

Abstract
Purpose
In a phase I trial for patients with refractory solid tumors, hedgehog pathway inhibitor vismodegib (GDC-0449) showed little decline in plasma concentrations over 7 days after a single oral dose and nonlinearity with respect to dose and time after single and multiple dosing. We studied the role of GDC-0449 binding to plasma protein alpha-1-acid glycoprotein (AAG) to better understand these unusual pharmacokinetics.
Experimental Design
Sixty-eight patients received GDC-0449 at 150 (n = 41), 270 (n = 23), or 540 (n = 4) mg/d, with pharmacokinetic (PK) sampling at multiple time points. Total and unbound (dialyzed) GDC-0449 plasma concentrations were assessed by liquid chromatography/tandem mass spectrometry, binding kinetics by surface plasmon resonance–based microsensor, and AAG levels by ELISA.
Results
A linear relationship between total GDC-0449 and AAG plasma concentrations was observed across dose groups (R2 = 0.73). In several patients, GDC-0449 levels varied with fluctuations in AAG levels over time. Steady-state, unbound GDC-0449 levels were less than 1% of total, independent of dose or total plasma concentration. In vitro, GDC-0449 binds AAG strongly and reversibly (KD = 13 μmol/L) and human serum albumin less strongly (KD = 120 μmol/L). Simulations from a derived mechanistic PK model suggest that GDC-0449 pharmacokinetics are mediated by AAG binding, solubility-limited absorption, and slow metabolic elimination.
Conclusions
GDC-0449 levels strongly correlated with AAG levels, showing parallel fluctuations of AAG and total drug over time and consistently low, unbound drug levels, different from previously reported AAG-binding drugs. This PK profile is due to high-affinity, reversible binding to AAG and binding to albumin, in addition to solubility-limited absorption and slow metabolic elimination properties.
Introduction
Aberrant hedgehog pathway activation has been implicated in a number of cancers (1–9). Vismodegib, GDC-0449; 2-chloro-N-[4-chloro-3-(pyridin-2-yl)phenyl]-4-[methylsulfonyl]benzamide, is a potent hedgehog signaling pathway inhibitor in development for treatment of various cancers (10–13). GDC-0449 binds to and inhibits smoothened (SMO), a 7-transmembrane hedgehog pathway signaling protein. Activity of GDC-0449 was first shown in vivo in preclinical models of medulloblastoma (11), colon, and pancreatic tumors (9). In a phase I study for patients with advanced malignancies, GDC-0449 was well-tolerated, with pharmacodynamic (PD) evidence of hedgehog pathway inhibition and tumor regressions in patients with basal cell carcinoma and medulloblastoma (10, 11, 13).
Preclinical pharmacokinetic (PK) properties of GDC-0449 were favorable, with low in vivo clearance and good oral bioavailability across animal species (14). In vitro studies in human hepatocytes suggested that GDC-0449 was very metabolically stable; nearly 100% of the compound remained intact following coincubations (14). At physiologic pH, GDC-0449 exhibits limited solubility in vitro (0.99 mg/mL, at pH 0.1, compared with 0.0001 mg/mL, at pH 6.5–7.4).
In a phase I study, an atypical PK profile was observed, with little decline in GDC-0449 plasma concentrations during a 7-day observation period following a single oral dose (10, 13). After continuous daily dosing, steady-state plasma concentrations were achieved earlier than expected (within 7–14 days); plasma concentrations did not increase with increasing dose levels, suggesting nonlinear pharmacokinetics with regard to dose and time.
Like many drugs, GDC-0449 binds to human serum albumin (HSA) but GDC-0449 also binds to alpha-1-acid glycoprotein (AAG) with high affinity. AAG is an acute-phase reactant protein and carrier of basic and neutrally charged lipophilic drugs (15–18). Binding to AAG results in clinically pertinent alterations in pharmacokinetics and/or pharmacodynamics for many classes of pharmacologic agents, including anticancer drugs (18) such as docetaxel (19), erlotinib (20), gefitinib (21), imatinib (22), and UCN-01 (23, 24). Previous in vitro experiments had shown that GDC-0449 is highly bound (>95%) to human plasma proteins at clinically relevant concentrations (14). In vitro equilibrium dialysis experiments with GDC-0449 concentrations of 5, 25, and 75 μmol/L and AAG concentrations of 0.5, 1, and 5 mg/mL showed that binding of GDC-0449 to AAG was saturable within a clinically relevant concentration range for GDC-0449 and physiologically relevant range for AAG. Specifically, binding was saturated by GDC-0449 at the low and medium concentrations of AAG when drug concentration was greater than 5 μmol/L. Using surface plasmon resonance (SPR) methodology, we found that the binding dissociation constant for AAG (KD AAG = 13 μmol/L), was lower than KD HSA (120 μmol/L), suggesting stronger binding to AAG than to HSA (18). Given this in vitro protein-binding data, we conducted a preliminary analysis of AAG and HSA concentrations in 40 patients on a phase I study who received GDC-0449 at 150, 270, or 540 mg/d. A single plasma sample from each patient was analyzed for AAG, HSA, and GDC-0449 21 days after initiation of daily dosing; a full AAG, HSA, and AAG PK profile was determined for 3 of these patients. Exploratory analyses indicated a strong correlation between clinical GDC-0449 plasma and AAG (but not HSA) concentrations, as well as parallel fluctuations in plasma GDC-0449 and AAG concentrations over time (18).
