

Abstract
The Signal Transducer and Activator of Transcription factor STAT4 is a critical regulator of Th1 differentiation and inflammatory disease. Yet, how STAT4 regulates gene expression is still unclear. In this report, we define a STAT4-dependent sequence of events including H3K4 methylation, Jmjd3 association with STAT4 target loci, and a Jmjd3-dependent decrease in H3K27 trimethylation and DNA methyltransferase (Dnmt) 3a association with STAT4 target loci. Dnmt3a has an obligate role in repressing Th1 gene expression, and in Th1 cultures deficient in both STAT4 and Dnmt3a, there is recovery in the expression of a subset of Th1 genes that is sufficient to increase IFNγ production. Moreover, although STAT4-deficient mice are protected from the development of EAE, mice deficient in STAT4 and conditionally-deficient in Dnmt3a in T cells develop paralysis. Th1 genes that are de-repressed in the absence of Dnmt3a have greater induction following the ectopic expression of the Th1-associated transcription factors T-bet and Hlx1. Together, these data demonstrate that STAT4 and Dnmt3a play opposing roles in regulating Th1 gene expression, and that one mechanism for STAT4-dependent gene programming is in establishing a de-repressed genetic state susceptible to transactivation by additional fate-determining transcription factors.
Keywords: T helper 1 cells, transcription factors, IL-12, STAT4, Dnmt3a, Jmjd3
Introduction
Th1 cells are critical regulators of inflammation and play obligate roles in immunity to intracellular pathogens and in the development of autoimmune inflammation (1, 2). Th1 development initiates when a T cell, activated by antigen in the context of MHC molecules, is stimulated by IL-12 and IFNγ (3–5). IL-12 and IFNγ stimulation result in the phosphorylation of STAT4 and STAT1 respectively, transcription factors that are required for optimal Th1 differentiation (4, 6, 7). The Tbx21 gene, encoding the T-box transcription factor T-bet, is a critical target for both factors, with STAT1 binding early in differentiation, and STAT4 binding later (8–10). STAT4 and T-bet are required for the expression of many genes expressed in Th1 cells, although both factors activate the expression of a subset of Th1 genes in the absence of the other factor (11). IFNγ production is the hallmark of Th1 cells, and both STAT4 and T-bet, in cooperation with other transcription factors including Hlx1 and Runx3, activate transcription from the Ifng locus (12, 13).
Epigenetic events that include DNA methylation and histone modification play a key role in T cell differentiation. DNA methyltransferase (DNMT) enzymes catalyze DNA methylation at CpG dinucleotides, resulting in gene repression in T helper cells (14–18). Histone modifications that occur at the tails of the core histones include methylation, acetylation, phosphorylation, sumoylation and ubiquitination. The addition and removal of histone marks alters chromatin into either an active or repressed state correlating with the amount of transcription at the locus. Tri-methylation of histone H3 lysine 4 (H3K4) correlates with active gene transcription (19). Acetylation of histones at specific lysine residues (H3K9, K18, K27 and K36) result in decreased association with DNA and greater access for trans-acting factors (19, 20). In contrast, tri-methylation of histone H3 lysine 27 (H3K27) is associated with gene repression (19, 20). Histone modifying enzymes are recruited to DNA by transcription factors and are able to recognize histone marks thus allowing the extension of histone modification across adjacent regions of the target locus (19).
STAT4 binds a multitude of DNA sequences throughout the genome, and genome-wide studies have begun to define targets in Th1 cells. Using a chromatin immunoprecipitation (ChIP)-on-chip approach, STAT4 was found to bind at least 1500 sites in a 10kb region spanning murine gene promoters, following acute IL-12 stimulation in differentiating T cells (21). Using a ChIP-seq approach in differentiated Th1 cells, STAT4 bound almost 4500 sites, with slightly over one-third located in promoter regions, providing a good concordance of the two studies (21, 22). Both studies found the STAT consensus sequence TTCN3GAA in the majority of bound sites. Despite this, numerous genes that bound STAT4 were not induced by IL-12 stimulation, and for genes that were induced, there was no correlation between the peak intensity of STAT4 binding to a site and the fold induction of the associated gene (21, 22). Thus, although we now have a detailed appreciation for STAT4 target genes in Th1 cells, it is still not clear how STAT4 activates long-lasting changes in Th1 gene expression.
Once STAT4 is bound to a locus, it can recruit other transcription factors and chromatin modifying enzymes. At the Il2ra locus, STAT4 is responsible for the recruitment of c-Jun-containing complexes and the histone acetyltransferase CBP (23). In the absence of STAT4, IL-12 does not induce histone acetylation at the Il2ra locus (23). At the Il18r1 locus, STAT4 is required for total histone acetylation, H3K9 acetylation, H3K4 di- and tri-methylation, and for limiting H3K27 tri-methylation (14, 24). Genome wide analysis of STAT4 binding sites also found a requirement for STAT4 in H3K4me3 (22). At the Ifng and IL12RB2 loci, STAT4 recruits BAF-containing SWI/SNF complexes that are required for nucleosome remodeling (25, 26). Globally, STAT4 is required for recruitment of p300 to enhancer elements active in Th1 cells (27).
STAT4 also limits DNA methylation of the Il18r1 locus by reducing the association of Dnmt3a, one of the two de novo DNA methyltransferases, with the Il18r1 promoter, and the promoters of several additional Th1 genes (14). T cells deficient in Dnmt3a, but not Dnmt3b, the other de novo DNA methyltransferase, have increased IFNγ production, and increased flexibility in their ability to switch from Th2, Th17 or Treg cells to an IFNγ-secreting phenotype (15, 28, 29). Dnmt3a also represses alternative lineage gene expression in Th1 cells. The Il13 locus is particularly sensitive to Dnmt3a-deficiency, and GATA3 can more effectively induce Il13 expression in Dnmt3a-deficient Th1 cells than in wild type Th1 cells (15). Thus, Dnmt3a is required for appropriate gene repression in T helper subsets.
The apparent opposing function of STAT4 and Dnmt3a raises the question whether STAT4 is required to activate gene expression, or whether it functions solely by eliminating negative regulators of gene expression from target loci. In this report we examine histone modifications associated with gene expression or repression at a subset of STAT4 target genes. We demonstrate STAT4-dependent chromatin modifying enzyme recruitment, and using mice deficient in STAT4 and conditionally deficient in Dnmt3a in T cells, demonstrate increased expression of Th1 genes in the absence of both a positive (STAT4) and negative (Dnmt3a) regulator of gene expression compared to cells lacking STAT4 alone.
Materials and Methods
Mice and institutional approval
C57BL/6 mice were purchased from Harlan Sprague Dawley (Indianapolis, IN, USA). Dnmt3afl/fl CD4-Crepositive mice (15) were mated with Stat4−/−(6) to generate Dnmt3afl/flStat4−/− CD4-Cre positive with Cre-negative littermates as control mice. Mice were maintained under specific pathogen-free conditions. All experiments were performed with the approval of the Indiana University Institutional Animal Care and Use Committee.
Induction of EAE and cytokine production analysis
Induction and scoring of EAE disease has been described (30). In brief, a cohort of 8–12 week old female wild type (WT), Dnmt3afl/fl CD4-Cre positive or Dnmt3afl/fl Stat4−/− CD4-Cre negative or positive mice (4–7 mice/group) were immunized subcutaneously with 100ug of MOGp35-55 peptide (Genemed Synthesis) in 150ul emulsion of CFA (Sigma Aldrich) on days 0 and 7. Mice also received 100ng of pertussis toxin (i.p. injection) (Sigma Aldrich) on days 0 and 2. The clinical signs were scored daily for 30 days. On day 14, some mice were sacrificed for cytokine production analysis. Mononuclear cells were isolated from brain, stimulated with PMA/Ionomycin and monensin (Sigma Aldrich) added for the last 3 h of a 5 h activation for intracellular staining analysis. To determine the cytokine response, spleen cells were cultured in the presence of 10 ug/ml MOGp35-55 for 48 h, and cytokine production was measured using ELISA.
