters were used in order to increase the prediction accuracy.
3.1. Screening of Deleterious nsSNPs by PolyPhen 2.0, SIFT, I-Mutant 3.0, and PANTHER Program
Out of 63 input polymorphic dataset, 42 nsSNPs were found to be “damaging” (0.5 to 1.000) to protein structure and function and the remaining 21 nsSNPs were characterized as benign by PolyPhen 2.0. Among these 42 deleterious nsSNPs, 15 SNPs G63S, W249G, C303G, Y522C, G300E, A380S, V189L, R153C, N132I, N435H, R374G, D343V, T262M, R326H, and R356Q were reported to be highly deleterious with PolyPhen score of 1.000 (Table 1). In SIFT, 34 mutations (G63S, M266T, R146W, W249G, A486T, R73W, T366M, C303G, Y522C, A380S, S305R, G309E, V189L, A409V, L7P, D123V, A31G, N132I, F383L, N96Y, N435H, R114C, G309R, R471W, D343V, G174L, A24T, T262M, R93H, R505C, V319G, R326H, R93C, and R356Q) were predicted to be deleterious with tolerance index ≥0.05 (Table 1). Among these, 17 mutations G63S, M266T, W249G, C303G, G309E, V189L, N132I, N96Y, N435H, R114C, G309R, R471W, D343V, G174L, T262M, R326H, and R356Q were reported to be highly deleterious with SIFT score of 0.00 (Table 1). Furthermore, 29 mutations were identified as deleterious and damaging in SIFT and PolyPhen 2.0 server (Table 1) which also shows a strong correlation between the prediction methodologies implemented by these two servers. SIFT and PolyPhen were shown to have better performance in identifying functional nsSNPs among other in silico tools [60]. The accuracy of SIFT and PolyPhen was further validated through our results, which makes these tools more suitable for the prediction [61]. All the nsSNPs submitted to PolyPhen 2.0 and SIFT were also submitted as input to the I-Mutant 3.0 server. 45 mutations were predicted to affect the stability of the protein structure by I-Mutant 3.0. Remaining out of 18 mutations, 16 mutations showed the neutral effect on protein structure and 2 mutations showed increased stability of the structure. To further validate these results we implemented HMM based statistical prediction method to identify the functionally significant point mutations using PANTHER server. The mutations with subPSEC score less than −3 have been reported to be probably deleterious. 39 mutations with subPSEC score less than or equal to −3 were characterized to be deleterious. We filtered 19 mutations (M266T, R146W, W249G, C303G, Y522C, G309E, V189L, A409V, F383L, N435H, G309R, D343V, G174L, T262M, R93H, V319G, R326H, R93C, and R356Q) which were commonly predicted to be deleterious and damaging by SIFT, PolyPhen 2.0, I-Mutant 3.0, and PANTHER servers (Table 1).
Table 1.
nsSNPs analyzed by four computational methods PolyPhen 2.0, SIFT, I-Mutant 3.0, and PANTHER in TYRP1 gene.
| SNP ID | Mutation | PolyPhen 2.0 | SIFT | I-MUTANT 3.0 | PANTHER | ||||
|---|---|---|---|---|---|---|---|---|---|
| PSIC | Prediction | Score | Prediction | DDG | Stability | subPSEC | Prediction | ||
| rs202189890 | S270R | 0.996 | Damaging | 0.18 | Tolerated | −0.43 | Neutral | −2.23829 | Tolerated |
| rs202126779 | T253M | 0.996 | Damaging | 0.08 | Tolerated | −0.32 | Decrease | −3.02723 | Deleterious |
| rs201899938 | S8F | 0.963 | Damaging | 0.7 | Tolerated | 0.20 | Neutral | −2.19231 | Tolerated |
| rs201789348 | E139K | 0.301 | Benign | 0.24 | Tolerated | −0.97 | Decrease | −3.62832 | Deleterious |
| rs201605146 | S470N | 0.134 | Benign | 0.36 | Tolerated | −0.48 | Neutral | −2.11901 | Tolerated |
| rs201457510 | G63S | 1.000 | Damaging | 0.00 | Deleterious | −1.17 | Neutral | −4.06048 | Deleterious |
| rs201345670 | M266T | 0.772 | Damaging | 0.00 | Deleterious | −1.63 | Decrease | −3.93879 | Deleterious |
| rs201293896 | R146W | 0.975 | Damaging | 0.02 | Deleterious | −0.36 | Decrease | −5.75453 | Deleterious |
| rs200882524 | L487F | 0.998 | Damaging | 0.23 | Tolerated | −0.98 | Decrease | −4.01429 | Deleterious |
| rs200754545 | W249G | 1.000 | Damaging | 0.00 | Deleterious | −2.43 | Decrease | −7.90178 | Deleterious |
| rs200607153 | A486T | 0.029 | Benign | 0.05 | Deleterious | −0.56 | Decrease | −2.56844 | Tolerated |
| rs199989943 | R73W | 0.999 | Damaging | 0.02 | Deleterious | −0.24 | Neutral | −5.69984 | Deleterious |
| rs199823942 | T366M | 0.014 | Benign | 0.01 | Deleterious | −0.43 | Decrease | −4.14092 | Tolerated |
| rs193035382 | C303G | 1.000 | Damaging | 0.00 | Deleterious | −1.49 | Decrease | −3.02248 | Deleterious |
| rs188236569 | Y522C | 1.000 | Damaging | 0.01 | Deleterious | −0.79 | Decrease | −4.39247 | Deleterious |
| rs187959351 | G300E | 1.000 | Damaging | 0.86 | Tolerated | −0.39 | Decrease | −3.66042 | Deleterious |
| rs184910238 | A380S | 1.000 | Damaging | 0.05 | Deleterious | −0.66 | Decrease | −2.88075 | Tolerated |
| rs183546444 | P476T | 0.000 | Benign | 1.00 | Tolerated | −1.13 | Decrease | −2.05148 | Tolerated |
| rs182508840 | T275A | 0.064 | Benign | 0.10 | Tolerated | −1.58 | Decrease | −2.92255 | Tolerated |
| rs181755026 | E524A | 0.001 | Benign | 0.22 | Tolerated | −0.45 | Decrease | −2.15096 | Tolerated |
| rs151091299 | S305R | 0.992 | Damaging | 0.01 | Deleterious | −0.22 | Neutral | −3.02846 | Deleterious |
| rs150899857 | R505H | 0.753 | Damaging | 0.07 | Tolerated | −1.32 | Decrease | −3.32609 | Deleterious |
| rs149856598 | G309E | 0.566 | Damaging | 0.00 | Deleterious | −0.80 | Decrease | −3.82367 | Deleterious |
| rs149324507 | V189L | 1.000 | Damaging | 0.00 | Deleterious | −0.79 | Decrease | −3.93115 | Deleterious |
| rs148777155 | S498P | 0.761 | Damaging | 0.19 | Tolerated | −0.27 | Neutral | −4.76166 | Deleterious |
| rs148248971 | A409V | 0.972 | Damaging | 0.01 | Deleterious | −0.02 | Decrease | −3.75227 | Deleterious |