On the basis of these preliminary protein-binding results, and the important role of AAG binding on the PK profile of a number of other drugs, the role of AAG binding on the clinical PK profile of GDC-0449 was investigated; results are presented herein. In addition, a mechanistic PK model was derived to further assess the role of AAG binding.
Methods
Study design
The phase I trial was an open-label multicenter trial evaluating escalating doses of GDC-0449 administered orally once daily. Descriptions of study design, patient eligibility, and assessments are provided in the accompanying article (13). Human investigations were conducted after approval by a local Human Investigations Committee in accordance with assurances approved by the Department of Health and Human Services. All patients provided written informed consent according to federal and institutional guidelines before study procedures began.
Trial enrollment occurred in 2 stages. Stage 1 consisted of dose escalations to estimate a maximum tolerated dose. Stage 2 consisted of 3 cohorts: (i) an expanded cohort, at the proposed phase II dose of 150 mg, for additional safety, and PK and PD data, (ii) an additional cohort of patients with locally advanced or metastatic basal cell carcinoma at 150 and 270 mg dose levels, and (iii) a cohort to evaluate pharmacokinetics of a new 150 mg phase II drug formulation (smaller particles with faster in vitro dissolution than the phase I formulation). Comparisons of GDC-0449 pharmacokinetics between each of these cohorts are beyond the scope of this article but are included in the accompanying article (13).
Treatment and biosampling
Serial blood samples were obtained as specified in the following text to determine plasma concentrations of AAG and of total and unbound GDC-0449:
Stage 1
Three increasing dose levels of GDC-0449 were used: 150, 270, and 540 mg. On day 1, each patient received GDC-0449 in capsule form at the assigned dose, followed by a 7-day (washout) observation period. Planned blood sampling included 5 samples within 24 hours after the first dose, and additional samples on days 2, 3, 4, and 8. Continuous daily dosing was initiated on day 8; samples were obtained predose on days 15, 22, 29, 36, and every 28 days thereafter during treatment.
Stage 2
Patients received 150 or 270 mg GDC-0449 daily. Samples were collected predose on days 8, 15, 22, 29, 36, and every 28 days thereafter during treatment. For patients receiving the phase II formulation, blood samples were additionally collected at 2, 8, and 24 hours following first dose.
HSA concentrations were determined at the same time points, except for days 2, 3, and 4 for stage 1 patients and 2, 8, and 24 hours after the first dose for stage 2 patients receiving the phase II formulation.
GDC-0449 was administered on an empty stomach; patients consumed only water for 1 hour pre- and postdose and took their dose more than 1 hour prior to the first meal of the day, around the same time each day.
Bioanalysis of GDC-0449 in plasma
GDC-0449 plasma concentrations were determined by Tandem Labs, using a validated solid-phase extraction liquid chromatography/tandem mass spectrometry (LC/MS-MS) method (25). Human plasma (K2EDTA) samples containing GDC-0449 were analyzed in 200 μL aliquots. GDC-0449 concentrations were calculated using a 1/x2 quadratic regression over a concentration range of 5.00 to 5,000 ng/mL (0.012–11.9 μmol/L) with GDC-0449-d5 as an internal standard. The API 3000 was operated in the selected reaction monitoring (SRM) mode under optimized conditions for detection of GDC-0449 and GDC-0449-d5 positive ions formed by electrospray ionization.
Equilibrium dialysis
Equilibrium dialysis was conducted by QPS, using a validated method, with a Single-Use Plate Rapid Equilibrium Dialysis device equipped with dialysis membranes of molecular weight cutoff of approximately 8,000 (Thermo Scientific). The dialysis plate was placed on an orbital shaker at approximately 500 rpm and incubated in a 5% CO2 humid incubator (Napco 5400) at 37°C. The dialysis of plasma samples (0.3 mL) was conducted against 0.133 mol/L isotonic potassium phosphate buffer (0.5 mL) at pH 7.4 for 6 hours. Protein-binding samples in mixed matrix (plasma/buffer = 1:1) were shipped to Tandem Labs for analysis, as described later.
Bioanalysis of GDC-0449 in plasma/buffer
Tandem Labs conducted analyses of human plasma (K2EDTA): PBS (50:50 v/v) samples containing GDC-0449 in 100 μL aliquots by a validated solid-phase extraction procedure followed by LC/MS-MS. GDC-0449 concentrations were calculated using a 1/x2 linear regression over a concentration range of 0.100 to 100 ng/mL (0.00024–0.24 μmol/L), using GDC-0449-d5 as an internal standard. The API 5000 was operated in the SRM mode under optimized conditions for detection of GDC-0449 and GDC-0449-d5 positive ions formed by electrospray ionization.