In vitro T cell differentiation
Naïve CD4+CD62L+ T cells were isolated from spleen and lymph nodes using a MACS isolation system (Miltenyi Biotec). CD4+ T cells were activated with plate-bound anti-CD3 (2 ug/ml 145-2C11 Bio X Cell) and soluble anti-CD28 (0.5 ug/ml BD Pharmingen for Th0, Th1 and Th17 or 1 ug/ml for iTreg) for non-skewing T cell conditions or with additional cytokines (all from PeproTech) and antibodies to generate Th1 (5 ng/ml IL-12; and 10 ug/ml anti-IL-4 11B11), Th17 (100 ng/ml IL-6; 10 ng/ml IL-23; 10 ng/ml IL-1β; 2 ng/ml TGF-β;10 ug/ml anti-IL-4, 11B11; and 10 ug/ml anti-IFNγ, XMG) or iTreg (2ng/ml TGF-β, and 10ug/ml anti-IL-4, 11B11) culture conditions. Non-polarized T cells and Th1 cells (expanded after 3 days in fresh medium) were harvested on days 3 and 5 respectively, and restimulated with 5ng/ml IL-12 for 1, 4, and 6 h for further analyses.Th17 and iTreg cells were expanded on day 3 with 50 U/ml human-IL-2 (iTreg) or half concentration of the original cytokines (Th17) in fresh medium. Cells were harvested on day 5 for analysis.
Retroviral expression vectors and retroviral transduction
Bicistronic retrovirus expressing EGFP only (MIEG), T-bet and EGFP (T-bet), MSCV-Thy1.1 (Thy1.1), and Runx3-Thy1.1 (Runx3) were previously described (31–33). Human HLX1 (Open Biosystems) cDNA was amplified, sub-cloned into the TOPO vector (Invitrogen), digested and sub-cloned into either MIEG-EGFP or MSCV-Thy1.1. Retroviral stocks were prepared as described previously (33). Purified CD4+ T cells were cultured under Th1 cell polarizing conditions. On day 2, cells were transduced with retrovirus expressing vector control or gene of interest by centrifugation at 2000 rpm at 25°C for 1 h in the presence of 8 ug/ml polybrene. Viral supernatant was replaced with media supplemented with 50 U/ml human IL-2. After spin infection, cells were expanded on day 3 and analyzed on day 5.
Cell sorting, analysis of gene expression, and flow cytometry
Transduced cells were collected on day 5, stained with anti-rat CD90/mouse CD90.1 APC (Biolegend), and sorted for single or doubly positive cells using a Reflection cell sorter (iCyt). Sorted cells were rested or re-stimulated with 2ug/ml anti-CD3 for 24 h for ELISA and qRT-PCR analyses (32). For cytokine staining, cells were stimulated with either 2ug/ml anti-CD3 with Golgi Plug inhibitor (BD Pharmingen) or PMA/Ionomycin and monensin (Sigma Aldrich)added for the last 3 h of a 5 h activation, fixed, permeabilized using 0.1% saponin, and stained for rat-anti mouse TNFα FITC and rat anti-mouse IFNγ APC (BD Pharmingen).For phospho-STAT1 and phospho-STAT4 analyses, cells were fixed, permeabilized using 100% ice cold methanol, and stained for anti-phospho-Stat1FITC and anti-phospho-Stat4PE (BD Pharmingen) before analysis. For viability staining, cells were washed twice with 1X PBS and stained for fixable viability dye eFluor 780 (eBioscience) for 30 min at 4°C. For Jmjd3 intracellular staining, cells were fixed, permeabilized using 0.1% saponin, and stained for biotinylated Jmjd3 (Novus Biologicals) or biotinylated rabbit IgG (BD Pharmingen) for 30 min at 4°C follows by streptavidin PE (BD Pharmingen) for an additional 30 min at 4°C.In vitro generated Treg or total splenocytes (for nTreg staining) were used for Foxp3 staining using Foxp3/Transcription factor staining kit (eBioscience). nTreg cells were stained for CD4 PeCy7, CD25 APC (BD Pharmingen) and Fopx3 FITC (eBioscience).
siRNAtransfection
Day 5 differentiated wild type Th1 cells were transfected with control or Jmjd3-specific siRNA (Santa Cruz) using Amaxa Nucleofector system for CD4+ T cells (Lonza). Transfected cells were supplemented with 50U/ml human IL-2 and rested overnight. Cells were harvested, restimulated with 2ug/ml anti-CD3 for 6 h for flow cytometry, 24h for ELISA or restimulated with 5ng/ml IL-12 for 4 h for qRT-PCR and ChIP experiments.
Chromatin Immunoprecipitation(ChIP)
ChIP assay was performed as described (11). In brief, resting or restimulated Th1 cells were cross-linked for 10 min with 1% formaldehyde and lysed by sonication. After pre-clearing with salmon sperm DNA, bovine serum albumin, and Protein A agarose bead slurry (50%), cell extracts were incubated with Abs to Stat4 C-20, T-bet 4B10, PEBP2β FL-182 (Santa Cruz), H3K27me3 (Millipore), H3K4me3 (Abcam), JMJD3 RB10082 (Abgent), Dnmt3a 64B1446 (IMGENEX) or normal rabbit IgG (Millipore) overnight at 4°C. For Dnmt3a ChIP, the immunocomplexes were incubated with rabbit anti-mouse antibody for an additional 2 h. The immunocomplexes were precipitated with protein A agarose beads at 4°C for 2 h, washed, eluted, and cross-links reversed at 65°C overnight. DNA was purified, resuspended in H20 and analyzed by quantitative PCR with Taqman or SYBR primers (14, 34, 35).
Results
STAT4-dependent association of chromatin modifying enzymes at Th1 gene loci
In previous work we defined STAT4 target genes in activated T cells (21). In developing a further understanding of how STAT4 activates gene expression, we examined STAT4 binding at several known STAT4 targets expressed in Th1 cells. Naïve CD4+ T cells were activated with anti-CD3 and anti-CD28, polarized towards the Th1 phenotype in the presence of IL-12 and anti-IL-4, and restimulated with IL-12 after 5 days differentiation. The Th1-associated genes display increased transcription (Fig. 1A), and STAT4 binding (Fig. 1B) over six hours following IL-12 stimulation. Thus, STAT4 binding correlates with IL-12-inducible gene expression.
Figure 1. Gene expression, histone modification, and chromatin modifying enzyme patterns in IL-12-stimulated WT and Stat4−/− Th1 cells.
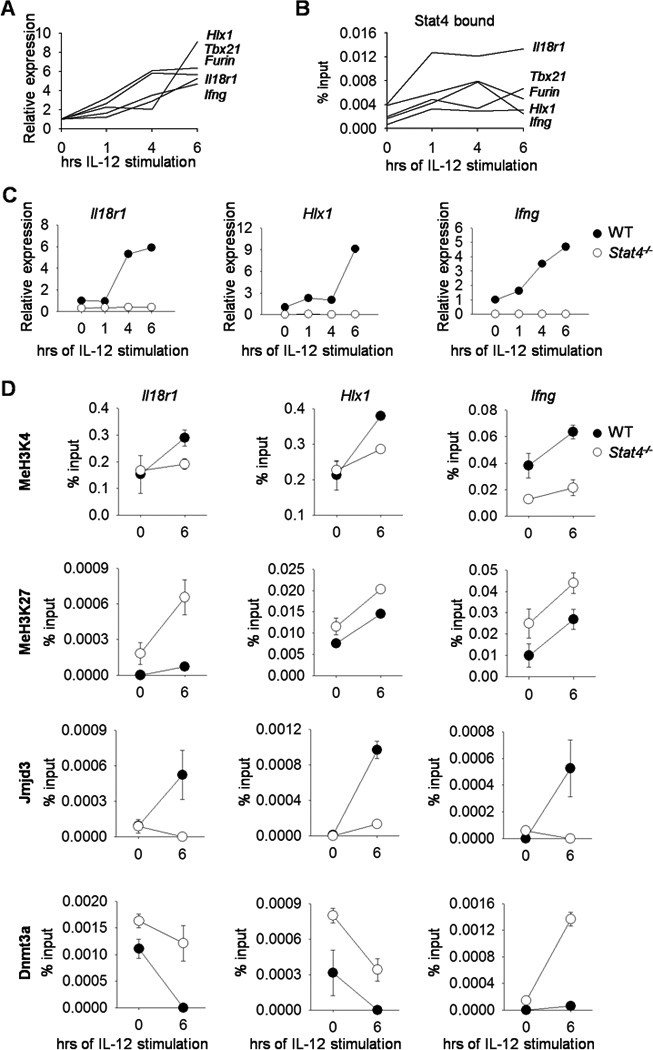
Naïve CD4+CD62L+ T cells were isolated from WT or Stat4−/− C57BL/6 mice and cultured under Th1 polarizing conditions. On day 5, cells were harvested, stimulated with IL-12 for the indicated time points, and gene expression was examined by qRT-PCR (A, C) or used for STAT4 binding, chromatin modifying enzymes and histone modification analyses by ChIP assay using qPCR primers specific for the promoters of the indicated genes (B, D). Data are average of replicate samples ± S.D. and representative of three independent experiments with similar results. Gene names are indicated adjacent to the corresponding line (A, B) representing mRNA expression or ChIP results at the promoter.