| rs147212712 | L7P | 0.924 | Damaging | 0.01 | Deleterious | −1.44 | Decrease | NA | NA |
| rs146962444 | D123V | 0.628 | Damaging | 0.03 | Deleterious | −0.34 | Neutral | −3.42156 | Deleterious |
| rs146838872 | V33A | 0.000 | Benign | 0.45 | Tolerated | −0.86 | Decrease | −2.4943 | Tolerated |
| rs146467307 | V373I | 0.013 | Benign | 0.54 | Tolerated | −0.61 | Decrease | −2.69258 | Tolerated |
| rs146027807 | R153C | 1.000 | Damaging | 0.05 | Tolerated | −0.90 | Decrease | −5.95016 | Deleterious |
| rs144438412 | A67T | 0.001 | Benign | 0.36 | Tolerated | −0.69 | Decrease | −2.56649 | Tolerated |
| rs144206983 | S207I | 0.968 | Damaging | 0.24 | Tolerated | 0.43 | Increase | NA | NA |
| rs143705229 | A31G | 0.159 | Benign | 0.02 | Deleterious | −1.39 | Decrease | −4.24808 | Deleterious |
| rs143610120 | N132I | 1.000 | Damaging | 0.00 | Deleterious | 0.82 | Increase | −6.81596 | Deleterious |
| rs143106698 | F383L | 0.999 | Damaging | 0.04 | Deleterious | −0.99 | Decrease | −3.52387 | Deleterious |
| rs143004036 | N96Y | 0.987 | Damaging | 0.00 | Deleterious | −0.20 | Neutral | −4.96986 | Deleterious |
| rs142019860 | Q520K | 0.067 | Benign | 0.05 | Tolerated | −0.16 | Decrease | −2.41741 | Tolerated |
| rs141949653 | N435H | 1.000 | Damaging | 0.00 | Deleterious | −0.72 | Decrease | −4.94266 | Deleterious |
| rs141834891 | R23W | 0.02 | Benign | 0.22 | Tolerated | 0.13 | Neutral | −2.04234 | Tolerated |
| rs141196295 | R114C | 0.019 | Benign | 0.00 | Deleterious | −0.68 | Neutral | −4.80738 | Deleterious |
| rs140233627 | S506G | 0.004 | Benign | 0.40 | Tolerated | −0.96 | Decrease | −2.00879 | Tolerated |
| rs139946740 | G309R | 0.754 | Damaging | 0.00 | Deleterious | −0.56 | Decrease | −5.11275 | Deleterious |
| rs139670838 | R55H | 0.771 | Damaging | 0.13 | Tolerated | −1.12 | Decrease | −3.93481 | Deleterious |
| rs138272660 | R471W | 0.986 | Damaging | 0.00 | Deleterious | −0.23 | Neutral | −5.61942 | Deleterious |
| rs138038972 | R125K | 0.000 | Benign | 0.21 | Tolerated | −1.00 | Neutral | −2.33412 | Tolerated |
| rs121912778 | R374G | 1.000 | Damaging | 0.06 | Tolerated | −1.48 | Decrease | −3.98421 | Deleterious |
| rs113146199 | D343V | 1.000 | Damaging | 0.00 | Deleterious | 0.14 | Decrease | −4.03201 | Deleterious |
| rs78071458 | G174L | 0.993 | Damaging | 0.00 | Deleterious | −0.29 | Decrease | −4.71688 | Deleterious |
| rs61758405 | A24T | 0.219 | Benign | 0.02 | Deleterious | −0.52 | Decrease | −3.03529 | Deleterious |
| rs61752939 | T262M | 1.000 | Damaging | 0.00 | Deleterious | −0.34 | Decrease | −5.00198 | Deleterious |
| rs61752937 | R93H | 0.998 | Damaging | 0.04 | Deleterious | −1.60 | Decrease | −3.09393 | Deleterious |
| rs61752864 | A70T | 0.859 | Damaging | 0.09 | Tolerated | −0.66 | Decrease | −2.58273 | Tolerated |
| rs41306053 | D308N | 0.000 | Benign | 0.70 | Tolerated | −1.12 | Decrease | −2.04757 | Tolerated |
| rs41305647 | Q518H | 0.000 | Benign | 0.23 | Tolerated | −0.43 | Decrease | −2.67099 | Tolerated |
| rs41305645 | Q530R | 0.029 | Benign | 0.43 | Tolerated | −0.07 | Neutral | −2.1177 | Tolerated |
| rs41303653 | G485A | 0.000 | Benign | 1.00 | Tolerated | −0.63 | Neutral | −3.35217 | Deleterious |
| rs41303651 | R505C | 0.861 | Damaging | 0.01 | Deleterious | −0.89 | Neutral | −2.64052 | Tolerated |
| rs35197549 | V319G | 0.997 | Damaging | 0.01 | Deleterious | −1.86 | Decrease | −3.20938 | Deleterious |
| rs16929374 | R326H | 1.000 | Damaging | 0.00 | Deleterious | −1.21 | Decrease | −4.86382 | Deleterious |
| rs3202399 | E413K | 0.976 | Damaging | 0.18 | Tolerated | −1.07 | Decrease | −2.76125 | Tolerated |
| VAR_068176 | R93C | 0.999 | Damaging | 0.01 | Deleterious | −1.24 | Decrease | −4.10005 | Deleterious |
| VAR_026828 | R356Q | 1.000 | Damaging | 0.00 | Deleterious | −1.49 | Decrease | −5.86235 | Deleterious |
SNPs highlighted in bold are predicted to be deleterious.
3.2. Prediction of Disease-Associated nsSNPs
We applied PhD-SNP which is based on support vector machine tool to further classify the predicted deleterious nsSNPs as disease associated. Total 19 nsSNPs which were commonly predicted in SIFT, PolyPhen 2.0, I-Mutant 3.0, and PANTHER tools were further used in PhD-SNP server. Out of 19 mutations, 16 of them (R146W, C303G, G309E, V189L, A409V, F383L, N435H, G309R, D343V, G174L, T262M, R93H, V319G, R326H, R93C, and R356Q) were predicted to be disease associated (Table 2). In SNP&GO, 19 nsSNPs were predicted to be disease associated. To verify this prediction, we further employed artificial neural network (ANN) based Pmut tool. Out of 19 nsSNPs, 10 mutations showed pathogenecity and remaining 9 nsSNP showed as neutral (Table 2). Particularly, R326H showed higher pathogenecity level with pathogenicity index of 0.9314 (Table 2). 8 mutations (R146W, G309E, G309R, D343V, T262M, R93H, R326H, and R356Q) were predicted as most disease associated by PhD-SNP, SNP&GO, and Pmut servers. These 8 mutations were further analysed by MutPred tool to predict the SNP disease-association probability and probable change in the molecular mechanism in the mutant. We found R326H to be highly deleterious with general probability (g) scores of 0.938 and was predicted to induce the loss of stability with (p) score of 0.0202, showing confident hypothesis. R356Q was found to be highly deleterious with general probability (g) scores of 0.801 and was predicted to induce the loss of catalytic residue at R356 with (p) score of 0.0446, showing confident hypothesis. At the end of so many mutations considered, we screened R326H and R356Q as the most deleterious and disease associated mutation in TYRP1 gene (Table 3). This prediction could be endorsed with the observed experimental data [62].