AAG and HSA analytic methods
Concentrations of AAG in human K2EDTA plasma were determined using a commercially available kit (Dade Behring Marburg, Germany) modified for assessment by 96-well ELISA. HSA concentrations were determined as part of each patient’s standard chemistry panel.
In vitro protein-binding interaction methods
The binding affinities and kinetic profiles of GDC-0449 with AAG and albumin were assessed using SPR-based optical biosensors (Biacore; GE Healthcare) by methods similar to those described by Rich and colleagues (26) and Frostell-Karlsson and colleagues (27).
PK and statistical analyses
Individual patient GDC-0449 PK parameter values for total and unbound plasma concentration–time data were derived using noncompartmental methods (WinNonlin version 5.2.1; Pharsight Corp.).
Summary statistics were tabulated for calculated and observed PK parameters. A linear mixed-effects model was used to examine relationships between total plasma drug concentration and protein (AAG and albumin) concentration.
A mechanism-based conceptual PK simulation approach was used to explore multiple hypotheses for the observed total and unbound GDC-0449 concentrations, as well as the relationship to levels of AAG. Multiple mathematical model structures were tested incorporating one or both of the principal factors, AAG binding and solubility-limited absorption, in various mathematical permutations. Parameter values were selected on the basis of in vitro or in vivo measurements and to best represent phase I PK observations. Simulations were conducted using Berkeley Madonna software (version 8.3.11). Results were evaluated on the basis of visual inspection of observed versus predicted PK profiles.
Results
Pharmacokinetics of GDC-0449 after single or multiple doses
PK data for total and unbound GDC-0449 in plasma following single-dose administration are summarized in Table 1. Concentration–time profiles at 150, 270, or 540 mg showed that maximum total or unbound plasma concentrations (Cmax) were achieved by day 2, with little decline in concentrations over the ensuing 6-day washout period (Table 1, Fig. 1A and B). Cmax increased with dose escalation from 150 to 270 mg. At 540 mg, the mean total and unbound plasma Cmax was similar to that observed at 270 mg as shown in Table 1.
Table 1.
Single-dose unbound and total GDC-0449 PK parameters from patients enrolled in stage 1
| Stage 1 patients by dose cohort |
Mean ± SD |
|||||
|---|---|---|---|---|---|---|
| Unbound GDC-0449 |
Total GDC-0449 |
|||||
| Tmax, d | Cmax, μmol/L | AUClast, μmol/L h | Tmax, d | Cmax, μmol/L | AUClast, μmol/L h | |
| 150 mg (n = 7) | 2.43 ± 2.22 | 0.0093 ± 0.0121 | 0.577 ± 0.769 | 2.43 ± 2.22 | 3.58 ± 1.34 | 322 ± 185 |
| 270 mg (n = 9) | 1.61 ± 1.16 | 0.0324 ± 0.0247 | 2.41 ± 1.37 | 2.11 ± 0.928 | 6.34 ± 3.40 | 839 ± 458 |
| 540 mg (n = 4) | 0.834 ± 0.871 | 0.0292 ± 0.0289 | 2.43 ± 1.45 | 2.04 ± 1.34 | 6.81 ± 2.69 | 1,010 ± 446 |
Figure 1.

Pharmacokinetics of GDC-0449 after single- and multiple-dose administration. Plasma concentrations of total (A) and unbound (B) GDC-0449 over time are shown after a single dose and after multiple daily doses (C, total; D, unbound). (For C and D, PK samples from a patient who discontinued from the study early were not collected after the initiation of multiple dosing.) Average steady-state concentrations of total (E) and unbound (F) GDC-0449 in all subjects in the 150, 270, 540, and 150 mg phase II formulation cohorts are also presented. In E and F, The line in the middle of the box represents the median, the top and bottom box limits represent the 25th and 75th percentiles, and the top and bottom bars represent 1.5 times the interquartile range.
For patients in stage 1 who received a single dose of GDC-0449 on day 1 and continuous daily dosing beginning on day 8, the observed time to reach steady-state plasma concentrations ranged from 7 to 14 days (corresponding to study days 14–21). Steady-state GDC-0449 plasma concentrations were calculated as an average of plasma concentrations from study day 21 (stage 2) or study day 28 (stage 1) onward. Similar levels (total and unbound) were observed across all dosing cohorts, including the 150 mg phase II capsule formulation (Fig. 1E and F).
On average, steady-state, unbound GDC-0449 concentrations in plasma were less than 1% of total GDC-0449 concentrations, and similar levels were detected regardless of dose or total plasma concentration (ranging from 5.46 to 56.0 μmol/L; Fig. 2).
Figure 2.
