

Abstract
Microtubule nucleation in all eukaryotes involves γ-tubulin small complexes (γTuSCs) that comprise two molecules of γ-tubulin bound to γ-tubulin complex proteins (GCPs) GCP2 and GCP3. In many eukaryotes, multiple γTuSCs associate with GCP4, GCP5 and GCP6 into large γ-tubulin ring complexes (γTuRCs). Recent cryo-EM studies indicate that a scaffold similar to γTuRCs is formed by lateral association of γTuSCs, with the C-terminal regions of GCP2 and GCP3 binding γ-tubulin molecules. However, the exact role of GCPs in microtubule nucleation remains unknown. Here we report the crystal structure of human GCP4 and show that its C-terminal domain binds directly to γ-tubulin. The human GCP4 structure is the prototype for all GCPs, as it can be precisely positioned within the γTuSC envelope, revealing the nature of protein-protein interactions and conformational changes regulating nucleation activity.
Earlier models of γ-tubulin ring complex assembly have suggested that γ-tubulin complex proteins GCP4, GCP5 and GCP6 serve as a scaffold for arranging multiple copies of γ-tubulin small complex into a microtubule template1. Obtaining a high-resolution structural model of the 2.2 MDa γTuRC is essential to our understanding of the mechanism of γ-tubulin-based microtubule nucleation. Although the cryo-EM structure of γTuSC oligomers2,3 and the crystal structure of γ-tubulin4 have provided important insights into the core organization of γTuRC2, a critical missing component has been the structural and functional role of the GCP proteins. All of the GCPs contain islands of distant but identifiable sequence similarity5,6, with GCP4 composed almost entirely of these homologous regions. We therefore reasoned that a structural and biochemical characterization of human GCP4 could serve as a model for understanding GCPs in general.
RESULTS
Crystal structure of human GCP4
The full-length human GCP4 (ref. 7) (666 residues) with a C-terminal histidine (His) tag was crystallized in a hexagonal form diffracting to 2.3 Å (Table 1). The structure was determined by SAD using selenomethionine (SeMet)-labeled protein. After refinement, the final model comprises 571 out of 677 amino acids, which leads to acceptable R factors and stereochemistry (Table 1). GCP4 forms a rather elongated and slightly curved structure, with a length of 140 Å and a width of 25–50 Å (Fig. 1a). Approximately 70% of the residues are in a helical conformation, and the newly discovered fold is composed of 4 short 310 helices and 21 α helices, each 11–47 Å in length. There are also six short strands, which contribute to three antiparallel β-sheets. Five solvent-exposed loop regions were absent from the electron density maps. One short missing loop (Gly601–Pro602) is located on the distal end of the C-terminal domain, and the remaining four are clustered halfway along the convex side of the molecule (Val66–Gln78; Pro209–Pro252; Gln289–Gly297; Lys423–Pro445). Additionally, the last 23 amino acids, including the 11 residues from the His tag, are missing.
Table 1.
Data collection, phasing and refinement statistics
| Native | Pk-Se | |
|---|---|---|
| Data collection | ||
| Space group | P 6322 | P6322 |
| Cell dimensions | ||
| a, b, c (Å) | 214.95, 214.95, 128.66 | 215.60, 215.60, 128.72 |
| α, β, γ (°) | 90.00, 90.00, 120.00 | 90.00, 90.00, 120.00 |
| Wavelength (Å) | 1.033 | 0.979 |
| Resolution (Å) | 2.30 (2.40–2.30)a | 3.50 (3.55–3.50) |
| R sym | 0.044 (0.564) b | 0.235 (0.737) |
| I / σ I | 18.7 (3.5) | 10.0 (4.1) |
| Completeness (%) | 99.4 (99.8) | 98.1 (97.0) |
| Redundancy | 5.4 (5.5) | 7.9 (7.8) |
| Refinement | ||
| Resolution (Å) | 20.0–2.30 | |
| No. unique reflections | 73,248 | |
| Rwork / Rfree | 0.227 / 0.260 | |
| No. atoms | ||
| Protein | 4,572 | |
| Ligand | 56 | |
| Water | 215 | |
| B-factors | ||
| Protein | 53.5 | |
| Ligand | 52.9 | |
| Water | 60.4 | |
| r.m.s. deviations | ||
| Bond lengths (Å) | 0.0248 | |
| Bond angles (°) | 2.105 |
Values in parentheses are for highest-resolution shell.