Table 2.
The disease associated SNPs are predicted from PHDsnp, SNP&GO, and Pmut servers.
| SNP IDs | Mutation | PHDsnp results | SNP&GO | Pmut | |
|---|---|---|---|---|---|
| Score | Prediction | ||||
| rs201345670 | M266T | Neutral | Disease | 0.4759 | Neutral |
| rs201293896 | R146W | Disease | Disease | 0.6728 | Pathological |
| rs200754545 | W249G | Neutral | Disease | 0.5638 | Pathological |
| rs193035382 | C303G | Disease | Disease | 0.2516 | Neutral |
| rs188236569 | Y522C | Neutral | Disease | 0.6752 | Pathological |
| rs149856598 | G309E | Disease | Disease | 0.8774 | Pathological |
| rs149324507 | V189L | Disease | Disease | 0.4793 | Neutral |
| rs148248971 | A409V | Disease | Disease | 0.4533 | Neutral |
| rs143106698 | F383L | Disease | Disease | 0.3564 | Neutral |
| rs141949653 | N435H | Disease | Disease | 0.1202 | Neutral |
| rs139946740 | G309R | Disease | Disease | 0.6503 | Pathological |
| rs113146199 | D343V | Disease | Disease | 0.5798 | Pathological |
| rs78071458 | G174L | Disease | Disease | 0.3075 | Neutral |
| rs61752939 | T262M | Disease | Disease | 0.6624 | Pathological |
| rs61752937 | R93H | Disease | Disease | 0.6320 | Pathological |
| rs35197549 | V319G | Disease | Disease | 0.2561 | Neutral |
| rs16929374 | R326H | Disease | Disease | 0.9314 | Pathological |
| VAR_068176 | R93C | Disease | Disease | 0.1465 | Neutral |
| VAR_026828 | R356Q | Disease | Disease | 0.6210 | Pathological |
Disease associated SNPs are displayed in bold.
Table 3.
The G score, P score, molecular variations, and prediction reliability calculated from MutPred server. Here the most disease associated mutations are displayed in bold.
| SNP ID | Mutation | MUTPred | |||
|---|---|---|---|---|---|
| G score | P score | Molecular Variation | Prediction reliability | ||
| rs201293896 | R146W | 0.524 | 0.0566 | Loss of disorder | No reliable Inference |
| rs149856598 | G309E | 0.563 | 0.0869 | Loss of catalytic residue | No reliable inference |
| rs139946740 | G309R | 0.611 | 0.0971 | Gain of solvent accessibility | No reliable inference |
| rs113146199 | D343V | 0.613 | 0.0676 | Loss of disorder | No reliable inference |
| rs61752939 | T262M | 0.599 | 0.079 | Loss of helix | No reliable inference |
| rs61752937 | R93H | 0.354 | 0.0986 | Loss of disorder | No reliable inference |
| rs16929374 | R326H | 0.938 | 0.0202 | Loss of stability | Confident hypothesis |
| VAR_026828 | R356Q | 0.801 | 0.0446 | Loss of catalytic residue at R356 | Confident hypothesis |
3.3. Modelling of Protein
The human Tyrp1 protein (between domain regions 190–385) was modelled by automated protein structure prediction program, I-TASSER [53]. The program used more than ten templates to model the protein. The top most template (PDB ID: 3nm8A) has covered 86% of the Tyrp1 protein query sequence. The best structure with high confidence score was collected and used for further investigations. The disease associated mutations of R326H and R356Q can probably alter the native conformation of the Tyrp1 protein structure. Hence we made a point mutation in native Tyrp1 protein at the position of 326 (arginine to histidine) and 356 (arginine to glutamine) to build the mutant structures. The quality of the modeled structure of native and mutant Tyrp1 protein was evaluated independently by the PROCHECK [54] and PROSA [55] programs, which showed good stereochemical properties of the modeled proteins. Native Tyrp1 protein showed 91.7% of residues in most favoured and allowed region and z-score value of −3.4. Mutant of R326H showed 92.3% of residues in most favoured and allowed region and z-score value of −2.13. Mutant of R356Q showed 92.3% of residues in most favoured and allowed region and z-score value of −2.81. Native and mutant (R326H and R356Q) Tyrp1 structures showed the g-factor in the ranges of 0.40, 0.45, and 0.46, respectively. The overall G-factors of native and mutant Tyrp1 protein structures (acceptable between 0 and 0.5) were produced by PROCHECK in the range of 0.40–0.46. These scores implicate high confidence level and hence the structures were selected for further MD analysis.
3.4. Molecular Dynamics Simulation
To understand the structural and functional behaviour of the prioritized disease associated mutations, we performed molecular dynamics simulation for native and mutant Tyrp1 proteins. The following seven factors, namely, tolerance index, PSIC score, DDG value, subPSEC score, disease-association study, pathogenecity index, general score (g), and property score (p), which correspond to conformational changes in protein residues due to the mutation, may lead to affect the functional behaviour of Tyrp1 protein. The results obtained from above analysis further inspired us to study the dynamic behaviour of native and mutant (R326H and R356Q) Tyrp1 proteins. We studied RMSD, RMSF, Rg, SASA, and NH bond variations, DSSP, density plot, and ED analysis between the native and mutant (R326H and R356Q) Tyrp1 protein structures. Further, the RMSD for all Cα atoms from the initial structure was examined to study the convergence of the protein system. In Figure 2(a), native and both mutant (R326H and R356Q) structures showed similar way of deviation till 3050 ps from their starting structure, resulting in a backbone RMSD of ~0.14 to 0.52 nm during the simulations. After this, native structure retained the maximum deviation till the end of the simulation resulting in the backbone RMSD of ~0.51 to ~0.66 nm, respectively. R326H and R356Q mutant structures showed the minimum deviation till the end of the simulation, resulting in the backbone RMSD of ~0.37 to ~0.51 nm and ~0.38 to ~0.54 nm, respectively.
Figure 2.