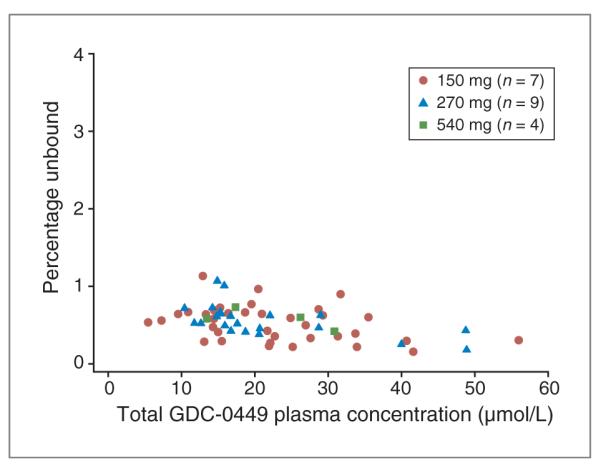
The percentage of unbound GDC-0449 is low and consistent across the range of GDC-0449 concentrations achieved within the study.
Relationship between AAG and GDC-0449 pharmacokinetics
A linear mixed model with subjects as a random effect was used to study the relationship between total GDC-0449 plasma concentration and AAG concentration. All GDC-0449 and AAG plasma samples collected after day 21 for each patient were included; data were pooled across cohorts and dose groups for the analysis. A strong relationship was observed (R2 = 0.73). The population estimate describing the relationship between GDC-0449 and AAG (the fixed-effect part of the model) was as follows:
where the value +4.658 was the population intercept and 0.482 was the population slope of the regression line (Fig. 3). There was no significant correlation between HSA levels and GDC-0449 total plasma concentration (shown in Fig. 4 for representative patients).
Figure 3.

Total GDC-0449 and AAG plasma concentrations are highly correlated. The scatter plot includes all data from each patient after day 21 of daily dosing and is reflective of steady-state conditions.
Figure 4.
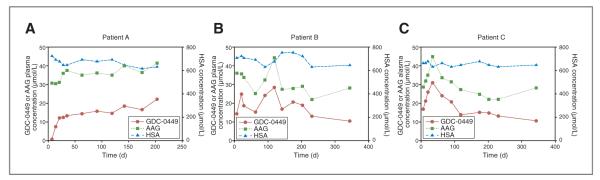
Concentration-time profiles in three representative patients (A), (B), and (C). Left y-axis reflects plasma concentration of total GDC-0449 and AAG, whereas right y-axis reflects serum concentration of HSA.
In individual patients, GDC-0449 concentrations varied with AAG concentrations over the study period. Concentration–time plots for representative patients are shown in Figure 4. Patient A received 150 mg daily for up to 200 days and showed fairly consistent plasma GDC-0449 and AAG concentrations after initial dosing. Over a similar time period, patients B and C received 270 mg GDC-0449 daily; plasma concentrations of GDC-0449 fluctuated over time in parallel with AAG concentrations, whereas HSA levels remained stable. On a molar basis, AAG concentrations remained higher than total GDC-0449 concentrations in each of these patients, consistent with overall results (Fig. 3).
Development of a mechanistic PK model
A mechanism-based conceptual PK model was developed to provide a quantitative description of the observed GDC-0449 concentration data. Figure 5A illustrates the factors considered in model development that are most likely relevant for GDC-0449 PK and pharmacologic interactions at the target tumor site: GDC-0449 absorption, distribution, elimination, and binding to AAG. In this model, plasma drug exists in 3 possible forms: unbound (Du), drug–AAG complex (D-AAG), and drug–HSA complex (D-HSA; Fig. 5A). Kinetics of protein binding are described using a rapid equilibrium binding equation with the dissociation constants designated as KD AAG and KD HSA. The final model assumed that drug was absorbed from the gastrointestinal (GI) tract into vasculature at a constant zero-order rate (k0) due to limited solubility in the GI tract. In addition, drug can be absorbed only during a limited period of GI transit time (T0), which limits its bioavailability, especially at higher doses and after multiple doses. Drug elimination from plasma is incorporated into the model as a first-order elimination of the unbound drug with rate constant (kel).
Figure 5.

Mechanistic PK model and associated simulations. A, schematic depicting the physiologic processes that likely dictate GDC-0449 pharmacokinetics. The model assumes that the unbound drug and the drug–HSA binding complex are both available for binding to AAG in order to reproduce the strong correlation between GDC-0449 and AAG plasma concentrations. Unbound drug elimination from plasma is incorporated into the model as a first-order rate constant (kel) via metabolism and/or parent excretion. Unbound drug can bind to and inhibit SMO activity, leading to blockade of hedgehog signaling at the target tumor site. The final model assumed that drug was absorbed from the GI tract into vasculature at a constant zero-order rate (k0) due to limited solubility in the GI tract. In addition, drug can be absorbed only during a limited period of GI transit time (T0), which limits its bioavailability, especially at higher doses and after multiple doses. Model simulations for total GDC-0449 (B), unbound GDC-0449 (C), and the correlation between GDC-0449 and AAG concentrations (D) are shown.
Simulations suggested that solubility-limited absorption could primarily explain the observed nonlinearity with respect to dose and time whereas a protein-binding component is necessary to explain the tight correlation between total GDC-0449 and AAG, as well as the low, unbound fraction. Incorporation of solubility-limited absorption, slow elimination, and protein-binding components into a single model provides an explanation for the observed PK characteristics of GDC-0449.