RmergeF values21 for Pk-Se are 0.162 (0.446).
Figure 1.

The crystal structure of GCP4 reveals a previously undescribed fold. (a) Topology diagram (left) and ribbon representation (right). The first helix bundle is in light blue, the second in purple, the third in orange, the fourth in light pink and the fifth in blue. All structural elements excluded from helix bundles are in green. Helices and beta strands are numbered. Stretches of missing residues are represented by dashed lines (left) and by colored spheres (right). Residues preceding and following missing loops are labeled. (b) Ribbon representation colored according to sequence similarity over orthologous GCP4 proteins as shown in Supplementary Figure 1. Residues with similarity <80% are in white; conserved areas with similarity in the range 80–100% are colored light red to bright red.
The structure of GCP4 reveals an original protein fold
The structure can be viewed as five successive layers or bundles of roughly aligned helices (Fig. 1a). The first three bundles and their appendages constitute the N-terminal diagonal of the V-shaped structure of GCP4. The C-terminal diagonal comprises two additional helical bundles, which are flanked on one side by a small domain and on the other side by the C-terminal helix. The five helix bundles are organized around relatively conserved hydrophobic cores (Fig. 1b and Supplementary Fig. 1) and also involve polar and electrostatic interactions for connecting the participating helices (described in detail in the Supplementary Discussion and Supplementary Fig. 2). Our preliminary small-angle X-ray scattering (SAXS) experiments strongly suggested that GCP4 in solution behaves at low concentration as an elongated monomer compatible with the crystal structure. The fold of GCP4 has no apparent structural homolog in the Protein Data Bank (Supplementary Table 1).
The GCP4 structure is the prototype for all GCPs
Sequence alignment of human GCPs (hGCPs) based on the GCP4 structure helped delineate conserved regions in the N- and C-terminal domains (Supplementary Fig. 3). These regions extend the boundaries previously defined for the grip1 and grip2 motifs5,6. As GCP4 is composed almost entirely of these common regions, its crystal structure likely represents the structure of the core of all GCPs. Consistently, the five loops that cannot be traced in the electron density map of GCP4 are located outside the conserved motifs and are often missing in other hGCPs (Supplementary Fig. 3a).
Conserved residues from the N-terminal domain are mostly buried, contributing to the integrity of the N-terminal half of the GCP fold. This is especially the case in the central part where almost all strictly conserved residues (Trp190, Gly194, Asp198, Glu202, Phe203 and Gly280) are clustered (Supplementary Fig. 2c). The potential importance of this region is reflected in the anisotropic growth phenotype observed in Arabidopsis thaliana upon G305R mutation in GCP2 (ref. 8) (corresponding to Gly280 in GCP4). Conserved residues from the C-terminal domain of the GCP core structure are also mostly buried. It is also noteworthy that practically all strictly conserved residues (Leu350, Leu362, Gly366, Tyr456, Val458, Pro461, Tyr474, Phe478, His560 and Leu564) form a patch that helps insure the cohesion between the small domain, the third bundle and the fourth bundle (Supplementary Fig. 2e,g).
The C-terminal domain of GCP4 binds directly to γ-tubulin
Structure-based sequence analysis also revealed the existence of an exposed surface area conserved in all hGCPs and in GCP4 orthologs. This area is located in the C-terminal domain of GCP4 (Fig. 2a), and we confirmed in vitro that it can bind directly to γ-tubulin. Stable complexes of Flag-tagged γ-tubulin and V5-tagged GCP4 were isolated by immunoprecipitation with anti-Flag affinity beads (Fig. 2b,c). We determined a dissociation constant around 10–8 M for the binding of full-length GCP4 to γ-tubulin (Supplementary Fig. 4). In a qualitative binding assay, this very stable complex resisted treatment with high salt or nonionic detergent and eluted only in denaturing buffer containing SDS (Fig. 2c). In deletion experiments, we confirmed that the C-terminal domain of GCP4 is essential and sufficient for binding to γ-tubulin (Fig. 2d,e). A fragment of GCP4 comprising amino acids 349–637 in the C-terminal domain interacted with γ-tubulin, although it was sensitive to detergent treatment (Fig. 2f). This sensitivity was due to removal of the C-terminal helix (α21) from GCP4, which may have a stabilizing role in γ-tubulin binding (compare Fig. 2e,f and Fig. 2c,g). Direct binding to γ-tubulin through the C-terminal domain of GCP4 is consistent with previous reports that in γTuSC, GCP2 and GCP3 bind γ-tubulin through their C-terminal domains3,9,10, and this suggests that all of the GCP proteins bind γ-tubulin in an equivalent manner.