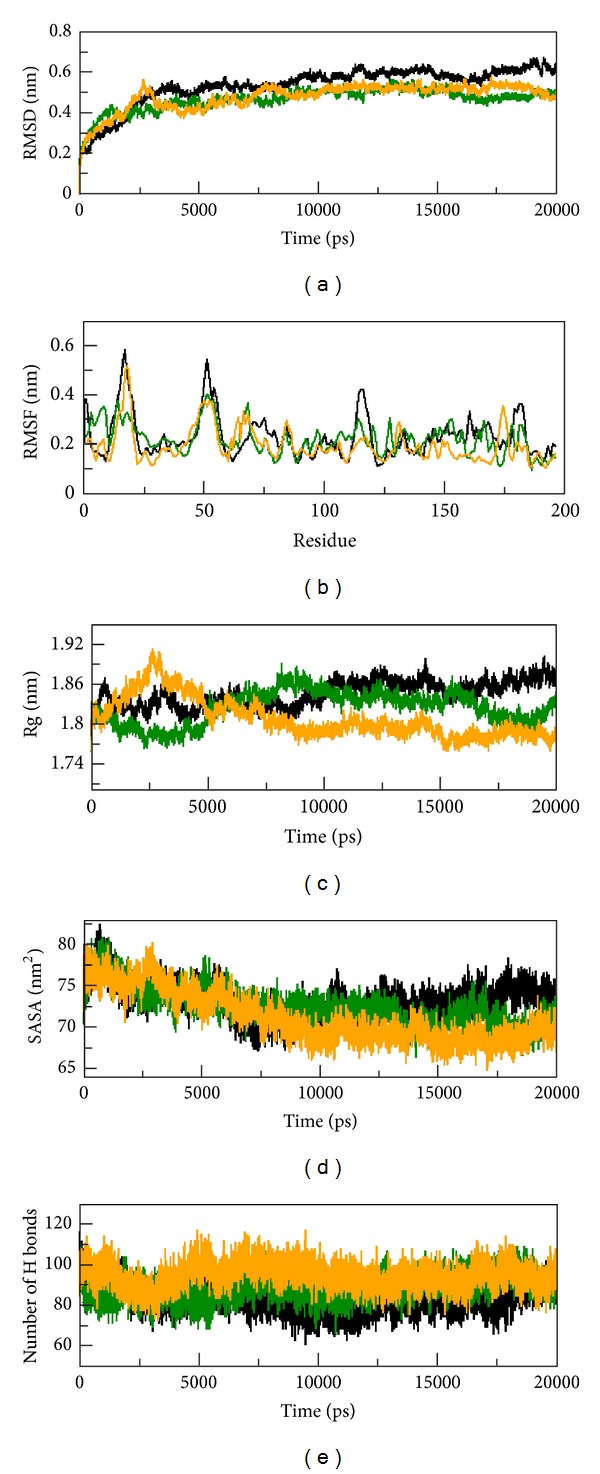
RMSD, RMSF, Rg, SASA, and NH bond of native and mutant Tyrp1 proteins versus time at 300 K. Native is shown in black, mutant (R326H) in green, and mutant (R356Q) in yellow.
This magnitude of fluctuations, together with very small difference between the average RMSD values after the relaxation period (~0.52 nm), leads to produce stable trajectories in simulation, and it provided an appropriate basis for further analysis. The average value of RMSD during the simulation time period both native and mutant (R326H and R356Q) structures is signified in Table 4. Through the aim of determining RMSF we predicted whether the mutation disturbs the dynamic behaviour of residues. The RMSF values of native and mutant (R325H and R356Q) structures were collected and shown in Figure 2(b). Analysis of fluctuation score depicted that the higher degree of flexibility was observed in native structure than mutant (R326H and R356Q) Tyrp1 protein structures. The radius of gyration (Rg) is defined as the mass-weight root-mean-square distance of collection of atoms from their common center of mass. Therefore it provides an insight into the overall dimension of the protein. Radius of gyration plot for Cα atoms of protein versus time at 300 K is shown in Figure 2(c).
Table 4.
Average values of RMSD, Rg, SASA, NH-bonds, trace of co-variance, and density value of native and mutant (R326H and R356Q) structures.
| Native | Mutant (R326H) | Mutant (R356Q) | |
|---|---|---|---|
| RMSD | 0.62 | 0.50 | 0.52 |
| Rg | 1.84 | 1.81 | 1.79 |
| SASA | 74.5 | 71.2 | 69.1 |
| NH-bonds | 84.7 | 93.2 | 92.4 |
| Density value | 41.9 | 52.2 | 52.3 |
| Trace of Co-variance | 11.56 | 10.74 | 8.16 |
RMSD: root-mean-square deviation; Rg: radius of gyration; SASA: solvent accessible surface area; NH bonds: number of hydrogen bonds. The value of RMSD, Rg and SASA, co-variance, and total helicity are given in nm, whereas density value is given in nm−3.
In Figure 2(c), at the end of the simulation native structures showed greater Rg value than mutant (R326H and R356Q) structures. The native structure showed Rg value of ~1.76 nm at 0 ps, ~1.82 nm at 4000 ps, ~1.82 nm at 9500 ps, ~1.85 nm at 11800 ps, ~1.86 nm at 14600 ps, ~1.86 nm at 17500 ps, and ~1.87 nm at 20000 ps. R326H mutant structure showed Rg value of ~1.76 nm at 0 ps, ~1.78 nm at 4000 ps, ~1.86 nm at 9500 ps, ~1.84 nm at 11800 ps, ~1.82 nm at 14600 ps, ~1.81 nm at 17500 ps, and ~1.83 nm at 20000 ps. R356Q mutant structure showed Rg value of ~1.77 nm at 0 ps, ~1.84 nm at 4000 ps, ~1.79 nm at 9500 ps, ~1.79 nm at 11800 ps, ~1.80 nm at 14600 ps, ~1.77 nm at 17500 ps, and 1.78 nm at 20000 ps, respectively. The average Rg value was 1.84 nm in native, whereas the mutant R326H and R356Q structures showed average Rg value of 1.81 and 1.79 nm, respectively, signified in Table 4. A notable change was observed in both mutant (R326H and R356Q) structures as compared to the native. The change of SASA for native and mutant (R326H and R356Q) proteins with time is shown in Figure 2(d). Solvent accessibility surface area accounts for bimolecular surface area that is assessable to solvent molecules. Decreased value of SASA in mutant structures denotes its relatively shrunken nature as compared to the native structure. The change of SASA of native and mutant proteins with time is shown in Figure 2(d). Native and mutant (R326H and R356Q) structures showed similar fashion of deviation till 12000 ps from the initial structure, but after this native structure showed greater value of SASA than mutant (R326H and R356Q) structures. The average SASA value was 74.5 in native, whereas the mutant (R326H and R356Q) structures showed average SASA value of 71.2 and 69.1, respectively, as depicted in Table 4. We also observed notable differences in NH bond pattern during simulation, whereas the native structure showed less participation in NH bonds formation with other amino acid, while in mutant (R326H and R356Q) structures there was greater number of NH bonds (Figure 2(e)). The average value of NH bond was signified in Table 4.
