

Abstract
The activity-regulated cytoskeletal protein Arc/Arg3.1 is required for long-term memory formation and synaptic plasticity. Arc expression is robustly induced by activity, and Arc protein localizes both to active synapses and the nucleus. While its synaptic function has been examined, it is not clear why or how Arc is localized to the nucleus. We found that murine Arc nuclear expression is regulated by synaptic activity in vivo and in vitro. We identified distinct regions of Arc that control its localization, including a nuclear localization signal, a nuclear retention domain, and a nuclear export signal. Arc localization to the nucleus promotes an activity-induced increase in promyelocytic leukemia nuclear bodies, which decreases GluA1 transcription and synaptic strength. Finally, we show that Arc nuclear localization regulates homeostatic plasticity. Thus, Arc mediates the homeostatic response to increased activity by translocating to the nucleus, increasing promyelocytic leukemia levels, and decreasing GluA1 transcription, ultimately downscaling synaptic strength.
INTRODUCTION
The molecular basis of learning and memory is the modification of neuronal synapses in response to electrical activity, a process termed synaptic plasticity. The activity-regulated cytoskeletal protein Arc/Arg3.1 is critical for long-term memory formation and essentially every form of plasticity, including long-term potentiation (LTP)1, 2, long-term depression (LTD)3, 4, and homeostatic scaling5, 6. In Arc/Arg3.1 knock-out (Arc−/−) mice, short-term learning is not affected, but consolidation and maintenance of memory are lost7.
Arc/Arg3.1 (hereafter referred to as Arc) is extremely highly regulated. It is an immediate-early gene, and basal levels of its mRNA and protein are low in neurons8, 9. Excitatory activity regulates Arc transcription9–11, mRNA localization12, 13, translation14, 15 and protein degradation16. Considerable work has focused on the localization of Arc mRNA to the synapse, but it is unknown whether activity independently regulates Arc protein localization. Arc protein is found at the synapse17, 18, but is even more highly localized to the nucleus19, and nothing is known about the mechanisms controlling Arc nuclear localization.
At the synapse, Arc regulates synaptic strength in several ways. It regulates spine number and type20 and decreases synaptic strength by promoting internalization of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs)21, 22. Such mechanisms may explain Arc’s involvement in synapse-specific forms of plasticity (e.g., LTP and LTD)4. However, they cannot as easily explain how Arc mediates the cell-wide changes in homeostatic plasticity that require regulation of the total levels of AMPAR subunits, such as GluA123. In addition, it is unclear how Arc switches from mediating responses to short-term stimuli that result in LTP and LTD to mediating responses to long-term changes in activity that result in homeostatic scaling.
While nothing is known about how Arc nuclear localization affects synaptic plasticity, Arc may have an important role in the nucleus. It promotes formation of promyelocytic leukemia tumor suppressor protein nuclear bodies (PML-NBs)19. These protein complexes are in most mammalian cell nuclei and regulate nuclear functions, such as transcription24, 25. They are found in the brain, are required for neural development26 and associate with dysfunctional proteins in neurodegenerative diseases27–30. However, if or how they function in neurons is not clear.
Here we examined Arc nuclear expression in vitro and in vivo and identified stimuli and domains that control nuclear localization. In the nucleus, Arc regulates synaptic strength and GluA1 transcription via an activity-induced increase in PML-NBs to promote downscaling of synaptic strength.
RESULTS
Arc is expressed in the nucleus after novel environment
We first determined if Arc is expressed in the nucleus in vivo after a physiological stimulus. Because proteins are synthesized in the cytoplasm and must translocate to the nucleus, we induced Arc expression by exposing mice to a novel environment for varying times. Mouse brains sections were stained for Arc with a highly specific antibody (Supplemental Fig. 1a) and the nuclear marker Hoechst. To quantify Arc expression, we calculated the ratio of nuclear to cytoplasmic Arc expression in the hippocampus and somatosensory cortex.
All regions showed a time-dependent increase in Arc localization to the nucleus. Under basal conditions, few cells expressed Arc. Those that did were in dentate gyrus and had Arc in both the nucleus and cytoplasm. The number of neurons expressing Arc increased rapidly after novel environment exposure (Supplemental Fig. 1b). After 30 minutes of novel environment exposure, Arc was primarily in the cytoplasm in all hippocampal regions. Nuclear expression was seen by 2 hours and increased over time. By 8 hours, more Arc was in nuclei than elsewhere in cells (Fig. 1a–c, Supplemental Fig. 1c–e). In dentate gyrus, the increase was gradual over 0.5–8 hours, whereas in CA3 and CA1, nuclear expression increased by 2 hours but increased no further until 8 hours (Supplemental Fig. 1f–h). In addition, we examined the somatosensory cortex. Arc was not detected until 2 hours, and its localization was considerably more heterogeneous (supplemental Fig. 1i–k). However, Arc still showed a time-dependent increase in nuclear localization, similar to that in the hippocampus.
Figure 1. Arc becomes enriched in neuronal nuclei in mice after novel environment exposure.

(a) Immunohistochemical staining of Arc and the Hoechst nuclear stain in coronal sections of mouse hippocampus after exposure to novel environment for 0–8 h.
(b) Higher magnification images of boxed regions from a.
(c) Quantification of the ratio of nuclear to cytoplasmic Arc expression in all regions of the hippocampus shows a time-dependent increase in Arc localization to the nucleus. N=50 at 0 h, 238 at 0.5 h, 345 at 2 h, 278 at 4 h, 215 at 6 h, and 78 at 8 h from two mice for each time point.
***, p<0.001 by ANOVA. Error bars show 95% confidence intervals. Scale bars=10 μm.
Two domains in Arc increase nuclear localization
Although Arc is small enough to possibly diffuse into the nucleus, the time-dependent concentration of Arc in nuclei suggests Arc nuclear localization is a regulated process. With the program PSORT, we determined that Arc had a possible Pat7 nuclear localization signal (NLS) at amino acids 331–335. We deleted this region from GFP-tagged Arc (GFP-Arc-ΔPat7) and found GFP-Arc-ΔPat7 predominantly in the cytoplasm (Fig. 2a, Supplemental 2a), indicating the Pat7 is required for nuclear localization. Deletion of the Pat7 from untagged Arc resulted in subtler decreases in nuclear localization (Supplemental Fig. 2b–f), measurable by the more sensitive quantification of nuclear:cytoplasmic ratio. Without the GFP tag, Arc is smaller and may diffuse into the nucleus. We mutated several of the potentially important amino acids in the Pat7 region. A single amino acid mutation of the first proline of the Pat7 (Arc-Pat7m1) similarly decreased in Arc localization to the nucleus while mutations of other basic amino acids in the Pat7 (Arc-Pat7m4 and Arc-Pat7m5) did not (Supplemental Fig. 2d).
Figure 2. Arc has Two regions that regulate nuclear enrichment.

(a) Representative images of hippocampal neurons transfected with full-length GFP-Arc or GFP-Arc containing a deletion of the C-terminal Pat7 NLS region show the Pat7 is necessary for nuclear localization.
(b) Representative images of Myc staining of neurons transfected with Myc-tagged regions of Arc show the N-terminal 77 amino acids are necessary for nuclear localization.
(c) Quantification of the percentage of neurons with Myc-Arc enriched in the nucleus. N=30 neurons from 3 independent cultures (10 neurons per culture).
(d) Representative images of Arc staining of neurons transfected with Arc constructs containing different 10 amino acid deletions in the N-terminal region show amino acids 29–68 are required for nuclear localization.
(e) Quantification of the percentage of neurons with Arc enriched in the nucleus. N=30 neurons from 3 independent cultures (10 neurons per culture).
*, p<0.05; ***; p<0.001; n.s., not significant, by ANOVA. Error bars show 95% confidence intervals. Scale bars=10 μm.
As an alternative approach, we used deletional analysis to find additional regulatory regions in Arc. We added a Myc epitope tag to Arc to assay subcellular distribution without significantly increasing its size. The first 77 amino acids of Arc (Myc-Arc-1–77) were expressed throughout the cell, as expected for a small protein. However, deletion of the N-terminal of Arc (Myc-Arc-77–396) decreased Arc in the nucleus (Fig. 2b, c). We then made a series of smaller deletions within the N-terminal of Arc abutting the boundaries of the coiled-coil domain. Each of the deletions between amino acids 29 and 68 depleted Arc from the nucleus (Fig. 2fd e), despite the presence of the Pat7 NLS.
To learn if either the Pat7 or N terminal region was sufficient to promote nuclear localization, we put them into a heterologous protein, GFP-tagged β-galactosidase (GFP-β-gal) (Supplemental Fig. 2g, h). GFP-β-gal is too large to diffuse into the nucleus and provides a stringent test of NLS function. GFP-Pat7-β-gal was expressed in nuclei significantly more than GFP-β-gal and to the same degree as GFP-Arc-β-gal, indicating that the Pat7 NLS is sufficient to actively target Arc to the nucleus. However, the N-terminal region was not a bona fide NLS because it failed to target GFP-β-galactosidase to the nucleus.