Figure 2.
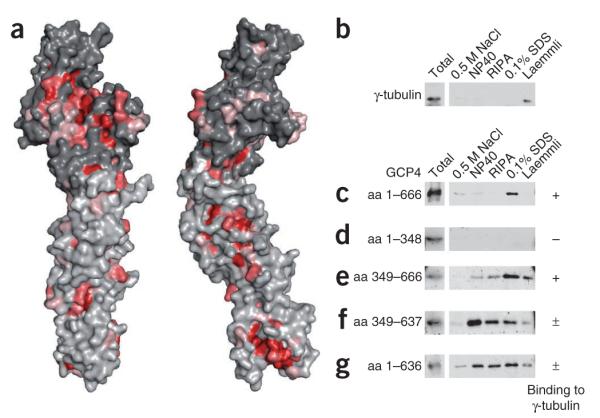
GCP4 binds to γ-tubulin through its C-terminal domain. (a) View down the convex face and side view of the molecular surface of GCP4. Conserved residues are colored as in Figure 1b. Nonconserved residues of the N- and C-terminal domains are colored light- and dark gray, respectively. (b–g) Co-transcription–translation of V5-tagged GCP4 and Flag-tagged γ-tubulin in reticulocyte lysate, followed by immunoprecipitation with anti-Flag affinity beads. Immunoblots of the eluted fractions from beads rinsed with HEPES buffer, followed by consecutive treatments with 0.5 M NaCl, 0.1% NP40, RIPA buffer, 0.1% SDS and gel loading buffer according to Laemmli22 are shown, probed with antibody against γ-tubulin (b) or against the V5 tag (c–g). Full-length (c) and deletion constructs (d–g) of GCP4 were tested as indicated on the figure. Amino acid, aa.
Construction of a pseudo atomic model of γTuSC
A molecular model of γTuSC was constructed by fitting models of GCP2 and GCP3 derived from the GCP4 crystal structure (Supplementary Fig. 5), along with the γ-tubulin crystal structure (PDB code 1Z5V)4, into the 8-Å cryo-EM reconstruction of Saccharomyces cerevisiae γTuSC2. The overall fit of the models is quite good (Fig. 3a,b), and unfilled EM density can be accounted for by insertions in GCP2 (Fig. 3a, black arrow) or at the N termini of GCP2 and GCP3. The kink in the GCP4 structure between helical bundles 3 and 4 closely matches the bent shape of GCP3 in the EM density. The best fit for the straighter GCP2 subunit, however, was achieved by straightening the structure at the kink point by about 7°. The resultant pseudo atomic model provides unique insight into the interactions among γTuSC components. The C-terminal regions of GCP2 and GCP3 bind γ-tubulin, primarily through helices α16 and α20 in the fifth bundle. Notably, helix α16 is lined by the conserved depression of the molecular surface described above (Fig. 3c). The sequence and structural conservation suggest that this cavity is the primary interaction site with γ-tubulin, common to all GCPs. Regions of γ-tubulin that interact with GCP2 and GCP3 correspond to the minus end of the protein, opposite to the nucleotidebinding pocket, and is likely to include the following patches conserved within the γ-tubulin sequences: the long peptide segment between helix H1 and strand S2, the H7–H8 (or T7) loop, helices H8 and H10, strand S9, and the connecting loop between H10 and S9 (ref. 11). The γTuSC model also provides a more detailed understanding of the interactions between GCP2 and GCP3, both within and between γTuSCs (Supplementary Fig. 6). Dimerization of GCP2 and GCP3 is predominantly achieved by contacts between their first and second bundles, although minor contacts are made between the third bundles. In γTuSC oligomers, the lateral interactions between γTuSCs are very similar to the packing within the γTuSC.
Figure 3.