To further define how STAT4 is required for IL-12-induced gene expression we examined the expression of Th1 genes in wild type and Stat4−/− cultures following IL-12 stimulation. In the absence of STAT4, Th1 cells have lower basal expression of Th1 genes, and IL-12-induced expression is completely absent (Fig. 1C). Since H3K4 tri-methylation and H3K27 tri-methylation are correlated with active and repressed gene expression respectively (36), we wanted to examine these modifications at Th1 gene loci in Stat4−/− Th1 cells. H3K4 methylation is induced by IL-12, and the induction is attenuated in Stat4−/− Th1 cells (Fig. 1D). In contrast, H3K27 methylation is increased in STAT4-deficient Th1 cultures, compared to wild type cultures, and the amounts of methylation increased upon IL-12 stimulation (Fig. 1D). This was concomitant with decreased association of the H3K27 demethylase Jmjd3 and increased association of Dnmt3a (Fig. 1D). Dnmt3a associates with methylated H3K27, and genomic localization is inversely correlated with trimethyl-H3K4 (14, 37–40). Although our previous results suggested that Dnmt3a demonstrated increased association with Th1 loci in the absence of STAT4, these results suggest that IL-12-induced STAT4 reciprocally, and perhaps sequentially, modulates Jmjd3 and Dnmt3a association at Th1 gene loci.
Jmjd3 facilitates IL-12-induced gene expression
To determine if Jmjd3 contributes to IL-12-induced gene expression in Th1 cells, we transfected wild type Th1 cells with control orJmjd3-specific siRNA. We used intracellular staining to assess Jmjd3 expression and observed decreased mean fluorescence intensity without any effects on cell viability (Fig. 2A-B). Reducing Jmjd3 expression resulted in diminished IFNγ production after anti-CD3 or IL-12 stimulation (Fig. 2C-D). Moreover, Jmjd3 siRNA reduced IL-12-induced Th1 gene expression (Fig. 2D). Transfection of Th1 cells with Jmjd3-specific siRNA resulted in decreased Jmjd3 associated with Th1 cytokine loci, and a corresponding decrease in H3K4 tri-methylation and increase in H3K27 tri-methylation (Fig. 2E). Decreasing Jmjd3 expression also increased Dnmt3a association with Th1 gene loci (Fig. 2E). STAT4 binding at Th1 gene loci were not altered by Jmjd3-specific siRNA (Fig. 2E). These results support a pathway in which IL-12 induces STAT4-dependent H3K4 tri-methylation and Jmjd3 association with Th1 gene loci, which then decreases H3K27 tri-methylation, and limits Dnmt3a association with target loci.
Figure 2. IL-12-induced gene expression requires Jmjd3.

(A-F) Naïve CD4+CD62L+ T cells were isolated from C57BL/6 mice and cultured under Th1 polarizing conditions. (A-D) On day 5, cells was harvested, transfected with control or Jmjd3-specific siRNA, rested overnight and stimulated with anti-CD3 for 6 h for Jmjd3 (with average of mean fluorescence intensity) and viability analyses by intracellular staining (A-B) or 24 h to measure cytokine production by ELISA (C). Transfected cells were stimulated with IL-12 for gene expression analysis by qRT-PCR (D) or chromatin modifying enzymes, histone modification and STAT4 binding by ChIP assay using primers specific for the promoters of the indicated genes (E). Data are average of three mice ± S.D. (A-C) or are average of replicated samples ± S.D. and representative of three independent experiments with similar results (D-E) *p<0.05. NS not significant
Dnmt3a negatively regulates Th1 gene expression
Based on the increased association of Dnmt3a with Th1 gene loci in the absence of STAT4, we tested whether elimination of a negative regulator, Dnmt3a, would rescue Th1 differentiation in Stat4−/− T cells. We mated Stat4−/− mice with Dnmt3afl/fl CD4-Cre mice to generate compound mutant mice. Naïve CD4+ T cells were isolated from wild type, Dnmt3afl/fl CD4-Cre positive and Stat4−/−Dnmt3afl/fl mice that were Cre-negative (STAT4-deficient, but expressing Dnmt3a) or Cre-positive (STAT4- and Dnmt3a-deficient) and differentiated under Th1, Th17 and regulatory T cells (iTreg) polarizing conditions. STAT4-deficient Th1 cells had diminished IFNγ production compared to wild type cultures when stimulated with anti-CD3 or PMA and Ionomycin, although there was no significant effect on TNFα production (Fig. 3A-C). We have previously seen that Dnmt3a-deficiency resulted in modest increases in IFNγ production (15)(Fig. 3C). However, Th1 cells deficient in both STAT4 and Dnmt3a demonstrated greater production of IFNγ than Stat4−/− cells, assessed early using intracellular staining (Fig. 3A-B). Recovery of IFNγ production was comparable to wild type cells in response to PMA and Ionomycin, but was only partially restored when cells were stimulated with anti-CD3 (Fig. 3A-C). This is consistent with the ability of PMA and Ionomycin stimulation to overcome some of the effects of STAT4-deficiency (4, 21). Th17 cell differentiation and ex vivo regulatory T cells (nTreg) were normal in cells deficient in both STAT4 and Dnmt3a compared to wild type cells in terms of IL-17A production (Th17) and the percentage of CD25+Fopx3+ cells (nTreg) (Fig. 3D-E). In vitro derived Treg cells from single Dnmt3a-deficient or double STAT4-and Dnmt3a-deficient naïve T cells demonstrated increased differentiation to Foxp3-expressing cells, compared to wild type cells (Fig. 3F).
Figure 3. Dnmt3a is a negative regulator of Th1 genes.
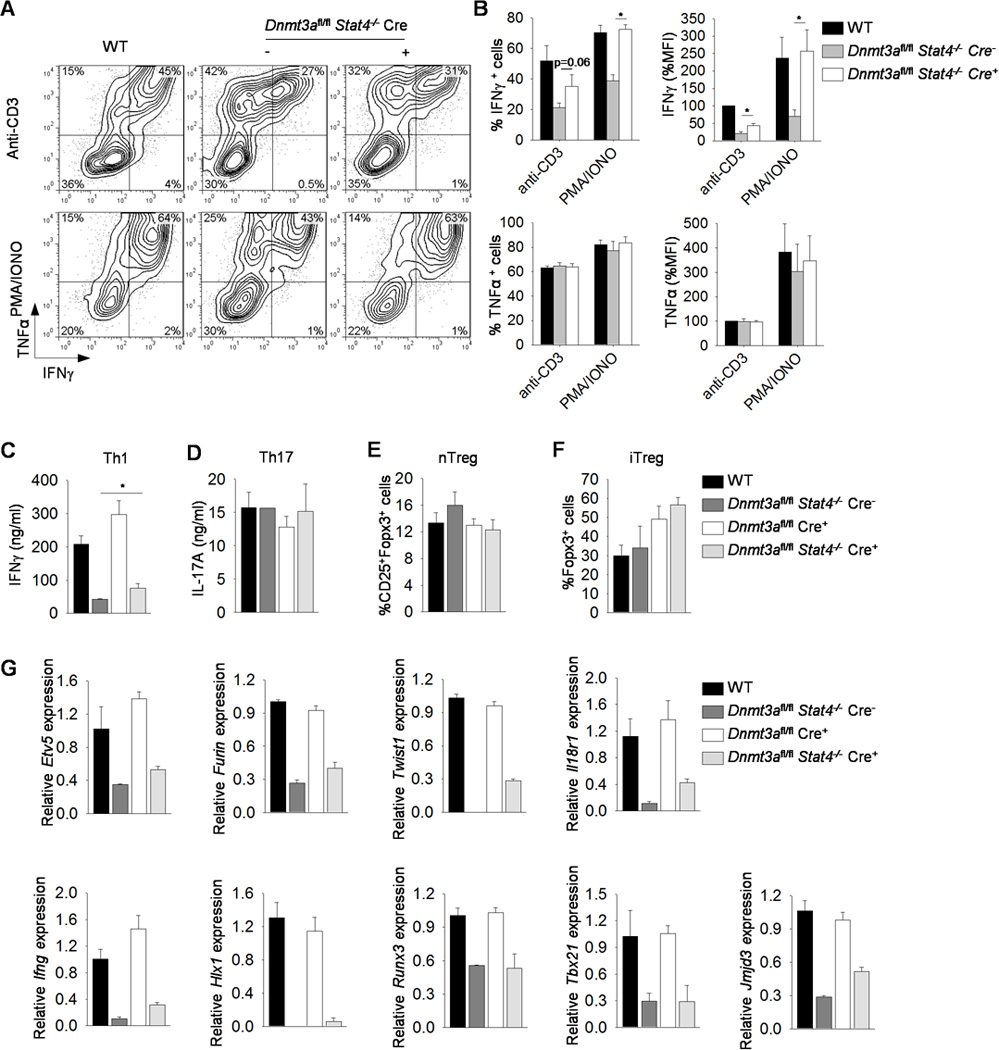
(A-G) Naïve CD4+CD62L+ T cells were isolated from WT, Dnmt3afl/fl CD4-Cre positive or Dnmt3afl/fl Stat4−/− CD4-Cre negative (−) or positive (+) mice and cultured under Th1, Th17 or iTreg polarizing conditions. (A-B) On day 5, Th1 cells were harvested, activated with anti-CD3 or PMA and Ionomycin for 6 h before assessing cytokine production by ICS, B, Averages of percent positive cells and mean fluorescence intensity for data in (A). (C-D) Day 5 Th1 and Th17 cells were harvested and activated with anti-CD3 for 24 h before assessing cytokine production by ELISA. (E-F) Splenocytes isolated from mice with indicated genotypes or day 5 in vitro generated regulatory T (Treg) cells were assessed for natural (nTreg, CD25+Foxp3+) (E) or inducible (Foxp3+) (F) Treg cells by intracellular staining. Data is gated on CD4+ cells. (G) Day 5 Th1 cells were used to examine gene expression by qRT-PCR before (Etv5, Furin, Twist1, Il18r1, Hlx1, Runx3, Tbx21 and Jmjd3) or after (Ifng) anti-CD3 reactivation. Data are average ± S.D. of three independent experiments (A-F) or average of replicated samples ± S.D. and representative of three independent experiments with similar results (G). *p<0.05
We then examined several Th1 genes to determine if Dnmt3a had a similar negative effect on expression. Stat4−/− Th1 cells had diminished Th1 gene expression while Dnmt3a-deficiency resulted in modest increases in IFNγ production but minimal effects on the expression of other Th1 genes (15)(Fig. 3G). We observed partial recovery of Etv5, Furin, Twist1, Il18r1 and Jmjd3 expression in Th1 cells deficient in both STAT4 and Dnmt3a, compared to wild type and Stat4−/− Th1 cultures (Fig. 3G). However, there was no recovery of Hlx1, Runx3, or Tbx21 expression (Fig. 3G).
