

Abstract
We have investigated the molecular geometries of a series of dicoordinated d10-transition-metal complexes ML2 (M=Co−, Rh−, Ir−, Ni, Pd, Pt, Cu+, Ag+, Au+; L=NH3, PH3, CO) using relativistic density functional theory (DFT) at ZORA-BLYP/TZ2P. Not all complexes have the expected linear ligand–metal–ligand (L–M–L) angle: this angle varies from 180° to 128.6° as a function of the metal as well as the ligands. Our main objective is to present a detailed explanation why ML2 complexes can become bent. To this end, we have analyzed the bonding mechanism in ML2 as a function of the L–M–L angle using quantitative Kohn–Sham molecular orbital (MO) theory in combination with an energy decomposition analysis (EDA) scheme. The origin of bent L–M–L structures is π backdonation. In situations of strong π backdonation, smaller angles increase the overlap of the ligand’s acceptor orbital with a higher-energy donor orbital on the metal-ligand fragment, and therefore favor π backdonation, resulting in additional stabilization. The angle of the complexes thus depends on the balance between this additional stabilization and increased steric repulsion that occurs as the complexes are bent.
Keywords: bond theory, density functional calculations, energy decomposition analysis, molecular geometry, transition-metal complexes, π backdonation
Introduction
Dicoordinated d10-transition-metal complexes ML2 occur in numerous catalytic reaction mechanisms.1 These complexes, in general, have a linear geometry2–5 with a ligand–metal–ligand (L–M–L′) angle (or bite angle) of 180°, although exceptions6, 7 have been observed. This geometrical preference can be easily understood for a closed-shell d10 configuration. In most cases, the dominant bonding orbital interaction is σ donation from the ligand’s lone-pair orbitals into the empty metal (n+1)s atomic orbital (AO), which has a ligand–metal bond overlap that is independent of the L–M–L′ angle (see Figure 1).8 At the same time, the steric repulsion associated with a L⋅⋅⋅L′ overlap between the lone pairs (and other closed shells) of the two ligands yields a force that maximizes their mutual distance and thus yields the well-known linear L–M–L′ arrangement.
Figure 1.
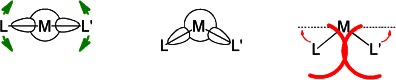
σ Donation has no preference (left, middle) whereas sterics favor linear L–M–L (right).
The same conclusion is obtained if one uses valence shell electron pair repulsion (VSEPR) theory adapted for treating transition-metal complexes,9, 10 or more sophisticated methods based on molecular orbital (MO) theory. Proceeding from the latter, one can deduce the preference for linear over bent ML2 complexes from the number of electrons in the valence orbitals and the dependence of the orbital energies on the geometrical parameter of interest (here, the L–M–L angle) in Walsh diagrams.8 These diagrams show again that dicoordinate d10-transition-metal complexes, for example, Ag(NH3)2+, adopt a linear geometry due to the significant destabilization of the metal dxz AO by the ligand’s lone-pair orbitals in combination with steric repulsion between the latter upon bending (see below). Nearly all instances with substantial deviations of the L–M–L bite angle from linearity are complexes in which this distortion is imposed by the structural constraints in bidentate ligands in which a bridge or scaffold forces the two coordinating centers L towards each other.1b–d
In this work, we show that d10-ML2 complexes are not necessarily linear and may even have a pronounced intrinsic preference to adopt a nonlinear equilibrium geometry. To this end, we have investigated the molecular geometries and electronic structure of a series of d10-ML2 complexes (M=Co−, Rh−, Ir−, Ni, Pd, Pt, Cu+, Ag+, Au+; L=NH3, PH3, CO) using relativistic density functional theory (DFT). Simple d10-ML2 complexes are found with substantial deviations from linearity, featuring bite angles as small as 131° or even less. All that is necessary for bent d10-ML2 complexes to occur is sufficiently strong π backdonation. This emerges from our detailed metal–ligand bonding analyses in the conceptual framework of quantitative MO theory contained in Kohn–Sham DFT. The analyses explain the phenomenon and provide a tool for rationally tuning the bite angle. Based on our analyses, we can augment the text-book Walsh diagram for bending ML2 complexes involving only σ donation with an extended Walsh diagram that also includes π backbonding.
Theoretical Methods
Computational details: All calculations were carried out using the Amsterdam Density Functional (ADF) program developed by Baerends and co-workers11–13 The numerical integration was performed using the procedure developed by te Velde et al.14 The molecular orbitals (MOs) were expanded in a large uncontracted set of Slater-type orbitals (STOs): TZ2P (no Gaussian functions are involved). The TZ2P basis set15 is of triple-ζ quality for all atoms and has been augmented with two sets of polarization functions, that is, 2p and 3d on H, 3d and 4f on C, N, O and P, 4p and 4f on Co, Ni, Cu, 5p and 4f on Rh, Pd and Ag and 6p and 5f on Ir, Pt and Au. An auxiliary set of s, p, d, f and g STOs was used to fit the molecular density and to represent the Coulomb and exchange potentials accurately in each self-consistent field (SCF) cycle. All electrons are included in the variational treatment (no frozen-core approximation used).
Equilibrium structures were obtained by optimizations using analytical gradient techniques.16 Geometries and energies were calculated at the BLYP level of the generalized gradient approximation (GGA): exchange is described by Slater’s Xα potential,17 with nonlocal corrections due to Becke18 added self-consistently, and correlation is treated using the gradient-corrected functional of Lee, Yang and Parr.19 Scalar relativistic effects were accounted for using the zeroth-order regular approximation (ZORA).20 This approach has been extensively tested and was shown to agree well with high-level coupled-cluster reference data.21 Energy minima have been verified through vibrational analysis.22 All minima were found to have zero imaginary frequencies. The PyFrag program was used to facilitate the analyses of the bonding mechanism as a function of the L–M–L angle.23
Bond energy analysis: The bond energy ΔE is decomposed into the strain energy ΔEstrain, that is associated with the geometrical deformation of the fragments as the bond formation takes place, plus the actual interaction energy ΔEint between the deformed fragments [Equation (1)].
| (1) |
The interaction energy ΔEint(ζ) between two molecular fragments is analyzed as a function of the bite angle ζ in the conceptual framework provided by the Kohn–Sham MO method.24 To this end, it is decomposed in three physically meaningful terms [Eq. (2)] using a quantitative energy decomposition scheme developed by Ziegler and Rauk.25
| (2) |
The term ΔVelstat corresponds to the classical electrostatic interaction between the unperturbed charge distributions ρA(r)+ρB(r) of the prepared or deformed fragments A and B (see below for definition of the fragments) that adopt their positions in the overall molecule AB, and is usually attractive. The Pauli repulsion term ΔEPauli comprises the destabilizing interactions between occupied orbitals and is responsible for the steric repulsion. This repulsion is caused by the fact that two electrons with the same spin cannot occupy the same region in space. It arises as the energy change associated with the transition from the superposition of the unperturbed electron densities ρA(r)+ρB(r) of the geometrically deformed but isolated fragments A and B, to the wavefunction Ψ0=N Â [ΨA ΨB], that properly obeys the Pauli principle through explicit antisymmetrization (Â operator) and renormalization (N constant) of the product of fragment wavefunctions (see Ref. 24 for an exhaustive discussion). The orbital interaction ΔEoi accounts for charge transfer (interaction between occupied orbitals on one fragment with unoccupied orbitals on the other fragment, including the HOMO–LUMO interactions) and polarization (empty-occupied orbital mixing on one fragment due to the presence of another fragment). It can be further divided into contributions from each irreducible representation Γ of the interacting system [Eq. (3)].
| (3) |
Results and Discussion
Structure and energetics
Structural and energetic data emerging from our ZORA-BLYP/TZ2P computations are collected in Tables 1–4. Most ML2 complexes have a linear L–M–L angle, which leads to either D3h-symmetric complexes M(NH3)2 and M(PH3)2 or D∞h-symmetric complexes M(CO)2. However, numerous significantly smaller angles appear throughout Table 1 as well, where the symmetry of the complexes is lowered to C2v. For instance, the complexes become increasingly bent when the ligands are varied along NH3 (a strong σ donor), PH3 (a σ donor and π acceptor) and CO (a strong π acceptor). This is most clearly seen for the group 9 complexes, where, for example, the angle decreases along Rh(NH3)2−, Rh(PH3)2− and Rh(CO)2− from 180.0° to 141.2° and 130.8° (Figure 2). In a later section, we will show that the π-backbonding properties of the complexes constitute a prominent part of the explanation of why d10-ML2 complexes can adopt nonlinear geometries. The increasingly strong π backbonding along this series also results in stronger metal–ligand bonds (see Table 2 for bond dissociation energies (BDEs) and Table 3 for energy decomposition analyses (EDA) results for ML complexes).