A molecular model of γTuSC. (a) Models of GCP2 and GCP3 generated from the GCP4 crystal structure and the γ-tubulin crystal structure were manually fit into the cryo-EM γTuSC reconstruction. The inset shows the region of GCP3 previously shown by EM to act as a hinge, corresponding to the interface between helical bundles 3 (blue) and 4 (cyan); the position of Trp460 in GCP4 is indicated with a red sphere. Empty regions of the EM map include density corresponding to an insert in GCP2 (black arrowhead) and the density of a bound adaptor protein (Spc110) in the EM structure (gray arrowheads). (b) The EM map sharpened with a 500-Å2 B-factor has α-helical features in good agreement with bundles 2 and 3 of GCP3. (c) Semi-transparent molecular surface of the C-terminal domain of GCP4 colored according to sequence similarity over hGCPs as shown in Supplementary Figure 1. Enlarged stereo view (right) of the conserved cavity is colored with respect to atom type (C, gray; N, blue; O, red) and sequence conservation of important residues (given in the hGCP2-hGCP3-hGCP4-hGCP5-hGCP6 order) that delineate the cavity.
Conformational changes regulating nucleation activity
In the EM structures of both the free and assembled γTuSCs2,3, the two γ-tubulins within each γTuSC are positioned in a manner inconsistent with lateral spacing in microtubules, suggesting that these structures represent a default nucleation-incompetent state. Analysis of isolated γTuSCs indicated the existence of a hinge point within GCP3 (refs. 3,10). A rotation of 20° about this hinge would be sufficient to bring all the γ-tubulins within the assembled complex into optimal positions for microtubule nucleation, suggesting that allosteric modulation and/or post translational modifications may be used to regulate the ability of preassembled GCP complexes to nucleate microtubule formation2,12. The GCP4 structure reveals that the hinge region corresponds to the interface between helical bundles 3 and 4, with the sharp kink between helices α11 and α12 providing the flex point (Fig. 3a). Rearrangement of packing interactions between the two bundles may serve to stabilize different conformations. Indeed, normal mode analysis predicts flexing at this interface to be the largest motion in the GCP4 structure (Supplementary Fig. 7). The wellconserved residues Trp460 and Pro461 form part of this interface, making contacts with the α9–α10 loop. An aromatic residue (tryptophan, tyrosine or phenylalanine) and a proline are found at those positions in all GCP2s, whereas in GCP3s the proline is conserved but the aromatic residue is replaced by a smaller residue (glycine, valine, proline, serine). The presence of a smaller residue at this position may result in weaker packing at the interface, explaining the observed flexibility in GCP3. What might mediate the proposed change in GCP3 bend angle remains a mystery, but a putative phosphorylation site within human GCP3 has been identified at Ser512 (ref. 13), which corresponds to Ala309 on helix α10 of the third bundle of GCP4, near the hinge interface.
DISCUSSION
The structural and functional conservation of GCP4 with the γTuSC subunits GCP2 and GCP3 has strong implications for the role of GCP4 in the γTuRC. It seems very likely that GCP4, and by extension GCP5 and GCP6, is incorporated directly into the core helical ring of γTuRC, using the same packing surfaces as GCP2 and GCP3. Such a model is in direct contrast with earlier γTuRC models suggesting that GCP4–GCP6 form a scaffold for organizing a γTuSC ring1, but it is consistent with an earlier report stating that all GCPs directly bind γ-tubulin in vitro5 and with the more recent observation that γTuSCs have an intrinsic propensity to form ring structures2. In the new unified model, it remains an open question whether GCP4–GCP6 act as a substitute for γTuSC components, form unique, alternative γTuSC-like structures or are incorporated into γTuRCs as a kind of unusual half γTuSC. The GCPs are not functionally redundant, as depletion of any single member leads to loss of γTuRC stability on sucrose gradients14–18, suggesting that GCP4–GCP6 are important for initiating or terminating ring assembly or for stabilizing the ends of the ring. The low abundance of GCP4–GCP6 in purified γTuRCs6,19 is consistent with this idea. In this regard, GCP4 includes two highly charged inserts at lateral assembly contact positions that could potentially interfere with assembly in one direction, allowing GCP4 to function as a ring initiator or terminator (Supplementary Fig. 6). In S. cerevisiae, where homologs of GCP4–GCP6 are missing, γTuRC-like complexes are nevertheless formed from γ-TuSCs alone, both in vitro2 and in vivo20. Although these complexes appear less stable, they may be sufficient to ensure nucleation of the small number of microtubules in this organism. Alternatively, proteins different from GCPs may be involved in stabilizing γTuRCs in S. cerevisiae20.