Arc contains an NES
How could a region (Arc-29–78) be required for nuclear localization without functioning as an NLS? We hypothesized that it retains Arc in the nucleus, and that Arc also contains a nuclear export signal (NES) to return Arc to the cytoplasm in the absence of the retention sequence. To determine if Arc-29–78 functions as a nuclear retention domain (NRD), we inserted it into a fusion protein that shuttles into and out of the nucleus (GFP-NLS-NES). The NES dominates over an NLS, so the GFP is predominantly cytoplasmic. When we replaced the NLS with Arc-29–78 (GFP-Arc29–78-NES), the fusion protein was partly retained in the nucleus, despite the strong NES, suggesting that it diffused into the nucleus and became bound (Supplemental Fig. 3b, c). We used fluorescence recovery after photobleaching (FRAP) to measure the effect of this region on the mobility of Arc. GFP-Arc has a lower mobile fraction than GFP alone, and GFP-Arc29–78-NES is lower still (Supplemental Fig. 3d–f). Diffusion of GFP through the bleach region is so rapid that over the brief period of bleaching, a degree of equilibration between the bleach region and the rest of the cell occurs, resulting in bleaching of the entire cell, which is taken into account in the equation measuring mobility. These data show the N-terminal region decreases mobility and retains it in the nucleus, most likely by binding to one or more nuclear constituents.
The reasoning that led to the discovery of the NRD also predicts Arc contains an NES. We used leptomycin B to inhibit active export of proteins from the nucleus. Leptomycin B treatment for 2 hours increased the ratio of nuclear to cytoplasmic Arc (Fig. 3a, b, Supplemental Fig. 3a). This suggests Arc is actively exported from nuclei, though only slowly as expected due to the NRD. The response of Arc to leptomycin B was lost with either of 2 deletions between amino acids 121–154 (Fig. 3a, b), indicating this region contains an NES. We tested whether the region is sufficient to mediate active export by fusing Arc-121–154 to GFP. Arc-121–154 decreased the nuclear localization of GFP, and leptomycin B treatment reversed the effect (Fig. 3c, d). Arc therefore has at least three regions that control its localization to the nucleus: a Pat7 NLS, an N-terminal NRD, and an NES at amino acids 121–154 (Fig. 3e).
Figure 3. Arc contains an NES.

(a) Representative images of Arc staining of neurons transfected with full-length or deletion mutants of Arc with or without a 2-h treatment with leptomycin B (LeptB) show Arc has an NES between amino acids 121 and 154.
(b) Quantification of nuclear to cytoplasmic ratio of Arc with LeptB treatments. N=46 for Arc, N=40 for Arc+LeptB, N=42 for ArcΔ121–140, N=40 for ArcΔ121–140+LeptB, N=40 for ArcΔ141–154, N=45 for ArcΔ141–154+LeptB, from three independent cultures.
(c) Representative images of neurons transfected with GFP alone, or GFP fused to Arc 121–154 with or without LeptB treatment show Arc 121–154 is sufficient for active export.
(d) Quantification of the nuclear to cytoplasmic ratio of GFP expression. N=63 for GFP, N=20 for GFP+LeptB, N=46 for GFP-Arc-NES, N=47 for GFP-ArcNES+LeptB from three independent cultures.
**, p<0.01; ***, p<0.001, by ANOVA. Error bars show 95% confidence intervals. Scale bars=10 μm.
Arc nuclear localization is regulated by activity
The intricate mechanisms controlling Arc subcellular localization in vitro and in vivo suggest that Arc translocation to the nucleus is regulated by extracellular stimuli. To test this, we expressed Arc constitutively to provide a system to assay effects of stimuli on Arc localization without the confounding effects on Arc expression,. We treated neurons with BDNF, which is used extensively as a model for physiologically relevant synaptic stimulation8, 31–33. While neurons vary in Arc localization (Supplemental Fig. 4a), short-term BDNF treatment drastically decreased both the ratio of nuclear to cytoplasmic Arc expression (Figs. 4a, c, Supplemental Fig. 4a–c) and the absolute levels of Arc in the nucleus (Supplemental Fig. 4c). Longer BDNF treatments had an opposite effect; Arc began to return to the nucleus within 2 hours of BDNF stimulation. By 8 hours, the ratio of nuclear to cytoplasmic Arc was increased well over baseline (Fig. 4a, c).
Figure 4. Activity regulates Arc nuclear import and export.

(a, e) Representative images of neurons transfected with Arc and treated with BDNF (a) or bicuculline (Bic) (e).
(c, g) Quantification of Arc expression with BDNF (c) or bicuculline (g). For 0h BDNF N=100, 0.5h N=42, 2h N=51, 4h N=40, 6h N=40, 8h N=102. For Bic 0h N=62, 0.5h N=50, 8h N=41, three biological replicates.
(b, f) Representative images of staining of endogenous Arc in neurons treated with BDNF (b) or bicuculline (f).
(d, h) Quantification of endogenous Arc expression with BDNF (d) or bicuculline (h). For BDNF 0h N=86, 0.5h N=35, 2h N=68, 4h N=35, 6h N=34, 8h N=165. For Bic 0h N=59, 0.5h N=82, 8h N=77, three independent cultures.
(i, j) Quantification of ArcΔ121–140 (i) or ArcΔPat7 (j) expression in response to BDNF. ArcΔ121–140 N=38, ArcΔ121–140+0.5 h BDNF N=40, ArcΔ121–140+8 h BDNF N=29, ArcΔPat7 N=40, ArcΔ Pat7+0.5 h BDNF N=46, ArcΔ Pat7+8 h BDNF N=31 three independent cultures.
(l, m) Quantification of Arc localization with 0.5 (l) or 8 hours (m) of BDNF and inhibitors of downstream pathways. For 0.5h BDNF, control N=49, BDNF N=58, Rap N=40, AKTX N=21, U0126 N=40, PD N=41, ET N=35, U73122 N=31 from three independent cultures. For 8h BDNF, control N=100, BDNF N=102, Rap N=29, AKTX N=33, U0126 N=50, PD N=32, ET N=27, U7 N=30, three independent cultures. *, p<0.05; **, p<0.01; ***, p<0.001 vs untreated, by ANOVA. Error bars show 95% confidence intervals. Scale bars=10 μm.
To ensure the effects were not an artifact of overexpression, we examined the few neurons that express endogenous Arc under basal conditions and the effect of BDNF on endogenous Arc. The temporal pattern of endogenous Arc localization confirmed that of ectopic Arc (Fig. 4b, d) and both were remarkably similar to that seen in vivo in response to a novel environment. In addition, the same effect was observed with BDNF treatment of neurons transfected with GFP-tagged Arc (Supplemental Fig. 4e, f), indicating that the effect is not an artifact of antibody staining. To determine if this effect occurs with other manipulations of activity levels, we used bicuculline, a GABAA receptor antagonist that increases activity levels. Like BDNF, bicuculline initially decreased and then strongly increased nuclear localization for ectopic and endogenous Arc (Fig. 4e–h). Thus, ectopic Arc expression in vitro is a relevant model system to study Arc localization, and Arc localization to the nucleus is bidirectionally regulated by increased activity.
We hypothesized that the bidirectional changes in Arc localization depend on active import and export of Arc across the nuclear membrane. To test this, we asked whether BDNF or bicuculline causes rapid nuclear extrusion of versions of Arc that lack the NES. ArcΔ-121–140 did not exhibit the 30-minute decrease in Arc nuclear localization but did show the increase at 8 hours (Fig. 4i, Supplemental Fig. 4g). In addition, leptomycin B inhibited the 30-minute decrease in Arc nuclear localization (Supplemental Fig. 4i), confirming that active export is required for this effect. Conversely, Arc that lacks the NLS (ArcΔPat7) showed the expected 30-minute decrease but did not have a significant increase in Arc nuclear localization at 8 hours (Fig. 4j, Supplemental Fig. 4h). In addition, we used FRAP to confirm that long-term BDNF treatment increases Arc transport into the nucleus. We bleached the nucleus of GFP-Arc transfected neurons and used the rate of recovery of GFP fluorescence in the nucleus as a measurement of Arc transport into the nucleus. In neurons treated with BDNF for at least 4 hours, Arc returned to the nucleus faster then in untreated neurons (Supplemental Fig. 4j). Finally, we examined whether continuous activity was required for Arc translocation to the nucleus. We treated neurons with continuous periods of bicuculline stimulation or 30 minutes of bicuculline, followed by washout and subsequent TTX treatment until fixation at 8 hours after initial treatment to ensure no lasting increase in activity. We found that continuous activity was required for increased Arc localization to the nucleus (Supplemental Fig. 4k). Thus, short periods of increased activity result in the active export of Arc from the nucleus and long periods of increased activity result in active import of Arc into the nucleus.
To better understand the mechanisms that control Arc localization, we examined the signaling pathways downstream of BDNF to determine which are necessary for regulating Arc nuclear import and export. We inhibited each of the three pathways downstream of BDNF with two inhibitors and then treated neurons with BDNF for either 30 minutes or 8 hours. We found that different signaling pathways control Arc localization in response to short or long BDNF stimulation. Inhibiting the mTOR pathway blocked the export of Arc from the nucleus in response to a short BDNF stimulation but did not affect the response to a long BDNF stimulation. Conversely, inhibition of the MEK/ERK or PLC pathways blocked import of Arc into the nucleus in response to a long BDNF treatment but did not affect BDNF-induced export (Fig. 4k, l, Supplemental Fig. 4l, m). These data are the first demonstration that Arc protein localization is directly regulated by activity. In addition, the existence of three cis-acting elements within Arc and the identification of stimuli that utilize those domains to regulate Arc localization suggest that Arc plays important and distinct functions in the nucleus.