Table 1.
L–M–L angle [°] and linearization energy ΔElin [kcal mol−1] in dicoordinate d10-ML2 complexes[a]
| Group 9 | Group 10 | Group 11 | ||||||
|---|---|---|---|---|---|---|---|---|
| L–M–L | ΔElin[b] | L–M–L | ΔElin[b] | L–M–L | ΔElin[b] | |||
| Co(NH3)2− | 180.0 | 0 | Ni(NH3)2 | 180.0 | 0 | Cu(NH3)2+ | 180.0 | 0 |
| Co(PH3)2− | 131.8 | 6.4 | Ni(PH3)2 | 180.0 | 0 | Cu(PH3)2+ | 180.0 | 0 |
| Co(CO)2− | 128.6 | 19.9 | Ni(CO)2 | 144.5 | 2.1 | Cu(CO)2+ | 180.0 | 0 |
| Rh(NH3)2− | 180.0 | 0 | Pd(NH3)2 | 180.0 | 0 | Ag(NH3)2+ | 180.0 | 0 |
| Rh(PH3)2− | 141.2 | 2.0 | Pd(PH3)2 | 180.0 | 0 | Ag(PH3)2+ | 180.0 | 0 |
| Rh(CO)2− | 130.8 | 10.2 | Pd(CO)2 | 155.6 | 0.5 | Ag(CO)2+ | 180.0 | 0 |
| Ir(NH3)2− | 180.0 | 0 | Pt(NH3)2 | 180.0 | 0 | Au(NH3)2+ | 180.0 | 0 |
| Ir(PH3)2− | 144.1 | 2.4 | Pt(PH3)2 | 180.0 | 0 | Au(PH3)2+ | 180.0 | 0 |
| Ir(CO)2− | 134.2 | 13.4 | Pt(CO)2 | 159.0 | 0.6 | Au(CO)2+ | 180.0 | 0 |
[a] Computed at ZORA-BLYP/TZ2P. [b] Relative energy of the linear ML2 complex relative to its equilibrium geometry.
Table 4.
Ligand orbital energies ε [eV] and proton affinities [kcal mol−1][a]
| ε(LP) | ε(π*) | PA | |
|---|---|---|---|
| NH3 | −6.05 | +1.42 | +201.4 |
| PH3 | −6.63 | −0.24 | +185.2 |
| CO | −8.93 | −1.92 | +141.5 |
[a] Computed at ZORA-BLYP/TZ2P. LP: lone pair, π*: acceptor orbital. Proton affinities (PA) from enthalpies at 298.15 K and 1 atm.
Figure 2.
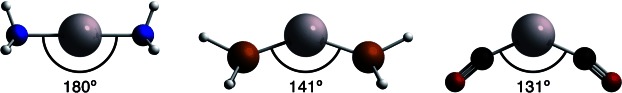
Equilibrium geometries computed at ZORA-BLYP/TZ2P. From left to right: Rh(NH3)2−, Rh(PH3)2− and Rh(CO)2−.
Table 2.
M–L bond length [Å] and BDE [kcal mol−1] in monocoordinate d10-ML and dicoordinate d10-ML2 complexes[a]
| M–L | BDE | M–L | BDE | M–L | BDE | |||
|---|---|---|---|---|---|---|---|---|
| CoNH3−[b,c] | 1.845 | 217.1 | NiNH3[c] | 1.827 | 77.0 | CuNH3+ | 1.911 | 70.0 |
| CoPH3−[b,c] | 1.971 | 240.6 | NiPH3[c] | 1.979 | 88.0 | CuPH3+ | 2.163 | 68.7 |
| CoCO−[b,c] | 1.630 | 280.6 | NiCO[c] | 1.663 | 109.3 | CuCO+ | 1.833 | 50.2 |
| Co(NH3)2−[b,c] | 1.908 | 24.0 | Ni(NH3)2[c] | 1.888 | 36.2 | Cu(NH3)2+ | 1.919 | 61.1 |
| Co(PH3)2−[c] | 2.051 | 48.2 | Ni(PH3)2[c] | 2.108 | 36.3 | Cu(PH3)2+ | 2.232 | 48.0 |
| Co(CO)2−[c] | 1.715 | 76.3 | Ni(CO)2[c] | 1.765 | 48.6 | Cu(CO)2+ | 1.882 | 45.0 |
| RhNH3−[c] | 2.001 | 55.5 | PdNH3 | 2.115 | 21.6 | AgNH3+ | 2.212 | 48.7 |
| RhPH3−[c] | 2.068 | 89.9 | PdPH3 | 2.172 | 39.4 | AgPH3+ | 2.415 | 47.9 |
| RhCO−[c] | 1.750 | 122.0 | PdCO | 1.861 | 47.4 | AgCO+ | 2.137 | 28.4 |
| Rh(NH3)2−[c] | 2.089 | 22.6 | Pd(NH3)2 | 2.106 | 28.6 | Ag(NH3)2+ | 2.172 | 45.2 |
| Rh(PH3)2−[c] | 2.196 | 38.2 | Pd(PH3)2 | 2.287 | 28.6 | Ag(PH3)2+ | 2.444 | 38.1 |
| Rh(CO)2−[c] | 1.866 | 58.1 | Pd(CO)2 | 1.949 | 34.7 | Ag(CO)2+ | 2.113 | 30.7 |
| IrNH3−[c] | 1.967 | 85.0 | PtNH3[c] | 1.981 | 50.1 | AuNH3+ | 2.085 | 71.4 |
| IrPH3−[c] | 2.056 | 126.5 | PtPH3[c] | 2.095 | 77.3 | AuPH3+ | 2.240 | 84.2 |
| IrCO−[c] | 1.734 | 166.3 | PtCO[c] | 1.776 | 87.9 | AuCO+ | 1.927 | 55.0 |
| Ir(NH3)2−[b,c] | 2.071 | 23.6 | Pt(NH3)2[c] | 2.061 | 41.6 | Au(NH3)2+ | 2.088 | 64.6 |
| Ir(PH3)2−[c] | 2.190 | 44.1 | Pt(PH3)2[c] | 2.249 | 38.7 | Au(PH3)2+ | 2.351 | 52.6 |
| Ir(CO)2−[c] | 1.854 | 66.3 | Pt(CO)2[c] | 1.911 | 47.1 | Au(CO)2+ | 2.002 | 40.4 |
[a] Computed at ZORA-BLYP/TZ2P. Bond dissociation energies (BDEs) are given for the complexes in the electronic configuration corresponding to a d10s0 electron configuration and relative to closed-shell d10s0 metal atoms. [b] The d10s0-type configuration is an excited state of the complex. [c] The d10s0 configuration is an excited state of the atomic metal fragment.
Table 3.