The solution of the GCP4 crystal structure has led to the generation of a pseudo atomic model of γTuSC and the core of γTuRC, and it provides a new framework for understanding γ-tubulin complex assembly and function. It also provides a much more sophisticated approach to addressing important unanswered questions: what are the unique roles of the different GCPs; where are they positioned in the γTuRC ring; and how do GCP4–GCP6 mediate noncentrosomal localization of γTuRC?
ONLINE METHODS
Plasmids
cDNA sequences encoding full-length γ-tubulin and full-length GCP4, as well as partial sequences, were amplified by PCR and cloned into pET26b (+) (Novagen). The following tags and linker sequences were fused to the respective C termini: γ-tubulin-VD-Flag, GCP4-CGRLE-His6 and GCP4-CGRLESRGPFE-V5.
Protein expression and purification
For expression of full-length GCP4, pET-GCP4-His6 was transformed into Escherichia coli strain Rosetta(DE3)pLysS. The transformed cells were grown in LB medium at 25 °C, with the addition of 0.2% (w/v) sorbitol in the medium. Protein expression was induced with 0.4 mM IPTG when the optical density at 600 nm reached 0.5–0.8. After 4 h, cells were harvested by centrifugation (2,500g, 10 min), washed in PBS and stored at −80 °C for later use.
For expression of SeMet-labeled protein, we transformed B834(DE3) cells with pETGCP4-His6 and pLysS-RARE2 plasmids. Cells were inoculated in LB medium and then grown in LeMaster medium23. Protein expression was induced with 0.4 mM IPTG at optical density 0.5–0.8, and after further incubation for 7 h, cells were harvested by centrifugation (5,000g, 1 h), washed with PBS buffer and stored at −80 °C for later use.
Purification of native and SeMet protein was carried out at 4 °C. Cells were resuspended in lysis buffer (50 mM sodium phosphate, pH 8.0, 300 mM NaCl, 10 mM imidazole, 5% glycerol) with a protease inhibitor cocktail, 2.5 mM tris-(2-carboxyethyl)phosphine (TCEP) and benzonase (5 U ml−1 culture) for 1 h and were lysed by sonication by applying five 30-s pulses. Cell debris was pelleted by centrifugation at 20,000g for 40 min. The supernatant was diluted 1/5 in phosphate buffer (50 mM sodium phosphate, pH 8.0, 150 mM NaCl, 5% glycerol and 2.5 mM DTT) supplemented with 10 mM imidazole and was then loaded onto a 5-ml HisTrap FF column (GE Healthcare). The column was washed first with phosphate buffer supplemented with 10 mM imidazole until the absorbance at 280 nm reached zero, then with 50 mM imidazole in phosphate buffer to nonspecifically elute proteins bound to the column. Recombinant protein was eluted at 150 mM imidazole in phosphate buffer. The recombinant proteins were further purified by size-exclusion chromatography using a Superdex 200 16/60 column (GE Healthcare), equilibrated in gel filtration buffer (20 mM Tris, pH 8.0, 150 mM NaCl and 2 mM DTT).
Flag pulldown assay
Flag-γ-tubulin and GCP4-V5, or V5-tagged domains of GCP4, were transcribed and translated together in a coupled reticulocyte lysate system, according to the manufacturer (TNT, Promega), using 500 ng cDNA plasmid of each partner as a template in 50 μl of reaction mixture. The sample was then incubated with 10 μl of anti-Flag M2 agarose beads (Sigma-Aldrich) for 2 h at 4 °C and then washed three times with 30 μl of HEPES buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 1 mM EGTA and 2 mM MgCl2). The bound proteins were eluted by increasing buffer stringency: HEPES 500 (HEPES buffer with 500 mM NaCl), NP40 (HEPES buffer supplemented with 1% Igepal CA-630 (Sigma-Aldrich)), RIPA (HEPES buffer supplemented with 1% Igepal CA-630 and 0.25% deoxycholic acid), SDS (HEPES buffer supplemented with 0.1% SDS) and 1× SDS gel loading buffer22. Samples were analyzed on 7.5% SDS-polyacrylamide gels and then transferred to nitrocellulose membranes. The blots were incubated with anti-V5 (clone V5-10) from Sigma-Aldrich and rabbit serum R75 against γ-tubulin24. The dissociation constant of the GCP4-γ-tubulin complex was determined from Scatchard plot analysis after quantifying bound and free GCP4 from immunoblots using polyclonal antibody against GCP4. Experiments were conducted using increasing amounts of GCP4 bound to a fixed amount of γ-tubulin-loaded beads, ranging from 100 ng to 400 ng of GCP4 purified from bacteria.