Figure 5B–D show concentration profiles from PK simulations for single-dose GDC-0449 with a 7-day washout, followed by continuous daily dosing. Total GDC-0449 concentrations (Fig. 5B), unbound GDC-0449 concentrations (Fig. 5C), and the correlation of the AAG concentration with the total GDC-0449 concentration (Fig. 5D) are shown. The model prediction represented most of the key phase 1 observations robustly, including dose-proportional exposure increase from 150 to 270 mg after single dose but no further increase at 540 mg, similar steady-state total GDC-0449 concentrations across all doses, and consistently low, unbound fraction. The observed linear relationship between total GDC-0449 plasma concentrations and AAG was also predicted by the simulation (Fig. 5D).
Discussion
In a phase I clinical trial, GDC-0449 was shown to have a unique PK profile characterized by (i) little decline in plasma concentrations over a 7-day washout period following a single dose and (ii) nonlinearity with regard to dose and time after continuous daily dosing (10, 13). To explain these observations, we considered in vitro and preliminary clinical protein-binding data for GDC-0449 and the established importance of in vivo AAG binding to a number of drugs.
The most remarkable characteristic of GDC-0449 pharmacokinetics that we observed in this study was the strong correlation between GDC-0449 and AAG plasma concentrations. We further investigated the role of AAG binding on the pharmacokinetics of GDC-0449, including derivation of a semimechanistic PK model and measurement of AAG and unbound GDC-0449 levels in all phase I patient samples. The results from this report suggest that the PK profile of GDC-0449 is largely dictated by 3 factors: solubility-limited absorption, associated with dose and time nonlinearity; a slow rate of systemic elimination, associated with the long half-life; and binding to AAG (the focus of this report), which resulted in the surprisingly tight correlation between in vivo levels of GDC-0449 and AAG. The binding of AAG strongly impacted the pharmacokinetics of GDC-0449, as evidenced by intrapatient parallel fluctuation in AAG and GDC-0449 levels.
Binding of AAG impacts pharmacokinetics and/or pharmacodynamics of a number of drugs across a broad spectrum of classes. In the oncology setting (28, 29), AAG binds to chemotherapeutic agents such as docetaxel (19) and several small-molecule tyrosine and protein kinase inhibitors, including erlotinib (20), gefitinib (21), imatinib (22), and UCN-01 (23, 24). Plasma concentrations of AAG are normally around 0.28 to 0.92 g/L but increase during inflammatory or stress reactions (28, 29). Levels of AAG in patients with malignant tumors are elevated up to 5-fold (30). In addition, a mixture of 2 or 3 genetic variants of AAG exists in most individuals. Although many drugs have similar binding constants for AAG, other drugs (such as imatinib) have shown differences in binding to genetic variants (30, 31). These properties of AAG largely explain the alterations or interpatient differences in pharmacokinetics and/or pharmacodynamics for a variety of drugs (19–24).
The reported half-lives of most AAG-binding drugs (e.g., more than 20 tyrosine kinase inhibitors in a recent report; ref. 32) are much shorter than the reported half-life of AAG (~5 days; ref. 33), suggesting that factors other than AAG binding also determine their PK characteristics. UCN-01 is an investigational anticancer drug with a binding affinity for AAG that is stronger by approximately 4 orders of magnitude (reported KA of 8.0 × 108 L/mol, which corresponds to a KD of 1.25 nmol/L; ref. 24) than GDC-0449 and a slower dissociation rate (18). This is associated with a very prolonged elimination phase after intravenous (IV) administration; UCN-01 total plasma concentrations approximate plasma concentrations of AAG (23). The pharmacokinetics of UCN-01 seem to essentially reflect the strong binding to AAG, with drug taking on the kinetics of the protein. In comparison, the elimination half-life of GDC-0449 after IV administration is also long (34); however, for GDC-0449, this is not strictly a result of AAG binding, as indicated by the complete reversibility of GDC-0449 binding to AAG and a much lower binding affinity compared with UCN-01.
In addition to binding kinetics, we considered whether nonlinear binding to AAG was an important contributing factor to the PK profile of GDC-0449. For example, imatinib–AAG binding results in a nonlinear relationship between total and unbound imatinib plasma concentrations (35) and levels of unbound imatinib correlate with PD responses to treatment. Similarly to GDC-0449, imatinib binds strongly to AAG (KA of 1.7 × 106 L/mol, which corresponds to a KD of 0.6 μmol/L) and less strongly to HSA (KA of 3.0 × 104 L/mol, which corresponds to a KD of 33 μmol/L; ref. 32). However, for imatinib, high AAG levels result in low available plasma concentrations of unbound drug and lower clearance, associated with hematologic toxicity and imatinib resistance in leukemia (36, 37). Unlike imatinib, the concentration of unbound GDC-0449 in plasma remained low and a relatively constant proportion of total steady-state plasma concentration across the observed range (~5.46–56.0 μmol/L); our data and PK modeling strongly suggest that this can be explained only by the binding of GDC-0449 to both AAG and HSA. Once AAG binding becomes saturated by GDC-0449, the unbound GDC-0449 fraction remains low due to binding to HSA, which serves as a high-capacity drug-binding protein relative to AAG due to its high level in plasma. Therefore, saturation of overall protein binding is not achieved by GDC-0449 at clinically relevant drug concentrations. Notably, the relative difference in binding affinity of imitinib for AAG and HSA is 2 orders of magnitude but only 1 order of magnitude for GDC-0449, which could explain why there is a nonlinear relationship between total and unbound imatinib plasma concentrations but not GDC-0449 plasma concentrations.