Energy decomposition analyses [kcal mol−1] and orbital energies ε [eV] for the metal–ligand bonds in monoligated transition-metal complexes M–L[a]
| ML | ΔE | ΔEint | ΔVelstat | ΔEPauli | ΔEoi | Δ
|
Δ [b] [b]
|
ε[dσ] | ε[dπ] | ε[dδ] |
|---|---|---|---|---|---|---|---|---|---|---|
| CoNH3− | −217.1 | −218.4 | −110.0 | 166.3 | −274.7 | −241.8 | −32.9 | +1.84 | +2.91 | +3.99 |
| CoPH3− | −240.6 | −241.7 | -197.9 | 204.5 | −248.2 | −123.9 | −124.4 | +1.67 | +1.81 | +3.38 |
| CoCO− | −280.6 | −286.4 | −233.4 | 274.5 | −327.5 | −141.7 | −185.8 | +1.34 | +1.17 | +3.20 |
| RhNH3− | −55.5 | −56.2 | −143.2 | 202.1 | −115.1 | −110.8 | −4.3 | +1.72 | +1.83 | +2.53 |
| RhPH3− | −89.9 | −90.3 | −269.7 | 311.7 | −132.3 | −61.7 | −70.6 | +1.49 | +0.91 | +2.20 |
| RhCO− | −122.0 | −126.0 | −273.3 | 364.1 | −216.8 | −96.7 | −120.1 | +1.05 | −0.09 | +1.56 |
| IrNH3− | −85.0 | −85.8 | −196.9 | 268.9 | −157.8 | −142.9 | −14.9 | +1.54 | +2.16 | +2.91 |
| IrPH3− | −126.5 | −127.2 | −349.2 | 396.0 | −174.1 | −85.9 | −88.2 | +1.18 | +0.73 | +2.28 |
| IrCO− | −166.3 | −171.3 | −353.5 | 461.5 | −279.2 | −129.6 | −149.7 | +0.63 | −0.26 | +1.68 |
| NiNH3 | −77.0 | −77.3 | −116.2 | 139.8 | −100.8 | −94.5 | −6.3 | −3.28 | −2.99 | −2.21 |
| NiPH3 | −88.0 | −88.7 | −161.3 | 173.3 | −100.7 | −50.8 | −49.9 | −3.79 | −3.93 | −2.90 |
| NiCO | −109.3 | −110.4 | −171.6 | 210.3 | −149.1 | −60.4 | −88.7 | −4.89 | −5.40 | −4.14 |
| PdNH3 | −21.6 | −21.7 | −88.0 | 105.1 | −38.8 | −34.5 | −4.4 | −3.46 | −3.81 | −3.47 |
| PdPH3 | −39.4 | −39.8 | −166.2 | 190.3 | −63.8 | −35.3 | −28.5 | −4.49 | −5.29 | −4.56 |
| PdCO | −47.4 | −47.8 | −161.4 | 213.3 | −99.7 | −48.0 | −51.8 | −5.28 | −6.48 | −5.53 |
| PtNH3 | −50.1 | −50.4 | −170.1 | 211.4 | −91.7 | −82.0 | −9.7 | −4.19 | −4.46 | −3.72 |
| PtPH3 | −77.3 | −78.9 | −273.9 | 310.3 | −115.3 | −70.5 | −44.8 | −4.92 | −5.72 | −4.53 |
| PtCO | −87.9 | −88.7 | −271.6 | 356.9 | −174.0 | −91.6 | −82.4 | −5.97 | −7.28 | −5.77 |
| CuNH3+ | −70.0 | −70.1 | −104.5 | 86.0 | −51.7 | −41.9 | −9.8 | −11.80 | −12.13 | −12.02 |
| CuPH3+ | −68.7 | −73.5 | −101.7 | 94.0 | −65.8 | −51.8 | −14.0 | −11.99 | −12.44 | −12.15 |
| CuCO+ | −50.2 | −50.3 | −89.8 | 100.7 | −61.2 | −38.8 | −22.4 | −13.7 | −14.28 | −13.90 |
| AgNH3+ | −48.7 | −48.7 | −73.3 | 58.8 | −34.2 | −28.5 | −5.8 | −12.56 | −13.60 | −13.57 |
| AgPH3+ | −47.9 | −51.8 | −84.3 | 81.3 | −48.8 | −39.9 | −8.9 | −12.41 | −13.67 | −13.85 |
| AgCO+ | −28.4 | −28.6 | −59.1 | 67.2 | −36.7 | −26.2 | −10.6 | −14.08 | −15.07 | −14.86 |
| AuNH3+ | −71.4 | −71.6 | −124.8 | 123.2 | −70.0 | −60.3 | −9.7 | −12.49 | −13.32 | −12.92 |
| AuPH3+ | −84.2 | −91.0 | −177.9 | 187.2 | −100.3 | −80.9 | −19.4 | −12.52 | −13.70 | −13.06 |
| AuCO+ | −55.0 | −55.1 | −149.0 | 188.4 | −94.5 | −64.9 | −29.7 | −14.20 | −15.53 | −14.73 |
[a] Computed at ZORA-BLYP/TZ2P. See [Eqs. (1)–(3)]. [b] Also includes small contributions from δ orbital interactions, which can only be separated for C∞v-symmetric MCO complexes. There, the δ term amounts at most to 3.5 % of the π term.
The extent of bending systematically decreases when the π-backbonding capability of the metal center decreases from the group 9 anions, via neutral group 10 atoms, to the group 11 cations. This is clearly displayed by the series of isoelectronic complexes Rh(CO)2−, Pd(CO)2 and Ag(CO)2+ along which the L–M–L angle increases from 130.8° to 155.6° to 180° (Table 1). The data in Table 3 for the corresponding monocoordinate RhCO−, PdCO and AgCO+ nicely show how along this series the distortive π-orbital interactions Δ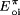 indeed become weaker, from −120 to −51 to −11 kcal mol−1, respectively. In the case of group 9 metals, both phosphine and carbonyl complexes are bent, whereas, for group 10 metals, only the carbonyl complexes deviate from linearity. Complexes with a metal center from group 11 all have a linear L–M–L configuration. The reduced π backbonding also leads to weaker metal–ligand bonds. For the cationic metal centers, for which π backdonation plays a much smaller role, the metal–ligand BDEs decrease in the order NH3>PH3>CO (see Table 2). This trend originates directly from the σ-donating capabilities of the ligands as reflected by the energy of the lone-pair orbital ε(LP), which decreases in this order (see Table 4). Note that, for the same reason, the basicity of the ligand as measured by the proton affinity (PA) decreases along NH3>PH3>CO.26 For the anionic group 9 metal centers, the opposite order is found, that is, metal–ligand BDEs decrease in the order CO>PH3>NH3, following the π-accepting capabilities of the ligands.
indeed become weaker, from −120 to −51 to −11 kcal mol−1, respectively. In the case of group 9 metals, both phosphine and carbonyl complexes are bent, whereas, for group 10 metals, only the carbonyl complexes deviate from linearity. Complexes with a metal center from group 11 all have a linear L–M–L configuration. The reduced π backbonding also leads to weaker metal–ligand bonds. For the cationic metal centers, for which π backdonation plays a much smaller role, the metal–ligand BDEs decrease in the order NH3>PH3>CO (see Table 2). This trend originates directly from the σ-donating capabilities of the ligands as reflected by the energy of the lone-pair orbital ε(LP), which decreases in this order (see Table 4). Note that, for the same reason, the basicity of the ligand as measured by the proton affinity (PA) decreases along NH3>PH3>CO.26 For the anionic group 9 metal centers, the opposite order is found, that is, metal–ligand BDEs decrease in the order CO>PH3>NH3, following the π-accepting capabilities of the ligands.
Linearity also increases if one descends in a group. For example, from Ni(CO)2 to Pd(CO)2 to Pt(CO)2, the L–M–L angle increases from 144.5° to 155.6° to 159.0°. Interestingly, this last trend is opposite to what one would expect proceeding from a steric model. If one goes from a larger to a smaller metal center, that is, going up in a group, the ligands are closer to each other and thus experience stronger mutual steric repulsion. But instead of becoming more linear to avoid such repulsion, the complexes bend even further in the case of the smaller metal. For example, when the palladium atom in Pd(CO)2 is replaced by a smaller nickel atom, the L–M–L angle decreases from 155.6° in Pd(CO)2 to 144.5° in Ni(CO)2. Later on, we show that this seemingly counterintuitive trend also originates from enhanced π backbonding which dominates the increased steric repulsion.