Crystallization
The purified GCP4 protein (MW 77,399.7 Da) was concentrated to approximately 3.0 mg ml−1 in 20 mM Tris-HCl, 150 mM NaCl, 2 mM DTT, pH 8.0. Crystals were obtained by mixing equal volumes of the protein solution and of a reservoir solution composed of 8–10% MPD (2-methyl-2,4-pentanediol), 0–20% glycerol (w/v), 300–400 mM Tris-HCl, pH 8.0. GCP4 crystals were cryoprotected in reservoir solutions made up with 20% glycerol (w/v) and were soaked for 1 h. Crystals were then frozen under the cryogenic stream and stored at 100 K. These crystals belong to space group P6322 and diffracted to 2.3-Å resolution at ESRF or SOLEIL synchrotron beamlines. SeMet-labeled protein crystals were obtained and cryoprotected under similar conditions. The unit cell contained one molecule per asymmetric unit and about 80% solvent.
Data collection and structure determination
A native dataset was measured at the SOLEIL beamline PROXIMA-1 to a resolution of 2.3 Å at 100 K and λ = 1.033 Å (Table 1, native). A 3.5-Å SeMet dataset was recorded at the same beamline at the K edge of selenium (λ = 0.979 Å) (Table 1, Pk-Se). The images were processed using XDS25 and the data were scaled with XSCALE25. The structure of the SeMet protein was solved using the SAD method. Nine selenium sites over 11 SeMet residues introduced in the GCP4 sequence were located using the program SHELXD26. The remaining two selenium sites are located in mobile loop regions not visible in the final structure. Phases were calculated with the program PHASER27, which also found nine selenium sites. The correct enantiomorph was found by visual inspection of the electron density map after solvent flattening with DM28.
Model building and refinement
Quality of the experimentally derived solvent-flattened electron density map allowed automatic building by Buccaneer29 of up to 79% of the total of 674 amino acids in the recombinant GCP4 sequence. Model building was then continued with the graphics program Coot30. The refined model of SeMet GCP4 included 535 residues, of which 417 residues were correctly sequenced, with Rwork and Rfree of 0.331 and 0.370, respectively. This structure was used as the molecular replacement search model for the higher resolution native dataset using the program Phaser31, giving one highly correlated solution that showed no overlap with crystal neighbors. The automatic water molecule search and refinement of the high-resolution model was carried out with Refmac5 (ref. 32). The quality of the final model was checked with PROCHECK33. Figures depicting the structural features of GCP4 were prepared using using TopDraw from the CCP4 graphical user interface34 and PyMOL (http://www.pymol.org).
Bioinformatic sequence analysis
Amino acid sequences were obtained from the National Center for Biotechnology Information (NCBI) and from The Arabidopsis Information Resource (http://www.arabidopsis.org/). Alignments were done using the Clustal W2 program35 and manually adjusted in Seaview36; the figures were shaped using ESPript37.
Building the pseudo atomic model
The cryo-EM reconstruction of S. cerevisiae γTuSC and the fitting of γ-tubulin into that structure were previously described2. Homology models of S. cerevisiae GCP2 (Spc97) and GCP3 (Spc98) were generated from the GCP4 crystal structure and manually fit into the corresponding density in the cryo-EM map. Manual fitting was done and figures generated using the UCSF Chimera package38. Normal mode analysis of GCP4 was done using elNemo39.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Wright and J.-E. Gairin (Centre de Recherche en Pharmacologie-Santé, Centre National de la Recherche Scientifique Pierre Fabre, Toulouse) for their advice and support; the staff of synchrotron beamlines PROXIMA 1 at SOLEIL Synchrotron, ID14-1, ID14-2, ID23-2 and ID29 at the European Synchrotron Radiation Facility; P. Legrand from the PROXIMA 1 beamline for his help during data collection and preliminary calculation of experimental phases; and F. Viala for her help in preparing the figures. This project was financed in part with grant 08-BLAN-0281 from the French ‘Agence Nationale de la Recherche’ (A.M. and L.M.) and by the Howard Hughes Medical Institute and the US National Institutes of Health grant GM31627 (J.M.K. and D.A.A.).