Arc in to the nucleus regulates synaptic strength
Arc expression leads to reduced synaptic strength, an effect attributed to its actions at the synapse21, 22. That Arc localization to the nucleus is governed by synaptic activity raises the intriguing possibility that Arc acts in the nucleus to regulate the response to synaptic strength. Arc expression decreases surface levels of the AMPA receptor subunit GluA1 (Fig. 5a)21. However, we found that versions of Arc that are expressed in the cytoplasm and not in the nucleus (ArcΔ29–38, ArcΔ59–68, or deletion of the coiled-coil domain in the NRD, ArcΔCC) do not significantly decrease surface GluA1 levels (Fig. 5b). We used deletions in the NRD instead of the Pat7 NLS deletion because NRD deletions have much more potent effects on untagged forms of Arc (Fig. 2). These findings suggest Arc functions in the nucleus to regulate surface GluA1 levels.
Figure 5. Nuclear Arc regulates synaptic strength.

(a) Representative images of surface GluA1 staining of neurons transfected with GFP and a control plasmid or Arc.
(b) Quantification of surface GluA1 in neurons transfected with GFP and control, Arc, or Arc deletion constructs ArcΔ29–38, ArcΔ59–68, and ArcΔCC that do not localize to the nucleus shows that Arc in the nucleus regulates surface GluA1. Control N=113, Arc N=106, ArcΔ29–38 N=41, ArcΔ59–68 N=60, ArcΔCC N=65, from at least four independent cultures.
(c) Representative images of Arc tagged with a strong NES to prevent nuclear localization or a mutant form of the NES (mNES) that does not affect localization.
(d) Quantification of surface GluA1 in neurons transfected with a control plasmid, Arc-mNES, or Arc-NES shows Arc in the nucleus regulates surface GluA1. Control N=85, Arc-mNES N=64, Arc-NES N=64, from six independent cultures.
(e) Representative traces of mEPSCs from neurons transfected with control or Arc constructs show Arc decreases mEPSC amplitude.
(f, g) Quantification of mEPSC amplitude of neurons transfected with Arc constructs (f) or Arc-NES constructs (g) shows Arc in the nucleus regulates synaptic strength.
(h, i) mEPSC frequency with Arc constructs (h) or Arc-NES constructs (i) shows that Arc does not affect AMPAR frequency. For deletion constructs, control N=40, Arc N=25, and ArcΔ59–68 N=14 from six independent cultures. For tagged constructs, control N=39, Arc-mNES N=11, Arc-NES N=23 from six independent cultures.
*, p<0.05; **, p<0.01; ***, p<0.001, by ANOVA. Error bars show 95% confidence intervals. Scale bars=10 μm.
To ensure this result is not due to off-target effects of deleting regions of the protein, we took a complementary approach. We made a version of Arc that was excluded from the nucleus by appending a 14–amino acid, extremely strong NES to Arc (Arc-NES). To control for the additional amino acids, we made three amino acid substitutions in the NES (Arc-mNES) that prevent its export function and result in a subcellular distribution identical to endogenous Arc (Fig. 5c). Expressing the version of Arc with the same localization as endogenous Arc (Arc-mNES) resulted in the typical decrease in GluA1 surface levels. However, excluding Arc from the nucleus (Arc-NES) significantly impaired its ability to reduce surface GluA1 levels (Fig. 5d). The effects of manipulating Arc expression in the nucleus cannot be due to unintended changes in Arc expression elsewhere in the cell as all Arc constructs had equivalent levels outside of the nucleus (Supplemental Fig. 5a–d) and localized similarly within dendrites as demonstrated by equivalent colocalization to a synaptic marker PSD-95 (Supplemental Fig. 5e, f). Finally, all constructs also functioned similarly outside of the nucleus in regulating the endocytosis of AMPA receptors as measured by a GluA1 internalization assay (Supplemental Fig. 5g).
Having shown that nuclear Arc regulates surface GluA1 levels, we next wanted to know if these changes had consequences for AMPAR function at the synapse. We recorded mini excitatory post-synaptic currents (mEPSCs) mediated by AMPARs. Arc expression decreased mEPSC amplitude with no significant effect on frequency (Fig. 5e–i), indicating Arc decreases the number of functional AMPARs at synapses without significantly changing synapse number, similar to previous findings22. In agreement with our findings on surface GluA1 levels, versions of Arc that did not localize to the nucleus had significantly less effect on mEPSC amplitudes (Fig. 5f, g). Some of Arc’s effects on mEPSCs and AMPAR expression remain, despite loss of Arc in the nucleus, likely because Arc also functions at the synapse to regulate synaptic strength21, 22.
Arc regulates synaptic strength via GluA1 transcription
Arc expression decreases AMPAR surface expression in vitro21, 22 and in vivo20. This was previously attributed to a role of Arc in AMPAR endocytosis, but Arc localization to the nucleus clearly contributes to this effect, too (Fig. 5). Arc also decreases total levels of GluA121, which cannot be explained solely by effects of Arc on endocytosis. Arc must have unknown roles in regulating the production or clearance of GluA1, a function we hypothesized might be mediated by Arc in the nucleus. To test this, we measured total levels of GluA1 and found Arc expression decreased them, similar to what has been shown21. However, versions of Arc that do not localize to the nucleus did not decrease total GluA1 levels (Fig. 6a–c). Thus, Arc acts in the nucleus to regulate total levels of GluA1.
Figure 6. Nuclear Arc regulates GluA1 transcription.
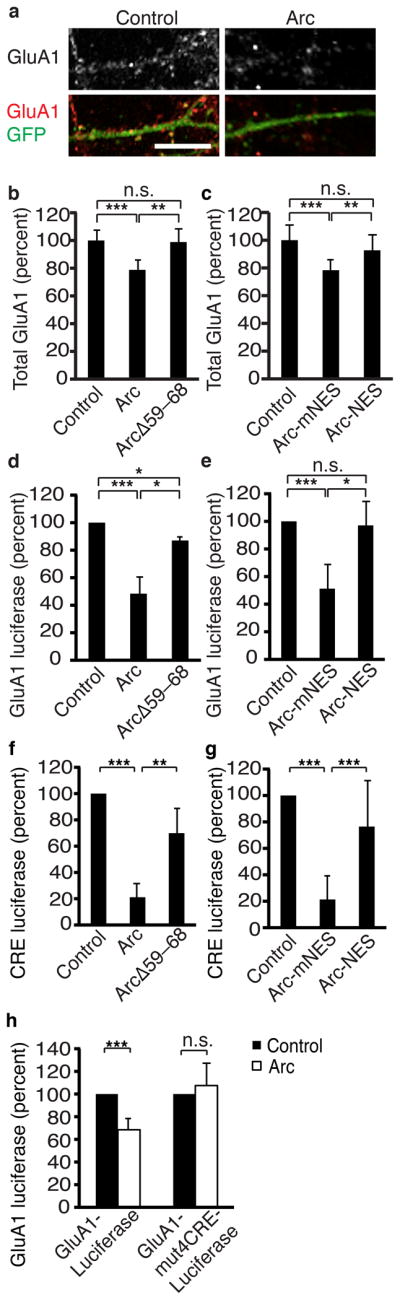
(a) Representative images of total GluA1 staining of neurons transfected with GFP and a control plasmid or Arc.
(b, c) Quantification of total GluA1 in neurons transfected with Arc or a mutant of Arc (ArcΔ59–68) that abrogates nuclear localization (b) or a version of Arc directed to the cytoplasm (Arc-NES) (d) shows that Arc in the nucleus regulates total GluA1. For deletion constructs, control N=59, Arc N=58, ArcΔ59–68 N= 46 from 6 independent cultures. For tagged constructs, control N=54, Arc-mNES N=53, Arc-NES N=46 from 6 independent cultures.
(d, e) GluA1 luciferase levels in neurons tranfected with versions of Arc from b, c show that Arc in the nucleus regulates GluA1 transcription. Levels were normalized to activity of a co-transfected enzyme Seap to control for transfection efficiency. N=5 with triplicate technical measurements for each replicate.
(f, g) CRE luciferase levels in neurons transfected with Arc deletion constructs (f) or NES constructs (g) show that Arc in the nucleus regulates CRE-mediated transcription. N=3 with triplicate technical measurements for each replicate.
(h) GluA1 luciferase levels measured from the full GluA1 promoter or the GluA1 promoter with all CRE sites mutated (GluA1-mut4CRE-Luciferase) show that Arc in the nucleus regulates GluA1 transcription through CRE sites. Levels were normalized to activity of a co-transfected enzyme Seap to control for transfection efficiency. N=4 with triplicate technical measurements for each replicate.
*, p<0.05; **, p<0.01; ***, p<0.001; n.s., not significant, by ANOVA. Error bars show 95% confidence intervals. Scale bars=10 μm.
Since GluA1 transcription occurs in the nucleus, and GluA1 clearance presumably occurs in the cytoplasm, we hypothesized that Arc lowers GluA1 levels by suppressing its transcription. We used a GluA1-luciferase reporter gene in which luciferase expression is controlled by the regulatory regions of the GluA1 promoter34. Arc expression drastically decreased luciferase activity (Fig. 6d, e), indicating Arc suppresses GluA1 transcription. This is the first evidence that Arc regulates GluA1 production and is consistent with the immunocytochemistry findings of reduced total GluA1 levels in neurons with Arc.