General bonding mechanism
The bending of our model complexes can be understood in terms of a monocoordinate complex to which a second ligand is added either in a linear or a bent arrangement, ML+L→ML2 (see below). Using Pd(CO)2 as an example, we start from a PdCO fragment, and consider the addition of the second CO ligand both at a 180° angle and a 90° angle. Our Kohn–Sham MO analyses show that, in PdCO, the degeneracy of the five occupied d orbitals on palladium is lowered by interactions with the ligand (see Figure 3). Choosing the M–L bond along the z axis, the dxz and dyz orbitals act as donor orbitals for π backdonation into the two π*-acceptor orbitals on the CO ligand, resulting in two stabilized “dπ” orbitals at −6.5 eV (value not shown in Figure 3). The dxy and d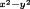 (or “dδ”) orbitals at −5.5 eV do not overlap and interact with the ligand. The d
(or “dδ”) orbitals at −5.5 eV do not overlap and interact with the ligand. The d orbital is destabilized due to the antibonding overlap with the lone pair on the ligand, resulting in a “dσ” orbital that is relatively high in energy, at −5.3 eV.
orbital is destabilized due to the antibonding overlap with the lone pair on the ligand, resulting in a “dσ” orbital that is relatively high in energy, at −5.3 eV.
Figure 3.
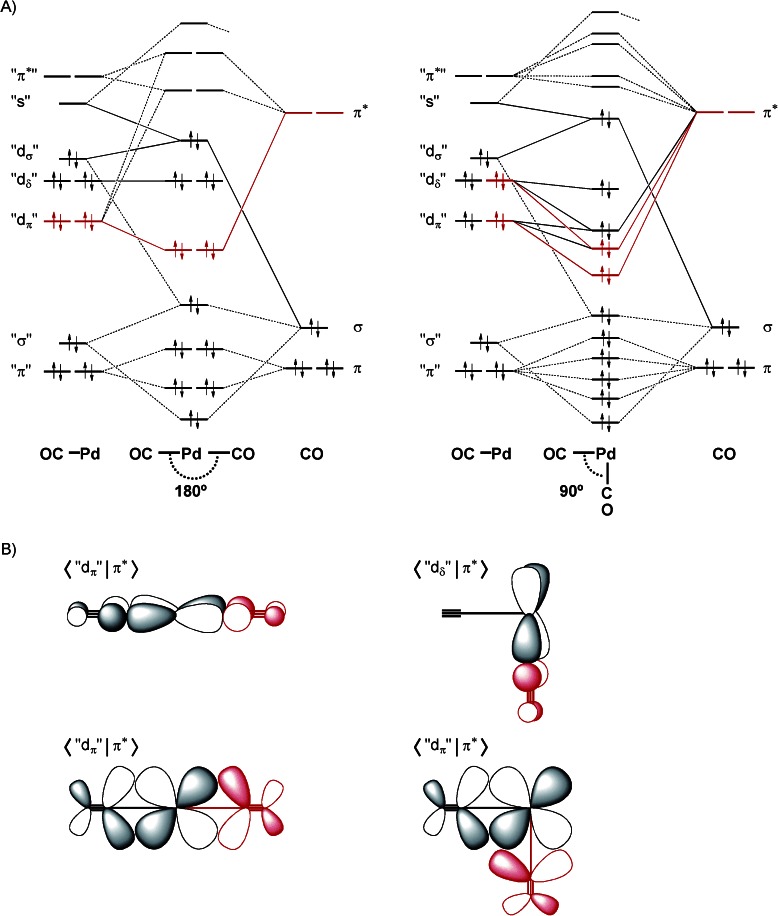
A) Schematic MO diagrams for the bonding mechanism between PdCO and CO in linear Pd(CO)2 (left) and at a L–M–L angle of 90° (right): dominant interactions (—), other interactions (- - - -), π backbonding (—). B) Schematic representation of the bonding overlaps of the donating orbital on PdCO (black) with the π-accepting orbital on the second CO ligand (red).
When the second CO ligand coordinates opposite the first one (i.e., in a linear L–M–L arrangement), its π*-acceptor orbitals interact with the dπ orbitals on the PdCO fragment. The latter are already considerably stabilized by π backdonation to the first CO ligand (Figure 3 B, left). When, instead, the second ligand is added at an angle of 90°, its π* orbitals overlap with only one dπ orbital, and with one dδ orbital (Figure 3 B, right). This dδ orbital is essentially a pure metal d orbital that has not yet been stabilized by any coordination bond. Consequently, this orbital has a higher energy and is, therefore, a more capable donor orbital for π backdonation into the π* orbital of the second CO ligand. This results in a stronger, more stabilizing donor–acceptor interaction of this pair of orbitals in the 90° (Figure 3 A, right) than in the 180° ML2 geometry (Figure 3 A, left: cf. red-highlighted π interactions). σ-Donation interactions are affected less by bending. It is therefore π backdonation that favors bending. The more detailed energy decomposition analyses in the following sections consolidate this picture.
Bonding mechanism: Variation of ligands
To understand the trends in nonlinearity of our ML2 complexes (see above and Table 1), we have quantitatively analyzed the metal–ligand bonding between ML and the second ligand L as a function of the L–M–L angle. The results are collected in Table 2 and displayed in Figure 4–7. Most of our model complexes have a d10-type ground-state configuration but not all of them, as indicated in detail in Table 2. Yet, all model systems discussed here have been kept in d10-configuration, to achieve a consistent comparison and because, on the longer term, we are interested in understanding more realistic dicoordinated d10-transition-metal complexes that feature, for example, as catalytically active species in metal-mediated bond activation. We start in all cases from the optimal linear ML2 structure (i.e., the complex optimized in either D∞h or D3h symmetry) and then analyze the bonding between ML and L′ as a function of the L–M–L angle, from 180° to 90°, while keeping all other geometry parameters frozen. The analyses were done in Cs symmetry, bending the complexes in the mirror plane, with the out-of-plane hydrogen atoms of M(NH3)2 and M(PH3)2 towards each other. Thus, we are able to separate the orbital interactions symmetric to the mirror plane (A′ irrep) from the orbital interactions asymmetric to the mirror plane (A″ irrep): ΔEoi(ζ)= 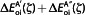 [Eq. (3)]. The use of frozen fragment geometries allows us to study purely how the interaction energy changes as the angle is varied, without any perturbation due to geometrical relaxation. Therefore, any change in ΔE stems exclusively from a change in ΔEint=ΔVelstat+ΔEPauli+Δ
[Eq. (3)]. The use of frozen fragment geometries allows us to study purely how the interaction energy changes as the angle is varied, without any perturbation due to geometrical relaxation. Therefore, any change in ΔE stems exclusively from a change in ΔEint=ΔVelstat+ΔEPauli+Δ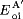 +Δ
+Δ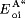 . Note that rigid bending of the linearly optimized L–M–L complexes causes minima on the energy profiles to shift to larger angles than in fully optimized complexes, but this does not alter any relative structural or energy order.
. Note that rigid bending of the linearly optimized L–M–L complexes causes minima on the energy profiles to shift to larger angles than in fully optimized complexes, but this does not alter any relative structural or energy order.
Figure 4.
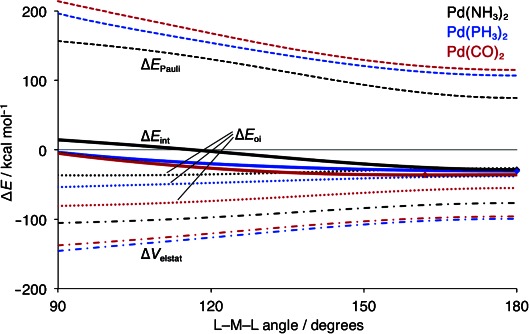
Energy decomposition analysis [Eq. (2)] of the interaction between PdL and L in dicoordinated palladium complexes PdL2 as a function of the L–M–L angle (L=NH3, PH3, CO).
Figure 7.
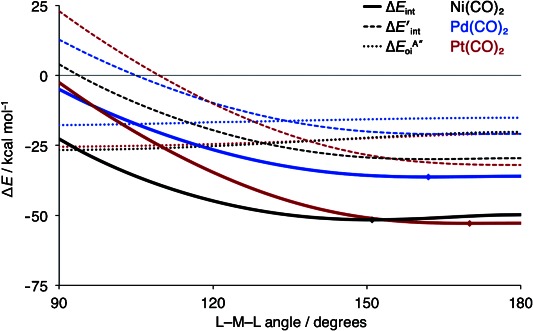
Energy decomposition analysis [Eq. (4)] of the interaction between MCO and CO in dicarbonyl-transition-metal complexes M(CO)2 as a function of the L–M–L angle (M=Ni, Pd, Pt).