A hypothesis to explain the unique interpatient and intrapatient (over time) correlations between AAG and GDC-0449 levels is that due to the poor solubility of GDC-0449, the intestine acts as a continuous source of drug to the systemic circulation with a zero-order input rate whereas AAG in the systemic circulation acts as a sink for GDC-0449. In the event of an increase or decrease in plasma AAG, the absorption rate may transiently increase or decrease, respectively. Additional experiments and mathematical modeling are needed to further test this hypothesis.
To our knowledge, this is the first report in which plasma AAG levels explain most (>70%) of the observed PK variability. The ability of AAG to influence the PK profile of a particular drug depends on a balance of several factors, including level of affinity, binding kinetics, binding capacity, relative affinity to HSA compared with AAG, and other factors such as intrinsic hepatic clearance and oral absorption. Taken together, our study results, PK model simulations, and comparisons with other AAG-binding drugs suggest that the pharmacokinetics of GDC-0449 are dictated by its solubility-limited absorption following oral administration, slow rate of metabolic elimination, and interaction with AAG. Although the current PK model has been predictive of the key PK properties of the molecule, additional enhancement and validation of this model would permit the incorporation of elements that fully describe other PK properties, such as AAG and unbound GDC-0449 fluctuations, as they relate to total GDC-0449 over time. Ongoing studies of GDC-0449, including the assessment of absolute bioavailability (by IV and oral administration), alternative dose schedules, binding to different AAG genetic variants, and mechanistic and population PK modeling and simulations, will further define factors that contribute to the unique PK characteristics of GDC-0449.
Translational Relevance.
Vismodegib (GDC-0449),a small-molecule hedgehog pathway inhibitor, has shown encouraging antitumor activity in advanced basal cell carcinoma and medulloblastoma. In the first phase 1 study, GDC-0449 showed an unusual pharmacokinetic (PK) profile with an unexplained elimination half-life of more than 7 days and accumulation that unexpectedly plateaued within the first 14 days. This article is the first to describe some of the phenomena contributing to the GDC-0449 PK profile: high-affinity binding to alpha-1-acid glycoprotein (AAG) with tight correlation to plasma AAG levels over time and consistently low, unbound drug levels. This is unique compared with previously reported AAG-binding drugs, possibly due to unprecedented saturation of plasma AAG. These phenomena, in combination with binding to serum albumin, solubility-limited gastrointestinal absorption, and slow metabolic elimination, are captured in a biologically based GDC-0449 PK model, presented here for the first time. The model will be used to simulate alternate dose regimens and will be validated through ongoing clinical trials of GDC-0449.
Acknowledgments
The authors thank Vikram Malhi for clinical pharmacokinetic data analysis, Dr. Mark Dresser for his thoughtful review and input, Drs. Harvey Wong, Anthony Giannetti, and Alan Deng for bioanalytic contributions, Dr. Ilsung Chang for biostatistics input, and Lisa Beckel and Hilary Nelson for their work in clinical operations. We also thank Dr. Leslie Benet for his insightful input and discussion. Dr. Abie Craiu at Genentech, Inc., provided assistance with preparation of the manuscript.
Footnotes
Disclosure of Potential Conflicts of Interest D. D. Von Hoff received a grant to his institution for the phase I trial of GDC-0449; P.M. LoRusso has research funding from Genentech, is on Genentech’s speakers bureau (compensated) and has participated in Genentech Advisory Boards (compensated); CM Rudin has been a paid consultant to Genentech (not on projects related to Hedgehog inhibitors). R.A. Graham, B.L. Lum, S. Cheeti, J.Y. Jin, K. Jorga, J.C. Reddy, and J.A. Low are employees of Genentech, A Member of the Roche Group, and shareholders of Roche.