The NH bond results of native and mutant Tyrp1 structure according to the RMSD, RMSF, Rg, and SASA plot results depicted that the mutant (R326H and R356Q) structural conformation became rigid upon mutation which may lead to disturb the functional behaviour of the protein. This was further supported by the atomic density plot and PCA analysis. The consequences of these molecular changes were clearly observed in the atomic density distribution plot. There was a significant change in density distribution in mutant as compared to the native and it was depicted. Moreover the native structure shows highest atomic density distribution of 41.9 nm−3 but mutant (R326H and R356Q) structures showed 52.2 and 52.3 nm−3, respectively, (Table 4) (Figure S1a-c available online at: http://dx.doi.org/10.1155/2013/697051). It was further indicated that native structure has more flexibility than mutant (R326H and R356Q) structures.
The spectrum of the corresponding eigenvalues indicated the level of fluctuation and dynamic behaviour of protein molecule in the system and was basically confined within the first two eigenvectors. Both mutant (R326H and R356Q) structures covered a small region of phase space particularly along PC1 plane than native (Figure 3(a)). Overall flexibility of two proteins was further examined by the trace of the diagonalized covariance matrix of the Cα atomic positional fluctuations. We obtained the following values for native and mutant (R326H and R356Q) structures: 11.56 nm2, 10.74 nm2, and 8.16 nm2, respectively (Table 4), and again it was confirming the overall increased flexibility in native than mutant (R326H and R356Q) structures at 300 K.
Figure 3.
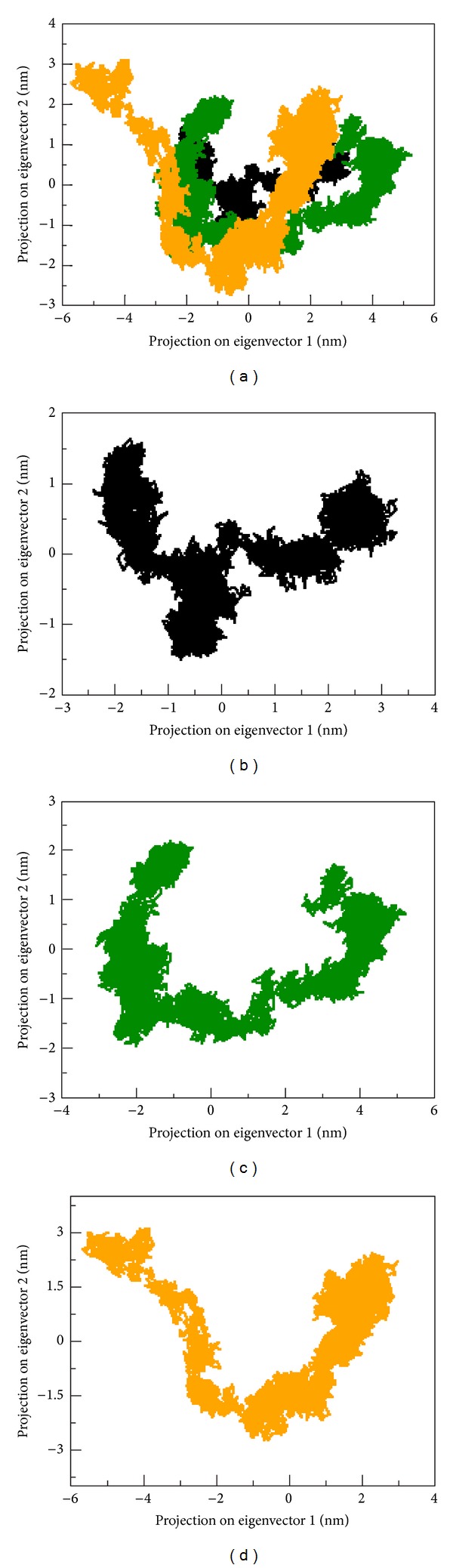
Projection of the motion of the protein in phase space along the first two principal eigenvectors at 300 K. (a) Native is shown in black, mutant (R326H) in green, and mutant (R356Q) in yellow. For clarity's sake, each trajectory is also shown separately in (b), (c), (d).
We applied the DSSP algorithm [63] to monitor changes in secondary structure during the simulations. As shown in Figures 4(a)–4(c), a difference is observed only at the level of Helix from the amino acid residual position of 150 to 175 and the level of sheet from the amino acid residual position of 5 to 20. In DSSP, the most significant structural change was an increase in helical content and absence in β-sheet, which was observed in both mutant (R326H and R356Q) structures.
Figure 4.
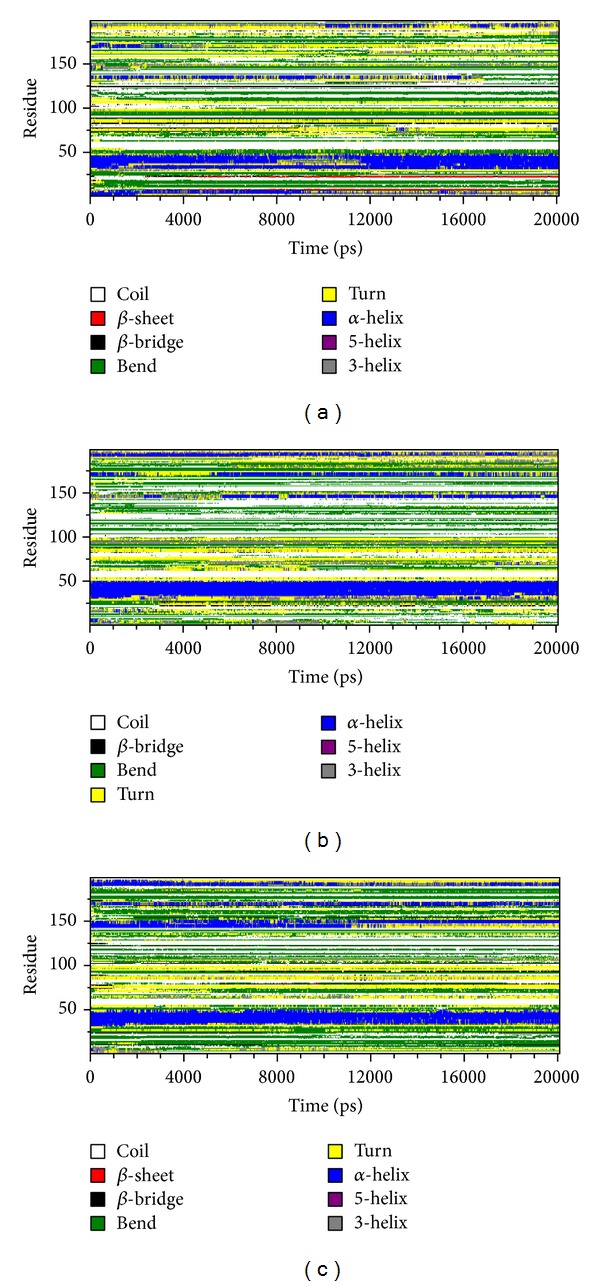
Time evolution of the secondary structural elements of the native and mutant (R326H and R356Q) Tyrp1 proteins at 300 K (DSSP classification). (a) Native, (b) mutant R326H, and (c) mutant R356Q.