Footnotes
Accession codes. Protein Data Bank: Atomic coordinates and structure factors have been deposited under the accession number 3RIP.
Note: Supplementary information is available on the Nature Structural & Molecular Biology website.
AUTHOR CONTRIBUTIONS V.G. helped optimize protein production and purification, grew the crystals and conducted diffraction data collection, analyzed the structure, prepared figures and participated in manuscript writing. M.K. optimized native and SeMet-labeled protein production and purification. L.G.-P. participated in data processing and did structure determination and refinement, analyzed the structure and helped prepare tables and figures. M.-H.R. made the constructs, produced and purified the proteins, carried out Flag pulldown experiments and prepared figures. C.C. made the constructs and carried out Flag pulldown experiments. B.R.-M. did initial purification studies. C.B. participated in protein characterization. J.M.K. analyzed structure-function relationships, carried out the fitting into the cryo-EM map of γTuSC and prepared figures. D.A.A. analyzed the data and revised the manuscript. A.M. devised the experiments, designed figures and wrote the manuscript. L.M. devised the experiments, participated in diffraction data collection, analyzed the structure, designed tables and figures and wrote the manuscript.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Moritz M, Agard DA. Gamma-tubulin complexes and microtubule nucleation. Curr. Opin. Struct. Biol. 2001;11:174–181. doi: 10.1016/s0959-440x(00)00187-1. [DOI] [PubMed] [Google Scholar]
- 2.Kollman JM, Polka JK, Zelter A, Davis TN, Agard DA. Microtubule nucleating gamma-TuSC assembles structures with 13-fold microtubule-like symmetry. Nature. 2010;466:879–882. doi: 10.1038/nature09207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kollman JM, et al. The structure of the gamma-tubulin small complex: implications of its architecture and flexibility for microtubule nucleation. Mol. Biol. Cell. 2008;19:207–215. doi: 10.1091/mbc.E07-09-0879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aldaz H, Rice LM, Stearns T, Agard DA. Insights into microtubule nucleation from the crystal structure of human gamma-tubulin. Nature. 2005;435:523–527. doi: 10.1038/nature03586. [DOI] [PubMed] [Google Scholar]
- 5.Gunawardane RN, et al. Characterization and reconstitution of Drosophila gamma-tubulin ring complex subunits. J. Cell Biol. 2000;151:1513–1524. doi: 10.1083/jcb.151.7.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy SM, et al. GCP5 and GCP6: two new members of the human gamma-tubulin complex. Mol. Biol. Cell. 2001;12:3340–3352. doi: 10.1091/mbc.12.11.3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fava F, et al. Human 76p: A new member of the gamma-tubulin-associated protein family. J. Cell Biol. 1999;147:857–868. doi: 10.1083/jcb.147.4.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakamura M, Hashimoto T. A mutation in the Arabidopsis gamma-tubulin-containing complex causes helical growth and abnormal microtubule branching. J. Cell Sci. 2009;122:2208–2217. doi: 10.1242/jcs.044131. [DOI] [PubMed] [Google Scholar]
- 9.Knop M, Pereira G, Geissler S, Grein K, Schiebel E. The spindle pole body component Spc97p interacts with the gamma-tubulin of Saccharomyces cerevisiae and functions in microtubule organization and spindle pole body duplication. EMBO J. 1997;16:1550–1564. doi: 10.1093/emboj/16.7.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choy RM, Kollman JM, Zelter A, Davis TN, Agard DA. Localization and orientation of the gamma-tubulin small complex components using protein tags as labels for single particle EM. J. Struct. Biol. 2009;168:571–574. doi: 10.1016/j.jsb.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inclán YF, Nogales E. Structural models for the self-assembly and microtubule interactions of gamma-, delta- and epsilon-tubulin. J. Cell Sci. 2001;114:413–422. doi: 10.1242/jcs.114.2.413. [DOI] [PubMed] [Google Scholar]