From these results, we could not tell if Arc regulates GluA1 transcription by affecting activity-dependent signaling pathways emanating from the synapse or at the level of the GluA1 promoter. Arc’s cytoplasmic actions on AMPAR endocytosis decrease synaptic strength and thereby could decrease activity-dependent transcription of genes, such as GluA1. If so, versions of Arc that stay in the cytoplasm should suppress GluA1-luciferase at least as potently as wild-type Arc. Instead, we found that preventing Arc from localizing to the nucleus blocked Arc suppression of GluA1-luciferase expression (Fig. 6d, e). We conclude that Arc in the nucleus decreases GluA1 transcription, thus decreasing levels of GluA1 protein and synaptic strength.
GluA1 transcription can be regulated through cyclic AMP response elements (CRE) within its promoter34. We found Arc expression in the nucleus suppresses expression of a minimal CRE reporter (Fig. 6f, g) gene. Arc also regulates another CRE-dependent gene c-Fos but does not appear to regulate transcription or levels of another AMPAR subunit GluA2 (Fig. S6a–e), which lacks known CRE sites, indicating Arc may regulate GluA1 transcription through its CRE sites. To test this, we mutated the four CRE sites in the GluA1 promoter of the GluA1-luciferase construct and found that Arc no longer decreased GluA1-luciferase levels (Fig. 6h). Finally, we examined whether Arc can regulate endogenous GluA1 when the ability to control CRE-mediated transcription is removed. We expressed a constitutively active form of CREB (CREB-VP16) in neurons with Arc and measured surface GluA1 levels. CREB-VP16 expression blocked the Arc-dependent decrease in GluA1 levels (Supplemental Fig. 6f) demonstrating that Arc regulates GluA1 levels by decreasing CRE-mediated transcription.
Arc decreases GluA1 transcription through an PML-NBs
Though Arc strongly suppresses GluA1 transcription, it has no known DNA binding regions. However, in HEK293 cells, Arc overexpression promotes formation of PML-NBs19, nuclear structures that mediate a range of functions, including transcriptional regulation. Notably, GluA1 transcription can be regulated through CRE sites within its promoter34, and PML-NBs regulate CRE-dependent transcription in other cell types35. However, whether PML-NBs link to endogenous Arc or serve a critical function in neurons is unknown.
To determine if endogenous Arc upregulates PML, we stimulated neurons with BDNF. Arc induction correlated highly to PML levels in individual neurons (Fig. 7a, b). In addition, sustained BDNF or bicuculline treatment often results in the formation of Arc nuclear puncta, which colocalize with PML, whereas sustained periods of decreased activity results in fewer Arc puncta (Supplemental Fig 7a–c). Arc co-immunoprecipitates with PML and CBP (Fig. 7c) and BDNF increased PML levels at the same time points that Arc localizes to the nucleus (Fig. 7d, e). Stimulating synaptic activity with bicuculline also increased PML levels (Supplemental Fig. 7d, e). To see if Arc was required for PML upregulation, we repeated the experiments in cultured neurons from wild-type and Arc−/− mice. Wild-type neurons showed a time-dependent increase in PML levels after BDNF treatment, but Arc−/− neurons did not (Fig. 7f–i). Finally, to determine if increased levels of PML protein resulted in increased PML-NB formation, we tracked neurons expressing EGFP-tagged PML (EGFP-PML) over time with a robotic microscope36. BDNF treatment resulted in a robust increase in formation of PML-NBs (Fig. 7j, Supplemental Fig. f). Our data show that Arc mediates activity-induced PML-NB formation and represent the first characterization of PML regulation in mature neurons.
Figure 7. Arc mediates the activity-induced upregulation of PML-NBs, which decreases GluA1 transcription.

(a) Representative images of Arc and PML staining.
(b) Intensity of PML and Arc staining shows a highly significant correlation. R=0.785. p< 0.001. N=180 neurons.
(c) Arc co-immunoprecipitates with PML and CBP in neurons treated for 6 h with bicuculline.
(d, f, h) Western blots of endogenous PML levels in BDNF-treated neurons from rats
(d), mice (f), or Arc−/− mice (h) show Arc mediates the BDNF-induced increase in PML. (e, g, i) Quantification of PML levels from western blots from rats (e), mice (g), or Arc−/− mice (i). N=5 independent cultures for each.
(j) Representative images of neurons transfected with GFP-PML and tracked over time after BDNF treatment.
(k, l) Representative images of surface (k) or total GluA1 (l) in neurons transfected with a control plasmid or PML show PML decreases GluA1 levels.
(m) Quantification of total GluA1 in neurons transfected with IE1Δ290–320, which does not disrupt PML-NBs, or IE1, which prevents PML-NB formation along with control or Arc plasmids. IE1Δ290–320+control N=37, IE1Δ290–320+Arc N=31, IE1+control N=34, and IE1+Arc N=37 from three independent cultures.
(n) Quantification of total GluA1 in neurons transfected with GFP or GFP-ICP0 and either control or Arc plasmids shows PML is required for Arc’s effects on total GluA1 levels. GFP+control N=34, GFP+Arc N=31, GFP-ICPO+control N=29, GFP-ICPO+Arc N=29 from three independent cultures. Full blots are shown in supplemental material.
*, p<0.05; **, p<0.01; ***, p<0.001, by ANOVA. Error bars show 95% confidence intervals. Scale bars=10 μm.
We next determined if PML is responsible for Arc’s effects on transcription. If Arc promotes formation of PML-NBs and these structures are responsible for Arc’s effects on GluA1 levels, then increasing PML levels should phenocopy the effects of increased Arc levels on GluA1. Ectopic PML expression decreased surface and total GluA1 levels and GluA1-luciferase activity (Fig. 7k, l, Supplemental Fig. g–i), similar to the effect of Arc expression in the nucleus. We conclude PML is sufficient to decrease GluA1 levels. Finally, we determined if PML-NBs are necessary for Arc regulation of GluA1 levels by disrupting PML-NB function with the cytomegalovirus immediate early gene 1 (IE1)37 (Supplemental Fig. 7j). We expressed IE1 or an inactive version of IE1 (IE1Δ290–320) with Arc and measured total levels of GluA1. In neurons with disrupted PML-NBs, Arc no longer decreased total GluA1 levels (Fig. 7m). Another viral protein, ICP0, that works through a different mechanism to disrupt PML-NBs38 has the same effect (Fig. 7n, Supplemental Fig. 7k). Thus, PML is both sufficient and necessary to mediate Arc suppression of total levels of GluA1.
Arc nuclear localization regulates homeostatic plasticity
We showed that prolonged periods of increased activity result in accumulation of Arc in the nucleus, which reduces synaptic strength globally by decreasing GluA1 transcription. Such a negative feedback mechanism to regulate activity encompasses the main elements of homeostatic synaptic scaling. In this form of plasticity, long periods of increased activity result in decreased synaptic strength, which allows neurons to maintain a stable firing rate.
Arc is critical for synaptic scaling in vitro and in vivo5, 39. We hypothesized that Arc functions in the nucleus to mediate synaptic scaling in response to increased activity. Bicuculline treatment, a known inducer of downscaling, significantly decreased surface GluA1 staining as expected (Fig. 8a, b). Ectopic expression of a version of Arc with normal nuclear localization (Arc-mNES) blocked downscaling induced by bicuculline. Increased Arc expression in the nucleus presumably occluded downscaling by increasing PML-NBs and decreasing GluA1 levels and synaptic strength, so they could not be lowered further by bicuculline. To confirm this, we tested the effects of a version of Arc that is excluded from the nucleus (Arc-NES) and found it failed to block downscaling (Fig. 8b). Since the downscaling mechanism has not yet been activated in these neurons, bicuculline can do so by inducing endogenous Arc.
Figure 8. Arc localization to the nucleus mediates homeostatic plasticity.

(a) Representative images of surface GluA1 staining of neurons transfected with a control plasmid, Arc-mNES or Arc-NES and treated with bicuculline for 48 h.
(b) Quantification of surface GluA1 staining in neurons transfected with a control plasmid, Arc-mNES or Arc-NES shows Arc localization to the nucleus prevents downscaling. N=65 for control, N=64 for bicuculline, N=37 for Arc-mNES, N=50 for Arc-mNES+bicuculline, N=50 for Arc-NES, N=50 for Arc-NES+bicuculline from four independent cultures.
(c) Quantification of Arc levels in untransfected neurons using Arc staining and neurons transfected with ABCDEFG-Arc-His-Flag-HI using Flag staining, with and without 8-h BDNF treatment. N=55 for endogenous Arc, N=37 for endogenous Arc + BDNF, N=40 for ABCDEFG-Arc-His-Flag-HI, N=37 for ABCDEFG-Arc-His-Flag-HI+BDNF from three independent cultures.
(d) Representative images of ABCDEFG-Arc-His-Flag-HI construct expression with and without BDNF treatment in neurons stained with anti-Flag antibody.