References
- 1.Scales SJ, de Sauvage FJ. Mechanisms of hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci. 2009;30:303–12. doi: 10.1016/j.tips.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 2.Hahn H, Christiansen J, Wicking C, Zaphiropoulos PG, Chidambaram A, Gerrard B, et al. A mammalian patched homolog is expressed in target tissues of sonic hedgehog and maps to a region associated with developmental abnormalities. J Biol Chem. 1996;271:12125–8. doi: 10.1074/jbc.271.21.12125. [DOI] [PubMed] [Google Scholar]
- 3.Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–71. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 4.Pietsch T, Waha A, Koch A, Kraus J, Albrecht S, Tonn J, et al. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer Res. 1997;57:2085–8. [PubMed] [Google Scholar]
- 5.Raffel C, Jenkins RB, Frederick L, Hebrink D, Alderete B, Fults DW, et al. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997;57:842–5. [PubMed] [Google Scholar]
- 6.Vorechovsky I, Tingby O, Hartman M, Strömberg B, Nister M, Collins VP, et al. Somatic mutations in the human homologue of Drosophila patched in primitive neuroectodermal tumours. Oncogene. 1997;15:361–6. doi: 10.1038/sj.onc.1201340. [DOI] [PubMed] [Google Scholar]
- 7.Fan L, Pepicelli CV, Dibble CC, Catbagan W, Zarycki JL, Laciak R, et al. Hedgehog signaling promotes prostate xenograft tumor growth. Endocrinology. 2004;145:3961–70. doi: 10.1210/en.2004-0079. [DOI] [PubMed] [Google Scholar]
- 8.Dierks C, Grbic J, Zirlik K, Beigi R, Englund NP, Guo GR, et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat Med. 2007;13:944–51. doi: 10.1038/nm1614. [DOI] [PubMed] [Google Scholar]
- 9.Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455:406–10. doi: 10.1038/nature07275. [DOI] [PubMed] [Google Scholar]
- 10.Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361:1164–72. doi: 10.1056/NEJMoa0905360. [DOI] [PubMed] [Google Scholar]
- 11.Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361:1173–8. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, et al. Smoothened mutation confers resistance to a hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326:572–4. doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LoRusso PM, Rudin CM, Reddy JC, Tibes R, Weiss GJ, Borad MJ, et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin Cancer Res. 2011;17:2502–11. doi: 10.1158/1078-0432.CCR-10-2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong H, Chen JZ, Chou B, Halladay JS, Kenny JR, La H, et al. Preclinical assessment of the absorption, distribution, metabolism and excretion of GDC-0449 (2-chloro-N-(4-chloro-3-(pyridin-2-yl) phenyl)-4-(methylsulfonyl)benzamide), an orally bioavailable systemic Hedgehog signalling pathway inhibitor. Xenobiotica. 2009;39:850–61. doi: 10.3109/00498250903180289. [DOI] [PubMed] [Google Scholar]
- 15.Israili ZH, Dayton PG. Human alpha-1-glycoprotein and its interactions with drugs. Drug Metab Rev. 2001;33:161–235. doi: 10.1081/dmr-100104402. [DOI] [PubMed] [Google Scholar]
- 16.Kremer JM, Wilting J, Janssen LH. Drug binding to human alpha-1-acid glycoprotein in health and disease. Pharmacol Rev. 1988;40:1–47. [PubMed] [Google Scholar]
- 17.Slaviero KA, Clarke SJ, Rivory LP. Inflammatory response: an unrecognized source of variability in the pharmacokinetics and pharmacodynamics of cancer chemotherapy. Lancet Oncol. 2003;4:224–32. doi: 10.1016/s1470-2045(03)01034-9. [DOI] [PubMed] [Google Scholar]
- 18.Giannetti AM, Wong H, Dijkgraaf GJP, Dueber E, Ortwine DF, Bravo BJ, et al. Identification, characterization, and implications of species-dependent plasma protein binding for the oral hedgehog pathway inhibitor vismodegib (GDC-0449) J Med Chem. 2011 Mar 25; doi: 10.1021/jm1008924. [DOI] [PubMed] [Google Scholar]
- 19.Urien S, Barre J, Morin C, Paccaly A, Montay G, Tillement JP, et al. Docetaxel serum protein binding with high affinity to alpha 1-acid glycoprotein. Invest New Drugs. 1996;14:147–51. doi: 10.1007/BF00210785. [DOI] [PubMed] [Google Scholar]
- 20.Lu JF, Eppler SM, Wolf J, Hamilton M, Rakhit A, Bruno R, et al. Clinical pharmacokinetics of erlotinib in patients with solid tumors and exposure-safety relationship in patients with non-small cell lung cancer. Clin Pharmacol Ther. 2006;80:136–45. doi: 10.1016/j.clpt.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 21.Li J, Brahmer J, Messersmith W, Hidalgo M, Baker SD. Binding of gefitinib, an inhibitor of epidermal growth factor receptor-tyrosine kinase, to plasma proteins and blood cells: in vitro and in cancer patients. Inv New drugs. 2006;24:291–7. doi: 10.1007/s10637-006-5269-2. [DOI] [PubMed] [Google Scholar]