In Figure 4, we show the energy decomposition analyses [Eq. (2)] and how they vary along the palladium complexes Pd(NH3)2, Pd(PH3)2 and Pd(CO)2. Upon bending the LM–L′ complex from 180° to 90°, the average distance between the electron density on LM and the nuclei of L′ decreases (the Pd–P distance however remains constant), which results in a more stabilizing electrostatic attraction ΔVelstat. Likewise, the Pauli repulsion ΔEPauli increases because of a larger overlap of the lone pair on L′ with the d -derived dσ orbital on the ML fragment. The latter is the antibonding combination of the metal d
-derived dσ orbital on the ML fragment. The latter is the antibonding combination of the metal d orbital and the ligand lone pair, with a fair amount of metal s character admixed in an L–M bonding fashion. The resulting hybrid orbital is essentially the d
orbital and the ligand lone pair, with a fair amount of metal s character admixed in an L–M bonding fashion. The resulting hybrid orbital is essentially the d orbital with a relatively large torus. The increase in Pauli repulsion that occurs as the L–M–L′ angle decreases stems largely from the overlap of the lone pair on the second ligand L′ with this torus. For Pd(CO)2 for example, the overlap of the L′ lone pair with the dσ hybrid orbital on ML increases from 0.05 to 0.28 upon bending from 180° to 90°. We note that this repulsion induces a secondary relaxation, showing up as a stabilizing Δ
orbital with a relatively large torus. The increase in Pauli repulsion that occurs as the L–M–L′ angle decreases stems largely from the overlap of the lone pair on the second ligand L′ with this torus. For Pd(CO)2 for example, the overlap of the L′ lone pair with the dσ hybrid orbital on ML increases from 0.05 to 0.28 upon bending from 180° to 90°. We note that this repulsion induces a secondary relaxation, showing up as a stabilizing Δ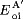 , by which it is largely canceled again. The mechanism through which this relief of Pauli repulsion happens is that, in the antibonding combination with the L′ lone pair, the dσ orbital is effectively pushed up in energy and (through its L′-lone-pair component) interacts in a stabilizing fashion with the metal s-derived LUMO on ML.
, by which it is largely canceled again. The mechanism through which this relief of Pauli repulsion happens is that, in the antibonding combination with the L′ lone pair, the dσ orbital is effectively pushed up in energy and (through its L′-lone-pair component) interacts in a stabilizing fashion with the metal s-derived LUMO on ML.
The aforementioned π backbonding that favors bending (see Figure 3) shows up in an increased stabilization in the asymmetric Δ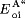 component as the L–M–L angle decreases. To more clearly reveal the role of the orbital interactions within A′′ symmetry, we separate the interaction energy ΔEint into the corresponding term
component as the L–M–L angle decreases. To more clearly reveal the role of the orbital interactions within A′′ symmetry, we separate the interaction energy ΔEint into the corresponding term 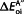 plus the remaining interaction energy ΔE′int, which combines the other interaction terms comprising electrostatic attraction ΔVelstat, Pauli repulsion ΔEPauli, and the symmetric orbital interactions Δ
plus the remaining interaction energy ΔE′int, which combines the other interaction terms comprising electrostatic attraction ΔVelstat, Pauli repulsion ΔEPauli, and the symmetric orbital interactions Δ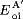 :
:
| (4) |
Thus, the interaction energy is split into two contributions which are both stabilizing along a large part of the energy profiles studied and which vary over a significantly smaller range. Therefore, this decomposition allows us to directly compare the importance of Δ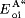 with respect to the combined influence of all other terms, contained in ΔE′int. The latter contains the aforementioned counteracting and largely canceling terms of strong Pauli repulsion between A′ orbitals and the resulting stabilizing relaxation effect Δ
with respect to the combined influence of all other terms, contained in ΔE′int. The latter contains the aforementioned counteracting and largely canceling terms of strong Pauli repulsion between A′ orbitals and the resulting stabilizing relaxation effect Δ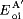 .
.
The results of this alternative decomposition appear in Figure 5, again for the series of palladium complexes Pd(NH3)2, Pd(PH3)2 and Pd(CO)2. In each of these complexes, bending begins at a certain point to weaken the ΔE′int energy term and, at smaller L–M–L angles, makes it eventually repulsive as the Pauli repulsion term becomes dominant (see also Figure 4). Numerical experiments, in which we consider the rigid bending process of a complex in which the metal is removed, show that steric repulsion between ligands does contribute to this repulsion, especially at smaller angles. Thus, direct Pauli repulsion between L and L′ in LM–L′ goes, upon bending from 180° to 90°, from 0.3 to 4.6 kcal mol−1 for Pd(NH3)2 and from 0.4 to 9.0 kcal mol−1 for Pd(CO)2 (data not shown in Figures). This finding confirms that ligands avoid each other for steric reasons, but it also shows that the effect is small as compared to the overall change in the ΔEint curves (see Figure 5). The dominant term that causes ΔEint to go up in energy upon bending is the increasing Pauli repulsion that occurs as the L′ lone pair overlaps more effectively with the LM dσ orbital.
Figure 5.
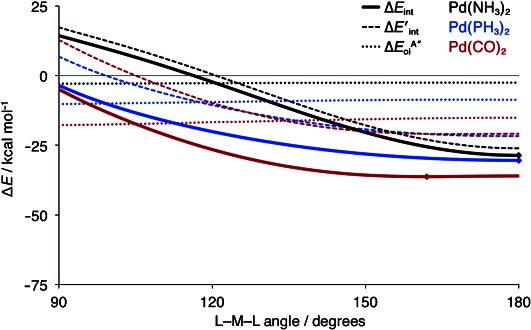
Energy decomposition analysis [Eq. (4)] of the interaction between PdL and L in dicoordinated palladium complexes PdL2 as a function of the L–M–L angle (L=NH3, PH3, CO).
In a number of cases, the stabilization upon bending from the asymmetric orbital interactions Δ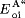 dominates the destabilization from the ΔE′int term. These cases are the complexes that adopt nonlinear equilibrium geometries. This
dominates the destabilization from the ΔE′int term. These cases are the complexes that adopt nonlinear equilibrium geometries. This 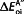 term gains stabilization upon bending LM–L′ because the π*-acceptor orbital on the ligand L′ moves from a position in which it can overlap with a ligand-stabilized LM dπ orbital to a more or less pure metal and, thus, up to 1 eV higher-energy dδ orbital (see Table 3), which leads to a more stabilizing donor–acceptor orbital interaction (see Figure 5). The gain in stabilization of Δ
term gains stabilization upon bending LM–L′ because the π*-acceptor orbital on the ligand L′ moves from a position in which it can overlap with a ligand-stabilized LM dπ orbital to a more or less pure metal and, thus, up to 1 eV higher-energy dδ orbital (see Table 3), which leads to a more stabilizing donor–acceptor orbital interaction (see Figure 5). The gain in stabilization of Δ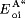 upon bending and, thus, the tendency to bend increases along NH3 to PH3 to CO. The reason is the increasing π-accepting ability of the ligands as reflected by the energy ε(π*) of the ligands′ π* orbital which is lowered from +1.42 to −0.24 to −1.92 eV, respectively (see Table 4). Thus, for Pd(NH3)2, where π backdonation plays essentially no role, the
upon bending and, thus, the tendency to bend increases along NH3 to PH3 to CO. The reason is the increasing π-accepting ability of the ligands as reflected by the energy ε(π*) of the ligands′ π* orbital which is lowered from +1.42 to −0.24 to −1.92 eV, respectively (see Table 4). Thus, for Pd(NH3)2, where π backdonation plays essentially no role, the 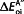 term is stabilized by less than 0.5 kcal mol−1 if we go from 180° to 90°. For PH3, known as a moderate π-accepting ligand, this energy term is stabilized by 1.5 kcal mol−1 from 180° to 90° and, for CO, this stabilization amounts to 2.5 kcal mol−1. Thus, in the case of palladium complexes, the energy profile for bending the complexes becomes progressively more flat as the ligands are better π acceptors, but only the carbonyl ligand generates sufficient stabilization through increased π-backbonding in Δ
term is stabilized by less than 0.5 kcal mol−1 if we go from 180° to 90°. For PH3, known as a moderate π-accepting ligand, this energy term is stabilized by 1.5 kcal mol−1 from 180° to 90° and, for CO, this stabilization amounts to 2.5 kcal mol−1. Thus, in the case of palladium complexes, the energy profile for bending the complexes becomes progressively more flat as the ligands are better π acceptors, but only the carbonyl ligand generates sufficient stabilization through increased π-backbonding in Δ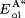 to shift the equilibrium geometry to an angle smaller than 180°.
to shift the equilibrium geometry to an angle smaller than 180°.