Since the recovery in STAT4-dependent IFNγ production and Th1 gene expression was only partial with simultaneous Dnmt3a deficiency, we wanted to determine if this effect was sufficient to enhance inflammation in vivo. To test this, we used the myelin oligodendrocyte glycoprotein (MOG)-induced EAE model to compare the level of disease in wild type, Dnmt3afl/flCre-positive Dnmt3afl/flStat4−/− Cre-negative and Dnmt3afl/flStat4−/−Cre-positive mice. Disease development in wild type and Dnmt3afl/flCre-positive mice was indistinguishable, consistent with a minimal effect of Dnmt3a-deficiency on Th1 development in vitro. In agreement with previous reports (41, 42), mice deficient in STAT4 had minimal disease that developed much later than disease in wild type mice (Fig. 4B). However, Dnmt3afl/flStat4−/−Cre-positive mice demonstrated onset and paralysis that was intermediate to disease in wild type and Stat4−/− mice (Fig. 4B).The result correlated with the increased CD4+IFNγ+ and CD4+IFNγ+IL-17+ mononuclear cells isolated from brain in Dnmt3afl/flStat4−/−Cre-positive mice compared to Stat4-deficient mice (Fig. 4C). MOG-stimulated Dnmt3afl/flStat4−/−Cre-positive splenocytes produced significantly more IFNγ and GM-CSF compared to Stat4-deficient cells (Fig. 4D). Both Dnmt3afl/flStat4−/−Cre-positive and Stat4-deficient mice had higher CD4+IL-17+ mononuclear cells compared to wild type mice (Fig. 4C). Similarly, MOG-stimulated Dnmt3afl/flStat4−/−Cre-positive and Stat4-deficient splenocytes produced more IL-17A compared to wild type cells (Fig. 4D).Thus, in the absence of Dnmt3a and STAT4, there is a partial recovery in inflammatory T cell function.
Figure 4. Mice with double deficiency in STAT4 and Dnmt3a had partial recovery in inflammatory T cell function.
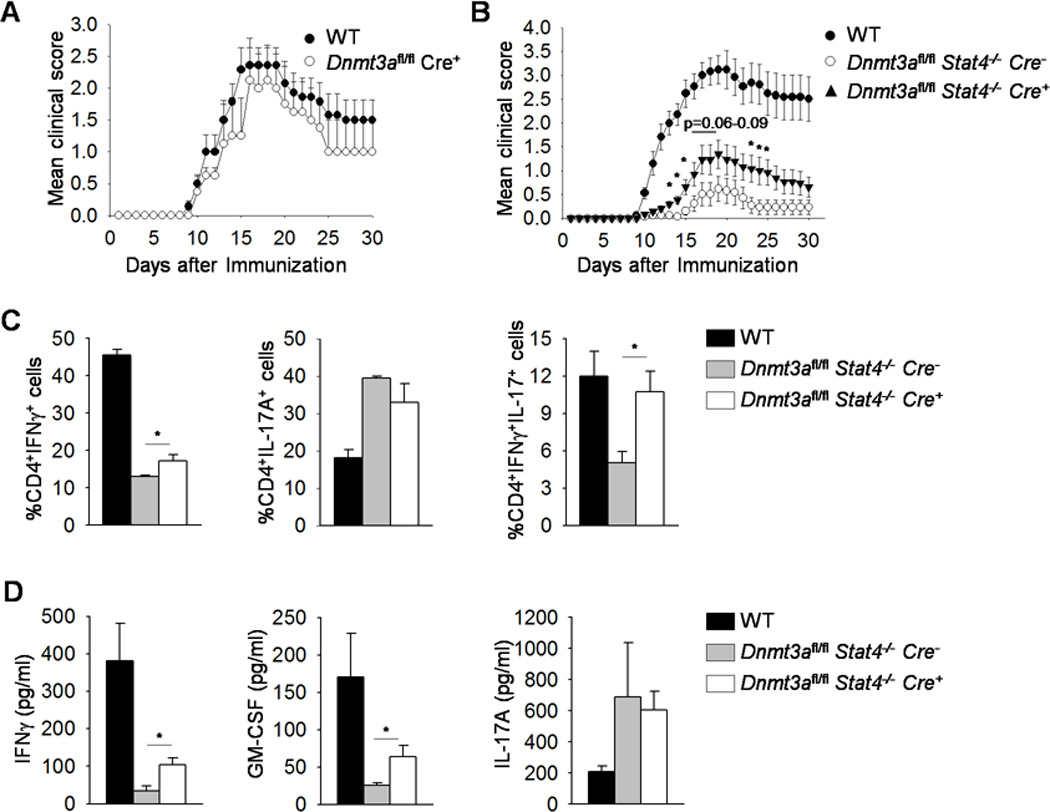
(A-B) Mean clinical score of MOG peptide (35–55)-induced EAE in WT, Dnmt3afl/fl CD4-Cre positive (A) or in WT, Dnmt3afl/fl Stat4−/− CD4-Cre negative or positive mice scored daily for 30 days (B). (C-D) Mice were sacrificed on day 14 and mononuclear cells were isolated from brain and stimulated with PMA and Ionomycin for 6 h before staining for cytokine production (C) or isolated splenocytes were stimulated with MOG peptide for 48 h and cytokine production was measured using ELISA (D). Data are average ± S.E.M. of two independent (A-B, n=6–10 mice/group/experiment) or average ± S.E.M. of 4 mice (C-D). *p<0.05 comparing Dnmt3afl/fl Stat4−/− CD4-Cre negative or positive samples.