To examine how the structure got damaged and leads to affect the functions upon mutation, we analysed the native and mutant (R326H and R356Q) structures at different time scales (Figure 5). It was clearly observed that there is continuous loss of helix in native structure than mutant structures till the end of the simulation. This was well supported by the DSSP analysis. It indicates that both mutant (R326H and R356Q) structures showed an increase in helical content and total absence in β-sheet in the amino acid residual position from 150 to 170 and 5 to 20, respectively. In general, helices are mostly rigid, whereas spanning loop regions are mostly flexible. [64–66]. Based on that here, both mutant structures (R326H and R356Q) showed more helical content which leads to more rigidity in the conformation. On the basis of DSSP analysis, it was confirmed that due to mutation the Tyrp1 structure became rigid. Therefore, it seems evident that both mutations (R326H and R356Q) have cruel damaging impact on protein structural orientation and its function. This prediction is also endorsed with the observed experimental data [62, 67]. This study provides an essential insight into the underlying molecular mechanism of Tyrp1 protein upon mutation and in future it may help to develop a personalized medicine for OCA3.
Figure 5.
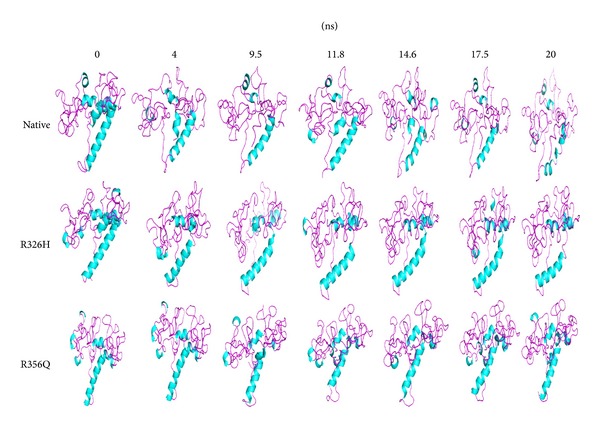
Snapshots of native and mutant (R326H and R356Q) Tyrp1 protein conformation at different simulation time steps.
4. Conclusion
Computational analysis has now become a roadmap to define a standard disease specific SNP at molecular level. In this study we screened two most disease associated mutations (R326H and R356Q) which are related to OCA3. Molecular dynamics simulation approaches have also been extensively used to report the structural consequences of the deleterious predicted point mutations. The flexibility loss is observed in RMSD, RMSF, Rg plot which is further supported by a decrease in SASA value in both mutant structures. This may produce a major impact on the structural conformation of Tyrp1 protein, which also affects the function of Tyrp1 protein. Due to mutation, the structure became more rigid which is also supported by NH bond, density plot, PCA, and DSSP analysis. Our result suggests a significant computational roadmap to detect the OCA3 associated SNPs from the large SNP dataset and reduce the expenses in experimental depiction of pathological nsSNPs. Further the predicted R326H and R356Q mutations can be further studied by wet lab scientist to investigate the evidence of Tyrp1 protein mutation in association to OCA3 and develop a potent drug target for OCA3.
Supplementary Material
The consequences upon mutations were clearly observed in the atomic density distribution plot. There was a significant change in density distribution in mutant as compared to the native. Native structure shows highest atomic density distribution of 41.9 nm−3 but in mutant (R326H & R356Q) structures showed 52.2 and 52.3 nm−3 respectively. It was further indicate that native has more flexibility than mutant (R326H & R356Q) structures.
{kind=link}
Acknowledgments
The authors gratefully acknowledge the management of Vellore Institute of Technology University for providing the facilities to carry out this work. They thank the anonymous reviewers for their helpful comments and critical reading of the paper.
Abbreviations
- nsSNPs:
Non-synonymous single nucleotide polymorphism
- OCA3:
Oculocutaneous albinism type3
- Tyrp1:
Tyrosinase-related protein-1
- MDS:
Molecular dynamics simulation
- RMSD:
Root-mean-square deviation
- RMSF:
Root-mean-square fluctuation
- Rg:
Radius of gyration
- SASA:
Solvent-accessible surface area
- NH bonds:
Number of hydrogen bonds
- PCA:
Principal component analysis
- DSSP:
Definition of secondary structure of proteins.
References
- 1.Murty VVVS, Bouchard B, Mathew S, Vijayasaradhi S, Houghton AN. Assignment of the human TYRP (brown) locus to chromosome region 9p23 by nonradioactive in situ hybridization. Genomics. 1992;13(1):227–229. doi: 10.1016/0888-7543(92)90228-k. [DOI] [PubMed] [Google Scholar]
- 2.Shibahara S, Taguchi H, Muller RM, et al. Structural organization of the pigment cell-specific gene located at the brown locus in mouse: its promoter activity and alternatively spliced transcripts. The Journal of Biological Chemistry. 1991;266(24):15895–15901. [PubMed] [Google Scholar]
- 3.Sturm RA, O’Sullivan BJ, Box NF, et al. Chromosomal structure of the human TYRP1 and TYRP2 loci and comparison of the tyrosinase-related protein gene family. Genomics. 1995;29(1):24–34. doi: 10.1006/geno.1995.1211. [DOI] [PubMed] [Google Scholar]
- 4.Box NF, Wyeth JR, Mayne CJ, O’Gorman LE, Martin NG, Sturm RA. Complete sequence and polymorphism study of the human TYRP1 gene encoding tyrosinase-related protein 1. Mammalian Genome. 1998;9(1):50–53. doi: 10.1007/s003359900678. [DOI] [PubMed] [Google Scholar]
- 5.Vijayasaradhi S, Doskoch PM, Houghton AN. Biosynthesis and intracellular movement of the melanosomal membrane glycoprotein gp75, the human b (Brown) locus product. Experimental Cell Research. 1991;196(2):233–240. doi: 10.1016/0014-4827(91)90256-t. [DOI] [PubMed] [Google Scholar]
- 6.Halaban R, Moellmann G. Murine and human b locus pigmentation genes encode a glycoprotein (gp75) with catalase activity. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(12):4809–4813. doi: 10.1073/pnas.87.12.4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prota G. Melanins and Melanogenesis. New York, NY, USA: Academic Press; 1992. [Google Scholar]
- 8.Hearing VJ., Jr. Mammalian monophenol monooxygenase (tyrosinase): purification, properties, and reactions catalyzed. Methods in Enzymology. 1987;142:154–165. doi: 10.1016/s0076-6879(87)42024-7. [DOI] [PubMed] [Google Scholar]
- 9.Prota G. Some new aspects of eumelanin chemistry. Progress in Clinical and Biological Research. 1988;256:101–124. [PubMed] [Google Scholar]
- 10.Alonso S, Izagirre N, Smith-Zubiaga I, et al. Complex signatures of selection for the melanogenic loci TYR, TYRP1 and DCT in humans. BMC Evolutionary Biology. 2008;8(1, article 74) doi: 10.1186/1471-2148-8-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ito S. A chemist’s view of melanogenesis. Pigment Cell Research. 2003;16(3):230–236. doi: 10.1034/j.1600-0749.2003.00037.x. [DOI] [PubMed] [Google Scholar]