Bonding mechanism: Variation of metals
Applying the same analysis along the series Rh(CO)2−, Pd(CO)2 and Ag(CO)2+, reveals a similar but clearer picture (Figure 6). Along this series of isoelectronic complexes, the equilibrium geometries have L–M–L angles of 130.8°, 155.6° and 180.0°. Similar to the results obtained for the series discussed above, we again find a ΔE′int term that is relatively shallow and eventually, at small angles, dominated by the Pauli repulsion. The ΔE′int term does not provide additional stabilization upon bending the complex. We do observe, however, a Δ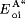 component that, from Rh(CO)2− to Pd(CO)2 to Ag(CO)2+, becomes more stabilizing and also gains more stabilization upon bending from 180° to 90°. That is, whereas for Ag(CO)2+ the
component that, from Rh(CO)2− to Pd(CO)2 to Ag(CO)2+, becomes more stabilizing and also gains more stabilization upon bending from 180° to 90°. That is, whereas for Ag(CO)2+ the 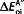 remains constant at a value of −5.4 kcal mol−1 as the complex is bent from 180° to 90°; the same component for Pd(CO)2 starts already at a more stabilizing value of −15.1 kcal mol−1 at 180° and is stabilized more than 2.5 kcal mol−1 as the complex is bent to 90°. For Rh(CO)2−, the effect of the additional stabilization upon bending is strongest, almost 10 kcal mol−1, as Δ
remains constant at a value of −5.4 kcal mol−1 as the complex is bent from 180° to 90°; the same component for Pd(CO)2 starts already at a more stabilizing value of −15.1 kcal mol−1 at 180° and is stabilized more than 2.5 kcal mol−1 as the complex is bent to 90°. For Rh(CO)2−, the effect of the additional stabilization upon bending is strongest, almost 10 kcal mol−1, as Δ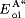 goes from −28.4 kcal mol−1 at 180° to −37.3 kcal mol−1 at 90°. The mechanism behind this trend is that the donor capability of the metal d orbitals increases as they are pushed up in energy from the cationic AgCO+ to the neutral PdCO to the negative RhCO− (see Table 3). This trend of increasing d orbital energies leads to a concomitant strengthening π backdonation and, thus, an increasing energy difference in the LM fragment between the pure metal dδ and the ligand-stabilized dπ orbitals. Thus, the ”fresh“ dδ orbitals are higher in energy than the ligand-stabilized dπ orbitals by 0.21 to 0.96 to 1.65 eV along AgCO+, PdCO and RhCO−, respectively (see Table 3). Consequently, the LM–L′ complexes benefit progressively along this series from increasing the overlap of L′ π* with the higher-energy dδ orbitals in the bent geometry.
goes from −28.4 kcal mol−1 at 180° to −37.3 kcal mol−1 at 90°. The mechanism behind this trend is that the donor capability of the metal d orbitals increases as they are pushed up in energy from the cationic AgCO+ to the neutral PdCO to the negative RhCO− (see Table 3). This trend of increasing d orbital energies leads to a concomitant strengthening π backdonation and, thus, an increasing energy difference in the LM fragment between the pure metal dδ and the ligand-stabilized dπ orbitals. Thus, the ”fresh“ dδ orbitals are higher in energy than the ligand-stabilized dπ orbitals by 0.21 to 0.96 to 1.65 eV along AgCO+, PdCO and RhCO−, respectively (see Table 3). Consequently, the LM–L′ complexes benefit progressively along this series from increasing the overlap of L′ π* with the higher-energy dδ orbitals in the bent geometry.
Figure 6.
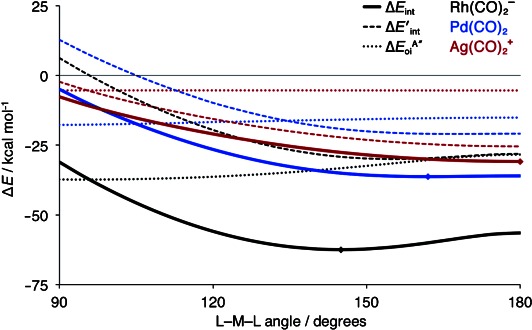
Energy decomposition analysis [Eq. (4)] of the interaction between MCO and CO in dicarbonyl-transition-metal complexes M(CO)2 as a function of the L–M–L angle (M=Rh−, Pd, Ag+).
Variation of the metal down a group goes with a less pronounced increase of the L–M–L angle that originates from more subtle changes in the bonding mechanism. The largest variation in bite angle is observed along the group 10 complexes Ni(CO)2, Pd(CO)2 and Pt(CO)2 which show L–M–L angles of 144.5°, 155.6° and 159.0°, respectively (see Table 1). Two factors lie behind this trend: (1) a weakening in π backbonding as the metal orbital energy decreases from nickel 3d to palladium 4d; (2) a steeper increase upon bending in Pauli repulsion between PtCO dσ (that has a large torus due to strong admixture of the relativistically stabilized Pt 6s AO) and the lone pair of the other CO ligand. As shown in Figure 7, the π-backbonding stabilization of Δ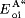 upon bending is indeed stronger for Ni(CO)2 than for Pd(CO)2 and Pt(CO)2. The difference between the latter is small because the greater (more favorable) overlap of the π* orbitals on the ligand with the more extended platinum d orbitals on PtCO compensates for the lower (less favorable) platinum d orbital energy. Figure 7 also shows how the ΔE′int term containing the aforementioned Pauli repulsion becomes more rapidly destabilizing at smaller angles for Pt(CO)2 than for Ni(CO)2 and Pd(CO)2. Likewise, in the case of group 9 complexes, the more steeply increasing Pauli repulsion of the ligand lone pair with the large iridium dσ torus pushes the equilibrium L–M–L angle of Ir(CO)2− (134.2°) to a larger value than for Rh(CO)2− (130.8°; see Table 1). Interestingly, here, the linearization energy ΔElin is nevertheless higher for the less bent Ir(CO)2− (13.4 kcal mol−1) than for Rh(CO)2− (10.2 kcal mol−1) because of the more favorable π-backbonding overlap between IrCO− and CO (see Table 1). This illustrates the subtlety of the interplay between the two features in the bonding mechanism.
upon bending is indeed stronger for Ni(CO)2 than for Pd(CO)2 and Pt(CO)2. The difference between the latter is small because the greater (more favorable) overlap of the π* orbitals on the ligand with the more extended platinum d orbitals on PtCO compensates for the lower (less favorable) platinum d orbital energy. Figure 7 also shows how the ΔE′int term containing the aforementioned Pauli repulsion becomes more rapidly destabilizing at smaller angles for Pt(CO)2 than for Ni(CO)2 and Pd(CO)2. Likewise, in the case of group 9 complexes, the more steeply increasing Pauli repulsion of the ligand lone pair with the large iridium dσ torus pushes the equilibrium L–M–L angle of Ir(CO)2− (134.2°) to a larger value than for Rh(CO)2− (130.8°; see Table 1). Interestingly, here, the linearization energy ΔElin is nevertheless higher for the less bent Ir(CO)2− (13.4 kcal mol−1) than for Rh(CO)2− (10.2 kcal mol−1) because of the more favorable π-backbonding overlap between IrCO− and CO (see Table 1). This illustrates the subtlety of the interplay between the two features in the bonding mechanism.