To further define how Dnmt3a was affecting Th1 gene expression, we examined acute IL-12 induced gene expression and histone modification. Although deficiency in Dnmt3a increased the basal level of gene expression in STAT4-deficient T cells following Th1 differentiation (Fig. 3A-C, 3G and 5A), we did not observe a rescue of IL-12-induced gene expression in the absence of Dnmt3a (Fig. 5A), suggesting that other STAT family members were not compensating for the function of STAT4. Histone modifications associated with activated or repressed genes were also altered at five regulatory elements across the Ifng locus, and at the Hlx1 promoter. The amount of H3K27me3 at each site of the Ifng locus, and the Hlx1 promoter was increased in the absence of STAT4 (Fig. 5B-C). At three of the five sites in the Ifng locus and at the Hlx1 promoter, deficiency of Dnmt3a and STAT4 decreased H3K27me3 to amounts close to those in wild type cells (Fig. 5B-C). Conversely, H3K4me3 was decreased at the Ifng locus and the Hlx1 promoter in the absence of STAT4. In Th1 cultures deficient in Dnmt3a and STAT4, three of the five sites in the Ifng locus, but not the Hlx1 promoter, showed H3K4me3 amounts increased from Stat4−/− cells, although not to amounts seen in wild type cells. The results are consistent with previous reports of STAT4 binding to the Ifng locus (22) (Fig. 5A-C).
Figure 5. Gene expression, histone modification, and transcription factor binding in the absence of Dnmt3a.
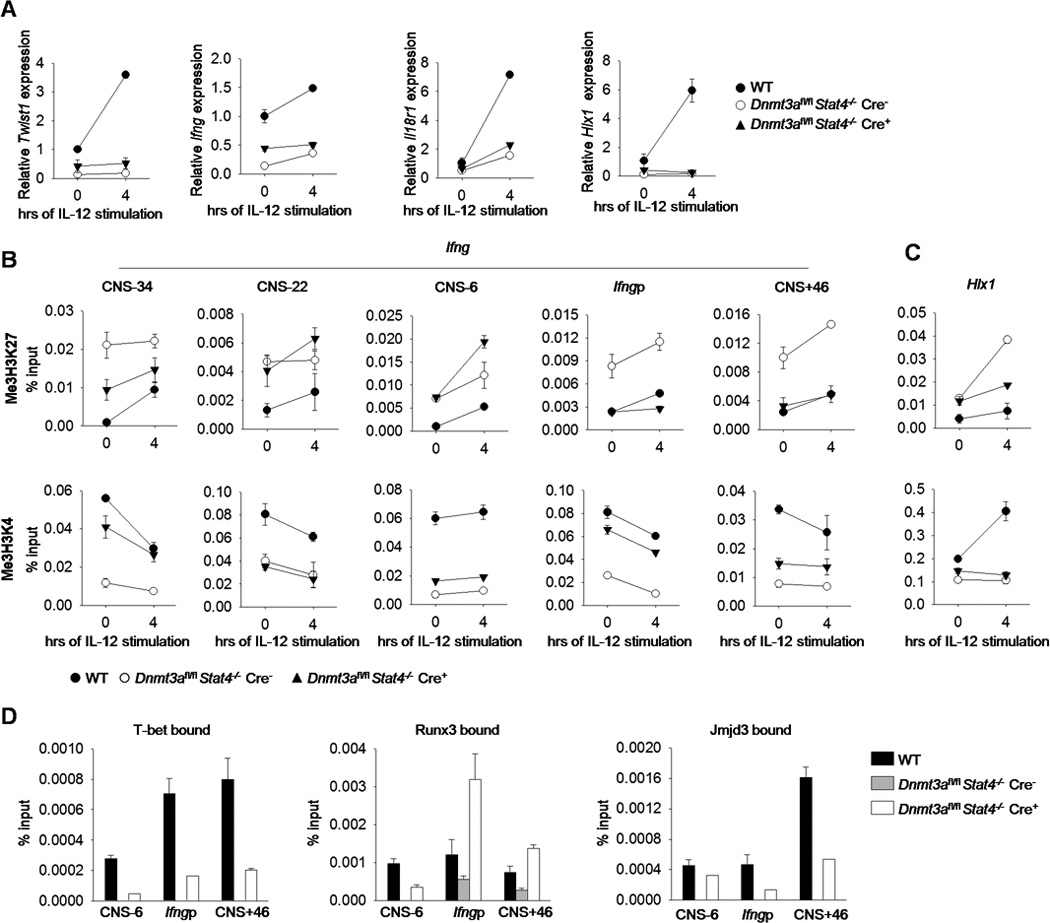
(A-D) Naïve CD4+CD62L+ T cells were isolated from WT, Dnmt3afl/fl Stat4−/− CD4-Cre negative or positive mice and cultured under Th1polarizing conditions. On day 5, cells were harvested and stimulated with IL-12 for 4 hours before gene expression analysis by qRT-PCR (A) or histone modification analysis by ChIP assay at Ifng regulatory elements (B) or the Hlx1 promoter (C). (D)T-bet, Runx3 and Jmjd3 bound to the Ifng locus was analyzed by ChIP assay in Th1 cells. Data are average of replicated samples ± S.D. and representative of three independent experiments with similar results.
Transcription factor regulation of Ifng in the absence of STAT4 and Dnmt3a
We then wanted to test whether Dnmt3a deficiency affected transcription factor binding at the Ifng locus using chromatin immunoprecipitation. At three sites that are known T-bet and Runx3 binding regions, we observed diminished or absent binding in Stat4−/− Th1 cells, compared to wild type cells (Fig. 5D). However, in Th1 cultures of cells lacking both STAT4 and Dnmt3a there was a partial recovery of binding by T-bet, and complete recovery of Runx3 binding at two of the three sites (Fig. 5D). Since the T-bet-Jmjd3 interaction is required for Ifng remodeling in differentiated Th1 cells (43), we then examined the binding of Jmjd3 at the Ifng locus. Paralleling T-bet binding, the binding of Jmjd3 at the Ifng locus was partially recovered in Th1 cultures of cells lacking both STAT4 and Dnmt3a, compared to Stat4−/− cells (Fig. 5D). This suggested that at least some of the recovery of Ifng expression in Dnmt3afl/flStat4−/−Cre-positive Th1 cells was due to increased binding of Ifng-inducing transcription factors and histone modifying enzymes.
In addition to decreased binding of factors to the Ifng locus, there was also decreased expression of several factors required for Ifng expression including Hlx1, Runx3 and Tbx21 in the absence of STAT4 (Fig. 3G). To determine if a combination of decreased Dnmt3a function and ectopic Th1 transcription factor expression would completely rescue IFNγ production, we used retroviral transduction to introduce transcription factor expression into Dnmt3afl/flStat4−/−Cre-positive and Cre-negative Th1 cultures. Transduction of either Hlx1 or Tbx21 had modest effects on IFNγ production and no effects of TNFα production (Fig. 6A-B). Transduction of Runx3 resulted in some recovery of IFNγ production from Dnmt3afl/flStat4−/−Cre-negative Th1 cells, but only modest effects on IFNγ production from Dnmt3afl/flStat4−/−Cre-positive Th1 cells (Fig. 6C-D). We reasoned that since Hlx1 expression showed no recovery in Dnmt3afl/flStat4−/−Cre-positive Th1 cells compared to Dnmt3afl/flStat4−/−Cre-negative Th1 cells (Fig. 3G), and since T-bet binding was still diminished in Dnmt3afl/flStat4−/−Cre-positive Th1 cells compared to Dnmt3afl/flStat4−/−Cre-negative Th1 cells (Fig. 5D), double-deficient Th1 cells might be especially sensitive to ectopic expression of these two factors. Thus, we transduced Dnmt3afl/flStat4−/− Cre-positive and -negative Th1 cells with retroviruses expressing T-bet and Hlx1. We observed that Dnmt3afl/flStat4−/−Cre-negative Th1 cells demonstrated induction of IFNγ production, but that Dnmt3afl/flStat4−/−Cre-positive Th1 cells demonstrated an even greater fold induction in IFNγ production (Fig. 6E-F). These results demonstrate that Dnmt3a represses gene expression in the absence of STAT4, and that in the absence of both factors, the Ifng locus is de-repressed and more sensitive to induction by additional Ifng trans-activators. Together, these studies reveal the negative regulatory role of Dnmt3a in Th1 gene expression.
Figure 6. Ectopic Th1 transcription factor expression rescues IFNγ production.