- 12.Jiménez-Cervantes C, Solano F, Kobayashi T, et al. A new enzymatic function in the melanogenic pathway. The 5,6- dihydroxyindole-2-carboxylic acid oxidase activity of tyrosinase-related protein-1 (TRP1) The Journal of Biological Chemistry. 1994;269(27):17993–18000. [PubMed] [Google Scholar]
- 13.Kobayashi T, Urabe K, Winder A, et al. Tyrosinase related protein 1 (TRP1) functions as a DHICA oxidase in melanin biosynthesis. The EMBO Journal. 1994;13(24):5818–5825. doi: 10.1002/j.1460-2075.1994.tb06925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kobayashi T, Vieira WD, Potterf B, Sakai C, Imokawa G, Hearing VJ. Modulation of melanogenic protein expression during the switch from eu- to pheomelanogenesis. Journal of Cell Science. 1995;108(6):2301–2309. doi: 10.1242/jcs.108.6.2301. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi T, Imokawa G, Bennett DC, Hearing VJ. Tyrosinase stabilization by Tyrp1 (the brown locus protein) The Journal of Biological Chemistry. 1999;273(48):31801–31805. doi: 10.1074/jbc.273.48.31801. [DOI] [PubMed] [Google Scholar]
- 16.Hirobe T, Abe H. Genetic and epigenetic control of the proliferation and differentiation of mouse epidermal melanocytes in culture. Pigment Cell Research. 1999;12(3):147–163. doi: 10.1111/j.1600-0749.1999.tb00508.x. [DOI] [PubMed] [Google Scholar]
- 17.Johnson R, Jackson IJ. Light is a dominant mouse mutation resulting in premature cell death. Nature Genetics. 1992;1(3):226–229. doi: 10.1038/ng0692-226. [DOI] [PubMed] [Google Scholar]
- 18.Fang D, Tsuji Y, Setaluri V. Selective down-regulation of tyrosinase family gene TYRP1 by inhibition of the activity of melanocyte transcription factor, MITF. Nucleic Acids Research. 2002;30(14):3096–3106. doi: 10.1093/nar/gkf424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sarangarajan R, Boissy RE. Tyrp1 and oculocutaneous albinism type 3. Pigment Cell Research. 2001;14(6):437–444. doi: 10.1034/j.1600-0749.2001.140603.x. [DOI] [PubMed] [Google Scholar]
- 20.Boissy RE, Sakai C, Zhao H, Kobayashi T, Hearing VJ. Human tyrosinase related protein-1 (TRP-1) does not function as a DHICA oxidase activity in contrast to murine TRP-1. Experimental Dermatology. 1998;7(4):198–204. doi: 10.1111/j.1600-0625.1998.tb00324.x. [DOI] [PubMed] [Google Scholar]
- 21.Zhao H, Zhao Y, Nordlund JJ, Boissy RE. Human TRP-1 has tyrosine hydroxylase but no dopa oxidase activity. Pigment Cell Research. 1994;7(3):131–140. doi: 10.1111/j.1600-0749.1994.tb00040.x. [DOI] [PubMed] [Google Scholar]
- 22.Urabe K, Aroca P, Hearing VJ. From gene to protein: determination of melanin synthesis. Pigment Cell Research. 1993;6(4):186–192. doi: 10.1111/j.1600-0749.1993.tb00601.x. [DOI] [PubMed] [Google Scholar]
- 23.Olivares C, Jiménez-Cervantes C, Lozano JA, Solano F, García-Borrón JC. The 5,6-dihydroxyindole-2-carboxylic acid (DHICA) oxidase activity of human tyrosinase. Biochemical Journal. 2001;354(1):131–139. doi: 10.1042/0264-6021:3540131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manga P, Sato K, Ye L, Beermann F, Lynn Lamoreux M, Orlow SJ. Mutational analysis of the modulation of tyrosinase by tyrosinase-related proteins 1 and 2 in vitro. Pigment Cell Research. 2000;13(5):364–374. doi: 10.1034/j.1600-0749.2000.130510.x. [DOI] [PubMed] [Google Scholar]
- 25.Carvalho MA, Marsillac SM, Karchin R, et al. Determination of cancer risk associated with germ line BRCA1 missense variants by functional analysis. Cancer Research. 2007;67(4):1494–1501. doi: 10.1158/0008-5472.CAN-06-3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carvalho M, Pino MA, Karchin R, et al. Analysis of a set of missense, frameshift, and in-frame deletion variants of BRCA1. Mutation Research. 2009;660(1-2):1–11. doi: 10.1016/j.mrfmmm.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldgar DE, Easton DF, Deffenbaugh AM, Monteiro ANA, Tavtigian SV, Couch FJ. Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. American Journal of Human Genetics. 2004;75(4):535–544. doi: 10.1086/424388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karchin R. Next generation tools for the annotation of human SNPs. Briefings in Bioinformatics. 2009;10(1):35–52. doi: 10.1093/bib/bbn047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Balu K, Purohit R. Mutational analysis of TYR gene and its structural consequences in OCA1A. Gene. 2013;513(1):184–195. doi: 10.1016/j.gene.2012.09.128. [DOI] [PubMed] [Google Scholar]
- 30.Purohit R, Rajendran V, Sethumadhavan R. Relationship between mutation of serine residue at 315th position in M. tuberculosis catalase-peroxidase enzyme and Isoniazid susceptibility: an in silico analysis. Journal of Molecular Modeling. 2011;17(4):869–877. doi: 10.1007/s00894-010-0785-6. [DOI] [PubMed] [Google Scholar]
- 31.Purohit R, Rajendran V, Sethumadhavan R. Studies on adaptability of binding residues and flap region of TMC-114 resistance HIV-1 protease mutants. Journal of Biomolecular Structure and Dynamics. 2011;29(1):137–152. doi: 10.1080/07391102.2011.10507379. [DOI] [PubMed] [Google Scholar]
- 32.Kumar A, Rajendran V, Sethumadhavan R, Purohit R. In silico prediction of a disease-associated STIL mutant and its affect on the recruitment of centromere protein J (CENPJ) FEBS Open Biology. 2012;2:285–293. doi: 10.1016/j.fob.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar A, Rajendran V, Sethumadhavan R, Purohit R. Relationship between a point mutation S97C in CK1δ protein and its affect on ATP-binding affinity. Journal of Biomolecular Structure & Dynamics. 2013 doi: 10.1080/07391102.2013.770373. [DOI] [PubMed] [Google Scholar]
- 34.Rajendran V, Sethumadhavan R. Drug resistance mechanism of PncA in Mycobacterium tuberculosis . Journal of Biomolecular Structure and Dynamics. 2013 doi: 10.1080/07391102.2012.759885. [DOI] [PubMed] [Google Scholar]
- 35.Rajendran V, Purohit R, Sethumadhavan R. In silico investigation of molecular mechanism of laminopathy caused by a point mutation (R482W) in lamin A/C protein. Amino Acids. 2012;43(2):603–615. doi: 10.1007/s00726-011-1108-7. [DOI] [PubMed] [Google Scholar]
- 36.Balu K, Purohit R. In-silico analysis of Betaine Aldehyde Dehydrogenase2 of Oryza sativa and significant mutations responsible for fragrance. Journal of Plant Interactions. 2012:1–13. [Google Scholar]