(e) Representative images of surface GluA1 staining of Arc−/− neurons transfected with a control plasmid, ABCDEFG-Arc-mNES-HI or ABCDEFG-Arc-NES-HI and treated with bicuculline for 48 h.
(f) Quantification of surface GluA1 staining in neurons transfected with a control plasmid, ABCDEFG-Arc-mNES or ABCDEFG-Arc-NES shows Arc localization to the nucleus rescues downscaling in Arc knockout neurons. N=58 for control, N=47 for control+bicuculline, N=59 for ABCDEFG-Arc-mNES-HI, N=45 for ABCDEFG-Arc-mNES-HI+bicuculline, N=40 for ABCDEFG-Arc-NES-HI, N=30 for ABCDEFG-Arc-NES-HI+bicuculline from at least three independent cultures.
***, p<0.001, n.s., not significant, by ANOVA. Error bars show 95% confidence intervals. Scale bars=10 μm.
To ensure that these effects are not an artifact of overexpression, we developed an inducible form of Arc that is expressed in response to activity similarly to endogenous Arc and still allows for manipulation of Arc localization. Previous work elucidated the portions of the Arc promoter required to mediate activity-dependent Arc induction9, 11. We created constructs that used these regions (termed ABCDEFG for downstream regions and HI for upstream regions as described by Pintchovski et al. 2009), to regulate Arc expression. To validate this approach, we put the Arc coding sequence under the control of these promoter regions and tagged Arc with a His-Flag tag (ABCDEFG-Arc-His-Flag-HI) to allow for simultaneous staining of endogenous Arc and the tagged Arc in neighboring neurons. We then transfected this construct into rat neurons and induced Arc with BDNF. We quantified endogenous Arc induction using an Arc antibody and quantified ABCDEFG-Arc-His-Flag-HI induction with a flag antibody. Endogenous Arc and ABCDEFG-Arc-His-Flag-HI had the same fold increase in response to BDNF (Fig. 8c) and ABCDEFG-Arc-His-Flag-HI has the same localization pattern in response to BDNF as endogenous Arc (Fig. 8d). To allow for manipulation of Arc localization, we replaced the His-Flag tag with either the NES or mNES tag, which were previously described (Fig. 5c). These constructs, or a control plasmid were transfected in Arc−/− neurons. Scaling was induced with bicuculline and, as expected, Arc−/− neurons with a control plasmid did not undergo homeostatic downscaling of GluA1. However, ABCDEFG-Arc-mNES-HI, which has normal localization to the nucleus, rescued homeostatic scaling. ABCDEFG-Arc-NES-HI, which does not localize to the nucleus, did not (Fig. 8e, f). In the absence of stimuli, both ABCDEFG-Arc-mNES and ABCDEFG-Arc-NES-HI result in a small, nonsignificant decrease in surface GluA1 levels. This is likely due to the high level of basal activity in Arc−/− cultures, which will result in some Arc induction and subsequent endocytosis of GluA1 at the synapse. However, downscaling only occurs when Arc can localize to the nucleus. We conclude Arc must act in the nucleus to regulate homeostatic plasticity, and Arc’s functions in dendrites and synapses are not sufficient for downscaling.
To further demonstrate that Arc regulates transcription in response to downscaling stimuli, we also examined GluA1 mRNA. We treated wild-type or Arc knockout mouse neurons with bicuculline for 24 hours and used quantitative real-time PCR to measure changes in levels of mRNA levels. We found that wild-type mouse neurons decrease GluA1 mRNA levels in response to bicuculline, whereas Arc knockout neurons do not (Supplemental Fig. 8a). Another receptor subunit GluN2A does not show similar changes (Supplemental Fig. 8b). Finally, we examined GluA1 levels by western blot to provide an additional measurement of total GluA1 levels. Total GluA1 was decreased with 48 hours of bicuculline treatment in wild-type but not Arc knockout mouse neurons (Supplemental Fig. 8c, d), demonstrating that Arc is required for the activity-induced decrease in GluA1 levels. In summary, prolonged periods of increased activity lead to Arc accumulation in the nucleus, which triggers PML-NB formation, suppression of GluA1 transcription and downscaling of synaptic strength (Supplemental Fig. 8e).
DISCUSSION
Here we demonstrated that Arc becomes enriched in neuronal nuclei after a novel experience and in response to prolonged increased activity. Arc has multiple regions that govern its levels in nuclei, including a Pat7 NLS, an NES, and an NRD, which regulate import, export, and retention within the nucleus, respectively. Short periods of activity decrease and long periods of activity increase nuclear localization. In the nucleus, Arc regulates global GluA1 levels and thereby regulates synaptic strength. This occurs through the activity-dependent formation of PML-NBs, which suppress GluA1 transcription. Through this mechanism, Arc localization to the nucleus regulates activity-dependent downscaling in homeostatic plasticity.
Regulating Arc localization
An early study noted Arc localization to the soma40, and a recent study reported levels of Arc can be higher in the nucleus than the cytoplasm19. However, the significance and regulation of nuclear Arc was not known. We found the first evidence that localization of Arc protein is regulated by activity, complementing the extensive literature showing that synaptic activity regulates the transcription, mRNA localization, translation, and degradation of Arc. We know of no other gene that is so highly regulated by synaptic activity. The extent to which the CNS controls Arc expression spatially and temporally underscores its importance.
Other plasticity-related proteins reportedly shuttle in and out of the nucleus, but the bidirectional regulation of Arc nuclear localization by activity is novel. We speculate that this may couple Arc’s effects on transcription to the immediate needs of the neuron. After short periods of stimulation, such as those resulting in LTP, synapses undergo potentiation, which involves increased AMPAR expression. Having Arc in the nucleus during these short-term stimulations would inhibit GluA1 transcription, so Arc is exported from the nucleus. In cases when stimulation is prolonged and the neuron must reduce its overall excitability, downscaling is activated by importing Arc into the nucleus.
The activity dependence of Arc localization may explain the different patterns we observed after novel environment stimulation in different hippocampal regions. Because these regions receive different activity patterns and inputs, it is not surprising that they have a different time course of Arc translocation to the nucleus.
We also found that the total number of neurons expression Arc decreases after long periods of novel environment. This fits with our model of Arc’s function in homeostatic scaling. After long periods of novel environment exposure, presumably some aspects of the environment begin to lose their novelty. Thus, many neurons will not be continually activated for the duration of the experiment and will eventually shut off Arc expression. The few neurons that are continually activated are also those that need to undergo downscaling and send Arc to the nucleus.
PML-NBs in neurons
PML regulates cell fate in the embryonic neocortex26, and Arc promotes formation of PML-NBs by interacting with the nuclear matrix protein Spectrin βIV19. However, PML-NBs mediate multiple functions in different cell types (e.g., mRNA export, protein degradation, and transcription), and little was known about them in neurons. Elucidating the function of PML in mature neurons is of particular importance because PML-NBs are disrupted in multiple neurodegenerative diseases27–30. We showed PML is both necessary and sufficient for mediating Arc’s nuclear functions. In addition, we provide the first evidence that PML regulates transcription in neurons and that activity regulates PML-NBs.
PML-NBs regulate transcription through multiple mechanisms, including the availability of transcription factors and the activity of HDACs24, 25, 41. PML-NBs likely regulate GluA1 through CREB binding protein (CBP), which promotes CRE-dependent transcription. CBP is a well-established binding partner of PML, and PML may sequester and degrade CBP35. Arc-induced upregulation of PML-NBs may result in accumulation and degradation of CBP at PML-NBs. Availability of CBP would decline, thereby decreasing CRE-dependent transcription of multiple activity-regulated genes, such as GluA1.
Arc in plasticity
Our data are the first to assign a nuclear function to Arc: homeostatic downscaling. Arc is critical for synaptic scaling in vitro and in vivo. Our proposed mechanism parsimoniously explains how Arc might regulate global synaptic strength without requiring that Arc act at each synapse individually to mediate cell-wide downscaling through endocytosis. In addition, our mechanism generally fits well with what is known about synaptic downscaling. Multiple studies found that, in response to a stimulus that increases activity, both surface and total levels of AMPARs are decreased23, 42. The decrease in total AMPARs suggests that downscaling involves a mechanism that regulates the amount of protein available cell-wide, possibly by changing levels of transcription. In addition, scaling occurs on a timescale similar to the turnover of AMPAR protein and is a process that requires transcription and translation of new proteins43, 44. Downscaling in response to increased activity is a cell-autonomous process44. Finally, our proposed mechanism does not exclude the possibility of synaptic mechanisms acting in concert with transcriptional regulation or models proposing that Arc contributes to synaptic downscaling through AMPAR endocytosis22, 45. Much of the previous research into mechanisms underlying scaling has focused on GluA2. While most of this previous work examined upscaling rather then downscaling, which is mediated by different mechanisms, our data do not exclude the possibility that GluA2 is also required for downscaling.
A role for nuclear Arc might also explain how Arc mediates functions that appear opposed to one another, such as consolidating LTP and LTD while also regulating scaling1–5. Arc regulates AMPAR endocytosis, and this function of Arc is likely required for some forms of LTD4. Arc also regulates spine structure and number20 and Notch signaling46 which may underlie its ability to regulate the late phase of LTP. Our data do not contradict these proposed functions for Arc. In fact, preventing Arc localization to the nucleus resulted in a partial but not a full return to baseline levels of surface GluA1 levels. This result is consistent with a function for Arc outside the nucleus, most likely at the synapse regulating GluA1 endocytosis. However, if Arc is prevented from localizing to the nucleus, total GluA1 and GluA1-luciferase levels return nearly to baseline. These data indicate that Arc localization to the nucleus is fully responsible for its ability to decrease GluA1 transcription, while both a nuclear and a dendritic function contribute to the decrease in surface GluA1 levels.