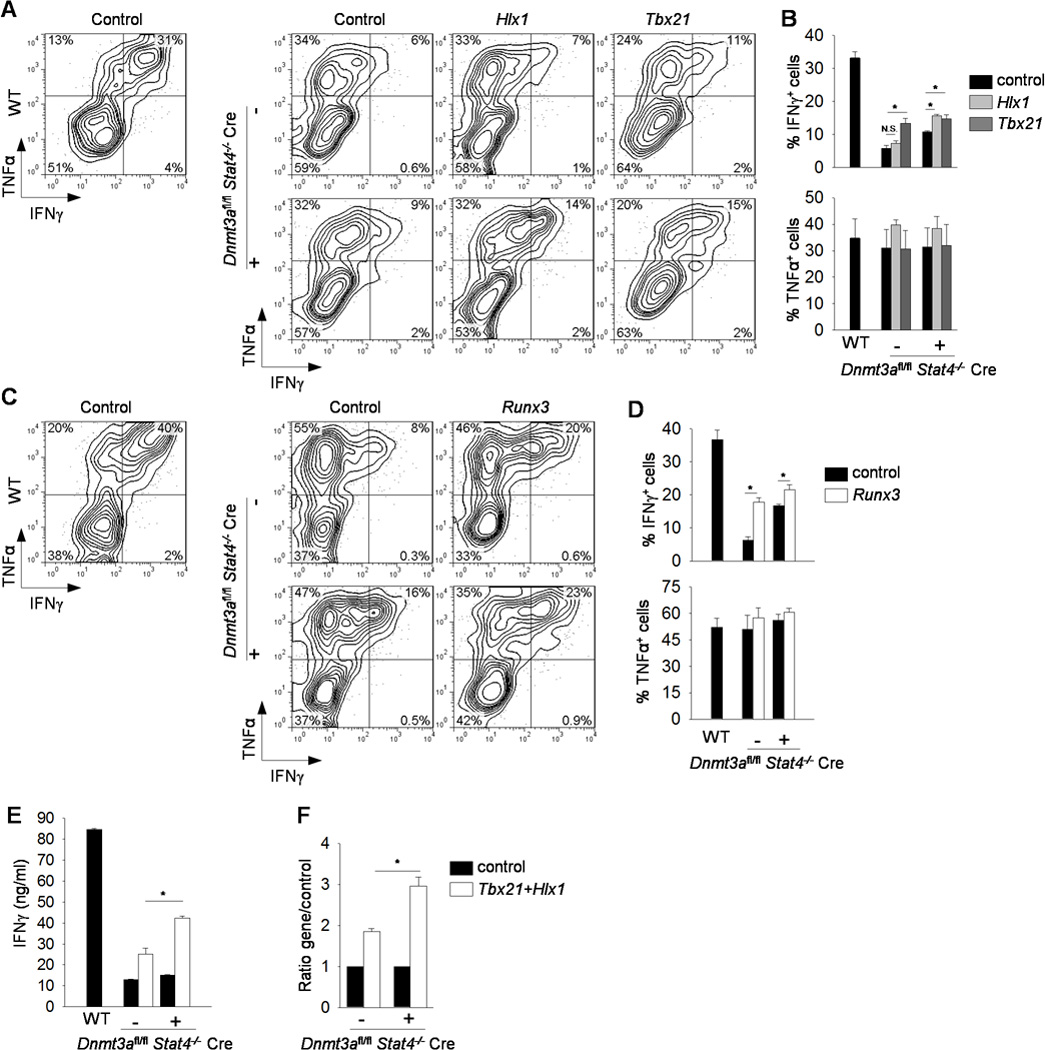
Naïve CD4+CD62L+ T cells were isolated from WT, Dnmt3afl/fl Stat4−/− CD4-Cre negative (−) or positive (+) mice and cultured under Th1polarizing conditions. On day 2, cells were transduced with retrovirus expressing MIEG-EGFP (MIEG), MIEG Hlx1-EGFP (Hlx1), or MIEG T-bet-EGFP (Tbx21) (A-B), or control vector Thy1.1 or Runx3-thy1.1 (Runx3) (C-D), or both MIEG T-bet-EGFP (Tbx21) and MSCV Hlx1-Thy1.1 (Hlx1) or matching controls (E-F). After five days of differentiation, Th1 cells were activated with anti-CD3 for 6 h before measuring cytokine production by ICS. Representative plots gated on transduced cells (A, C) with average percentage of positive cells indicated in bar graphs (B, D). (E-F) After five days of differentiation, doubly transduced Th1 cells were sorted, reactivated with anti-CD3 for 24 h, and cytokine production was measured by ELISA (E) with the fold induction in cytokine production between Dnmt3afl/fl Stat4−/− CD4-Cre negative and positive transduced Th1 cells compared to its control cells (F). Data are average ± S.D. of three independent experiments.*p<0.05.
Discussion
Although STAT4 is a critical factor in the development of Th1 cells and inflammatory immunity, a detailed understanding of how STAT4 programs gene expression has not been well documented. In this report we define a pathway for the STAT4-dependent induction of Th1 gene expression. STAT4 binds to target loci and recruits histone acetyltransferases that mediate total histone acetylation (14, 23) and acetylaton of specific histone residues, including H3K9/18, H3K36 and H3K27 (data not shown). STAT4 is required for the IL-12-inducible H3K4 methylation, and association of Jmjd3 with target loci (Fig. 1). Diminished Jmjd3 expression results in decreased Th1 gene induction increased H3K27 methylation, and increased Dnmt3a association with target loci (Fig. 2). This parallels data from Th2 cells that display decreased STAT4 expression accompanied by even greater Dnmt3a association and DNA methylation (14, 24). STAT4 is required for the expression of several other transcription factors that contribute to Th1 gene expression including Hlx1. Despite decreased expression of several of these factors in Stat4−/− cells, double deficiency of STAT4 and Dnmt3a results in a partial increase in Th1 gene expression compared to Stat4−/− Th1 cultures. These results demonstrate that Dnmt3a plays an obligate role in repressing Th1 gene expression that is attenuated by the activity of STAT4. Thus, STAT4 functions by facilitating increased histone acetylation and H3K4 methylation to induce gene expression, and by decreasing the association of DNA methyltransferases, in a Jmjd3-dependent mechanism, that repress gene expression.
Disease models can be complex and rely upon the balance of pro- and anti-inflammatory effector cells. EAE requires the function of Th1 and Th17 cells, and is inhibited by Treg cells (44). Partial recovery of the Th1 phenotype in Dnmt3afl/flStat4−/−Cre-positive cultures was recapitulated in an EAE disease model where Dnmt3afl/flStat4−/−Cre-positive mice had increased clinical disease and IFNγ production compared to Stat4−/− mice. The contribution of Th17 cells to EAE in Dnmt3afl/flStat4−/−Cre-positive mice is not limiting since there were no changes in IL-17-producing mononuclear cells, and Dnmt3afl/flStat4−/−Cre-positive T cells had similar in vitro Th17 differentiation to Stat4−/− cultures. Although DNA methyltransferases have been reported to play an important role in the generation of regulatory T cells (45), and in vitro derived Dnmt3afl/fl Cre-positive iTreg cultures had increased percentages of Fopx3+ cells compared to wild type cultures, Dnmt3afl/fl Cre-positive mice displayed normal EAE development (Fig. 4A). IL-12 has also been shown to limit the development of iTreg cells (46), although in the EAE inflammatory environment, there are many other cytokines that can modulate Foxp3 and compensate for the lack of STAT4. Moreover, if Tregs were increased in Dnmt3afl/flStat4−/−Cre-positive mice, we would not expect to observe increased disease. Together, these observations suggest the primary compensatory role of Dnmt3a-deficiency in Stat4−/− mice is recovery of Th1 function.
STAT4 is a multi-functional transcription factor with the ability to acutely activate gene expression in a short time span (within hours) and to program genes for lineage expression (lasting days). These intertwined functions likely involve both the chromatin remodeling mechanisms we have described here with the direct induction of genes via RNA polymerase II-dependent transcription. Although STAT4 can activate reporter genes in transient assays, suggesting a direct effect on rate of transcription, how it mediates these functions is not yet known. Importantly, despite the ability of IL-12 to activate several STAT proteins (47, 48), none can substitute for STAT4 in the acute IL-12-induction of gene expression (Fig. 1) or in IL-12-induced Th1 gene programming (21). This suggests that STAT4 specifically interacts with other transcription factors at target loci to mediate gene induction. The specificity of these interactions might also distinguish the effects of STAT4 on the IFN-inducible genes, where there is only transient gene induction, from genes that are programmed for expression in committed Th1 cells. The identity of some of these interacting factors is still unclear. STAT4 can interact with Jun family members (23, 49) and co-operation with AP-1 complexes is possible. Previous work defining STAT4-interacting proteins identified a LIM domain-containing protein that regulated STAT4 stability and a tyrosine phosphatase that regulated STAT4 phosphorylation (50, 51). Other interacting transcription factors have not been identified.