Walsh diagrams
Based on detailed Kohn–Sham MO analyses of individual complexes, we have constructed generalized Walsh diagrams corresponding to bending the ML2 complexes from 180° to 90°. This choice comes down to an alternative perspective on the same problem, and the emerging electronic mechanism, why bending may occur, is fully equivalent to the one obtained in the above analyses based on two interacting fragments LM+L′, namely: Bending ML2 to a nonlinear geometry enables ligand π* orbitals (if they are available on L) to overlap with and stabilize metal d orbitals that are not stabilized in the linear arrangement. The spectrum of different bonding situations has been summarized in two simplified diagrams that correspond to two extreme situations: weakly π-accepting ligands (Figure 8 A) and strongly π-accepting ligands (Figure 8 B). In these diagrams, we position the d orbital in linear ML2 above the other d orbitals, a situation that occurs, for example, for Pd(PH3)2. The relative position of the d
orbital in linear ML2 above the other d orbitals, a situation that occurs, for example, for Pd(PH3)2. The relative position of the d may change, and in some complexes, such as, Rh(NH3)2−, it is located below the other d orbitals. These variations do not affect the essential property of the orbitals, namely, their change in energy upon bending the ML2 complex. Furthermore, we speak about weakly π-accepting ligands, not just about (purely) σ-donating ligands, because it turns out that none of our model ligands has negligible π-accepting capability. The resulting Walsh diagrams summarize our results in a more easy to use pictorial manner which, in particular for the situation with strongly π-accepting ligands, is novel.
may change, and in some complexes, such as, Rh(NH3)2−, it is located below the other d orbitals. These variations do not affect the essential property of the orbitals, namely, their change in energy upon bending the ML2 complex. Furthermore, we speak about weakly π-accepting ligands, not just about (purely) σ-donating ligands, because it turns out that none of our model ligands has negligible π-accepting capability. The resulting Walsh diagrams summarize our results in a more easy to use pictorial manner which, in particular for the situation with strongly π-accepting ligands, is novel.
Figure 8.
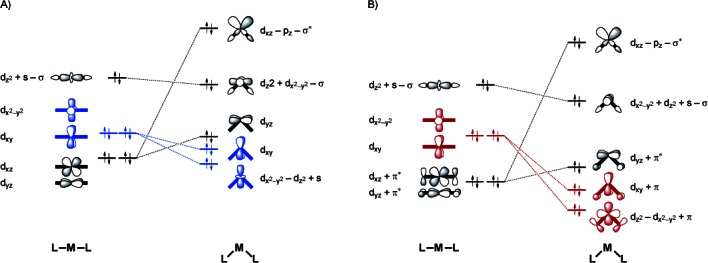
Simplified Walsh diagrams for bending ML2 complexes that emerge from our Kohn–Sham MO analyses (+/− indicate bonding/antibonding) A) without and B) with π backbonding.
We first examine the diagram with weakly π-accepting ligands (Figure 8 A). Bending ML2 from linear to nonlinear significantly destabilizes the dxz orbital because of turning on overlap with the out-of-phase combination of ligand lone pairs. This effect is related to the overlap between the LM dσ torus and the L′ lone pair in the fragment approach (see above). At small angles, direct ligand–ligand antibonding becomes important. The d orbital is slightly stabilized in the nonlinear situation due to a decreasing antibonding overlap with the in-phase combination of ligand lone pairs, augmented by admixing with the d
orbital is slightly stabilized in the nonlinear situation due to a decreasing antibonding overlap with the in-phase combination of ligand lone pairs, augmented by admixing with the d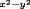 orbital (see a detailed scheme of this intermixing in Figure S1 of the Supporting Information). Note that if our model ligands would have been purely σ donating, the dxz, dyz and dxy levels would not be affected by L–M–L bending. Yet, they are, although only slightly so. This is a manifestation of some π backbonding, which is discussed in more detail below for the strongly π-accepting ligands.
orbital (see a detailed scheme of this intermixing in Figure S1 of the Supporting Information). Note that if our model ligands would have been purely σ donating, the dxz, dyz and dxy levels would not be affected by L–M–L bending. Yet, they are, although only slightly so. This is a manifestation of some π backbonding, which is discussed in more detail below for the strongly π-accepting ligands.
In the case of strongly π-accepting orbitals (Figure 8 B), bending ML2 from linear to nonlinear still goes with significant destabilization of dxz and slight stabilization of d (for the same reasons as discussed above for weakly π-accepting ligands). π Backbonding stabilizes both dxz and dyz in the linear L–M–L arrangement; bending reduces π overlap which causes also dyz to increase in energy. A striking phenomenon in the ML2 Walsh diagram with strongly π-accepting ligands is the significant stabilization of the d
(for the same reasons as discussed above for weakly π-accepting ligands). π Backbonding stabilizes both dxz and dyz in the linear L–M–L arrangement; bending reduces π overlap which causes also dyz to increase in energy. A striking phenomenon in the ML2 Walsh diagram with strongly π-accepting ligands is the significant stabilization of the d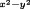 and dxy orbitals that occurs as bending moves ligand π* orbitals in the right orientation for π-accepting overlap with these orbitals. The resulting stabilization, if strong enough, can overcome the destabilization of the dxz orbital and accounts for the observed bent complexes described in this work. This effect is related to the overlap between the LM dδ orbital and the L′ π* in the fragment approach (see above). The same effect also nicely accounts for the nonlinear structures observed in earlier studies for d0 metal complexes with π-donating ligands.27–31 For these complexes, a π-bonding mechanism has been proposed in which bending is favorable because it effectively increases the number of d orbitals that have non-zero overlap with the π-donating orbitals on the ligands.28
and dxy orbitals that occurs as bending moves ligand π* orbitals in the right orientation for π-accepting overlap with these orbitals. The resulting stabilization, if strong enough, can overcome the destabilization of the dxz orbital and accounts for the observed bent complexes described in this work. This effect is related to the overlap between the LM dδ orbital and the L′ π* in the fragment approach (see above). The same effect also nicely accounts for the nonlinear structures observed in earlier studies for d0 metal complexes with π-donating ligands.27–31 For these complexes, a π-bonding mechanism has been proposed in which bending is favorable because it effectively increases the number of d orbitals that have non-zero overlap with the π-donating orbitals on the ligands.28
Conclusion
Dicoordinated d10-transition-metal complexes ML2 can very well adopt nonlinear geometries with bite angles that deviate significantly from the usually expected 180°. This follows from our relativistic density functional theory (DFT) computations on a broad range of archetypal d10-ML2 model systems. The smallest bite angle encountered in our exploration among 27 model systems amounts to 128.6° for Co(CO)2−.
Nonlinear geometries appear to be a direct consequence of π backbonding. The geometry of d10-ML2 complexes results from two opposing features in the bonding mechanism, which we have analyzed in terms of the interaction between ML and L as a function of the L–M–L angle using quantitative molecular orbital (MO) theory and energy decomposition analyses: Bending destabilizes the interaction ΔEint between ML and L through increasing steric (Pauli) repulsion between the ligands’ lone-pair orbital lobes as well as a destabilization, by the latter, of the ML dσ hybrid orbital; however, bending can also stabilize ΔEint because of enhanced π backdonation. The reason is that the π-accepor orbital on the ligand L (e.g., CO π*) interacts in the linear arrangement with an already stabilized ML dπ hybrid orbital, whereas in the bent geometry, it enters into a more favorable donor–acceptor orbital interaction with an unstabilized, that is, higher-energy metal dδ orbital.
Our analyses complement the existing text-book Walsh diagram for bending ML2 complexes8 with a variant that includes metal–ligand π backbonding. Our findings also contribute to a more rational design of catalytically active and selective ML2 complexes.1, 32
Acknowledgments
We thank the National Research School Combination-Catalysis (NRSC-C) and the Netherlands Organization for Scientific Research (NWO-CW and NWO-EW) for financial support.