Our data support a model in which Arc’s function depends on its location47, 48. With short periods of stimulation, Arc is predominately in the cytoplasm where it regulates the spine cytoskeletal matrix or the endocytic machinery. With long periods of stimulation, such as those that result in downscaling, Arc localizes to the nucleus where it regulates synaptic strength by acting through PML-NBs. The delay in activity-induced Arc localization to the nucleus may allow for a temporal separation of these functions of Arc. This suggests that Arc is a master regulator of memory, mediating different forms of synaptic plasticity such as LTP, LTD, and homeostatic scaling as needed depending on the nature of the stimulation.
METHODS
Behavior and tissue preparation
We used 3-month-old male mice (C57/BL6 strain) kept on a light/dark cycle for novel environment testing. Mice were added in pairs to a large cage (45 × 25 × 20 cm) in a new room with novel bedding, odors, food, and multiple novel objects during the light cycle. Mice were allowed to explore the cage for varying times and then deeply anesthetized and perfused transcardially with saline. Hemibrains were drop-fixed in 4% phosphate-buffered paraformaldehyde at 4°C for 48 h. Brains were then rinsed in PBS, transferred to 30% sucrose in PBS at 4°C for 24 h, and coronally sectioned (30 μm) with a sliding microtome. All experiments were approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco.
Cell culture
Long Evans rat hippocampal or cortical cells were dissected and dissociated from day 20 embryos and cultured for 10–14 days before transfections and treatments. Hippocampal neurons were used for all experiments except for luciferase assays, western blots, and quantitative real-time PCR experiments where larger numbers of neurons were required and cortical neurons were used. Constructs were typically transfected by the calcium phosphate method. Luciferase constructs were transfected with Lipofectamine 2000 (Invitrogen).
Mouse cortical cells were dissected at P0 from either wild-type C57/B6 strain (Charles River) or Arc-d2EGFP knock-in mice49, which contain d2EGFP, followed by a Neo cassette inserted after the Arc ATG start codon.
Pharmacology
BDNF (100 ng/mL) was from Amgen. U0126 (1 μM), U73122 (1 μM), Edelfosine (10 μM), D-2-amino-5 phosphonovaleric acid (APV) and bicuculline (10 μM) were from Tocris. AKT X (5 μM) and Tetrodotoxin (1 μM) were from Calbiochem/EMD. Leptomycin B (20 ng/mL) and PD0325901 (2 μM) were from Sigma. Rapamycin (0.13 μM) was from Cayman Chemical.
Constructs
GW1-Arc20 contains the rat coding sequence of Arc flanked by the rat 3′ and 5′UTR regions, followed by a polyA tail. GW1-Arc20 was used to create Arc constructs. The Pat7 NLS was found by PSORT (http://psort.hgc.jp/), a program for subcellular localization prediction based on protein sequences. Arc deletion constructs were made using the Strategene QuikChange Site-Directed Mutagenesis Kit. GFP-Arc includes GFP at the N terminus of Arc. Myc-tagged Arc constructs contain Myc on the N terminus of Arc cloned into the HindIII and Kpn1 sites of pGW1. Arc-NES and Arc-mNES constructs were made by adding a tag between Age1 and Mfe1 sites on the C terminus.
GFP-β-galactosidase has been described50. Regions of Arc were cloned in between the SacII and XbaI sites. The construct containing two regions of Arc had amino acids 29–78 cloned in between Nhe1 and Afl1 site. The NLS-NES-GFP construct has been previously described51. The coding sequence for Arc amino acids 29–78 was cloned into the construct between the Pst1 and BamH1 sites in place of the Rev-NLS. The NES region of Arc was cloned in between the BamH1 and Age1 sites of a NES-GFP construct in place of the NES. The PML construct has been described52.
Immunohistochemistry and immunocytochemistry
For immunohistochemistry, microtome slices were permeabilized with 0.5% Triton X-100 in PBS for 30 min and blocked in PBS with 5% donkey serum and 0.1% Triton X-100 for 1 h. Slices were incubated in primary antibody or antisera for 2 days at 4°C, washed and incubated in secondary antibody overnight at 4°C.
For immunocytochemistry, cells were fixed in 4% PFA with 4% sucrose, permeabilized with 0.1% Triton X-100 in PBS for 10 min, and blocked in PBS with 2% donkey serum, 3% BSA, and 0.1% Triton for 1 h. For surface staining, primary antibody was applied to live cells for 30 min at 37°C before fixation, cells were not permeabilized, and detergent was omitted from the blocking step. For all other staining, cells were incubated in primary antibody overnight at 4°C after blocking. Surface and total GluA1 staining were performed 30 h after transfection, the half-life of GluA1. Secondary antibodies were applied for 1 h at room temperature. For co-staining with surface GluA1 and Arc, cells were first stained for surface GluA1 as described, then permeabilized, blocked and stained for Arc.
Primary antibody or antisera concentrations were: Myc (mouse, Cell Signaling: 1:2000), Flag (mouse, Sigma 1:1000), PML (mouse, Sigma: 1:100), surface GluA1 (mouse, Millipore: 1:300), GluA2 antibody (mouse, Chemicon: 1:300) and total GluA1 (rabbit, Abcam 1:40). For Arc staining, polyclonal rabbit antisera was used that has been described20, or in cases where costaining with antisera raised in rabbit was required (for surface and total GluA1), a commercial Arc antibody (mouse, Santa Cruz: 1:100) was used. All secondary antibodies were used at 1:250 (Jackson).
Luciferase assays
Cortical neurons were transfected with a luciferase construct, a secreted embryonic alkaline phosphatase (Seap) construct, which is constitutively expressed, and a control construct or an Arc construct. At 12 h after transfection, 50 μL of medium was collected from each well, and a Seap assay (Applied Biosystems) was performed to assess transfection efficiency. At 30 h after transfection, lysates were collected and assayed with a Lucifease Kit (Promega) and a luminometer (Thermo Electron). To control for transfection efficiency, luciferase activity was normalized by Seap activity for each well. Each set of constructs were transfected in triplicate in each independent culture and values were averaged to obtain one value for each replicate. In addition, a set of coverslips were transfected in parallel for all Arc constructs used and stained for Arc to ensure neurons demonstrated normal Arc localization and similar levels of Arc for each construct used.
Western blotting
Assays were performed between 12 and 14 days in vitro as described8. Band intensities were quantified with ImageJ. Primary antibody or antisera concentrations were PML (mouse, Sigma: 1:1000), Arc20 (rabbit 1:5000), and GAPDH (chicken, Millipore: 1:5000). Secondary antibodies conjugated to horseradish peroxidase were imaged by enhanced chemiluminescence (Amersham Bioscience).
Co-Immunoprecipitation
Neurons were lysed in RIPA buffer and incubated for 1.5 h with magnetic beads (ThermoScientific) blocked in 4% BSA at 4°C. Additional blocked beads were incubated with Arc antiserum (in house, described in supplemental experimental procedures) or IgG for 30 min. Lysates were transferred to antibody-bound beads and incubated for 1 h at 4°C. Beads were washed three times with 1% Triton and then boiled for 5 min in loading buffer to elute proteins.
Electrophysiology
Hippocampal neurons were transfected at 12–14 days in vitro, and EGFP-expressing cells were recorded at 2–4 days after transfection in voltage clamp mode. The external solution (pH 7.35, 235 mOsm) contained APV (100 μM), TTX (1 μM), and Gabazine (10 μM) to isolate AMPAR currents. Patch recording pipettes (3–6 MΩ) were filled with an internal solution (pH 7.2, 223 mOsm). Recordings were only used if at least 300 mEPSCs were obtained. Data from each day were normalized to the average amplitude from control transfected neurons from that day.
The external solution was kept at 25°C and contained 119 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1.5 mM CaCl2, 10 mM HEPES, and 15 mM glucose. The internal solution contained 120 mM K-gluconate, 6 M NaCl, 1 mM MgCl2, 10 mM HEPES, and 0.2 mM EGTA. Data were acquired with an Axon MultiClamp amplifier and Clampex acquisition software and filtered at 2 kHz. AxoGraph software was used to correct for series resistance post hoc, identify and measure the amplitude and frequency of mEPSCs. Data were discarded if the series resistance increased over 30 MΩ or if there was a change in series resistance >10% over the course of the recording or if the holding current increased above 100 pAmps. One-way ANOVA with Fisher’s post hoc test was used to compare groups.