Although there is no evidence yet for physical interactions with other Th1-inducing transcription factors, it is clear that STAT4 functions in a network with these factors in Th1 cells. T-bet is the most obvious, where STAT4 induces expression of Tbx21 and cooperates with T-bet in the induction of a subset of Th1 genes (8, 10, 11). Among the genes that both STAT4 and T-bet are required for, Runx3 and Hlx1 also contribute to expression of Th1 genes (12, 13). In this report we tested the concept that in the absence of STAT4 acting as a positive regulator, eliminating a negative regulatory factor would lead to recovery of gene expression and differentiation. We observed a partial recovery of phenotype in vitro and function in vivo (Fig. 4-6), suggesting that STAT4 has functions in addition to elimination of Dnmt3a association. Among those functions, STAT4 regulates chromatin remodeling in the absence of T-bet (26), and the induction of a negative regulatory loop that requires the transcription factor Twist1 (33, 52). Ectopic expression of Runx3 or a combination of Tbx21 and Hlx1 were able to induce Th1 gene expression in Dnmt3afl/flStat4−/−Cre-positive Th1 cultures to a greater extent than in Stat4−/− Th1 cultures, although still not to wild type expression (Fig. 6). It is possible that reconstitution of all three factors would lead to complete recovery. However, it is also likely that STAT4 plays an indispensible role for initiating transcription at many Th1 target loci (21), and not simply as an initial activator of the Th1 transcriptional network.
The cooperation of STAT4 and T-bet may be required for some of the effects observed in this report, since T-bet is also required for Jmjd3 recruitment to target loci (43, 53). Indeed, we observed association of T-bet and Jmjd3 at the Ifng locus was increased in Dnmt3afl/flStat4−/−Cre-positive Th1 cells, compared to Stat4−/− Th1 cultures (Fig. 5). However, Dnmt3a association with Th1 loci was increased more in Stat4−/− Th1 cells than in Tbx21−/− Th1 cells, suggesting that STAT4 has T-bet/Jmjd3-independent mechanisms to limit Dnmt3a association (11). Since Dnmt3a interacts with unmethylated H3K4 and tri-methyl-H3K36, an additional possibility is STAT4-dependent recruitment of H3K4 methylases and H3K36 acetylases or demethylases (14, 37–40). Ultimately, STAT4 likely limits DNA methylation by recruiting several chromatin modifying enzyme complexes.
In this report, we have demonstrated that Dnmt3a plays an obligate role in the repression of Th1 genes that is attenuated by STAT4. Even in the absence of STAT4 and other transcriptional activators, genetic loss of Dnmt3a results in de-repression of a subset of Th1 genes, and a partial increase in expression that is sufficient to observe a modest recovery of STAT4-dependent inflammatory disease. In the de-repressed state, the Ifng locus becomes more responsive to ectopic expression of other transcription factors, providing an additional mechanism through which STAT4 cooperates with other factors in the appropriate expression of Th1 genes.
Acknowledgements
We thank En Li for permission to obtain the Dnmt3a-conditonal mutant mice.
This work was supported by NIH R01 AI045515. DP was supported by NIH T32 grant HL007910; QY was supported by NIH T32 grants CA111198 and HL091816.
References
- 1.Kaplan MH. STAT4: A critical regulator of inflammation in vivo. Immunologic Research. 2005;32:231–241. doi: 10.1385/IR:31:3:231. [DOI] [PubMed] [Google Scholar]
- 2.Watford WT, Hissong BD, Bream JH, Kanno Y, Muul L, O'Shea JJ. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol Rev. 2004;202:139–156. doi: 10.1111/j.0105-2896.2004.00211.x. [DOI] [PubMed] [Google Scholar]
- 3.Hsieh C-S, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Development of Th1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 4.Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, Murphy TL, Murphy KM. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol. 2002;3:549–557. doi: 10.1038/ni794. [DOI] [PubMed] [Google Scholar]
- 5.Wenner CA, Güler ML, Macatonia SE, O'Garra A, Murphy KM. Roles of IFN-γ and IFN-α in IL-12-induced T helper cell-1 development. J Immunol. 1996;156:1442–1447. [PubMed] [Google Scholar]
- 6.Kaplan MH, Sun Y-L, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 7.Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, Sangster MY, Vignali DAA, Doherty PC, Grosveld GC, Ihle JN. Requirement for Stat4 in interleukin-12 mediated responses of natural killer and T cells. Nature. 1996;382:171–174. doi: 10.1038/382171a0. [DOI] [PubMed] [Google Scholar]
- 8.Yang Y, Ochando JC, Bromberg JS, Ding Y. Identification of a distant T-bet enhancer responsive to IL-12/Stat4 and IFNgamma/Stat1 signals. Blood. 2007;110:2494–2500. doi: 10.1182/blood-2006-11-058271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lighvani AA, Frucht DM, Jankovic D, Yamane H, Aliberti J, Hissong BD, Nguyen BV, Gadina M, Sher A, Paul WE, O'Shea JJ. T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proc Natl Acad Sci U S A. 2001;98:15137–15142. doi: 10.1073/pnas.261570598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schulz EG, Mariani L, Radbruch A, Hofer T. Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-gamma and interleukin-12. Immunity. 2009;30:673–683. doi: 10.1016/j.immuni.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 11.Thieu VT, Yu Q, Chang HC, Yeh N, Nguyen ET, Sehra S, Kaplan MH. Signal transducer and activator of transcription 4 is required for the transcription factor T-bet to promote T helper 1 cell-fate determination. Immunity. 2008;29:679–690. doi: 10.1016/j.immuni.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, Ansel KM. Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat Immunol. 2007;8:145–153. doi: 10.1038/ni1424. [DOI] [PubMed] [Google Scholar]
- 13.Mullen AC, Hutchins AS, High FA, Lee HW, Sykes KJ, Chodosh LA, Reiner SL. Hlx is induced by and genetically interacts with T-bet to promote heritable T(H)1 gene induction. Nat Immunol. 2002;3:652–658. doi: 10.1038/ni807. [DOI] [PubMed] [Google Scholar]
- 14.Yu Q, Thieu VT, Kaplan MH. Stat4 limits DNA methyltransferase recruitment and DNA methylation of the IL-18Ralpha gene during Th1 differentiation. EMBO J. 2007;26:2052–2060. doi: 10.1038/sj.emboj.7601653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu Q, Zhou B, Zhang Y, Nguyen ET, Du J, Glosson NL, Kaplan MH. DNA methyltransferase 3a limits the expression of interleukin-13 in T helper 2 cells and allergic airway inflammation. Proc Natl Acad Sci U S A. 2012;109:541–546. doi: 10.1073/pnas.1103803109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pekowska A, Benoukraf T, Zacarias-Cabeza J, Belhocine M, Koch F, Holota H, Imbert J, Andrau JC, Ferrier P, Spicuglia S. H3K4 tri-methylation provides an epigenetic signature of active enhancers. EMBO J. 2011;30:4198–4210. doi: 10.1038/emboj.2011.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kouzarides T. SnapShot: Histone-modifying enzymes. Cell. 2007;131:822. doi: 10.1016/j.cell.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 18.Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, Pillus L, Reinberg D, Shi Y, Shiekhattar R, Shilatifard A, Workman J, Zhang Y. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–636. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 19.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 20.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annual review of biochemistry. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 21.Good SR, Thieu VT, Mathur AN, Yu Q, Stritesky GL, Yeh N, O'Malley JT, Perumal NB, Kaplan MH. Temporal induction pattern of STAT4 target genes defines potential for Th1 lineage-specific programming. J Immunol. 2009;183:3839–3847. doi: 10.4049/jimmunol.0901411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei L, Vahedi G, Sun HW, Watford WT, Takatori H, Ramos HL, Takahashi H, Liang J, Gutierrez-Cruz G, Zang C, Peng W, O'Shea JJ, Kanno Y. Discrete roles of STAT4 and STAT6 transcription factors in tuning epigenetic modifications and transcription during T helper cell differentiation. Immunity. 2010;32:840–851. doi: 10.1016/j.immuni.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Sullivan A, Chang HC, Yu Q, Kaplan MH. STAT4 is required for interleukin-12-induced chromatin remodeling of the CD25 locus. J Biol Chem. 2004;279:7339–7345. doi: 10.1074/jbc.M309979200. [DOI] [PubMed] [Google Scholar]