Supplementary material
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
References
- 1a.Hartwig JF. Organotransition Metal Chemistry: From Bonding to Catalysis, 1st ed. Sausalito: University Science Books; 2010. See, for example. [Google Scholar]
- 1b.van Leeuwen PWNM, Kamer PCJ, Reek JNH, Dierkes P. Chem. Rev. 2000;100:2741. doi: 10.1021/cr9902704. [DOI] [PubMed] [Google Scholar]
- 1c.Birkholz née Gensow M-N, Freixa Z, van Leeuwen PWNM. Chem. Soc. Rev. 2009;38:1099. doi: 10.1039/b806211k. [DOI] [PubMed] [Google Scholar]
- 1d.van Zeist W-J, Bickelhaupt FM. Dalton Trans. 2011;40:3028. doi: 10.1039/c0dt01550d. [DOI] [PubMed] [Google Scholar]
- 2.Otsuka S. J. Organomet. Chem. 1980;200:191. [Google Scholar]
- 3.Carvajal MA, Novoa JJ, Alvarez S. J. Am. Chem. Soc. 2004;126:1465. doi: 10.1021/ja038416a. [DOI] [PubMed] [Google Scholar]
- 4.King RB. Coord. Chem. Rev. 2000;197:141. [Google Scholar]
- 5.Ziegler T. Inorg. Chem. 1985;24:1547. [Google Scholar]
- 6.Otsuka S, Yoshida T, Matsumoto M, Nakatsu K. J. Am. Chem. Soc. 1976;98:5850. [Google Scholar]
- 7.Dinjus E, Leitner W. Appl. Organomet. Chem. 1995;9:43. [Google Scholar]
- 8.Albright TA, Burdett JK, Whangbo MH. Orbital Interactions in Chemistry. New York: John Wiley & Sons; 1985. [Google Scholar]
- 9a.Gillespie RJ, Nyholm RS. Q. Rev. Chem. Soc. 1957;11:339. [Google Scholar]
- 9b.Gillespie RJ. J. Chem. Educ. 1963;40:295. [Google Scholar]
- 10a.Gillespie RJ. Chem. Soc. Rev. 1992;21:59. [Google Scholar]
- 10b.Gillespie RJ. Coord. Chem. Rev. 2008;252:1315. [Google Scholar]
- 11.te Velde G, Bickelhaupt FM, Baerends EJ, Fonseca Guerra C, van Gisbergen SJA, Snijders JG, Ziegler T. J. Comput. Chem. 2001;22:931. [Google Scholar]
- 12.Fonseca Guerra C, Snijders JG, te Velde G, Baerends EJ. Theor. Chem. Acc. 1998;99:391–403. [Google Scholar]
- 13.Baerends EJ, Ziegler T, Autschbach J, Bashford D, Bérces A, Bickelhaupt FM, Bo C, Boerrigter PM, Cavallo L, Chong DP, Deng L, Dickson RM, Ellis DE, van Faassen M, Fan L, Fischer TH, Fonseca Guerra C, Ghysels A, Giammona A, van Gisbergen SJA, Götz AW, Groeneveld JA, Gritsenko OV, Grüning M, Gusarov S, Harris FE, van den Hoek P, Jacob CR, Jacobsen H, Jensen L, Kaminski JW, van Kessel G, Kootstra F, Kovalenko A, Krykunov MV, van Lenthe E, McCormack DA, Michalak A, Mitoraj M, Neugebauer J, Nicu VP, Noodleman L, Osinga VP, Patchkovskii S, Philipsen PHT, Post D, Pye CC, Ravenek W, Rodríguez JI, Ros P, Schipper PRT, Schreckenbach G, Seldenthuis JS, Seth M, Snijders JG, Solà M, Swart M, Swerhone D, teVelde G, Vernooijs P, Versluis L, Visscher L, Visser O, Wang F, Wesolowski TA, van Wezenbeek EM, Wiesenekker G, Wolff SK, Woo TK, Yakovlev AL. ADF2010, SCM, Theoretical Chemistry; Vrije Universiteit, Amsterdam, The Netherlands; http://www.scm.com/
- 14a.Boerrigter PM, te Velde G, Baerends EJ. Int. J. Quantum Chem. 1988;33:87. [Google Scholar]
- 14b.te Velde G, Baerends EJ. J. Comp. Physiol. 1992;99:84. [Google Scholar]
- 15.van Lenthe E, Baerends EJ. J. Comput. Chem. 2003;24:1142. doi: 10.1002/jcc.10255. [DOI] [PubMed] [Google Scholar]
- 16.Versluis L, Ziegler T. J. Chem. Phys. 1988;88:322. [Google Scholar]
- 17.Slater JC. Quantum Theory of Molecules and Solids, Vol. 4. New York: McGraw-Hill; 1974. [Google Scholar]
- 18a.Becke AD. J. Chem. Phys. 1986;84:4524. [Google Scholar]
- 18b.Becke AD. Phys. Rev. A. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 19a.Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 19b.Johnson BG, Gill PMW, Pople JA. J. Chem. Phys. 1993;98:5612. [Google Scholar]
- 19c.Russo TV, Martin RL, Hay PJ. J. Chem. Phys. 1994;101:7729. [Google Scholar]
- 20a.van Lenthe E, Baerends EJ, Snijders JG. J. Chem. Phys. 1994;101:9783. [Google Scholar]
- 20b.van Lenthe E, van Leeuwen R, Baerends EJ, Snijders JG. Int. J. Quantum Chem. 1996;57:281. [Google Scholar]
- 21a.de Jong GT, Solà M, Visscher L, Bickelhaupt FM. J. Chem. Phys. 2004;121:9982. doi: 10.1063/1.1792151. [DOI] [PubMed] [Google Scholar]
- 21b.de Jong GT, Geerke DP, Diefenbach A, Bickelhaupt FM. Chem. Phys. 2005;313:261. doi: 10.1002/jcc.20233. [DOI] [PubMed] [Google Scholar]
- 21c.de Jong GT, Geerke DP, Diefenbach A, Solà M, Bickelhaupt FM. J. Comput. Chem. 2005;26:1006. doi: 10.1002/jcc.20233. [DOI] [PubMed] [Google Scholar]
- 21d.de Jong GT, Bickelhaupt FM. J. Phys. Chem. A. 2005;109:9685. doi: 10.1021/jp053587i. [DOI] [PubMed] [Google Scholar]
- 21e.de Jong GT, Bickelhaupt FM. J. Chem. Theory Comput. 2006;2:322. doi: 10.1021/ct050254g. [DOI] [PubMed] [Google Scholar]
- 22a.Bérces A, Dickson RM, Fan L, Jacobsen H, Swerhone D, Ziegler T. Comput. Phys. Commun. 1997;100:247. [Google Scholar]
- 22b.Jacobsen H, Bérces A, Swerhone D, Ziegler T. Comput. Phys. Commun. 1997;100:263. [Google Scholar]
- 22c.Wolff SK. Int. J. Quantum Chem. 2005;104:645. [Google Scholar]
- 23.van Zeist WJ, Fonseca Guerra C, Bickelhaupt FM. J. Comput. Chem. 2008;29:312. doi: 10.1002/jcc.20786. [DOI] [PubMed] [Google Scholar]
- 24.Bickelhaupt FM, Baerends EJ. In: Reviews in Computational Chemistry, Vol. 15. Lipkowitz KB, Boyd DB, editors. Weinheim: Wiley-VCH; 2000. pp. 1–86. [Google Scholar]
- 25.Ziegler T, Rauk A. Inorg. Chem. 1979;18:1755. [Google Scholar]
- 26.Swart M, Rösler E, Bickelhaupt FM. J. Comput. Chem. 2006;27:1486. doi: 10.1002/jcc.20431. [DOI] [PubMed] [Google Scholar]
- 27.Jolly CA, Marynick DS. Inorg. Chem. 1989;28:2893. [Google Scholar]
- 28.Kaupp M. Angew. Chem. 2001;113:3642. [Google Scholar]
- Angew. Chem. Int. Ed. 2001;40:3534. [Google Scholar]
- 29.Gillespie RJ, Bytheway I, DeWitte RS, Bader RFW. Inorg. Chem. 1994;33:2115. [Google Scholar]
- 30.Kaupp M, Schleyer PvonR. J. Am. Chem. Soc. 1992;114:491. [Google Scholar]
- 31.Kaupp M. Chem. Eur. J. 1999;5:3631. [Google Scholar]
- 32a.van Zeist W-J, Visser R, Bickelhaupt FM. Chem. Eur. J. 2009;15:6112. doi: 10.1002/chem.200900367. See, for example. [DOI] [PubMed] [Google Scholar]
- 32b.van Zeist W-J, Bickelhaupt FM. Org. Biomol. Chem. 2010;8:3118. doi: 10.1039/b926828f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.