Microscopy and image processing
Images of fixed primary neurons were acquired with a LSM510 confocal microscope system (Zeiss) and a 63× oil immersion lens (numerical aperture 1.4). Confocal microscope settings were kept the same for all scans where protein levels were compared. The first 10 transfected neurons or separate fields of view observed were imaged to ensure random image collection. To achieve blinded image collection, transfected neurons were chosen for imaging using only the transfection marker fluorescence or the nuclear stain without examination of the channel of interest (such as Arc or GluA1). When neurons were counted for Arc expression or localization, slide labels were covered and only revealed after data collection. To measure nuclear and cytoplasmic levels of Arc, ImageJ was used to measure the average intensity in a region within the nucleus, a region immediately outside of the nucleus, and a region outside of the cell to use for background subtraction. To quantify the percentage of neurons with nuclear Arc localization, neurons with a ratio of nuclear to cytoplasmic Arc greater than 1 were counted as positive for nuclear Arc expression. To measure surface and total GluA1 levels, z stacks of four 0.5-μM spaced images were collected and projected into a single image. Using ImageJ, the GFP signal was used to threshold the image and create a region encompassing the transfected neuron and the average intensity within the region was measured. Values from each neuron were background subtracted and normalized to the average intensity of control transfected neurons from that day. Representative mages in each figure were processed identically.
For FRAP experiments, the LSM510 confocal microscope was used with a 63× immersion lens (numerical aperture 0.9). LSM510 software was used to obtain values for the bleach region and whole cell body before bleaching, immediately after bleaching, and after recovery. For data shown in Supplemental Fig. 3, the experiment was designed to measure the overall mobility of the protein throughout the cell and was not affected by the location of the bleach region. For data shown in Supplemental Fig. 4, the bleach region encompassed the entire nucleus because the experiment was designed to measure the ability of cytoplasmic protein to enter the nucleus. The mobility fraction was calculated according to Brough and Irvine53 by the equation Mf=(Fprecell/Fasym.cell)X[(Fasymp−F0)/(Fpre−F0)]. Fprecell indicates the total fluorescence of the entire cell, Fasym.cell indicates the total fluorescence of the entire cell after bleach and recover, Fasymp indicates the asymptote of fluorescence after recovery in the bleach region, F0 indicates the fluorescence of the bleach region immediately after bleaching, and Fpre indicates the initial fluorescence of the bleach region before bleaching.
For tracking EGFP-PML, we used a robotic imaging system as described36. Briefly, images were obtained with an inverted microscope (Nikon) with a 40× objective lens (numerical aperture 0.6). After imaging, neurons were treated and then returned to the incubator. The same neurons were then imaged again 8 h later. The mean fluorescence intensity of the cell body of transfected neurons was measured over time with Metamorph software (Universal Imaging), and the fold change in fluorescence was calculated.
To compare the cytoplasmic levels of the different Arc constructs used (Arc, ArcΔ29-38, ArcΔ59-68, ArcΔCC, Arc-mNES, and Arc-NES) transfected neurons were stained with polyclonal rabbit antisera raised against Arc. Images of fixed primary neurons were acquired with a LSM510 confocal microscope system (Zeiss) and a 63× oil immersion lens (numerical aperture 1.4) and settings were kept the same for all scans. To quantify the levels of Arc in the cytoplasm, Metamorph or ImageJ was used to threshold the image based on the mCherry image and create a region encompassing the transfected neuron. The average intensity of Arc expression within the region was measured. To measure the levels of Arc in the nucleus, Hoechst staining was used to select a region within the nucleus, and the average intensity of Arc expression within the region was measured. Values from each neuron were normalized to the average intensity of control transfected neurons from that day.
To measure internalized GluA1 levels in neurons expression the Arc constructs used, transfected neurons were incubated with GluA1 antibody at 37°C for 30 min and then fixed with 4% PFA with sucrose. Neurons were blocked and stained for with a secondary antibody to stain the surface population of GluA1. Neurons were then washed, permeabilized, and stained with a different secondary antibody to stain the GluA1 containing AMPARs that were internalized. z stacks of four 0.5-μm spaced images of internalized GluA1 were collected and projected into a single image. Settings were kept the same for all scans. To quantify the levels of Arc in the cytoplasm, Metamorph or ImageJ was used to threshold the image based on the image of the transfection marker and create a region encompassing the transfected neuron. The average intensity of internalized GluA1 was measured and normalized to control neurons for each experiment. Background subtraction was used for all image analysis.
Statistical analysis
Student’s 2-tailed t test was used when two groups were compared. 1-way ANOVA with a post hoc Bonferroni correction was performed with Prism (GraphPad Software) when more then two groups were compared. Arc expression was determined to have a normal distribution based on the Anderson-Darling normality test. All graphs show 95% confidence intervals. In general, experimental Ns were based on similar assays performed in high-profile papers examining Arc expression and function, such as examining Arc expression and function by immunohistochemistry21 or electrophysiology techniques22. In addition, a power analysis based on the standard deviations and effect sizes from data collected in Figure 1 indicated a sample size of 17 would be sufficient using a Cohen’s d of 1 for a two-tailed Student’s t-tests and a sample size of 25 would be sufficient when comparing multiple groups. Experiments typically measure at least 30 neurons from three independent cultures, whenever possible with each independent culture obtained from a single animal. Experimenters were blinded for microscopy experiments as described above. For other experiments, data collection and analysis were not performed blind to the conditions of the experiments. Mice were randomly assigned to groups for novel environment experiments. Cultures were divided randomly into groups for different stimulation conditions, with each grouping containing the same number of wells on the inside, outside, and corners of a plate.
Quantitative RT-PCR
Wild-type or Arc knockout neurons were lysed 2 h after NMDA stimulation, and RNA was collected with the RNAeasy qiagen kit. Equal amounts of RNA were used to generate cDNA from each replicate with the Applied Biosystems kit. SYBR Green (Applied Biosystems) and primers for GAPDH (Forward: CCCCAATGTGTCCGTCGT, Reverse: GCCTGCTTCACCACCTTCT), GluA1 (Forward: TCCTGAAGAACTCCTTAGTG, Reverse: ATCATGTCCTCATACACAGC), or GluN2A (Forward: GCCTGAGAATGTGGACTTCC, Reverse: TTCTGTGACCAGTCCTGC) were used with a 7900HT Fast Real-Time PCR system. Each reaction was performed in triplicate and averaged. The Ct method was used to measure the effects of NMDA treatment for each replicate. To compare between replicates, each control sample was normalized to 100 for both wild-type and knockout neurons.
Supplementary Material
Acknowledgments
We thank R. Truant for the GFP-β-galactosidase construct, B. Henderson for the GFP-NLS-NES construct, R. Dingledine for the GluA1-luciferase and GluA2-luciferase constructs, P. Pandolfi for the EGFP-PML construct, D. Bredt for the EGFP-PSD95 construct, R. Everett for the PML and ICP0 construct and J. Ahn for the IE1 constructs. We thank members of the Finkbeiner lab, L. Jan, R. Edwards, and N. Krogan for helpful suggestions and G. Howard for editorial input. E.K. was supported by a Ruth L. Kirschstein Fellowship (5 F31 MH087009). Primary support for this work was provided by the National Institute of Neurological Disease and Stroke (2 R01 NS39074), the National Institute on Aging (2 P01 AG022074), and the J. David Gladstone Institutes (S.F.) as well as the Keck Foundation (S.F.). The animal care facility was partly supported by an NIH Extramural Research Facilities Improvement Project (RR018928).
Footnotes
AUTHOR CONTRIBUTIONS
E.K. and S.M.F. designed the experiments. E.K. performed experiments. C.L.W. found the Pat7 and created the Arc antiserum. R.D. contributed to luciferase assays. K.L. contributed to FRAP experiments. S.M.F. supervised the project. E.K. and S.M.F. wrote the manuscript.