- 12.Masuda H, Sevik M, Cande WZ. In vitro microtubule-nucleating activity of spindle pole bodies in fission yeast Schizosaccharomyces pombe: cell cycle-dependent activation in Xenopus cell-free extracts. J. Cell Biol. 1992;117:1055–1066. doi: 10.1083/jcb.117.5.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oppermann FS, et al. Large-scale proteomics analysis of the human kinome. Mol. Cell. Proteomics. 2009;8:1751–1764. doi: 10.1074/mcp.M800588-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L, Keating TJ, Wilde A, Borisy GG, Zheng Y. The role of Xgrip210 in gamma-tubulin ring complex assembly and centrosome recruitment. J. Cell Biol. 2000;151:1525–1536. doi: 10.1083/jcb.151.7.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vérollet C, et al. Drosophila melanogaster gamma-TuRC is dispensable for targeting gamma-tubulin to the centrosome and microtubule nucleation. J. Cell Biol. 2006;172:517–528. doi: 10.1083/jcb.200511071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vogt N, Koch I, Schwarz H, Schnorrer F, Nusslein-Volhard C. The gammaTuRC components Grip75 and Grip128 have an essential microtubule-anchoring function in the Drosophila germline. Development. 2006;133:3963–3972. doi: 10.1242/dev.02570. [DOI] [PubMed] [Google Scholar]
- 17.Izumi N, Fumoto K, Izumi S, Kikuchi A. GSK-3beta regulates proper mitotic spindle formation in cooperation with a component of the gamma-tubulin ring complex, GCP5. J. Biol. Chem. 2008;283:12981–12991. doi: 10.1074/jbc.M710282200. [DOI] [PubMed] [Google Scholar]
- 18.Xiong Y, Oakley BR. In vivo analysis of the functions of gamma-tubulin-complex proteins. J. Cell Sci. 2009;122:4218–4227. doi: 10.1242/jcs.059196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi YK, Liu P, Sze SK, Dai C, Qi RZ. CDK5RAP2 stimulates microtubule nucleation by the gamma-tubulin ring complex. J. Cell Biol. 2010;191:1089–1095. doi: 10.1083/jcb.201007030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vinh DB, Kern JW, Hancock WO, Howard J, Davis TN. Reconstitution and characterization of budding yeast gamma-tubulin complex. Mol. Biol. Cell. 2002;13:1144–1157. doi: 10.1091/mbc.02-01-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diederichs K, Karplus PA. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat. Struct. Biol. 1997;4:269–275. doi: 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]
- 22.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 23.LeMaster DM, Richards FM. 1H-15N heteronuclear NMR studies of Escherichia coli thioredoxin in samples isotopically labeled by residue type. Biochemistry. 1985;24:7263–7268. doi: 10.1021/bi00346a036. [DOI] [PubMed] [Google Scholar]
- 24.Julian M, et al. γ-Tubulin participates in the formation of the midbody during cytokinesis in mammalian cells. J. Cell Sci. 1993;105:145–156. doi: 10.1242/jcs.105.1.145. [DOI] [PubMed] [Google Scholar]
- 25.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 1993;26:795–800. [Google Scholar]
- 26.Sheldrick GM. A short history of SHELX. Acta Crystallogr. A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 27.McCoy AJ, et al. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cowtan K, Main P. Miscellaneous algorithms for density modification. Acta Crystallogr. D Biol. Crystallogr. 1998;54:487–493. doi: 10.1107/s0907444997011980. [DOI] [PubMed] [Google Scholar]
- 29.Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 30.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 31.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr. D Biol. Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 32.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 33.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 34.Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 35.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 36.Galtier N, Gouy M, Gautier C. SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny. Comput. Appl. Biosci. 1996;12:543–548. doi: 10.1093/bioinformatics/12.6.543. [DOI] [PubMed] [Google Scholar]
- 37.Gouet P, Courcelle E, Stuart D, Metoz F. ESPript: multiple sequence alignments in PostScript. Bioinformatics. 1998;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 38.Pettersen EF, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 39.Suhre K, Sanejouand YH. ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004;32:W610–W614. doi: 10.1093/nar/gkh368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.