- 37.Balu K, Rajendran V, Sethumadhavan R, Purohit R. Investigation of binding phenomenon of NSP3 and p130Cas mutants and their effect on cell signalling. Cell Biochemistry and Biophysics. 2013:1–11. doi: 10.1007/s12013-013-9551-6. [DOI] [PubMed] [Google Scholar]
- 38.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nature Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature Protocols. 2009;4(7):1073–1082. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 40.Capriotti E, Fariselli P, Rossi I, Casadio R. A three-state prediction of single point mutations on protein stability changes. BMC Bioinformatics. 2008;9(2, article S6) doi: 10.1186/1471-2105-9-S2-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas PD, Campbell MJ, Kejariwal A, et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Research. 2003;13(9):2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Capriotti E, Calabrese R, Casadio R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics. 2006;22(22):2729–2734. doi: 10.1093/bioinformatics/btl423. [DOI] [PubMed] [Google Scholar]
- 43.Calabrese R, Capriotti E, Fariselli P, Martelli PL, Casadio R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Human Mutation. 2009;30(8):1237–1244. doi: 10.1002/humu.21047. [DOI] [PubMed] [Google Scholar]
- 44.Ferrer-Costa C, Gelpí JL, Zamakola L, Parraga I, de la Cruz X, Orozco M. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005;21(14):3176–3178. doi: 10.1093/bioinformatics/bti486. [DOI] [PubMed] [Google Scholar]
- 45.Li B, Krishnan VG, Mort ME, et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 2009;25(21):2744–2750. doi: 10.1093/bioinformatics/btp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amberger J, Bocchini CA, Scott AF, Hamosh A. McKusick’s Online Mendelian Inheritance in Man (OMIM®) Nucleic Acids Research. 2009;37(1):D793–D796. doi: 10.1093/nar/gkn665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sherry ST, Ward M-H, Kholodov M, et al. DbSNP: the NCBI database of genetic variation. Nucleic Acids Research. 2001;29(1):308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yip YL, Scheib H, Diemand AV, et al. The Swiss-Prot variant page and the ModSNP database: a resource for sequence and structure information on human protein variants. Human Mutation. 2004;23(5):464–470. doi: 10.1002/humu.20021. [DOI] [PubMed] [Google Scholar]
- 49.Yip YL, Famiglietti M, Gos A, et al. Annotating single amino acid polymorphisms in the UniProt/Swiss-Prot knowledgebase. Human Mutation. 2008;29(3):361–366. doi: 10.1002/humu.20671. [DOI] [PubMed] [Google Scholar]
- 50.Boeckmann B, Bairoch A, Apweiler R, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Research. 2003;31(1):365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaplan W, Littlejohn TG. Swiss-PDB viewer (Deep View) Briefings in Bioinformatics. 2001;2(2):195–197. doi: 10.1093/bib/2.2.195. [DOI] [PubMed] [Google Scholar]
- 52.Hess B, Kutzner C, van der Spoel D, Lindahl E. GRGMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. Journal of Chemical Theory and Computation. 2008;4(3):435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9, article 40 doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. Journal of Biomolecular NMR. 1996;8(4):477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 55.Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Research. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berendsen HJC, Postma JPM, van Gunsteren WF, Dinola A, Haak JR. Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics. 1984;81(8):3684–3690. [Google Scholar]
- 57.Cheatham TE, III, Miller JL, Fox T, Darden TA, Kollman PA. Molecular dynamics simulations on solvated biomolecular systems: the particle mesh Ewald method leads to stable trajectories of DNA, RNA, and proteins. Journal of the American Chemical Society. 1995;117(14):4193–4194. [Google Scholar]
- 58.Turner PJ. XMGRACE, Version 5.1.19. Beaverton, Ore, USA: Center For Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology; 2005. [Google Scholar]
- 59.Amadei A, Linssen ABM, Berendsen HJC. Essential dynamics of proteins. Proteins: Structure, Function and Genetics. 1993;17(4):412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- 60.Thusberg J, Vihinen M. Pathogenic or not? and if so, then how? Studying the effects of missense mutations using bioinformatics methods. Human Mutation. 2009;30(5):703–714. doi: 10.1002/humu.20938. [DOI] [PubMed] [Google Scholar]
- 61.Hicks S, Wheeler DA, Plon SE, Kimmel M. Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Human Mutation. 2011;32(6):661–668. doi: 10.1002/humu.21490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang K-H, Li Z, Lei J, et al. Oculocutaneous Albinism type 3 (OCA3): analysis of two novel mutations in TYRP1 gene in two Chinese patients. Cell Biochemistry and Biophysics. 2011;61(3):523–529. doi: 10.1007/s12013-011-9234-0. [DOI] [PubMed] [Google Scholar]
- 63.Livesay DR, Dallakyan S, Wood GG, Jacobs DJ. A flexible approach for understanding protein stability. FEBS Letters. 2004;576(3):468–476. doi: 10.1016/j.febslet.2004.09.057. [DOI] [PubMed] [Google Scholar]
- 64.Verma D, Jacobs DJ, Livesay DR. Changes in lysozyme flexibility upon mutation are frequent, large and long-ranged. PLoS Computational Biology. 2012;8(3) doi: 10.1371/journal.pcbi.1002409.100240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ribeiro AA, de Alencastro RB. Mixed Monte Carlo/molecular dynamics simulations of the prion protein. Journal of Molecular Graphics and Modelling. 2013;42:1–6. doi: 10.1016/j.jmgm.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 66.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22(12):2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 67.Simeonov DR, Wang X, Wang C, et al. DNA variations in oculocutaneous Albinism: an updated mutation List and current outstanding issues in molecular diagnostics. Human Mutation. 2013;34(6):827–835. doi: 10.1002/humu.22315. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The consequences upon mutations were clearly observed in the atomic density distribution plot. There was a significant change in density distribution in mutant as compared to the native. Native structure shows highest atomic density distribution of 41.9 nm−3 but in mutant (R326H & R356Q) structures showed 52.2 and 52.3 nm−3 respectively. It was further indicate that native has more flexibility than mutant (R326H & R356Q) structures.