- 22.Delbaldo C, Chatelut E, Ré M, Deroussent A, Séronie-Vivien S, Jambu A, et al. Pharmacokinetic-pharmacodynamic relationships of imatinib and its main metabolite in patients with advanced gastrointestinal stromal tumors. Clin Cancer Res. 2006;12:6073–8. doi: 10.1158/1078-0432.CCR-05-2596. [DOI] [PubMed] [Google Scholar]
- 23.Fuse E, Tanii H, Takai K, Asanome K, Kurata N, Kobayashi H, et al. Altered pharmacokinetics of a novel anticancer drug, UCN-01, caused by specific high affinity binding to alpha1-acid glycoprotein in humans. Cancer Research. 1999;59:1054–60. [PubMed] [Google Scholar]
- 24.Fuse E, Kuwabara T, Sparreboom A, Sausville EA, Figg WD. Review of UCN-01 development: a lesson in the importance of clinical pharmacology. J Clin Pharmacol. 2005;45:394–403. doi: 10.1177/0091270005274549. [DOI] [PubMed] [Google Scholar]
- 25.Ding X, Chou B, Graham RA, Cheeti S, Percey S, Matassa LC, et al. Determination of GDC-0449, a small molecule inhibitor of the Hedgehog signaling pathway, in human plasma by solid phase extraction-liquid chromatographic-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:785–90. doi: 10.1016/j.jchromb.2010.01.039. [DOI] [PubMed] [Google Scholar]
- 26.Rich RL, Day YS, Morton TA, Myszka DG. High-resolution and high-throughput protocols for measuring drug/human serum albumin interactions using BIACORE. Anal Biochem. 2001;296:197–207. doi: 10.1006/abio.2001.5314. [DOI] [PubMed] [Google Scholar]
- 27.Frostell-Karlsson A, Remaeus A, Roos H, Andersson K, Borg P, Hämäläinen M, et al. Biosensor analysis of the interaction between immobilized human serum albumin and drug compounds for prediction of human serum albumin binding levels. J Med Chem. 2000;43:1986–92. doi: 10.1021/jm991174y. [DOI] [PubMed] [Google Scholar]
- 28.Fournier T, Medjoubi NN, Porquet D. Alpha-1-acid glycoprotein. Biochim Biophys Acta. 2000;1482:157–71. doi: 10.1016/s0167-4838(00)00153-9. [DOI] [PubMed] [Google Scholar]
- 29.Hochepied T, Berger FG, Baumann H, Libert C. Alpha(1)-acid glycoprotein: an acute phase protein with inflammatory and immunomodulating properties. Cytokine Growth Factor Rev. 2003;14:25–34. doi: 10.1016/s1359-6101(02)00054-0. [DOI] [PubMed] [Google Scholar]
- 30.Duche JC, Urien S, Simon N, Malaurie E, Monnet I, Barre J. Expression of the genetic variants of human alpha-1-acid glycoprotein in cancer. Clin Biochem. 2000;33:197–202. doi: 10.1016/s0009-9120(00)00048-5. [DOI] [PubMed] [Google Scholar]
- 31.Duche JC, Herve F, Tillement J-P. Study of the expression of the genetic variants of human α1-acid glycoprotein in healthy subjects using isoelectric focusing and immunoblotting. J Chromatogr B Biomed Sci Appl. 1999;17:103–9. doi: 10.1016/s0378-4347(98)00084-x. [DOI] [PubMed] [Google Scholar]
- 32.Fitos I, Visy J, Zsila F, Mády G, Simonyi M. Selective binding of imatinib to the genetic variants of human alpha1-acid glycoprotein. Biochim Biophys Acta. 2006;1760:1704–12. doi: 10.1016/j.bbagen.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 33.Zsila F, Fitos I, Bencze G, Kéri G, Orfi L. Determination of human serum alpha1-acid glycoprotein and albumin binding of various marketed and preclinical kinase inhibitors. Curr Med Chem. 2009;16:1964–77. doi: 10.2174/092986709788682191. [DOI] [PubMed] [Google Scholar]
- 34.Graham RA, Morrison GE, Chang I, Jorga K, Hop C, Shin Y, et al. Bioavailability of the hedgehog pathway inhibitor GDC-0449 in a phase 1 pharmacokinetic (PK) study in healthy female subjects. Presented at the American Society of Clinical Oncology annual meeting; Chicago, IL. 2010; Abstract e13009. [Google Scholar]
- 35.Widmer N, Decosterd LA, Csajka C, Leyvraz S, Duchosal MA, Rosselet A, et al. Population pharmacokinetics of imatinib and the role of alpha-acid glycoprotein. Br J Clin Pharmacol. 2006;62:97–112. doi: 10.1111/j.1365-2125.2006.02719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gambacorti-Passerini C, Barni R, le Coutre P, Zucchetti M, Cabrita G, Cleris L, et al. Role of alpha1 acid glycoprotein in the in vivo resistance of human BCR-ABL(+) leukemic cells to the abl inhibitor STI571. J Natl Cancer Inst. 2000;92:1641–50. doi: 10.1093/jnci/92.20.1641. [DOI] [PubMed] [Google Scholar]
- 37.Petain A, Kattygnarath D, Azurd J, Chatelut E, Delbaldo C, Geoerger B, et al. Population pharmacokinetics and pharmacogenetics of imatinib in children and adults. Clin Cancer Res. 2008;14:7102–9. doi: 10.1158/1078-0432.CCR-08-0950. [DOI] [PubMed] [Google Scholar]