- 24.Yu Q, Chang HC, Ahyi AN, Kaplan MH. Transcription factor-dependent chromatin remodeling of Il18r1 during Th1 and Th2 differentiation. J Immunol. 2008;181:3346–3352. doi: 10.4049/jimmunol.181.5.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Letimier FA, Passini N, Gasparian S, Bianchi E, Rogge L. Chromatin remodeling by the SWI/SNF-like BAF complex and STAT4 activation synergistically induce IL-12Rbeta2 expression during human Th1 cell differentiation. Embo J. 2007;26:1292–1302. doi: 10.1038/sj.emboj.7601586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang F, Boothby M. T helper type 1-specific Brg1 recruitment and remodeling of nucleosomes positioned at the IFN-gamma promoter are Stat4 dependent. J Exp Med. 2006;203:1493–1505. doi: 10.1084/jem.20060066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vahedi G, Takahashi H, Nakayamada S, Sun HW, Sartorelli V, Kanno Y, O'Shea JJ. STATs shape the active enhancer landscape of T cell populations. Cell. 2012;151:981–993. doi: 10.1016/j.cell.2012.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gamper CJ, Agoston AT, Nelson WG, Powell JD. Identification of DNA methyltransferase 3a as a T cell receptor-induced regulator of Th1 and Th2 differentiation. J Immunol. 2009;183:2267–2276. doi: 10.4049/jimmunol.0802960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas RM, Gamper CJ, Ladle BH, Powell JD, Wells AD. De novo DNA methylation is required to restrict T helper lineage plasticity. J Biol Chem. 2012;287:22900–22909. doi: 10.1074/jbc.M111.312785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mo C, Chearwae W, O'Malley JT, Adams SM, Kanakasabai S, Walline CC, Stritesky GL, Good SR, Perumal NB, Kaplan MH, Bright JJ. Stat4 isoforms differentially regulate inflammation and demyelination in experimental allergic encephalomyelitis. J Immunol. 2008;181:5681–5690. doi: 10.4049/jimmunol.181.8.5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang HC, Zhang S, Thieu VT, Slee RB, Bruns HA, Laribee RN, Klemsz MJ, Kaplan MH. PU.1 expression delineates heterogeneity in primary Th2 cells. Immunity. 2005;22:693–703. doi: 10.1016/j.immuni.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 32.Mathur AN, Chang HC, Zisoulis DG, Kapur R, Belladonna ML, Kansas GS, Kaplan MH. T-bet is a critical determinant in the instability of the IL-17-secreting T-helper phenotype. Blood. 2006;108:1595–1601. doi: 10.1182/blood-2006-04-015016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pham D, Vincentz JW, Firulli AB, Kaplan MH. Twist1 regulates ifng expression in Th1 cells by interfering with runx3 function. J Immunol. 2012;189:832–840. doi: 10.4049/jimmunol.1200854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang S, Aune TM. Dynamic changes in histone-methylation 'marks' across the locus encoding interferon-gamma during the differentiation of T helper type 2 cells. Nat Immunol. 2007;8:723–731. doi: 10.1038/ni1473. [DOI] [PubMed] [Google Scholar]
- 35.Schoenborn JR, Dorschner MO, Sekimata M, Santer DM, Shnyreva M, Fitzpatrick DR, Stamatoyannopoulos JA, Wilson CB. Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-gamma. Nat Immunol. 2007;8:732–742. doi: 10.1038/ni1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT, Schones DE, Peng W, Sun HW, Paul WE, O'Shea JJ, Zhao K. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30:155–167. doi: 10.1016/j.immuni.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li JY, Pu MT, Hirasawa R, Li BZ, Huang YN, Zeng R, Jing NH, Chen T, Li E, Sasaki H, Xu GL. Synergistic function of DNA methyltransferases Dnmt3a and Dnmt3b in the methylation of Oct4 and Nanog. Mol Cell Biol. 2007;27:8748–8759. doi: 10.1128/MCB.01380-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Otani J, Nankumo T, Arita K, Inamoto S, Ariyoshi M, Shirakawa M. Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX-DNMT3-DNMT3L domain. EMBO Rep. 2009;10:1235–1241. doi: 10.1038/embor.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen S, Ma J, Wu F, Xiong LJ, Ma H, Xu W, Lv R, Li X, Villen J, Gygi SP, Liu XS, Shi Y. The histone H3 Lys 27 demethylase JMJD3 regulates gene expression by impacting transcriptional elongation. Genes Dev. 2012;26:1364–1375. doi: 10.1101/gad.186056.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y, Jurkowska R, Soeroes S, Rajavelu A, Dhayalan A, Bock I, Rathert P, Brandt O, Reinhardt R, Fischle W, Jeltsch A. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 2010;38:4246–4253. doi: 10.1093/nar/gkq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chitnis T, Najafian N, Benou C, Salama AD, Grusby MJ, Sayegh MH, Khoury SJ. Effect of targeted disruption of STAT4 and STAT6 on the induction of experimental autoimmune encephalomyelitis. J Clin Invest. 2001;108:739–747. doi: 10.1172/JCI12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mo C, Chearwai W, O'Malley JT, Adams SM, Kanakasabai S, Walline CC, Stritesky GL, Good SR, Perumal NB, Kaplan MH, Bright JJ. Stat4 isoforms differentially regulate inflammation and demyelination in experimental allergic encephalomyelitis. J. Immunol. 2008;181:5681–5690. doi: 10.4049/jimmunol.181.8.5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller SA, Mohn SE, Weinmann AS. Jmjd3 and UTX play a demethylase-independent role in chromatin remodeling to regulate T-box family member-dependent gene expression. Mol Cell. 2010;40:594–605. doi: 10.1016/j.molcel.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunological reviews. 2012;248:205–215. doi: 10.1111/j.1600-065X.2012.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lal G, Zhang N, van der Touw W, Ding Y, Ju W, Bottinger EP, Reid SP, Levy DE, Bromberg JS. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol. 2009;182:259–273. doi: 10.4049/jimmunol.182.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Malley JT, Sehra S, Thieu VT, Yu Q, Chang HC, Stritesky GL, Nguyen ET, Mathur AN, Levy DE, Kaplan MH. Signal transducer and activator of transcription 4 limits the development of adaptive regulatory T cells. Immunology. 2009;127:587–595. doi: 10.1111/j.1365-2567.2008.03037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bacon CM, Petricoin EF, III, Ortaldo JR, Rees RC, Larner AC, Johnston JA, O'Shea JJ. Interleukin 12 induces tyrosine phosphorylation and activation of Stat4 in human lymphocytes. Proc Natl Acad Sci-USA. 1995;92:7307–7311. doi: 10.1073/pnas.92.16.7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jacobson NG, Szabo SJ, Weber-Nordt RM, Zhong Z, Schreiber RD, Darnell JE, Jr, Murphy KM. Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcripton (Stat)3 and Stat4. J Exp Med. 1995;181:1755–1762. doi: 10.1084/jem.181.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakahira M, Ahn HJ, Park WR, Gao P, Tomura M, Park CS, Hamaoka T, Ohta T, Kurimoto M, Fujiwara H. Synergy of IL-12 and IL-18 for IFN-gamma gene expression: IL-12-induced STAT4 contributes to IFN-gamma promoter activation by up-regulating the binding activity of IL-18-induced activator protein 1. J Immunol. 2002;168:1146–1153. doi: 10.4049/jimmunol.168.3.1146. [DOI] [PubMed] [Google Scholar]
- 50.Tanaka T, Soriano MA, Grusby MJ. SLIM is a nuclear ubiquitin E3 ligase that negatively regulates STAT signaling. Immunity. 2005;22:729–736. doi: 10.1016/j.immuni.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 51.Nakahira M, Tanaka T, Robson BE, Mizgerd JP, Grusby MJ. Regulation of signal transducer and activator of transcription signaling by the tyrosine phosphatase PTP-BL. Immunity. 2007;26:163–176. doi: 10.1016/j.immuni.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 52.Niesner U, Albrecht I, Janke M, Doebis C, Loddenkemper C, Lexberg MH, Eulenburg K, Kreher S, Koeck J, Baumgrass R, Bonhagen K, Kamradt T, Enghard P, Humrich JY, Rutz S, Schulze-Topphoff U, Aktas O, Bartfeld S, Radbruch H, Hegazy AN, Lohning M, Baumgart DC, Duchmann R, Rudwaleit M, Haupl T, Gitelman I, Krenn V, Gruen J, Sieper J, Zeitz M, Wiedenmann B, Zipp F, Hamann A, Janitz M, Scheffold A, Burmester GR, Chang HD, Radbruch A. Autoregulation of Th1-mediated inflammation by twist1. J Exp Med. 2008;205:1889–1901. doi: 10.1084/jem.20072468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miller SA, Huang AC, Miazgowicz MM, Brassil MM, Weinmann AS. Coordinated but physically separable interaction with H3K27-demethylase and H3K4-methyltransferase activities are required for T-box protein-mediated activation of developmental gene expression. Genes Dev. 2008;22:2980–2993. doi: 10.1101/gad.1689708. [DOI] [PMC free article] [PubMed] [Google Scholar]