References
- 1.Guzowski JF, et al. Inhibition of activity-dependent arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J Neurosci. 2000;20:3993–4001. doi: 10.1523/JNEUROSCI.20-11-03993.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Messaoudi E, et al. Sustained Arc/Arg3.1 synthesis controls long-term potentiation consolidation through regulation of local actin polymerization in the dentate gyrus in vivo. J Neurosci. 2007;27:10445–10455. doi: 10.1523/JNEUROSCI.2883-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park S, et al. Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron. 2008;59:70–83. doi: 10.1016/j.neuron.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waung MW, Pfeiffer BE, Nosyreva ED, Ronesi JA, Huber KM. Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron. 2008;59:84–97. doi: 10.1016/j.neuron.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shepherd JD, et al. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52:475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beique JC, Na Y, Kuhl D, Worley PF, Huganir RL. Arc-dependent synapse-specific homeostatic plasticity. Proc Natl Acad Sci U S A. 2010;108:816–821. doi: 10.1073/pnas.1017914108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plath N, et al. Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron. 2006;52:437–444. doi: 10.1016/j.neuron.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 8.Rao VR, et al. AMPA receptors regulate transcription of the plasticity-related immediate-early gene Arc. Nat Neurosci. 2006;9:887–895. doi: 10.1038/nn1708. [DOI] [PubMed] [Google Scholar]
- 9.Pintchovski SA, Peebles CL, Kim HJ, Verdin E, Finkbeiner S. The serum response factor and a putative novel transcription factor regulate expression of the immediate-early gene Arc/Arg3.1 in neurons. J Neurosci. 2009;29:1525–1537. doi: 10.1523/JNEUROSCI.5575-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramirez-Amaya V, et al. Spatial exploration-induced Arc mRNA and protein expression: evidence for selective, network-specific reactivation. J Neurosci. 2005;25:1761–1768. doi: 10.1523/JNEUROSCI.4342-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawashima T, et al. Synaptic activity-responsive element in the Arc/Arg3.1 promoter essential for synapse-to-nucleus signaling in activated neurons. Proc Natl Acad Sci U S A. 2009;106:316–321. doi: 10.1073/pnas.0806518106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steward O, Wallace CS, Lyford GL, Worley PF. Synaptic activation causes the mRNA for the IEG Arc to localize selectively near activated postsynaptic sites on dendrites. Neuron. 1998;21:741–751. doi: 10.1016/s0896-6273(00)80591-7. [DOI] [PubMed] [Google Scholar]
- 13.Huang F, Chotiner JK, Steward O. Actin polymerization and ERK phosphorylation are required for Arc/Arg3.1 mRNA targeting to activated synaptic sites on dendrites. J Neurosci. 2007;27:9054–9067. doi: 10.1523/JNEUROSCI.2410-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bloomer WA, Vandongen HM, Vandongen AM. Arc/Arg3.1 Translation Is Controlled by Convergent N-Methyl-D-aspartate and Gs-coupled Receptor Signaling Pathways. J Biol Chem. 2008;283:582–592. doi: 10.1074/jbc.M702451200. [DOI] [PubMed] [Google Scholar]
- 15.Panja D, et al. Novel translational control in Arc-dependent long term potentiation consolidation in vivo. J Biol Chem. 2009;284:31498–31511. doi: 10.1074/jbc.M109.056077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greer PL, et al. The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell. 2010;140:704–716. doi: 10.1016/j.cell.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodriguez JJ, et al. Long-term potentiation in the rat dentate gyrus is associated with enhanced Arc/Arg3.1 protein expression in spines, dendrites and glia. Eur J Neurosci. 2005;21:2384–2396. doi: 10.1111/j.1460-9568.2005.04068.x. [DOI] [PubMed] [Google Scholar]
- 18.Moga DE, et al. Activity-regulated cytoskeletal-associated protein is localized to recently activated excitatory synapses. Neuroscience. 2004;125:7–11. doi: 10.1016/j.neuroscience.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 19.Bloomer WA, VanDongen HM, VanDongen AM. Activity-regulated cytoskeleton-associated protein Arc/Arg3.1 binds to spectrin and associates with nuclear promyelocytic leukemia (PML) bodies. Brain Res. 2007;1153:20–33. doi: 10.1016/j.brainres.2007.03.079. [DOI] [PubMed] [Google Scholar]
- 20.Peebles CL, et al. Arc regulates spine morphology and maintains network stability in vivo. Proc Natl Acad Sci U S A. 2010;107:18173–18178. doi: 10.1073/pnas.1006546107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chowdhury S, et al. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron. 2006;52:445–459. doi: 10.1016/j.neuron.2006.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rial Verde EM, Lee-Osbourne J, Worley PF, Malinow R, Cline HT. Increased expression of the immediate-early gene arc/arg3.1 reduces AMPA receptor-mediated synaptic transmission. Neuron. 2006;52:461–474. doi: 10.1016/j.neuron.2006.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Brien RJ, et al. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21:1067–1078. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- 24.Borden KL. Pondering the promyelocytic leukemia protein (PML) puzzle: possible functions for PML nuclear bodies. Mol Cell Biol. 2002;22:5259–5269. doi: 10.1128/MCB.22.15.5259-5269.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nature reviews. 2007;8:1006–1016. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- 26.Regad T, Bellodi C, Nicotera P, Salomoni P. The tumor suppressor Pml regulates cell fate in the developing neocortex. Nat Neurosci. 2009;12:132–140. doi: 10.1038/nn.2251. [DOI] [PubMed] [Google Scholar]
- 27.Skinner PJ, et al. Ataxin-1 with an expanded glutamine tract alters nuclear matrix-associated structures. Nature. 1997;389:971–974. doi: 10.1038/40153. [DOI] [PubMed] [Google Scholar]
- 28.Chai Y, Koppenhafer SL, Shoesmith SJ, Perez MK, Paulson HL. Evidence for proteasome involvement in polyglutamine disease: localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Human molecular genetics. 1999;8:673–682. doi: 10.1093/hmg/8.4.673. [DOI] [PubMed] [Google Scholar]
- 29.Yamada M, et al. Widespread occurrence of intranuclear atrophin-1 accumulation in the central nervous system neurons of patients with dentatorubral-pallidoluysian atrophy. Annals of neurology. 2001;49:14–23. doi: 10.1002/1531-8249(200101)49:1<14::aid-ana5>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 30.Yamada M, et al. Interaction between neuronal intranuclear inclusions and promyelocytic leukemia protein nuclear and coiled bodies in CAG repeat diseases. The American journal of pathology. 2001;159:1785–1795. doi: 10.1016/S0002-9440(10)63025-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annual review of neuroscience. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- 32.Tyler WJ, Alonso M, Bramham CR, Pozzo-Miller LD. From acquisition to consolidation: on the role of brain-derived neurotrophic factor signaling in hippocampal-dependent learning. Learning & memory (Cold Spring Harbor, NY. 2002;9:224–237. doi: 10.1101/lm.51202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu B. BDNF and activity-dependent synaptic modulation. Learning & memory (Cold Spring Harbor, NY. 2003;10:86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Borges K, Dingledine R. Functional organization of the GluR1 glutamate receptor promoter. J Biol Chem. 2001;276:25929–25938. doi: 10.1074/jbc.M009105200. [DOI] [PubMed] [Google Scholar]
- 35.St-Germain JR, Chen J, Li Q. Involvement of PML nuclear bodies in CBP degradation through the ubiquitin-proteasome pathway. Epigenetics. 2008;3:342–349. doi: 10.4161/epi.3.6.7203. [DOI] [PubMed] [Google Scholar]
- 36.Arrasate M, Finkbeiner S. Automated microscope system for determining factors that predict neuronal fate. Proc Natl Acad Sci U S A. 2005;102:3840–3845. doi: 10.1073/pnas.0409777102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee HR, et al. Ability of the human cytomegalovirus IE1 protein to modulate sumoylation of PML correlates with its functional activities in transcriptional regulation and infectivity in cultured fibroblast cells. Journal of virology. 2004;78:6527–6542. doi: 10.1128/JVI.78.12.6527-6542.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Everett RD, et al. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. Journal of virology. 2006;80:7995–8005. doi: 10.1128/JVI.00734-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao M, et al. A specific requirement of Arc/Arg3.1 for visual experience-induced homeostatic synaptic plasticity in mouse primary visual cortex. J Neurosci. 2010;30:7168–7178. doi: 10.1523/JNEUROSCI.1067-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lyford GL, et al. Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron. 1995;14:433–445. doi: 10.1016/0896-6273(95)90299-6. [DOI] [PubMed] [Google Scholar]
- 41.Zhong S, Salomoni P, Pandolfi PP. The transcriptional role of PML and the nuclear body. Nature cell biology. 2000;2:E85–90. doi: 10.1038/35010583. [DOI] [PubMed] [Google Scholar]
- 42.Anggono V, Clem RL, Huganir RL. PICK1 loss of function occludes homeostatic synaptic scaling. J Neurosci. 2011;31:2188–2196. doi: 10.1523/JNEUROSCI.5633-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 44.Goold CP, Nicoll RA. Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron. 2010;68:512–528. doi: 10.1016/j.neuron.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun Q, Turrigiano GG. PSD-95 and PSD-93 play critical but distinct roles in synaptic scaling up and down. J Neurosci. 2011;31:6800–6808. doi: 10.1523/JNEUROSCI.5616-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alberi L, et al. Activity-induced Notch signaling in neurons requires Arc/Arg3.1 and is essential for synaptic plasticity in hippocampal networks. Neuron. 2011;69:437–444. doi: 10.1016/j.neuron.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bramham CR, et al. The Arc of synaptic memory. Experimental brain research. Experimentelle Hirnforschung. 2009;200:125–140. doi: 10.1007/s00221-009-1959-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Korb E, Finkbeiner S. Arc in synaptic plasticity: from gene to behavior. Trends in neurosciences. 2011;34:591–598. doi: 10.1016/j.tins.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang KH, et al. In vivo two-photon imaging reveals a role of arc in enhancing orientation specificity in visual cortex. Cell. 2006;126:389–402. doi: 10.1016/j.cell.2006.06.038. [DOI] [PubMed] [Google Scholar]
- 50.Sorg G, Stamminger T. Mapping of nuclear localization signals by simultaneous fusion to green fluorescent protein and to beta-galactosidase. Biotechniques. 1999;26:858–862. doi: 10.2144/99265bm12. [DOI] [PubMed] [Google Scholar]
- 51.Henderson BR, Eleftheriou A. A comparison of the activity, sequence specificity, and CRM1-dependence of different nuclear export signals. Experimental cell research. 2000;256:213–224. doi: 10.1006/excr.2000.4825. [DOI] [PubMed] [Google Scholar]
- 52.Boutell C, Orr A, Everett RD. PML residue lysine 160 is required for the degradation of PML induced by herpes simplex virus type 1 regulatory protein ICP0. Journal of virology. 2003;77:8686–8694. doi: 10.1128/JVI.77.16.8686-8694.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brough D, Bhatti F, Irvine RF. Mobility of proteins associated with the plasma membrane by interaction with inositol lipids. Journal of cell science. 2005;118:3019–3025. doi: 10.1242/jcs.02426. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.